

Abstract
Boron functional groups are often introduced in place of aromatic carbon–hydrogen bonds to expedite small-molecule diversification through coupling of molecular fragments1,2,3. Current approaches based on transition-metal-catalysed activation of carbon–hydrogen bonds are effective for the borylation of many (hetero)aromatic derivatives4,5 but show narrow applicability to azines (nitrogen-containing aromatic heterocycles), which are key components of many pharmaceutical and agrochemical products6. Here we report an azine borylation strategy using stable and inexpensive amine-borane7 reagents. Photocatalysis converts these low-molecular-weight materials into highly reactive boryl radicals8 that undergo efficient addition to azine building blocks. This reactivity provides a mechanistically alternative tactic for sp2 carbon–boron bond assembly, where the elementary steps of transition-metal-mediated carbon–hydrogen bond activation and reductive elimination from azine-organometallic intermediates are replaced by a direct, Minisci9-style, radical addition. The strongly nucleophilic character of the amine-boryl radicals enables predictable and site-selective carbon–boron bond formation by targeting the azine’s most activated position, including the challenging sites adjacent to the basic nitrogen atom. This approach enables access to aromatic sites that elude current strategies based on carbon–hydrogen bond activation, and has led to borylated materials that would otherwise be difficult to prepare. We have applied this process to the introduction of amine-borane functionalities to complex and industrially relevant products. The diversification of the borylated azine products by mainstream cross-coupling technologies establishes aromatic amino-boranes as a powerful class of building blocks for chemical synthesis.
Similar content being viewed by others
Main
Borylated aromatics are fundamental building blocks in synthetic chemistry10. They are the coupling partners in Suzuki–Miyaura cross-coupling3 and also serve as substrates in a variety of other processes like oxidation, Chan–Lam coupling11, fluorination12 and homologation13.
Historically, these materials have been prepared by aromatic halogenation followed by Grignard formation or Pd-catalysed Miyaura borylation14. A substantial advance in the field has come with the development of methods for direct arene borylation through C–H activation using Ir/Rh-catalysts and B2(pin)2/HB(pin) reagents (pin is pinacol)4,5. These methods are now widely adopted in both academia and industry because they obviate the need for aromatic pre-functionalization. However, while C–H borylation of many aromatic derivatives is a generalized strategy, application to azines (N-heteroaromatics constituting the core of many drugs and agrochemicals6,15) is less established, often unselective and limited to a small subset of systems16. These methodologies functionalize C–H bonds distal from the Lewis-basic azine nitrogen atoms because targeting of the proximal sites requires higher-energy pathways and, crucially, leads to products that are unstable under the reaction conditions5. This effectively hampers application for chemical space exploration, especially considering that α-N-functionalization is the most frequent substitution pattern in bio-active azines17. As a result, multi-step strategies are often the only viable synthetic option to access these high-value materials. However, many of these borylated azines, including C2-substituted pyridines and quinolines, are chemically unstable and undergo fast protodeboronation18, which impedes their application in fragment coupling3. Among the C2-borylated azines, MIDA-boronates (MIDA is methyliminodiacetic acid) are chemically stable but their preparation is less general19,20.
Here we describe an orthogonal strategy for C–H borylation of azines, where the sp2 C–B bonds are assembled by exploiting the reactivity of boryl radicals. This process uses a commercial and inexpensive amine-borane reagent and an organic photocatalyst, and enables the preparation of stable borylated materials that cannot be accessed using mainstream C–H activation technology and that can be further functionalized.
