

Abstract
Defects in DNA repair frequently lead to neurodevelopmental and neurodegenerative diseases, underscoring the particular importance of DNA repair in long-lived post-mitotic neurons1,2. The cellular genome is subjected to a constant barrage of endogenous DNA damage, but surprisingly little is known about the identity of the lesion(s) that accumulate in neurons and whether they accrue throughout the genome or at specific loci. Here we show that post-mitotic neurons accumulate unexpectedly high levels of DNA single-strand breaks (SSBs) at specific sites within the genome. Genome-wide mapping reveals that SSBs are located within enhancers at or near CpG dinucleotides and sites of DNA demethylation. These SSBs are repaired by PARP1 and XRCC1-dependent mechanisms. Notably, deficiencies in XRCC1-dependent short-patch repair increase DNA repair synthesis at neuronal enhancers, whereas defects in long-patch repair reduce synthesis. The high levels of SSB repair in neuronal enhancers are therefore likely to be sustained by both short-patch and long-patch processes. These data provide the first evidence of site- and cell-type-specific SSB repair, revealing unexpected levels of localized and continuous DNA breakage in neurons. In addition, they suggest an explanation for the neurodegenerative phenotypes that occur in patients with defective SSB repair.
Similar content being viewed by others


Main
An obligatory and characteristic step of DNA repair is gap filling, in which excised or missing nucleotides are replaced using the undamaged strand as a template3. If a sufficient number of nucleotides are incorporated, DNA repair synthesis can be used as a proxy for the extent and location of endogenous DNA damage4. In light of this, we developed a method to map sites of DNA repair synthesis by sequencing (synthesis associated with repair sequencing (SAR-seq)). We labelled post-mitotic glutamatergic neurons derived from induced pluripotent stem cells (iPS cells) (i3Neurons5,6) on day 6 after differentiation with ethynyl deoxyuridine (EdU) for 18 h, biotinylated the labelled DNA, reduced it to 150–200 bp by sonication, and then isolated the biotinylated DNA for high-throughput sequencing (Extended Data Fig. 1a). We identified more than 55,000 SAR-seq peaks at recurrent genomic locations in neurons, which were highly reproducible between different experiments (Fig. 1a, Extended Data Fig. 1b–d). Peaks were not caused by DNA synthesis during S phase7,8 because i3Neurons are post-mitotic (Extended Data Fig. 1e), and the SAR-seq peaks were unaffected by inhibition of the replicative DNA polymerase-α (Extended Data Fig. 2a–c). By contrast, and as expected, neuronal synthesis associated with repair (SAR) was largely prevented by hydroxyurea (HU), which reduces the availability of deoxyribonucleotides (Extended Data Fig. 2a, b).

a, Genome browser screenshots of SAR-seq (n = 3), ATAC–seq (n = 1), and ChIP–seq for H3K4me1 (n = 2), H3K27ac (n = 1), and MLL4 (n = 1) in i3Neurons. Below, expanded view of the indicated region to show overlapping peaks. b, Genome browser screenshot of SAR-seq performed in rat primary neurons (n = 1) as well as input. The culture was co-incubated with 5 μM aphidicolin to block DNA replication of S-phase glial cells. c, Representative images of i3Neurons with immunofluorescence staining for PAR (green) and the neuronal marker tubulin-β3 (red), counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (blue) (data are representative of three independent experiments). As a positive control, cells were treated with 0.1 mg ml−1 MMS for 15 min; NT, not treated. Boxed regions in top row are enlarged below. d, Heat maps of SAR-seq signal and ChIP–seq signals for XRCC1 and PAR for 1 kb on either side of SAR-seq peak summits in i3Neurons, ordered by SAR-seq intensity.
The SAR-seq peaks were 200–2,000 bp in width (mean 901 bp) (Extended Data Fig. 2d), with each peak presumably comprising multiple clustered sites of DNA repair (see below). The most prominent neuronal SAR-seq peaks were detectable by pulse labelling with EdU for just 1 h, and EdU incorporation approached saturation after labelling for 18 h (Extended Data Fig. 2e, f).
When iPS cells were differentiated into skeletal muscle cells (iMuscle), we did not detect incorporation of EdU despite labelling cells for 18 h (Extended Data Fig. 3a). Similarly, we did not detect EdU incorporation in G0-arrested pre-B cells, although we could detect EdU incorporation in pre-B cells after the induction of site-specific DNA double-strand breaks (DSBs) (Extended Data Fig. 3b). To rule out the possibility that the SAR-seq peaks were an artefact of iPS cell differentiation, we labelled bona fide rat neurons with EdU. Similar to i3Neurons, we detected robust peaks of EdU incorporation at 22,196 specific sites in rat neurons (Fig. 1b, Extended Data Fig. 5e). Thus, the high frequency of recurrent DNA synthesis appears to be a specific feature of post-mitotic neurons.
DNA repair sites enriched at enhancers
Neuronal SAR-seq peaks were enriched in intragenic regions (Extended Data Fig. 3c, d) and further within expressed genes (Extended Data Fig. 3d, e). However, SAR signal intensity did not correlate with transcript levels (Extended Data Fig. 3e). Moreover, the sites of EdU incorporation were not associated with strand specificity, as EdU was incorporated uniformly in both transcribed and non-transcribed strands (Extended Data Fig. 3f).
We next searched for specific DNA motifs among the strongest 5,000 SAR-seq peaks. More than 25% of the sites contained a motif similar to the ONECUT family of transcription factors, which was centred at SAR-seq peak summits (Extended Data Fig. 4a). As ONECUT1 can promote genomic accessibility in neurons9, we compared SAR-seq peaks with accessible regions using an assay for transposase-accessible chromatin using sequencing (ATAC–seq). Fifty-four per cent of all SAR-seq regions coincided with ATAC-seq peaks (Fig. 1a, Extended Data Fig. 4b, c), and the widths of SAR-seq and ATAC-seq peaks were correlated (Extended Data Fig. 4d), suggesting that open chromatin structure influences the extent of DNA synthesis.
Despite their localization in open chromatin, SAR-seq peaks were not enriched at promoters (Extended Data Fig. 4e–g), which exhibited only modest levels of DNA synthesis. However, we detected a strong correlation between locations of DNA synthesis and of neuronal enhancers, as measured by chromatin immunoprecipitation with sequencing (ChIP–seq) for methylation at lysine 4 of histone H3 (H3K4me1), acetylation at lysine 27 of histone H3 (H3K27ac) and the lysine methyltransferase MLL4 (Fig. 1a, Extended Data Fig. 5a, b). Nevertheless, SAR-seq peaks were not strongly correlated with other, non-enhancer-specific, epigenetic indicators of open chromatin or condensed chromatin (Extended Data Fig. 5a, c). Similar to i3Neurons, SAR-seq peaks in primary rat neurons overlapped and correlated with rat H3K4me1 ChIP–seq (Extended Data Fig. 5d–f).
We performed ultra-deep Hi-C in i3Neurons to examine chromosomal interactions associated with SAR-seq peaks within the context of topological associated domains (TADs) (Extended Data Fig. 6a). Enhancers with SAR-seq peaks had more intra-TAD interactions than enhancers without SAR-seq peaks. Consistently, H3K27ac—a mark of active enhancers—was significantly enriched at enhancers with SAR-seq peaks (Extended Data Fig. 6a) (P < 2.2 × 10−6). Chromatin looping interactions detected by promoter capture Hi-C (pcHi-C) enables distal enhancer elements to be linked to their target genes10. Compared to random sites, we observed an eightfold enrichment of SAR-seq sites among in vivo-validated enhancer elements11 that overlapped with neuronal H3K4me1 histone marks and a fourfold enrichment using a pcHi-C dataset generated in i3Neurons10 (Extended Data Fig. 6b). The latter includes enhancers whose activity was validated in human neurons using CRISPR techniques (Extended Data Fig. 6c, d). Thus, distal-acting regulatory enhancers are hotspots of neuronal SAR.
To determine whether the enhancers with SAR-seq peaks are specific to neurons, we compared sites of H3K4me1 in i3Neurons and iPS cells. Only 2% of the SAR-seq peaks overlapped with iPS cell-specific H3K4me1 peaks, whereas most of the SAR-seq peaks overlapped with either neuron-specific or shared H3K4me1 sites (Extended Data Fig. 6e). Thus, SAR is associated with enhancers that are active in differentiated neurons.
Gene Ontology (GO) analysis of the genes that contained SAR-seq peaks revealed that they were enriched in GO terms related to nervous system function (Extended Data Fig. 6f). We then further characterized the neuronal enhancers that were associated with SAR. Super-enhancers are a large collection of enhancers that drive the transcription of genes involved in cell identity. Using H3K27ac ChIP–seq, we found approximately 1,300 super-enhancers in i3Neurons. Notably, 90% of super-enhancers exhibited SAR-seq peaks, whereas fewer than 25% of conventional enhancers possessed SAR-seq peaks (Extended Data Fig. 6g, h). Collectively, these data identify enhancers and genes that are associated with neuronal function as hotspots of recurrent DNA synthesis.
PARP activity at neuronal enhancers
Given the close association between unrepaired DNA strand breaks and neurodegeneration1,2, we wondered whether the sites of EdU incorporation might reflect sites of DNA break repair. To test this idea, we measured the activity of poly(ADP-ribose) polymerases (PARPs) at the sites of DNA synthesis. Various types of DNA damage, including SSBs, DSBs and single-strand gaps, activate PARP1 and PARP212,13. PARP activity signals the presence of these lesions by modifying localized proteins with poly(ADP-ribose) (PAR)12,13. When we monitored ADP-ribosylation in individual neurons14, we detected focal sites of nuclear ADP-ribose. These focal sites, along with pan-nuclear staining, increased following treatment with the genotoxin methylmethansulfate (MMS) (Fig. 1c, Extended Data Fig. 7a). To determine whether the sites of endogenous ADP-ribosylation were localized to sites of neuronal DNA synthesis, we used ADP-ribose ChIP–seq. As a positive control, we first confirmed that we could detect ADP-ribosylation at site-specific DSBs (Extended Data Fig. 7b). In i3Neurons, the endogenous sites of ADP-ribosylation co-localized with SAR-seq peaks (Fig. 1d, Extended Data Fig. 7c), suggesting that recurrent sites of DNA synthesis are associated with DNA strand break repair.
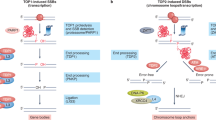
DNA repair is not associated with DSBs
Neuronal activity has been reported to cause DSBs generated by topoisomerase 2 (TOP2)15,16. TOP2-induced DSBs can promote the expression of early response genes15, associating these DNA breaks with regions of transcriptional activity17,18. Treatment of i3Neurons with etoposide to trigger TOP2-induced DSBs resulted in DNA synthesis within gene bodies (Extended Data Fig. 8a, b). However, most of the sites of etoposide-induced DNA synthesis were distinct from those detected in untreated neurons (Extended Data Fig. 8a, b). In addition, we did not detect DSBs in unchallenged i3Neurons as measured by immunostaining for either γH2AX or 53BP1 (Extended Data Fig. 8c) or by END-seq19 (which involves the ligation of a sequencing adapter to the ends of DSBs) (Extended Data Fig. 8d). Thus, sites of DNA synthesis in neuronal enhancers are independent of DSBs.
SSBs at sites of DNA synthesis
As well as DSBs, PARP1 and/or PARP2 are also activated at SSBs and subsequently recruit the XRCC1 protein complex, which accelerates SSB repair20,21. We therefore examined the genomic localization of XRCC1 by ChIP–seq. Similar to sites of PARP activity, XRCC1 co-localized with SAR-seq peaks, and the intensity of XRCC1 binding correlated with the intensity of EdU incorporation in both human i3Neurons and rat primary neurons (Fig. 1d, Extended Data Figs. 7c, 8e–g). Thus, sites of DNA synthesis colocalize with sites of PARP activation and XRCC1-associated SSB repair.
To directly map SSBs at nucleotide resolution, we treated agarose-embedded i3Neuron plugs with recombinant single strand-specific S1 nuclease in situ to convert SSBs into DSBs, which we then detected using END-seq. To test whether this method would successfully convert DNA nicks into DSBs, we first incubated the i3Neuron agarose plugs with the site-specific nicking endonuclease Nt.BspQI before treating them with S1 nuclease. Nt.BspQI nicks were readily converted into DSBs by S1 (Extended Data Fig. 9a). Moreover, S1 END-seq could detect nicks generated by an inducible CAS9 nickase expressed in G1-arrested MCF10 cells (Extended Data Fig. 9b, c). However, S1 END-seq alone did not detect endogenous SSBs in i3Neurons (Fig. 2a, Extended Data Fig. 9a).

a, Top, genome browser screenshots illustrating the overlap in i3Neurons between sites of SAR-seq and SSBs (S1 END-seq) detected in the presence of ddN (n = 1) to block DNA ligation, or in its absence (NT, n = 1). The S1 END-seq signal is separated for positive (black) and negative (grey) strands. Bottom, expanded views demonstrating the presence of multiple SSBs within one SAR-seq peak. b, Aggregate plots of S1 END-seq signal (red, positive strand; blue, negative strand) and END-seq signal in the presence of ddN (n = 1) (black, positive strand; grey, negative strand) for 500 bp on either side of SAR-seq peak summits. RPM, reads per million. c, Composite DNA sequence motif for 5 bp on either side of SSB summits on the positive strand (right) and on the negative strand (left) of the 10,000 most prominent S1 END-seq peaks.
We surmised that failure to detect endogenous SSBs in neurons could be due to their rapid repair. To increase the half-life of SSBs, we incubated i3Neurons with a mixture of chain-terminating dideoxynucleosides (ddA, ddC, ddT and ddG, denoted ddN). This led to robust SSB accumulation as detected by S1 END-seq (Fig. 2a), which co-localized extensively with SAR-seq peaks (Fig. 2a, b, Extended Data Fig. 9d, e). Many of the SAR-seq peaks contained multiple S1 END-seq peaks (Fig. 2a, inset). Each peak spanned about 25 nucleotides on average (Extended Data Fig. 9f), thus comprising multiple-clustered single-strand gaps. Notably, we also detected a very low level of DSBs at SAR-seq peaks following chain termination; these were independent of S1 nuclease treatment and probably reflected closely apposed SSBs on opposite DNA strands (Fig. 2b).
S1 END-seq enabled us to locate endogenous SSBs with much higher resolution than did SAR-seq, which is limited to the size of the sonicated fragments (150–200 nt). The improved resolution revealed a prevalence of C/G nucleotides at S1 END-seq peak summits on positive or negative strands (Fig. 2c), and showed that CpG dinucleotides were highly enriched at SAR sites (Extended Data Fig. 9g, h). Collectively, these data provide direct evidence for the site-specific formation of clustered SSBs in neuronal enhancers at or near C/G nucleotides.
SAR involves long-patch SSB repair
SSB repair comprises both short-patch and long-patch sub-pathways, in which single or multiple nucleotides, respectively, are replaced at the site of the SSB22,23. PARP1 and XRCC1 promote the repair of a wide spectrum of SSBs, primarily by short-patch repair. We therefore examined the effects of inhibiting and/or depleting these proteins on neuronal DNA synthesis. There was a reproducible increase in EdU incorporation at SAR sites if neurons were co-incubated with any of three independent inhibitors of PARP1, or if PARP1 was depleted using CRISPR interference (CRISPRi)24 (Fig. 3a, Extended Data Fig. 10a, b). Depletion of XRCC1 similarly led to a prominent increase in EdU incorporation at sites of SAR (Fig. 3b, Extended Data Fig. 10c–e). These data suggest that if PARP1- or XRCC1-dependent short-patch SSB repair is impeded, long-patch SSB repair is increased to compensate.

a, Bottom, heat maps of SAR-seq intensities for 1 kb on either side of SAR-seq peak summits for i3Neurons treated with the indicated PARP inhibitors (n = 3) or expressing non-targeted control (sgControl, n = 1) or PARP1-targeted (sgPARP1, n = 2) CRISPRi plasmids. Top, aggregate plots of SAR-seq intensity. Rep, replicate. b, Heat maps of SAR-seq intensities for 1 kb on either side of SAR-seq peak summits for i3Neurons expressing non-targeted control (sgControl, n = 2) or XRCC1-targeted (sgXRCC1, n = 2) CRISPRi plasmids. c, Heat maps of SAR-seq intensities for 1 kb on either side of SAR-seq peak summits for i3Neurons expressing non-targeted (sgControl, n = 2) or POLB-targeted (sgPOLB, n = 2) CRISPRi plasmids, either untreated or treated with 50 μM aphidicolin (Aph) for 24 h before and during EdU incorporation.
During short-patch SSB repair, a single nucleotide is replaced at the site of the break22,23,25. Nucleotide replacement typically requires DNA polymerase β (POLβ), which interacts directly with XRCC120. By contrast, during long-patch repair, alternative DNA polymerases such as POLε and POLδ can generate a 2–20-nucleotide DNA repair patch. Consistent with this, we found that depletion of POLβ resulted in a marked increase in DNA synthesis at neuronal enhancers (Fig. 3c, Extended Data Fig. 10f–h). By contrast, incubation with aphidicolin, an inhibitor of POLε and POLδ, greatly reduced SAR in both wild-type and POLβ-depleted neurons when used at a concentration (50 μM) that inhibits cellular repair synthesis4,26 (Fig. 3c, Extended Data Fig. 10i). Collectively, these data indicate that neuronal site-specific SSBs are repaired by both short-patch and long-patch repair, with the latter being the primary source of the SAR-seq signal.
SSBs and active cytosine demethylation
The detection of DNA repair synthesis at neuronal enhancers could indicate that the latter are sites of increased DNA damage or, alternatively, that they are sites of preferential DNA repair. Our observations using S1 END-seq, which showed that SSBs are enriched at neuronal enhancers, strongly support the first hypothesis. To test this further, we treated i3Neurons with MMS to introduce SSBs stochastically across the genome. We predicted that if DNA repair is targeted to enhancer sites, the induction of random SSBs across the genome should not affect the location of the SAR-seq peaks. However, despite the expected two- to fivefold increase in the overall level of EdU incorporation, MMS ablated the appearance of SAR-seq peaks at enhancers. This observation suggests that elevated levels of endogenous SSBs occur specifically at neuronal enhancers and account for the peaks of DNA repair synthesis at these sites, rather than it being a selective and site-specific repair process (Extended Data Fig. 11a).