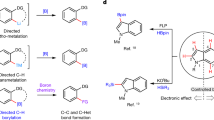
In approaching the development of an alternative strategy for azine borylation, we recognized the potential benefits that would come if boron functionalities could be introduced just like alkyl groups via the Minisci reaction9,21. Whereas current sp2 C–B bond-forming strategies utilize boron reagents as either electrophiles (Grignard and C–H borylation) or nucleophiles (Miyaura borylation), a ‘Minisci-style’ approach would necessitate the intermediacy of boron-centred radicals22,23,24,25. These reactive intermediates are underrepresented in synthetic chemistry but their engagement in azine borylation has the potential to target aromatic positions out of reach of C–H activation. This can be exemplified by looking at the functionalization of pyridine: while Ir-catalysed protocols cannot access C2-borylated products and usually lead to mixtures favouring C3 functionalization5,16,26,27,28,29, a Minisci-type approach should preferentially target the more activated but currently elusive α-N-site (Fig. 1a).

a, C–H borylation of azines is achieved using C–H activation with transition metals. This strategy uses boryl radicals mirroring the reactivity pattern observed in Minisci alkylations and displays orthogonal functionalization selectivity. b, Proposed mechanism and computational studies for the borylation of 1 using 2. c, The optimized radical coupling between 1 and 2 gives 3 in high yield. o/n, overnight.
The identification of an appropriate boryl radical precursor is crucial to the successful realization of this proposal as three main factors need to be simultaneously addressed. (i) The borylating reagent should be a stable, inexpensive and readily available chemical from which radical generation can occur under mild conditions. (ii) As Minisci reactions benefit from the use of nucleophilic alkyl radicals30, the boron substitution pattern is important to ensure the generation of an open-shell species with related philicity. (iii) Finally, the borylated azine products need to be chemically stable while still amenable to further divergent functionalization.
On considering the above-mentioned aspects, we preferred not to use the versatile NHC-boranes (NHC is N-heterocyclic carbene)23,31,32 mostly because the radical delocalization into the NHC ring lowers their nucleophilic character and this could thwart broad applicability in Minisci-type chemistry. Instead, we reasoned that the Lewis base adducts between tertiary amines and BH3, the amine-boranes, would be ideal reagents for our borylation blueprint. These species are stable and cost-effective feedstocks of low molecular weight, and are routinely used as polymer precursors and hydrogen storage materials7,33,34. Despite the fact that they have found limited applications in organic synthesis, our attention was drawn by pioneering studies from Roberts that demonstrated how amine coordination facilitates boryl radical generation by H-atom abstraction (HAT) and renders the corresponding open-shell species highly nucleophilic8,35,36,37,38.
A detailed description of our proposed strategy for the selective borylation of lepidine 1 is outlined in Fig. 1b, and would start with the visible-light irradiation of a photoredox catalyst (PC) to populate its long-lived excited state (*PC). As *PC are competent reductants, SET (single-electron transfer) with a persulfate A should be facile and generate the corresponding sulfate radical anion B. Roberts demonstrated that amine-boranes undergo efficient HAT from O-radicals (for example, the rate of HAT, kHAT, for the reaction of Me3N–BH3 (2) with t-BuO• is about 108 at −40 °C)8,37,38, so we speculated that B would generate the boryl radical C through a polarity matched HAT process (D) on 2. Boryl C is a metalloid σ-radical of a highly nucleophilic character (see Supplementary Information for details), hence we postulated it would undergo site-selective addition to protonated 124. This step would determine the selectivity of the borylation and, in line with the outcome of standard Minisci alkylations, deliver the C2-functionalized aminium radical intermediate E. In this way, we would have replaced reductive elimination from azine-organometallic intermediates with a simple radical addition as the C–B bond assembling step. The aminium species E can then lead to the formation of the borylated N-heteroaromatic 3-H+ by deprotonation followed by SET with the oxidized photocatalyst (PC•+) or, alternatively, by direct SET with the persulfate A (radical chain reactivity)39. In this mechanistic picture, a stoichiometric Brønsted acid is required to synergistically: (i) facilitate the SET reduction of A; (ii) amplify the electrophilic character of B, which will translate into a more efficient HAT on 240; (iii) increase the electrophilicity of the azine towards Minisci borylation by C; and (iv) insulate the resulting amine-borane product 3 from unwanted HAT from B. Indeed, according to our calculation the B–H bond in 3 is weaker than in 2 but N-protonation provides kinetic stability to the product, preventing any further HAT event that could lead to decomposition (see Supplementary Information for details). Regarding the key C–B forming step, it is interesting to note that although boryl radical C is isoelectronic with the neopentyl radical F, its addition to 1-H+ is considerably more facile both thermodynamically and kinetically (compare the energy parameters in G and H). We believe that the higher nucleophilic character of C compared to F (calculated using the local radical electrophilicity index41, ω+rc) leads to strong and stabilizing polar effects in the transition state for the borylation, which is further facilitated by the boryl radical’s reduced steric hindrance (the B–N bond in C is substantially longer than the C–C bond in F); see Supplementary Information for details.
The realization of this radical C–H borylation strategy was made possible by using the organic dye 4CzIPN as the photocatalyst, (NH4)2S2O8 as the oxidant and TFA as the Brønsted acid in CH3CN–H2O solvent under blue light irradiation at room temperature (Fig. 1c); the details of the reaction optimization including the control experiments are discussed in Supplementary Information. Under these conditions, 1 and 2 gave 3 in high yield and on a multigram scale. Useful amounts of 3 could also be obtained in the absence of 4CzIPN, but these conditions could not be translated to the following substrate scope (see Supplementary Information for details). The ‘boronate’ nature and high polarity of the amine-borane functionality rendered 3 a crystalline solid that is stable under ambient conditions for more than 6 months.
The Lewis-base borane complexes Ph3P–BH3 and NHC–BH3 have also been used in the literature for the formation of boryl radicals31,42. However, their utilization was not successful under our reaction conditions and led to quantitative recovery of 1 (see Supplementary Information). We believe this underscores the importance of the Me3N group in delivering a highly nucleophilic σ-boryl radical, which is a crucial feature that ensures efficient Minisci reactivity.
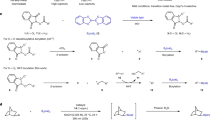
With an optimized set of conditions in hand, we sought to examine the range of azines compatible with this borylation strategy (Fig. 2). In tailoring the substrate scope, we collaborated with Janssen Research and Development to demonstrate applicability on industrially relevant building blocks (Fig. 2a).

Top, reaction studied. a, Azine scope. b, Application in late-stage borylation of pharmaceuticals. Yield in per cent is given under each compound. aReaction run using a compact fluorescent lamp. bReaction run using K2S2O8. cReaction run using (n-Bu4N)2S2O8. dReaction run using AcOH. eReaction run without acid in the presence of K2CO3 (2.0 equiv.). fReaction run without acid. gReaction run using Ru(bpy)3(PF6)2.
We began our investigation by testing C4-substituted quinolines that underwent C2-borylation and demonstrated compatibility with useful functionalities like chloride (4) and hydroxyl group (5). When the C2 was alkylated (that is, quinaldine), borylation took place at C4, albeit in lower yield (6). In analogy to Minisci alkylations, borylation of quinoline resulted in a mixture of C2 and C4 products with a marked preference for the more activated C2 site (7). Phenanthridine, the key aromatic unit of many DNA stains, underwent C6 functionalization in high yield and complete selectivity (8). Direct C1 borylation of isoquinoline and phthalazine is unprecedented but was feasible under our conditions, as demonstrated by the formation of 9–11, which contain a HAT-activated benzylic position and a free amino group. The reaction was also effective for the C2 functionalization of quinazolinol (12–14), an azine found in the core structure of many chemotherapeutic agents like cediranib.