We considered the source of the localized SSBs. Oxidative DNA lesions such as 8-oxoguanine have strongly been implicated as sources of DNA damage in the brain; however, unlike the sites of DNA repair synthesis detected here, such lesions have not been shown to preferentially accumulate in open chromatin or at enhancers27. By contrast, enhancers may be especially vulnerable to SSBs induced by TOP1, a topoisomerase enzyme that has been implicated in enhancer activation28,29. However, such SSBs are also unlikely to be the source of the localized SSBs, as depletion of the polynucleotide kinase 3′-phosphatase (PNKP)—which is recruited by XRCC1 to repair TOP1-induced SSBs30,31—had only a small effect on the SAR-seq peaks (Extended Data Fig. 11b, c). This was in contrast to the DNA repair synthesis of bona fide TOP1-induced SSBs that we triggered by treating neurons with the TOP1 poison camptothecin, which were increased by PNKP depletion and located in gene bodies rather than in enhancers (Extended Data Fig. 11d).
Given our finding that SSBs associated with SAR-seq peaks were enriched at C/G nucleotides (Fig. 2c, Extended Data Fig. 9g, h), we entertained the possibility that these SSBs were sites of cytosine demethylation (Fig. 4c). Consistent with this idea, active DNA demethylation of cytosine at CpG sites occurs preferentially at enhancers32, is tenfold more active in post-mitotic neurons than in peripheral cell types33, and generates SSBs that are intermediates of XRCC1-associated, but not PNKP-associated, base excision repair (BER)32,34. Active demethylation via ten-eleven translocation (TET) family enzymes (TET1, TET2, and TET3) is initiated through progressive oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), or 5-carbolxylcytosine (5caC), and steady-state levels of 5hmC account for approximately 40% of modified cytosines in the brain33. Using labelling methods to detect 5hmC and 5fC35,36, we mapped oxidized forms of 5-methylcytosine genome-wide in i3Neurons. We found that the sites of both DNA repair synthesis (measured by SAR-seq) and SSBs (measured by S1 END-seq) overlapped with peaks of 5hmC and 5fC, and the intensity of SSBs correlated with that of 5hmC and 5fC (Fig. 4a, b, Extended Data Fig. 11e, f). Although future studies will be needed to determine the precise source(s) of SSBs at neuronal enhancers, our data implicate cycles of cytosine methylation and demethylation at neuronal enhancers as a potential source of neuronal site-specific DNA single-strand breakage.

a, Genome browser screenshot illustrating the overlap in i3Neurons between sites of 5hmC (5hmC-Seal, n = 2)35,36, 5fC (5fC-Seal, n = 2)35,36, SSBs (ddN S1 END-seq), DNA repair synthesis (SAR-seq), and H3K4me1 ChIP–seq. Both positive (black) and negative (grey) S1 END-seq signal are shown. b, Heat maps of signals for DNA repair synthesis (SAR-seq), SSBs (ddN S1 END-seq), 5fC (5fC-Seal), and 5hmC (5hmC-Seal) in i3Neurons, for 1 kb on either side of the summits of the SAR-seq peaks and ordered by SAR-seq intensity. c, Model depicting the balance between short-patch and long-patch SSB repair at neuronal enhancers. Although our data suggest that SSBs arise during the base excision repair of modified cytosine residues within enhancers, we do not exclude the occurrence of other sites and sources of SSBs.
Conclusions
Our study reveals that human post-mitotic neurons are subject to an unexpected level of localized DNA synthesis that is associated with ongoing sites of SSB repair at neuronal enhancers. The scaffold protein XRCC1 is of particular importance during SSB repair because it is physically associated with a variety of SSB repair enzymes including TDP1, POLβ, PNKP, APTX, and DNA ligase III (LIG3)20, in some of which mutations result in hereditary neurodegenerative diseases1,2. Failure to repair the site-specific SSBs that we have discovered here may thus contribute to such diseases, although we do not rule out an involvement of other SSB sources. Unrepaired SSBs may trigger neuropathology in several ways, such as via excessive or aberrant activation of PARP137. In addition, our current data raise the possibility that increased dependency on DNA repair synthesis at specific sites of the genome increases the mutational burden in long-lived neurons38. For example, it is possible that an accrual of mutations at sites of recurrent DNA repair synthesis within enhancers could lead to aberrant gene expression, resulting in neurological dysfunction over time.
In summary, we describe methods that enable genome-wide mapping of endogenous sites of frequent DNA single-strand breakage and repair synthesis in post-mitotic neurons. Our findings identify enhancers as hotspots of DNA damage in human post-mitotic neurons, perhaps explaining why DNA SSB repair is important for neurological functionality during development and normal ageing. During the preparation of this manuscript, we became aware of the closely related work of D. Reid et al., which demonstrates recurrent DNA repair sites in embryonic stem cell-derived neurons39.
Methods
iPS cell culture
All iPS cell experiments used the WTC11 line, which was derived from a healthy human male participant and obtained from the Coriell cell repository. All policies of the NIH Intramural Research Program for the registration and use of this iPS cell line were followed. The WTC11 iPS cell line was validated to have a normal male karyotype, as expected, and was confirmed to be mycoplasma free based on the Lonza MycoAlert mycoplasma testing kit. iPS cell culture was performed as previously described5. Tissue culture-treated dishes were coated with human embryonic cell (hES cell)-qualified matrigel (Corning, ref 354277). Matrigel was removed and iPS cells were plated in Essential 8 Medium (E8; Thermo Fisher Scientific, cat. no. A1517001) and 10 μM ROCK inhibitor (RI; Y-27632; Selleckchem, cat. no. S1049). iPS cells were maintained in an incubator at 37 °C with 5% CO2 and fed every 1–2 days as needed. Cells were split using either accutase (Life Technologies, cat. no. A1110501) for enzymatic dissociation into single cells or EDTA (0.5 mM; Life Technologies, cat. no. 15575020) for routine passaging. Medium was supplemented with 10 μM RI to promote survival during passaging. As necessary, iPS cells were frozen in 90% ES cell-qualified fetal bovine serum (FBS) (Sigma Aldrich, cat. no. ES-009-B) and 10% DMSO (Mediatech, cat. no. 25-950-CQC), and then thawed rapidly at 37 °C, followed by removal of FBS/DMSO and plating in E8 + RI medium.
i3Neuron culture
The human iPS cells used in this study were previously engineered5,6 to express mouse neurogenin-2 (NGN2) under a doxycycline-inducible promoter integrated at the AAVS1 safe harbour in the WTC11 background, with or without CAG-dCas9-BFP-KRAB at the Clybl promoter24. For neuronal differentiation, 20–25 million iPS cells were plated on day 0 onto a 15-cm plate in N2 medium (knockout Dulbecco’s modified Eagle’s medium (DMEM)/F12 medium; Life Technologies Corporation, cat. no. 12660012) with N2 supplement (Life Technologies, cat. no. 17502048), 1× GlutaMAX (Thermofisher Scientific, cat. no. 35050061), 1× MEM nonessential amino acids (NEAA) (Thermofisher Scientific, cat. no. 11140050), 10 μM ROCK inhibitor (Y-27632; Selleckchem, cat. no. S1049), and 2 μg/ml doxycycline (Clontech, cat. no. 631311). N2 medium was changed once a day for two more days. On day 3, cells were replated onto freshly prepared dishes coated with poly-l-ornithine (PLO; 0.1 mg/ml; Sigma, cat. no. P3655-10MG) as follows. Cells were washed with PBS, dissociated with accutase for 10 min at 37 °C, washed and plated in i3Neuron culture medium: BrainPhys medium (STEMCELL Technologies, cat. no. 05790) supplemented with 1× B27 Plus Supplement (ThermoFisher Scientific, cat. no. A3582801), 10 ng/ml BDNF (PeproTech, cat. no. 450-02), 10 ng/ml NT-3 (PeproTech, cat. no. 450-03), 1 mg/ml mouse laminin (Sigma, cat. no. L2020-1MG), and 2 μg/ml doxycycline (Clontech, cat. no. 631311). For 10-cm plates used in SAR-seq and CHIP–seq, 12–15 million neurons were plated. For 15-cm plates, 30–45 million neurons were plated. For ibidi slides used in imaging experiments, 0.2 million neurons per well were plated. Unless otherwise noted, i3Neurons were fed on day 6 during a half-medium change and collected on day 7. For i3Neurons cultured beyond 7 days, half-medium changes were conducted three times per week. In some experiments, pre-differentiated i3Neurons were frozen on day 3 in 90% FBS (Sigma Aldrich, cat. no. ES-009-B) and 10% DMSO (Mediatech, cat. no. 25-950-CQC), and then thawed rapidly at 37 °C, followed by removal of FBS/DMSO and plating in i3Neuron culture medium. We did not detect any differences for experiments in which day 3 neurons were thawed or plated immediately after differentiation.
iMuscle culture
To generate skeletal muscle myoblasts (iMuscle) from human iPS cells, we engineered a doxycycline-inducible vector containing a MyoD1 transcription factor transgene immediately followed by a co-inducible short-hairpin RNA targeting OCT4 and a selection marker/fluorescent protein cassette (MyoD-O iPS cells), similar to previous methods40. We used the PiggyBac system to facilitate the delivery and genome integration of the transgene cassette. To increase transposase expression in iPS cells, we subcloned the PiggyBac transposase under a long version of the EEF1A1 (also known as eF1a) promoter, and co-transfected this transposase vector with the MyoD1 donor vector into iPS cells using lipofectamine stem (Thermofisher Scientific, cat. no. STEM00015). Transfection rates were approximately 55%, based on fluorescence of a co-expressed reporter gene, and we were able to obtain 100% MyoD1-O iPS cells after puromycin selection of iPS cells harbouring stable integration of the transcription factor cassette.