We next assessed pyridine, which is the most frequent N-heteroaromatic motif in drugs6 and its selective borylation is desirable for modular diversification. As C–H borylation is controlled by sterics and preferentially target C316,26,27,28,43, we were pleased that our approach enabled access to C2 borylated materials 15–17. Importantly, these products are stable during their preparation and displayed prolonged stability under ambient conditions (several months), implying that they are much less sensitive towards protodeborylation than the corresponding boronic acids44. When both C2 and C6 were alkylated (that is, lutidine), borylation occurred at C4 (18), while a 2:1 mixture of C2/C4 functionalized products was obtained in the case of a C5 nitrile-containing substrate (19) owing to enhanced C4 reactivity caused by both inductive and resonance effects.
Substituted pyridazines were targeted next because they are common motifs in bioactive molecules but their direct borylation is still underdeveloped and only one example has been reported by C–H activation16. Our Minisci strategy addressed this synthetic gap, as demonstrated by the selective C4 borylation of several industrially relevant substrates (20–27). These examples contain a C3 chlorine that inductively activates its α-position for radical addition and then enables additional diversification by nucleophilic aromatic substitution. Compatibility with pyrazole (21), free amino (23), methoxy (24) and ester (27) functionalities was demonstrated and we successfully accessed the congested tetra-substituted derivatives 25–27 in good yields, which would be very challenging by other methods.
The complementarity that this Minisci strategy can offer to mainstream C–H activation approaches was further shown by evaluating the borylation of pyrimidines. When a C2 substituent is present, C–H activation functionalizes C55 while this radical reactivity targeted the C4 site (28 and 29). In the case of a C4 alkylated substrate, borylation was diverted to C2 (30), which is also unprecedented.
Next, we questioned whether we could apply the process to complex annulated azines of industrial interest that have not been borylated before. We were able to functionalize building blocks based on the azaindazole (31 and 32), imidazo[1,2-b]pyridazine (33) and thieno[3,2-d]pyrimidine (34) cores, which are found in the structure of drugs like riociguat (treatment of pulmonary hypertension), ponatinib (treatment of leukaemia) and olmutinib (anticancer). In all cases, selective borylation of the electron-poor annulated six-membered ring was observed. So far, benzothiazole is the only system where functionalization of the annulated five-membered ring was possible (35).
Exploring the chemical space around lead structures is desirable in drug discovery programs so application in late-stage functionalization of complex pharmaceuticals and agrochemicals was pursued (Fig. 2b). Pleasingly, borylation of (iso)quinoline-based quinoxyfen (agrochemical), cinchonidine (alkaloid, also gram-scale) and fasudil (vasodilator) delivered 36–39 in moderate-to-good yields, proving additional compatibility with benzylic alcohol, tertiary amine, olefin and sulphonamide functionalities. Voriconazole, a structurally complex broad-spectrum antifungal agent, underwent selective borylation of its 5-fluoropyrimidine nucleus at C6 (40, also gram scale). Finally, we were able to engage the antiviral famciclovir that resulted in C6 functionalization of its purine core in the presence of labile O-acetate groups (41).
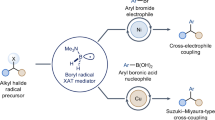
To realize the full potential of this strategy it is crucial to engage these borylated azines in mainstream organoboron chemistry and enable further functionalization. Since the reactivity of aromatic amine-boranes is largely unexplored, achieving this goal would render this borylation approach an effective gateway to access a variety of high-value materials, while establishing a novel class of stable borylated materials.
Aromatic borylation followed by oxidation is one of the most common approaches to prepare complex phenols, as direct sp2 C–H hydroxylation is mostly limited to enzymatic processes. As the amine-borane products obtained by this methodology are stable ‘boronate’ complexes, they do not display the required Lewis acidity for standard Brown oxidation by H2O2/NaOH. We found that simple treatment of 6 with oxone45 under acidic and aerobic conditions provided the desired oxidized product 42 in high yield (Fig. 3). This reactivity was extended to other borylated azines, as demonstrated by the formation of 43–47. The synthetic advantages offered by this borylation–oxidation approach can be realized by considering that there are no methods available for C2 oxidation of phenanthroline (44) and quinazolinol (46) while C4 oxidation of quinaldine (42)46 has been previously done in four chemical steps. Furthermore, the selectivity displayed by this borylation–oxidation sequence matches the one displayed by the activity of aldehyde oxidase, an enzyme involved in the metabolism of azine xenobiotics47. As such, this approach might also have the potential in streamlining the preparation of azine-based drug metabolites.

Details of products 42–74 are shown; yield in per cent is given under each product. Bold-font footnotes A to C refer to reaction conditions given in the box at the bottom of the figure; details in Supplementary Information. aReaction run with 50 mol% CuOAc. bReaction run using 4-CO2Me-C6H4–Br; cReaction conditions as in the Chan–Lam etherification. PMP, p-MeO-C6H4.
The most prominent use of aromatic organoborons in synthetic chemistry is their palladium-catalysed Suzuki–Miyaura cross-coupling with aryl halides1,3,48. However, to the best of our knowledge, amine-borane derivatives have not been demonstrated to participate in this reactivity. Indeed, the strong N–B coordination bond that makes these products very stable also prevents their hydrolysis to the corresponding boronic acids, which are generally the reactive intermediates in the coupling process. Moreover, Lewis base-borane complexes are known hydride donors able to reduce aryl halides under palladium catalysis49, which might represent a substantial competing pathway. Nonetheless, a survey of reaction conditions identified a mild protocol for the coupling between 6 and p-iodoanisole using Pd(dppf)2Cl2 as the catalyst at 40 °C (48); dppf is 1,1′-bis(diphenylphosphino)ferrocene. The scope of the process was investigated, showing tolerance of electron-neutral (49) and electron-poor aryl iodides (50 and 51) as well as a bromide (52).
In order to engage valuable but challenging C2-borylated azines, a more thorough survey of the reaction conditions was needed to overcome the otherwise prevalent protodeborylation (see Supplementary Information). Pleasingly, translation of Suzuki–Miyaura reactivity on quinoline 3 proved to be general, and the scope was demonstrated across a range of para- (54–57), meta- (58–60) and ortho- (61, 62) substituted aryl iodides and bromides of different electronic and steric properties as well as the Lewis-basic 3-Br-pyridine (63). Other borylated azines can be engaged in this Suzuki manifold, as demonstrated by the arylation of C4-borylated pyridazine 20 and 2-amino-pyrimidine 29, which gave 64 and 65 in useful to good yields. In a venture to establish the applicability of this Suzuki approach, we sought to explore the reactivity of complex aryl bromides as well as substrates resulting from the borylation of complex azines. We were able to apply the standard arylation conditions to 3-bromo-strychnine (66) and also engage the borylated quinoxyfen 36 and cinchonidine 37 in moderate to good yields (67 and 68).
Taken together, these results demonstrate that these amine-borane reagents are a useful class of heteroaryl boron donors for the modular assembly of heterobiaryls18 and that, through this novel borylation technology, they can enable ready access and exploration of chemical space in the core framework of bio-active materials.
To further demonstrate the utility of these borylated building blocks, we sought to identify conditions for application in Chan–Lam chemistry and downstream sp2 C–N and C–O bond formation11. We started by evaluating the reactivity of 6 with p-toluidine and allylamine using stoichiometric Cu(OAc) and obtained proof-of-concept results demonstrating that aminations at both C4 (69) and C2 (70) are feasible. Moreover, the use of these azines in Cham–Lam etherification with allyl alcohol was very effective, as demonstrated by the high-yielding formation of 71–73 and the successful translation to give the complex azine 74.