To induce myogenic differentiation, MyoD-O iPS cells were dissociated using accutase (37 °C for 10 min) and resuspended in myogenic induction medium (MIM) and plated with 3 × 106 iPS cells per 10-cm dish. The MyoD-O iPS cell differentiation was modified from a recent report41. The MIM contained DMEM/F12 HEPES (Gibco, cat. no. 11-330-032) supplemented to a final concentration of 1 mM sodium pyruvate, 1× MEM nonessential amino acids (NEAA) (Thermofisher Scientific, cat. no. 11140050), 0.1 mM 2-mercaptoethanol (Gibco, cat. no. 21985023), 10 μg/ml insulin (Roche, cat. no. 11376497001), 2 μg/ml doxycycline and 10 μM RI.
After plating the MyoD-O iPS cells in MIM on day 0, fresh medium was supplied daily until 80% confluency or more (day 6). Myoblast morphology was observed beginning at day 2 followed by myotube-like morphology at day 5. On day 6, the glycogen synthase kinase 3 (GSK-3) inhibitor, CHIR 99021, was administered for 48 h to enhance myoblast differentiation and stimulate fusion (3 μM, Tocris, cat. no. 4423) as reported by others42,43. At day 9 after doxycycline, the differentiating myoblasts were collected.
Lentivirus production for CRISPRi guide delivery
Lenti-X Human Embryonic Kidney (HEK) cells were plated onto dishes coated with poly-l-ornithine (0.1 mg/ml PLO) at a density of 10–15 million cells per 10-cm dish or 30–45 million cells per 15-cm dish in warm DMEM, high glucose GlutaMAX Supplement medium (Life Technologies, cat. no. 10566024) with 10% FBS (Sigma, cat. no. TMS-013-B) and then cultured overnight to achieve approximately 90% confluency. The next morning, cells were transfected using Lipofectamine 3000 (Life Technologies, cat. no. L3000150). For each transfection, 2.4 ml room temperature Opti-MEM medium (Fisher Scientific, cat. no. 31985062) and 60 μl Lipo 3K were combined and incubated at room temperature for 5–20 min. Then, in another tube, 2.4 ml room temperature Opti-MEM, 80 μl P3000, 13.3 μg psPAX2, 4.5 μg pMD2G, 1.8 μg pAdVantage, and 19.5 μg of the lenti-vector of interest were combined. The contents of the two tubes were combined and incubated at room temperature for 30 min. This mixture was then added dropwise to a plate of Lenti-X cells and cultured overnight. The next morning, the medium was changed with 36 ml of fresh warm DMEM + Glutamax + 10% FBS medium supplemented with 72 μl viral boost reagent (ALSTEM, cat. no. VB100). Then, 2–3 days later, the medium containing virus was collected and centrifuged to remove cell debris, and the supernatant was concentrated using Lenti-X concentrator, following the manufacturer’s protocol (Takara Bio, cat. no. 631231). The viral pellet was gently dissolved in PBS at either 1:10 or 1:100. Concentrated virus was then aliquoted and stored at −80 °C for future use.
CRISPRi plasmids
The sgRNAs used in this study were cloned into either the pU6-sgRNA EF1Alpha-puro-T2A-BFP vector (gift from J. Weissman; Addgene no. 60955)24,44 or the pMK1334 CROPSeq vector (gift from M. Kampmann; Addgene no. 127965)24. Guides were driven under the mouse U6 promoter. The sgRNA sequences were as follows: non-targeting controls, GTCCACCCTTATCTAGGCTA or GACCAGGATGGGCACCACCC; PARP1, GGGTGCGGCGTGTTCGGTGG; XRCC1, GGTAGAGTATGGGGTCCGAG; POLB sg1, GCGCCGGAGGGAGATCCCCA; POLB sg2, GCCAGCTTGAAGGAGGTACC; PNKP, GCCAGGGCTTGCCCGTCCGA.
Puromycin selection for CRISPRi knockdown i3Neurons
To perform CRISPRi knockdown, 1–4 million iPS cells were transduced with an aliquot of sgRNA-expressing virus immediately after an accutase split before cell attachment. The medium was changed the next day to remove residual virus. Two days after transduction, iPS cells were split into accutase and plated at low density in E8 + RI medium containing 10 μg/ml puromycin. The following morning, cells were washed with PBS and given fresh E8 or E8 + RI medium. Cells were then expanded for 1–2 days before inducing differentiation into neurons. sgRNA knockdown efficiency was tested at the iPS cell stage and confirmed in day 7 i3Neurons via quantitative PCR.
Immunofluorescence and Microscopy
For imaging, i3Neurons were plated onto 96-well plates (0.05 × 106 cells per well; Perkin Elmer, cat. no. 6055302) or on μ-Slide glass bottom IBIDI slides (0.2 × 106 cells per well; IBIDI). Before fixation, if indicated, cells were treated with 0.1 mg/ml MMS for 15 min, 10 μM PARG inhibitors (PARGi) for 20 min, or 50 μM etoposide (ETO) for 1 h. Cells were then washed with PBS and fixed in 4% paraformaldehyde in PBS for 15 min at room temperature. Cells were then washed three times in PBS, permeabilized in 0.5% Triton X-100 for 5 min at room temperature and blocked in 1% BSA/0.2% Triton in PBS for 1 h at room temperature before incubation with primary antibodies. Primary antibodies and dilutions used were as follows: anti-PAR (1:500, Sigma-Aldrich MABE1031), anti-53BP1 (1:1,000, Novus Biologicals, no. NB100-305), anti-phospho-histone H2AX (1:5,000, Millipore, no. JBW301), and anti-tubulin-β3 (TUBB3, 1:5,000, Biolegend no. 801201). Immunofluorescence was detected using fluorochrome-conjugated secondary antibodies as follows: rhodamine Red-X AffiniPure donkey anti-mouse IgG (1:1,000, Jackson Labs no. 715-295-151) for detection of TUBB3; Alexa Fluor 488 goat anti-rabbit (1:1,000, Invitrogen no. A11034) for detection of PAR or 53BP1; Alexa Fluor 555 goat anti-mouse (1:2,000, Invitrogen no. A21422) for detection of yH2AX. EdU was visualized using Click-iT Plus EdU Cell Proliferation Kit for Imaging, Alexa Fluor 488 dye (Life Technologies, cat. no. C10637), according to the manufacturer’s instructions. Finally, DNA was counterstained with DAPI (2 μg/ml, Thermo Fisher Scientific no. 62248). Images were acquired on an inverted Nikon spinning-disk confocal microscope (Nikon Eclipse T1), using a 60× 1.40 NA oil-immersion objective.
Flow cytometry
For cell cycle profiling, cells were incubated with 10 μM 5-ethynyl-2′-deoxyuridine for 30 min at 37 °C and stained using the Click-IT EdU Alexa Fluor 488 or 647 Flow Cytometry Assay Kit (ThermoFisher) according to the manufacturer’s instructions. DNA content was measured using DAPI (0.5 μg ml−1). Data were analysed using FlowJo v.10 software.
Rat primary neuron cell culture
All animal procedures were conducted according to the NIH Guide for the Care and Use of Laboratory Animals, under Animal Study Proposal no. 19-011 approved by the NICHD Animal Care and Use Committee. Ten-to-eleven-week-old pregnant albino rats were delivered to our facility on day 17 of gestation from Envigo. They were housed under a 12-h light–dark cycle for 24 h with access to food and water ad libitum. On the next day, the rats were killed by carbon dioxide inhalation followed by decapitation before embryos were extracted and neurons prepared. Neurons from embryos of the same litter were pooled for each experiment.