The results presented here demonstrate that Minisci reactivity can be broadly applied to engage metalloid boryl radicals in sp2 C–B bond formation. This provides a unique tool for the borylation of high-value azines with site selectivity orthogonal to that of current C–H activation strategies. The chemical stability of the resulting azine amine-borane products, combined with their ability to undergo oxidation, Suzuki–Miyaura arylation and Chan–Lam amination and etherification, suggests they can be considered as viable alternative to boronic acids, esters and trifluoroborates. We expect this chemistry to be relevant to medicinal chemistry, and that its borylated products will find broader synthetic application through the development of additional metal-catalysed processes.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
Roughley, S. D. & Jordan, A. M. The medicinal chemist’s toolbox: an analysis of reactions used in the pursuit of drug candidates. J. Med. Chem. 54, 3451–3479 (2011).
Blakemore, D. C. et al. Organic synthesis provides opportunities to transform drug discovery. Nat. Chem. 10, 383–394 (2018).
Blakemore, D. in Synthetic Methods in Drug Discovery Vol. 1 (eds Blakemore, D. C., Doyle, P. M. & Fobian, Y. M.) 1–69 (Royal Society of Chemistry, 2016).
Mkhalid, I. A. I., Barnard, J. H., Marder, T. B., Murphy, J. M. & Hartwig, J. F. C–H activation for the construction of C–B bonds. Chem. Rev. 110, 890–931 (2010).
Larsen, M. A. & Hartwig, J. F. Iridium-catalyzed C–H borylation of heteroarenes: scope, regioselectivity, application to late-stage functionalization, and mechanism. J. Am. Chem. Soc. 136, 4287–4299 (2014).
Taylor, R. D., MacCoss, M. & Lawson, A. D. G. Rings in drugs. J. Med. Chem. 57, 5845–5859 (2014).
Staubitz, A., Robertson, A. P. M., Sloan, M. E. & Manners, I. Amine− and phosphine−borane adducts: new interest in old molecules. Chem. Rev. 110, 4023–4078 (2010).
Baban, J. A., Marti, V. P. J. & Roberts, B. P. Ligated boryl radicals. Part 2. Electron spin resonance studies of trialkylamin–boryl radicals. J. Chem. Soc. Perkin Trans. 2 1723–1733 (1985).
Duncton, M. A. J. Minisci reactions: versatile CH-functionalizations for medicinal chemists. MedChemComm 2, 1135–1161 (2011).
Hall, D. G. in Boronic Acids (ed. Hall, D. G.) 1–99 (Wiley, 2005).
West, M. J., Fyfe, J. W. B., Vantourout, J. C. & Watson, A. J. B. Mechanistic development and recent applications of the Chan–Lam amination. Chem. Rev. 119, 12491–12523 (2019).
Furuya, T., Kaiser, H. M. & Ritter, T. Palladium-mediated fluorination of arylboronic acids. Angew. Chem. Int. Ed. 47, 5993–5996 (2008).
Bonet, A., Odachowski, M., Leonori, D., Essafi, S. & Aggarwal, V. K. Enantiospecific sp2–sp3 coupling of secondary and tertiary boronic esters. Nat. Chem. 6, 584–589 (2014).
Ishiyama, T., Murata, M. & Miyaura, N. Palladium(0)-catalyzed cross-coupling reaction of alkoxydiboron with haloarenes: a direct procedure for arylboronic esters. J. Org. Chem. 60, 7508–7510 (1995).
Vitaku, E., Smith, D. T. & Njardarson, J. T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 57, 10257–10274 (2014).
Sadler, S. A. et al. Iridium-catalyzed C–H borylation of pyridines. Org. Biomol. Chem. 12, 7318–7327 (2014).
Brown, D. G., Gagnon, M. M. & Boström, J. Understanding our love affair with p-chlorophenyl: present day implications from historical biases of reagent selection. J. Med. Chem. 58, 2390–2405 (2015).
Cook, X. A. F., de Gombert, A., McKnight, J., Pantaine, L. R. E. & Willis, M. C. The 2-pyridyl problem: challenging nucleophiles in cross-coupling arylations. Angew. Chem. Int. Ed. 60, 11068–11091 (2020).