Primary rat cortical neurons were prepared at embryonic day 18 (E18) as previously described45. Rats were killed at E18 by decapitation. The brain was collected and meninges were removed, after which cortices were isolated in sterile Hanks’ medium (Hanks’ balanced salt solution (HBSS), 20 mM HEPES, pH 7.5). Cortices were then collected and treated with 0.25% trypsin (Gibco), and 100 μg/ml DNase (Roche) for 15 min at 37 °C. One volume of adhesion medium (DMEM without phenol red, 4.5 g/l glucose, 25 mM HEPES, 10% heat-inactivated horse serum (Gibco), 100 U/ml penicillin and 100 mg/ml streptomycin) was added to stop trypsin enzymatic action. The tissue was then disrupted mechanically by pipetting it through a 10-ml serological pipette. Cells were then strained through a 70-μm nylon filter (Corning) and centrifuged at 700g for 10 min. The cell pellet was resuspended in 5 ml adhesion medium and cells were counted. Between 10 million and 25 million cells were plated on 10-cm culture dishes previously coated with poly-l-lysine (Sigma) and 5 μg/ml laminin (Roche). After 2 h, the neurons were adherent to the plate and the medium was changed to complete neurobasal medium (CNB) (neurobasal medium (Gibco), 1× B27 serum-free (Gibco), 4.5 g/l glucose, and 100 U/ml penicillin–streptomycin (Gibco)) and supplemented with 5 μM aphidicolin (Aph) to eliminate residual dividing cells. Primary neurons were then cultured for 7–8 days before being collected for CHiP–seq or SAR-seq.
SAR-seq
Neurons and iMuscle cells were incubated with 20 μM EdU for 18 h, unless otherwise noted. Cells were collected and fixed as follows. Cells were washed with PBS, incubated with accutase for 5–10 min, collected using a cell scrapper, pelleted at 200g for 5 min and resuspended in cold 0.1% BSA in PBS. Cold methanol was then added dropwise during slow vortexing to 80% final concentration. Samples were kept on ice for 20 min and then stored at −20 °C until processing.
Copper catalyses azide-alkyne click chemistry. For biotin labelling via Click-iT reaction, cells were first washed 1× in PBS, permeabilized with 0.2% Triton-X100/PBS for 10 min on ice, and then washed 1× in PBS. Then the following were added in order: 3 mM copper sulfate (Sigma), 50 μM biotin azide (ThermoFisher, cat. no. B10184), and 1× Click-iT additive (ThermoFisher, cat. no. C10424) for 2 h with shaking at room temperature. Cells were then washed 1× in PBS and lysed in 50 mM Tris pH 8.0 with 1% SDS and proteinase K overnight at 37 °C. DNA was extracted using UltraPure phenol:chloroform:isoamyl alcohol (25:24:1, v/v) (Invitrogen) according to the manufacturer’s instructions, followed by 2.5:1 volume ethanol and 1:10 volume sodium acetate precipitation. DNA pellets were resuspended in TE buffer and sheared to 150–200-bp fragments using a Covaris S220 sonicator at 10% duty cycle, 175 peak incident power, 200 cycles per burst, for 240 s. DNA was again precipitated in 2.5:1 volume ethanol and 1:10 volume sodium acetate and resuspended in TE buffer. Biotin–EdU fragments were pulled down using MyOne Streptavidin C1 Beads (ThermoFisher, cat. no. 650-01). Before pulldown, 35 μl of Dynabeads was washed twice with 1 ml 1× wash and binding buffer (1× W&B; 10 mM Tris-HCl pH 8.0, 1 mM EDTA, 1 M NaCl, 0.1% Tween20) on a DynaMag-2 magnetic separator (Invitrogen, cat. no. 12321D) and resuspended in 2× W&B (10 mM Tris-HCl pH8.0, 2 mM EDTA, 2 M NaCl, 0.2% Tween20). An equal volume of Dynabeads in 2× W&B was added to DNA in TE buffer and incubated at 24 °C with shaking in a ThermoMixer C at 800 rpm for 30 min. Dynabeads bound to biotin–EdU fragments were washed three times in 1 ml of 1× W&B, twice in 1 ml EB, and once in 1 ml 1× T4 DNA ligase buffer (NEB). Dynabeads were resuspended in 50 μl end-repair reaction mix (1X T4 DNA ligase buffer, 0.4 mM dNTPs, 2.7 U T4 DNA polymerase (NEB), 9 U T4 Polynucleotide Kinase (NEB), and 1 U Klenow fragment (NEB)) and incubated at 24 °C with shaking at 800 rpm for 30 min. Dynabeads were washed once in 1 ml 1× W&B, twice in 1 ml EB, and once in 1 ml NEBuffer 2 (NEB) and resuspended in 50 μl A-tailing reaction mix (1× NEB dA-tailing buffer and 20 U Klenow fragment exo- (NEB)), followed by incubation at 37 °C with shaking at 800 rpm for 30 min. Dynabeads were then washed again once in 1 ml NEBuffer 2 and resuspended in 115 μl ligation reaction mix (1× quick ligase buffer (NEB), 6,000 U quick ligase (NEB), 5 nM annealed TruSeq truncated adaptor) and incubated at 25 °C with shaking at 600 rpm for 20 min. The ligation reaction was stopped by adding 50 mM EDTA, and Dynabeads were washed three times in 1 mL 1× W&B, three times in 1 ml EB, and finally resuspended in 8 μl EB + 10 μl 2× Kapa HiFI HotStart Ready Mix (Kapa Biosciences). Primers (10 mM) 5′-CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGG AGTTCAGACGTGTGCTCTTCCGATC*T-3′ and 5′-AATGATACGGCGA CCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATC*T-3′ (asterisk indicates a phosphothiorate bond and a NNNNNN TruSeq index sequence) were added with 37 μl PCR reaction mix (20 μl 2× Kapa HiFI HotStart Ready Mix, 17 μl H2O) for a final volume of 60 μl. DNA was amplified using PCR programme: 98 °C, 45 s; 15 cycles of [98 °C, 15 s; 63 °C, 30 s; 72 °C, 30 s]; 72 °C, 5 min. PCR products were separated from DynaBeads and cleaned using 1.8× volume AMPure Beads XP. We isolated 150–200-bp bands on 2% agarose gel and purified them using QIA-quick Gel Extraction Kit (Qiagen). Before sequencing using Illumina NextSeq 550 (75 bp single read), library concentrations were calculated by KAPA Library Quantification Kit for Illumina Platforms (Kapa Biosystems).
To specifically sequence only EdU-incorporated strands of DNA (Strand-Specific SAR-seq), SAR-seq was followed exactly as above with additional steps before PCR amplification. After washing three times in 1 ml 1× W&B and three times in 1 ml EB post-ligation, Dynabeads were washed in 50 μl 1× SSC buffer and resuspended in 20 μl 0.15 M NaOH for 10 min at room temperature to denature DNA strands. Beads were placed back on the DynaMag-2 magnetic separator and washed once with 20 μl 0.1 M NaOH, once with 1 ml 1× W&B, and twice with 1 ml EB, and then resuspended in 8 μl EB + 10 μl 2× Kapa HiFI HotStart Ready Mix. Primers and PCR reaction mix were added as above.
In the indicated experiments, i3Neurons were treated with the following compounds: aphidicolin (Aph; 5 μM), etoposide (50 μM), olaparib (10 μM), velaparib (10 μM) or tazaparib (1 μM) were added along with EdU at 18 h before collection. The DNA polymerase-α inhibitor (POLαi) adarotene (1 μM) or Aph (50 μM) was added with EdU 14 h or 24 h before collection, respectively. Hydroxyurea (HU; 10 mM) powder was dissolved fresh into water to make a 1 M stock each time it was used. HU was added on day 3 of differentiation and again on day 6 at 18 h before collection along with EdU for a total of 4 days of treatment. For dideoxynucleoside (ddN) chain termination, 5 μM of each of ddA, ddT, ddG, and ddC chain-terminating nucleosides (20 μM total) were added to i3Neuron culture for 18 h before cell collecting for END-seq and S1 END-seq experiments.
END-seq and S1 END-seq
To dissociate i3Neurons for use in END-seq, we performed a modified papain dissociation protocol46. Papain (Worthington Biochemical Cat: LK003178) was dissolved into TrypLE Express Enzyme (1×) with no phenol red and warmed at 37 °C for 10 min. Then, day 7 i3Neurons on a 15-cm plate were washed with PBS and treated with 5 ml papain/TrypLE for 1 min at 37 °C. Papain was removed with gentle pipetting and 5 ml trituration solution was added (30 ml i3Neuron culture medium,10 μM ROCK inhibitor, and 1 vial freshly dissolved DNase). Cells were collected and gently pipetted 3–10 times in a conical tube using a wide-bore 10-ml pipette, being careful not to over-digest the sample. Cells were then washed with PBS, pelleted and resuspended in PBS containing 0.1% BSA and 0.5 mM EDTA, and kept on ice. Cells were processed for END-seq as previously described47. For S1 END-seq, cells were collected and embedded in 1% agarose plugs, lysed and digested with proteinase K (1 h at 50 °C, followed by 7 h at 37 °C), washed with TE buffer, and then treated with RNase A for 1 h at 37 °C. Plugs were then washed in EB and equilibrated in S1 nuclease buffer (40 mM sodium acetate pH 4.5, 300 mM NaCl, 2 mM ZnSO4) for 30 min. We added 1.8 U S1 nuclease to 100 μl S1 nuclease buffer per plug and incubated them on ice for 15 min to allow the enzyme to diffuse into the plug. The reaction mix was then placed at 37 °C for 20 min before addition of EDTA (10 mM final concentration) to terminate the reaction. Finally, plugs were processed through the standard END-seq protocol.
AsiSI induction in pre-B cells
Abelson-transformed mouse pre-B cells48 were retrovirally transduced with tetracycline-inducible ER-AsiSI49. Cells were arrested in G1 with 3 μM imatinib for 24 h, followed by addition of 3 μg/ml doxycycline for 24 h, and then further addition of 1 μM 4OHT for 18 h to induce AsiSI nuclear localization, as previously described19.
Construction of the Dox-Cas9-D10A nickase
Dox-inducible Cas9-D10A was constructed using isothermal assembly50. In brief, a plasmid encoding Dox-inducible Cas9 nuclease was obtained from Addgene. pCW-Cas9-Blast was a gift from M. Babu (Addgene plasmid no. 83481; http://n2t.net/addgene:83481; RRID: Addgene_83481). This plasmid was digested with NheI/BamHI and assembled with 2 PCR fragments (Nickase-P1, Nickase-P2) and transformed into competent cells. Gel-extracted PCR fragments of Nickase-P1 and Nickase-P2 were generated using the Q5 HotStart 2× mastermix with the primers as follows using Addgene-83481 as template: Nickase-P1 forward primer, GTCAGATCGCCTGGAGAATTG; Nickase-P1 reverse primer, tgC CAGGCCGATG CTGTACTTCT; Nickase-P2 forward primer, AGAAGTACAG CATCGGCCTG Gca ATCGGCACCAACTCTGTGGG; Nickase-P2 reverse primer, TGCCTTGGAAAAGGCGCAAC.
MCF10A Cas9D10A-inducible cell line
To produce the MCF10A Cas9-D10A inducible cell line, we infected MCF10A cells (sourced from ATCC, not aunthenticated, and not mycoplasma tested) with lentivirus containing Dox-Cas9-D10A and cells were selected with 10 μg/ml blasticidin. Cas9-D10A expression was induced using 3 μg/ml doxycycline and confirmed by western blotting. Three guide RNAs (sequences: 5′-TGGGGCGTTTATCCGATGTC-3′; 5′-GCACTAGCCGGCCCGGACGT-3′; 5′- CCAGCCTGGTAGCGCCCCCA-3′) were cloned into the Lenti-Guide-NLS–GFP vector51 and the MCF10A Cas9-D10A inducible cell line containing the three guide RNAs was selected using 2 μg/ml puromycin. To identify the nicks by S1 END-seq, cells were arrested in G1 for 48 h with 5 μM Palbociclib with doxycycline added during the last 24 h to induce Cas9D10A, followed by cell collection and S1 END-seq processing.
ChIP–seq and western blotting
Fifteen million i3Neurons or rat neurons were fixed in 1% formaldehyde at 37 °C for 10 min. The fixation reaction was quenched with glycine at a final concentration of 125 mM. Cells were spun down and washed twice with chilled PBS, and pellets were then snap frozen on dry ice and finally stored at −80 °C until sonication. Sonication, immunoprecipitation, and library preparation steps were done as previously reported17. All antibodies were pre-conjugated to 40 μl magnetic protein A beads before immunoprecipitation: H3K4me1 (5 μg, Abcam no. 8895); MLL4 (antibody courtesy of K. Ge); H3K27ac (5 μg, Abcam no. 4729); H3K27me3 (5 μg, Millipore no. CS200603); H3K4me3 (6 μl, Abcam no. 8580); H3K9me3 (10 μg, Active Motif, cat. no. 39765); CTCF (6 μl, Millipore, cat. no. 07-729); RNA polymerase II (8 μg, Abcam no. 26721); H3K36me3 (5 μg, Abcam cat. no. ab9050); anti-PAR (5 μg, Millipore-Sigma MABE1016); XRCC1 (2.6 μg, Novus, cat. no. NBP1-87154). For western blotting, cells were collected and lysed in a buffer containing 50 mM Tris-HCl (pH 7.5), 200 mM NaCl, 5% Tween-20, 0.5% NP-40, 2 mM PMSF, 2.5 mM β-glycerophosphate (all from Sigma-Aldrich) and protease inhibitor cocktail tablet (complete Mini, Roche Diagnostics). Equal amounts of protein were loaded into precast mini-gels (Invitrogen) and resolved by SDS–PAGE. Proteins were blotted onto a nitrocellulose membrane, blocked with Intercept (TBS) blocking buffer (LI-COR Biosciences) and incubated with the corresponding primary and secondary antibodies: anti-DNA polymerase β (1:1,000, Millipore no. ABE1408), anti-tubulin (1:10,000, Sigma-Aldrich no. T5168), IRDye 800 CW goat anti-rabbit (1:15,000, Li-Cor no. 926-32211), and IRDye 680 RD goat anti-mouse (1:15,000, Li-Cor no. 926-68070).
Selective chemical labelling and capture of 5hmC and 5fC
5hmC-Seal was performed as previously described52 with modifications. In brief, 80 μg genomic DNA was resuspended in TE buffer and sonicated to 200 bp fragments using a Covaris S220 sonicator. Fragmented DNA was precipitated using ethanol and sodium acetate and resuspended in TE buffer. The selective 5hmC chemical labelling was performed in 100 μl glucosylation buffer (50 mM HEPES buffer pH 8.0, 25 mM MgCl2) containing above fragmented DNA, β-GT (NEB, cat. no. M0357), and UDP-azide-glucose (Active Motif, cat. no. 55020), and incubated at 37 °C for 1.5 h. After the reaction, DNA was cleaned up with QIAquick Nucleotide Removal Kit. The labelled DNA was eluted with ddH2O, after which 1 mM DBCO-PEG4-Biotin (Click Chemistry Tools) was added and incubated at 37 °C for 2 h. Then, the biotin-labelled DNA was pulled down using C1 Streptavidin beads (ThermoFisher, cat. no. 650-01) for 30 min at room temperature. The captured DNA fragments were processed for library construction as described for SAR-seq (above).
For 5fC-Seal, we modified the previously described protocol10, which reduces 5fC to 5hmC using NaBH453. We labelled the newly generated 5hmC (derived from 5fC reduction with NaBH4) with an azide-modified glucose as described above for 5hmC-Seal. In brief, 80 μg of fragmented i3Neuron genomic DNA was incubated in 100 μl glucosylation buffer (50 mM HEPES buffer pH 8.0, 25 mM MgCl2) containing unmodified UDP-glucose (NEB, Cat No. M0357), and β-GT for 1.5 h at 37 °C. The labelled DNA was cleaned up with QIAquick Nucleotide Removal Kit. Then, an equal volume of freshly prepared NaBH4 (Aldrich, cat. no. 213462) solution was added to the glucose-blocked DNA solution. The reaction mixture was vortexed and incubated in a Thermomixer for 15 min at room temperature. The chemical labelling and capture were performed as described above for 5hmC-Seal.
In situ Hi-C from hiPS cell-derived i3Neurons
Two in situ Hi-C libraries were generated from 10 million cultured hiPS cell-derived i3Neurons as previously described54. In brief, in situ Hi-C consists of seven steps: (1) crosslinking cells with formaldehyde, (2) DNA digestion using MboI, (3) filling in and marking ends with biotin, (4) proximity ligation, (5) DNA shearing, (6) pulling down the biotinylated ligation junctions with streptavidin beads, and (7) paired-end sequencing. As quality control (QC), we confirmed efficient restriction, ligation and DNA shearing with an agarose DNA gel and for appropriate size selection in using Agilent 4200 TapeStation system after steps (5) and (6). For the final QC, we performed 100 paired-end sequencing on the Illumina Nextseq to assess the quality of the libraries based on the percentages of intra-chromosomal reads and long range (>20 kb) reads, and library complexity. The HiC libraries were sequenced on 150 paired-end sequencing using an Illumina Novaseq 6000.
RNA extraction, RNA-seq and quantitative real-time PCR
To extract RNA, cells were plated on six-well dishes and washed with PBS, and then 500 μl tri-reagent (Zymo Research, cat. no. R2050-1-200) was added directly to the cells. The lysed cells were collected using a cell scraper. To isolate RNA, we used a Direct-zol RNA miniprep kit (Zymo Research, cat. no. R2052), according to the manufacturer’s instructions including the optional DNase step. For quantitative PCR, total RNA was reverse-transcribed with iSCRIPT Advanced cDNA Synthesis Kit (Bio-Rad, cat. no. 1725037) according to the manufacturer’s instructions. The resulting cDNA was diluted tenfold and used for quantitative real-time PCR (qRT–PCR) analyses with specific primer and probe sets (Bio-Rad, ACTB qHsaCEP0036280; PARP1 qHsaCEP0052423; XRCC1 qHsaCIP0033686; POLB qHsaCEP0057881; FEN1 qHsaCEP0039485; PNKP qHsaCEP0057803 in a final volume of 20 μl, which contained 10 μl SsoAdvanced Universal Probes Supermix (Biorad, cat. no. 1725280) and 3 μl cDNA. qRT–PCR was performed in triplicate wells per sample on a CFX96 Real-Time System (Biorad). For RNA sequencing, six biological replicates were sequenced. Total RNA was enriched for polyA and sequenced 2 × 75 bp on a HiSeq machine.