Li, J. et al. Synthesis of many different types of organic small molecules using one automated process. Science 347, 1221–1226 (2015).
Dick, G. R., Woerly, E. M. & Burke, M. D. A general solution for the 2-pyridyl problem. Angew. Chem. Int. Ed. 51, 2667–2672 (2012).
Proctor, R. S. J. & Phipps, R. J. Recent advances in Minisci-type reactions. Angew. Chem. Int. Ed. 58, 13666–13699 (2019).
Taniguchi, T. Boryl radical addition to multiple bonds in organic synthesis. Eur. J. Org. Chem. 2019, 6308–6319 (2019).
Curran, D. P. et al. Synthesis and reactions of N-heterocyclic carbene boranes. Angew. Chem. Int. Ed. 50, 10294–10317 (2011).
Giles, J. R. M. & Roberts, B. P. An electron spin resonance study of the generation and reactions of borane radical anions in solution. J. Chem. Soc. Perkin Trans. 2 743–755 (1983).
Mills, H. A., Martin, J. L., Rheingold, A. L. & Spokoyny, A. M. Oxidative generation of boron-centered radicals in carboranes. J. Am. Chem. Soc. 142, 4586–4591 (2020).
Yang, L., Uemura, N. & Nakao, Y. Meta-selective C–H borylation of benzamides and pyridines by an iridium–Lewis acid bifunctional catalyst. J. Am. Chem. Soc. 141, 7972–7979 (2019).
Mkhalid, I. A. I. et al. Ir-catalyzed borylation of C–H bonds in N-containing heterocycles: regioselectivity in the synthesis of heteroaryl boronate esters. Angew. Chem. Int. Ed. 45, 489–491 (2006).
Preshlock, S. M. et al. A traceless directing group for C–H borylation. Angew. Chem. Int. Ed. 52, 12915–12919 (2013).
Chotana, G. A., Rak, M. A. & Smith, M. R. Sterically directed functionalization of aromatic C−H bonds: selective borylation ortho to cyano groups in arenes and heterocycles. J. Am. Chem. Soc. 127, 10539–10544 (2005).
Minisci, F. et al. Polar effects in free-radical reactions. Selectivity and reversibility in the homolytic benzylation of protonated heteroaromatic bases. J. Org. Chem. 51, 476–479 (1986).
Ueng, S.-H. et al. Complexes of borane and N-heterocyclic carbenes: a new class of radical hydrogen atom donor. J. Am. Chem. Soc. 130, 10082–10083 (2008).
Dai, W., Geib, S. J. & Curran, D. P. 1,4-Hydroboration reactions of electron-poor aromatic rings by N-heterocyclic carbene boranes. J. Am. Chem. Soc. 142, 6261–6267 (2020).
Hioe, J., Karton, A., Martin, J. M. L. & Zipse, H. Borane–Lewis base complexes as homolytic hydrogen atom donors. Chem. Eur. J. 16, 6861–6865 (2010).
Tehfe, M.-A. et al. N-heterocyclic carbene-borane radicals as efficient initiating species of photopolymerization reactions under air. Polym. Chem. 2, 625–631 (2011).
Roberts, P. B. Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev. 28, 25–35 (1999).
Wu, C., Hou, X., Zheng, Y., Li, P. & Lu, D. Electrophilicity and nucleophilicity of boryl radicals. J. Org. Chem. 82, 2898–2905 (2017).
Sheeller, B. & Ingold, K. U. Absolute rate constants for some reactions of the triethylamine-boryl radical and the borane radical anion. J. Chem. Soc. Perkin Trans. 2 480–486 (2001).
Lalevée, J., Tehfe, M. A., Allonas, X. & Fouassier, J. P. Boryl radicals as a new photoinitiating species: a way to reduce the oxygen inhibition. Macromolecules 41, 9057–9062 (2008).