ATAC–seq
ATAC–seq was performed as previously described55. i3Neurons were grown on 96-well plates (0.05 × 106 cells per well). Cells were washed gently with PBS. Then, 100 μl accutase per well was added and removed, and the plate was incubated at 37 °C for 5 min. Fifty microlitres of cold lysis buffer (10 mM Tris-HCl, pH 7.4, 10 mM NaCl, 3 mM MgCl2, 0.1% IGEPAL CA-630) was added directly to the well for 10 min. Cells were then pipetted 10–20 times to break clumps and centrifuged at 500g for 10 min at 4 °C. The nucleus pellet was resuspended in the Nextera transposition reaction mix (25 μl 2× TD buffer, 2.5 μl Nextera Tn5 transposase (Illumina, cat. no. FC-121-1030), and 22.5 μl nuclease-free H2O) on ice, then incubated for 30 min at 37 °C. The tagmented DNA was purified using the Qiagen MinElute kit and eluted with 10 μl EB. Following purification, library fragments were amplified using the Nextera index kit (Illumina, cat. no. FC-121-1011) under the following cycling conditions: 72 °C for 5 min, 98 °C for 30 s, followed by thermocycling at 98 °C for 10 s, 63 °C for 30 s, and 72 °C for 1 min for five cycles. To prevent saturation due to over-amplification, a 5-μl aliquot was then removed and subjected to qPCR for 20 cycles for calculation of the optimal number of cycles needed for the 45-μl reaction that remained. The number of additional cycles required was determined by calculating the cycle number that corresponded to a quarter of the maximum fluorescence intensity. Adding seven cycles to this estimate was found to yield optimal libraries. PCR reactions were subsequently cleaned with Agencourt AMPure XP beads (Beckman Coulter) and run on a 2% agarose gel, and a smear of 200–800 bp was cut and gel-purified using a QIAquick Gel Extraction Kit (QIAGEN). Library concentration was determined using a KAPA Library Quantification Kit for Illumina Platforms (Kapa Biosystems). Sequencing was performed on the Illumina Nextseq500 (75-bp paired-end reads).
Genome alignment
SAR-seq, END-seq, ChIP–seq and Seal reads were aligned to the reference genome (hg19 for human i3Neuron and iMuscle, mm10 for mouse pre B cells or rn6 for rat primary neurons) using bowtie (v.1.1.2)56 with parameters -n 3 -l 50 -k 1 for END-seq and -n 2 -l 50 -m 1 for the rest. ATAC–seq reads were aligned by bowtie (v.2.4.1)57. RNA-seq reads were aligned by STAR (v.2.7.6a)58. Functions ‘view’ and ‘sort’ of samtools (v.1.11)59 were used to convert and sort the aligned .sam files to sorted .bam files. .bam files were further converted to .bed files using the bedtools (v.2.29.2) bamToBed command60. Mitochondrial reads were removed in SAR-seq for intensity comparisons.
Peak calling
We used MACS (v.1.4.3)61 to call SAR-seq, XRCC1 ChIP–seq and ATAC–seq peaks. SAR-seq XRCC1, and CTCF ChIP–seq peaks with more than 10-fold enrichment over background were kept. Peaks of hisone modification determined by ChIP–seq peaks were called by SICER62 v.2-1.0.2 with default parameters. Peaks within blacklisted regions (https://sites.google.com/site/anshulkundaje/projects/blacklists) were filtered63. Overlapping SAR-seq peaks from three non-treated replicates shown in Extended Data Fig. 2b were used for most of the analyses. As peaks of ddN S1 END-seq are always clustered, subpeaks were called by the PeakSplitter tool of PeakAnalyzer with parameters -c 15 -v 0.664. Peak summits on both strands were identified, and the distance between the peak summit on the negative strand and its closed peak summit on the positive strand was calculated and represented in Extended Data Fig. 9f.
Quantification for sequencing data
For SAR-seq, Seal, and ChIP–seq, reads per kilobase of transcript per million mapped reads (RPKM) was calculated. For RNA-seq, fragments per kilobase of transcript per million mapped reads (FPKM) was calculated using cufflinks41 (v.2.2.1) based on the annotation from GENCODE v.3365. Mean values of replicates were used for analyses.
Enhancer enrichment and super-enhancer identification
Bedtools shuffle command with parameter (-chrom) was used to generate 1,000 random sets for SAR-seq peaks and ATAC–seq peaks, respectively, to estimate enrichments at genomic features. One-kilobase regions upstream of transcription start sites from GENCODE v33 annotation were defined as promoters in analyses. Super-enhancers were separated from conventional enhancers by identifying an inflection point of H3K27ac signal versus enhancer rank66.
Gene Ontology analysis
We used the DAVID web-tool67 to find the GO terms that were enriched for genes containing the top 2,000 SAR-seq peaks. The most significant Gene Ontology Biological Process terms and their associated false discovery rate (FDR) values are reported.
Motif analysis
MEME-ChIP68 of MEME suite was used to identify the common sequence motif of the nucleotide sequences from ± 500 bp around the summits for the top 5,000 SAR-seq peaks. The composite DNA sequence motif shown in Fig. 2c was plotted using ggseqlogo in R.
Kinetics model for EdU labelling
The rate of EdU labelling was estimated under the assumptions that the fraction of a synthesis event at a given site is fixed with different EdU incorporation times and that 18 h is enough time for EdU to incorporate into all synthesis events at a given site. The fraction of EdU-labelled synthesis event at h hours at a given site was denoted as [EdU]h = 1 – e−kh. k represents the rate of EdU labelling at a given site and [EdU]h was estimated by SAR-seq intensity at h hours divided by SAR-seq intensity at 18 h. The intensity values (RPKM) of SAR-seq at different time points (1, 2, 4, 8 and 18 h of EdU) was fitted to the function [EdU]h = 1 – e−kh using the nls package in R. The k value shown in Extended Data Fig. 2f was derived using the top 2,000 SAR-seq peaks.
Hi-C analysis
Using Juicer software69, .hic files were generated, and normalized contact matrices and observed over expected normalized contact matrices were obtained with a dump command. Loop or domain calls were also done using Juicer software69, and interaction matrices were visualized using Juicebox software. After two replicates had been processed, the final map was generated by merging these two replicates. The final bin resolution of the Hi-C map was 1 kb. From 2.1 billion raw reads generated together with QC and data-generation runs, we obtained 1.5 billion final total contacts whose read quality (Q) was over 30.
For compartment analysis, PC1 eigen vector values were extracted in 50k resolution by Juicer software, then the sign of the eigen vector in each chromosome was adjusted according to distribution patterns of the H3K27ac, H3K4me1 and ATAC–seq peaks in the whole chromosome. Among H3K4me1 peaks that did not overlap with transcription start sites within 1 kb, peaks that overlapped with SAR-seq peaks were extracted for the analysis (SAR-seq+H3K4me1+). Ten thousand random sequences were generated using the bedtools random program with 2,600 nt length, which is the average length of H3K4me1 peaks. While 40% of random sequences are located in A compartment, 63% of SAR-seq+H3K4me1+ peaks were located in A compartment.
Visualization
BedGraph files were generated using bedtools genomecov, normalized by reads per million (RPM) and then converted into .bigWig files using bedGraphToBigWig from UCSC pre-compiled utilities for visualization at the UCSC genome browser70,71.
Data matrices for heat maps were calculated using computeMatrix and plotted with plotHeatmap of deepTools suite72. Venn diagrams were plotted using VennDiagram package in R. Confocal images were processed using FIJI. Schematics were created using BioRender, and figures compiled in Adobe Illustrator.
Quantification of confocal Images
Confocal images were quantified using Nikon software. Images were first background subtracted. Then nuclei or EdU-positive cells were identified and counted using bright spot detection. The corresponding PAR signal intensity was measured per cell and reported as a mean. Data compilation and statistical analyses were performed using PRISM software.
Statistical analysis
Statistical analysis was performed using R version 3.6.2 (http://www.r-project.org). The statistical tests are reported in the figure legends and main text.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The SAR-seq, S1 END-seq, Hi-C, RNA-seq, Seal and ChIP–seq data have been deposited in the Gene Expression Omnibus (GEO) database under accession number GSE167259. iPS cell enhancers were identified using H3K4me1 ChIP–seq from an ENCODE iPS cell line (GSM2527632). Experimentally validated enhancers were from VISTA genome browser for enhancers: https://enhancer.lbl.gov/.
References
Caldecott, K. W. Single-strand break repair and genetic disease. Nat. Rev. Genet. 9, 619–631 (2008).
McKinnon, P. J. Genome integrity and disease prevention in the nervous system. Genes Dev. 31, 1180–1194 (2017).
Tubbs, A. & Nussenzweig, A. Endogenous DNA damage as a source of genomic instability in cancer. Cell 168, 644–656 (2017).