Jin, J. & MacMillan, D. W. C. Direct α-arylation of ethers through the combination of photoredox-mediated C–H functionalization and the Minisci reaction. Angew. Chem. Int. Ed. 54, 1565–1569 (2015).
Bietti, M. Activation and deactivation strategies promoted by medium effects for selective aliphatic C−H bond functionalization. Angew. Chem. Int. Ed. 57, 16618–16637 (2018).
De Vleeschouwer, F., Speybroeck, V. V., Waroquier, M., Geerlings, P. & Proft, F. D. Electrophilicity and nucleophilicity index for radicals. Org. Lett. 9, 2721–2724 (2007).
Baban, J. A. & Roberts, B. P. An electron spin resonance study of phosphine-boryl radicals; their structures and reactions with alkyl halides. J. Chem. Soc. Perkin Trans. 2 1717–1722 (1984).
Davis, H. J., Mihai, M. T. & Phipps, R. J. Ion pair-directed regiocontrol in transition-metal catalysis: a meta-selective C–H borylation of aromatic quaternary ammonium salts. J. Am. Chem. Soc. 138, 12759–12762 (2016).
Cox, P. A. et al. Base-catalyzed aryl-B(OH)2 protodeboronation revisited: from concerted proton transfer to liberation of a transient aryl anion. J. Am. Chem. Soc. 139, 13156–13165 (2017).
Molloy, J. J. et al. Chemoselective oxidation of aryl organoboron systems enabled by boronic acid-selective phase transfer. Chem. Sci. 8, 1551–1559 (2017).
Kuninobu, Y., Nagase, M. & Kanai, M. Benzylic C(sp3)–H perfluoroalkylation of six-membered heteroaromatic compounds. Angew. Chem. Int. Ed 54, 10263–10266 (2015).
Garattini, E. & Terao, M. Aldehyde oxidase and its importance in novel drug discovery: present and future challenges. Expert Opin. Drug Discov. 8, 641–654 (2013).
Brown, D. G. & Boström, J. Analysis of past and present synthetic methodologies on medicinal chemistry: where have all the new reactions gone? J. Med. Chem. 59, 4443–4458 (2016).
Chu, Q. et al. Ionic and organometallic reductions with N-heterocyclic carbene boranes. Chem. Eur. J. 15, 12937–12940 (2009).
Acknowledgements
D.L. thanks the EPSRC for a fellowship (EP/P004997/1) and the European Research Council for a research grant (758427); J.H.K. thanks Marie Curie Actions for a fellowship (842422). The Chemical Capabilities team of Janssen Research and Development (Toledo) is acknowledged for technical support.
Author information
Authors and Affiliations
Contributions
J.H.K. and D.L. designed the project; J.H.K., T.C., M.S. and J.L. performed the experiments; N.S.S. performed the computational studies; and all the authors analysed the results and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks the anonymous reviewers for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
This file contains Supplementary Information.
Rights and permissions
About this article

Cite this article
Kim, J.H., Constantin, T., Simonetti, M. et al. A radical approach for the selective C–H borylation of azines. Nature 595, 677–683 (2021). https://doi.org/10.1038/s41586-021-03637-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03637-6
- Springer Nature Limited
This article is cited by
-
Boryl radical-mediated halogen-atom transfer enables arylation of alkyl halides with electrophilic and nucleophilic coupling partners
Nature Synthesis (2024)
-
Transforming cyclopropanes to enamides via σ-C–C bond eliminative borylation
Nature Communications (2024)
-
Highly dispersive tetrahedral vanadium species on pure silica ZSM-12 zeolite boosting the selective oxidation of cyclohexane into cyclohexanone
Science China Chemistry (2024)
-
Electrochemical reactor dictates site selectivity in N-heteroarene carboxylations
Nature (2023)
-
Tunably strained metallacycles enable modular differentiation of aza-arene C–H bonds
Nature Communications (2023)