Miller, M. R. & Chinault, D. N. The roles of DNA polymerases alpha, beta, and gamma in DNA repair synthesis induced in hamster and human cells by different DNA damaging agents. J. Biol. Chem. 257, 10204–10209 (1982).
Fernandopulle, M. S. et al. Transcription factor-mediated differentiation of human iPSCs into neurons. Curr. Protoc. Cell Biol. 79, e51 (2018).
Wang, C. et al. Scalable production of iPSC-derived human neurons to identify tau-lowering compounds by high-content screening. Stem Cell Reports 9, 1221–1233 (2017).
Macheret, M. & Halazonetis, T. D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 555, 112–116 (2018).
Tubbs, A. et al. Dual roles of poly(dA:dT) tracts in replication initiation and fork collapse. Cell 174, 1127–1142.e19 (2018).
van der Raadt, J., van Gestel, S. H. C., Nadif Kasri, N. & Albers, C. A. ONECUT transcription factors induce neuronal characteristics and remodel chromatin accessibility. Nucleic Acids Res. 47, 5587–5602 (2019).
Song, M. et al. Mapping cis-regulatory chromatin contacts in neural cells links neuropsychiatric disorder risk variants to target genes. Nat. Genet. 51, 1252–1262 (2019).
Visel, A., Minovitsky, S., Dubchak, I. & Pennacchio, L. A. VISTA Enhancer Browser—a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92 (2007).
Gupte, R., Liu, Z. & Kraus, W. L. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes Dev. 31, 101–126 (2017).
Hanzlikova, H. & Caldecott, K. W. Perspectives on PARPs in S phase. Trends Genet. 35, 412–422 (2019).
Gibson, B. A., Conrad, L. B., Huang, D. & Kraus, W. L. Generation and characterization of recombinant antibody-like ADP-ribose binding proteins. Biochemistry 56, 6305–6316 (2017).
Madabhushi, R. et al. Activity-induced DNA breaks govern the expression of neuronal early-response genes. Cell 161, 1592–1605 (2015).
Suberbielle, E. et al. Physiologic brain activity causes DNA double-strand breaks in neurons, with exacerbation by amyloid-β. Nat. Neurosci. 16, 613–621 (2013).
Canela, A. et al. Topoisomerase II-induced chromosome breakage and translocation is determined by chromosome architecture and transcriptional activity. Mol. Cell 75, 252–266.e8 (2019).
Gómez-Herreros, F. et al. TDP2 suppresses chromosomal translocations induced by DNA topoisomerase II during gene transcription. Nat. Commun. 8, 233 (2017).
Canela, A. et al. DNA breaks and end resection measured genome-wide by end sequencing. Mol. Cell 63, 898–911 (2016).
Caldecott, K. W. XRCC1 protein; form and function. DNA Repair 81, 102664 (2019).
Hanzlikova, H., Gittens, W., Krejcikova, K., Zeng, Z. & Caldecott, K. W. Overlapping roles for PARP1 and PARP2 in the recruitment of endogenous XRCC1 and PNKP into oxidized chromatin. Nucleic Acids Res. 45, 2546–2557 (2017).
Caldecott, K. W. DNA single-strand break repair. Exp. Cell Res. 329, 2–8 (2014).
Caldecott, K. W. Mammalian DNA base excision repair: dancing in the moonlight. DNA Repair 93, 102921 (2020).
Tian, R. et al. CRISPR interference-based platform for multimodal genetic screens in human iPSC-derived neurons. Neuron 104, 239–255.e12 (2019).
Beard, W. A., Horton, J. K., Prasad, R. & Wilson, S. H. Eukaryotic base excision repair: new approaches shine light on mechanism. Annu. Rev. Biochem. 88, 137–162 (2019).
DiGiuseppe, J. A., Hunting, D. J. & Dresler, S. L. Aphidicolin-sensitive DNA repair synthesis in human fibroblasts damaged with bleomycin is distinct from UV-induced repair. Carcinogenesis 11, 1021–1026 (1990).
Poetsch, A. R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 18, 207–219 (2020).
Bansal, K., Yoshida, H., Benoist, C. & Mathis, D. The transcriptional regulator Aire binds to and activates super-enhancers. Nat. Immunol. 18, 263–273 (2017).
Puc, J. et al. Ligand-dependent enhancer activation regulated by topoisomerase-I activity. Cell 160, 367–380 (2015).
Kalasova, I. et al. Pathological mutations in PNKP trigger defects in DNA single-strand break repair but not DNA double-strand break repair. Nucleic Acids Res. 48, 6672–6684 (2020).
Whitehouse, C. J. et al. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 104, 107–117 (2001).
Lio, C. J. et al. TET methylcytosine oxidases: new insights from a decade of research. J. Biosci. 45, 21 (2020).
Kriaucionis, S. & Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930 (2009).
Steinacher, R. et al. SUMOylation coordinates BERosome assembly in active DNA demethylation during cell differentiation. EMBO J. 38, e99242 (2019).
Song, C. X. et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153, 678–691 (2013).
Szulwach, K. E. et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14, 1607–1616 (2011).
Hoch, N. C. et al. XRCC1 mutation is associated with PARP1 hyperactivation and cerebellar ataxia. Nature 541, 87–91 (2017).
Lodato, M. A. et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018).
Ried, D. A. et al. Incorporation of a nucleoside analog maps genome repair sites in post-mitotic human neurons. Science (in the press) (2021).
Watanabe, S. et al. MyoD gene suppression by Oct4 is required for reprogramming in myoblasts to produce induced pluripotent stem cells. Stem Cells 29, 505–516 (2011).
Akiyama, T. et al. Efficient differentiation of human pluripotent stem cells into skeletal muscle cells by combining RNA-based MYOD1-expression and POU5F1-silencing. Sci. Rep. 8, 1189 (2018).
Selvaraj, S. et al. Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. eLife 8, e47970 (2019).
Pawlowski, M. et al. Inducible and deterministic forward programming of human pluripotent stem cells into neurons, skeletal myocytes, and oligodendrocytes. Stem Cell Reports 8, 803–812 (2017).
Gilbert, L. A. et al. Genome-scale CRISPR-mediated control of gene repression and activation. Cell 159, 647–661 (2014).
Farías, G. G., Britt, D. J. & Bonifacino, J. S. Imaging the polarized sorting of proteins from the Golgi complex in live neurons. Methods Mol. Biol. 1496, 13–30 (2016).
Kirwan, P., Jura, M. & Merkle, F. T. Generation and characterization of functional human hypothalamic neurons. Curr. Protoc. Neurosci. 81, 3.33.1–3.33.24 (2017).
Wong, N., John, S., Nussenzweig, A. & Canela, A. END-seq: an unbiased, high-resolution, and genome-wide approach to map DNA double-strand breaks and resection in human cells. Methods Mol. Biol. 2153, 9–31 (2021).
Bredemeyer, A. L. et al. DNA double-strand breaks activate a multi-functional genetic program in developing lymphocytes. Nature 456, 819–823 (2008).
Santos, M. A. et al. DNA-damage-induced differentiation of leukaemic cells as an anti-cancer barrier. Nature 514, 107–111 (2014).
Gibson, D. G. et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat. Methods 6, 343–345 (2009).
Noordermeer, S. M. et al. The shieldin complex mediates 53BP1-dependent DNA repair. Nature 560, 117–121 (2018).
Cui, X. L. et al. A human tissue map of 5-hydroxymethylcytosines exhibits tissue specificity through gene and enhancer modulation. Nat. Commun. 11, 6161 (2020).
Dai, Q. & He, C. Syntheses of 5-formyl- and 5-carboxyl-dC containing DNA oligos as potential oxidation products of 5-hydroxymethylcytosine in DNA. Org. Lett. 13, 3446–3449 (2011).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014).
Buenrostro, J. D., Giresi, P. G., Zaba, L. C., Chang, H. Y. & Greenleaf, W. J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218 (2013).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Dobin, A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Zhang, Y. et al. Model-based analysis of ChIP–Seq (MACS). Genome Biol. 9, R137 (2008).
Zang, C. et al. A clustering approach for identification of enriched domains from histone modification ChIP-seq data. Bioinformatics 25, 1952–1958 (2009).
Amemiya, H. M., Kundaje, A. & Boyle, A. P. The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep. 9, 9354 (2019).
Salmon-Divon, M., Dvinge, H., Tammoja, K. & Bertone, P. PeakAnalyzer: genome-wide annotation of chromatin binding and modification loci. BMC Bioinformatics 11, 415 (2010).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 22, 1760–1774 (2012).
Whyte, W. A. et al. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319 (2013).
Huang, W., Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols 4, 44–57 (2009).
Machanick, P. & Bailey, T. L. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics 27, 1696–1697 (2011).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Kent, W. J., Zweig, A. S., Barber, G., Hinrichs, A. S. & Karolchik, D. BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207 (2010).
Kent, W. J. et al. The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002).
Ramírez, F. et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016).
Acknowledgements
We thank A. Rao, S. Wilson, S. Ruiz, N. Lopez-Bigas, D. Wilson III, Y. Sun, Y. Pommier, M. Adamowicz, P. Meltzer, and K. Ge for discussions and reagents; R. Chari for constructing the CAS9D10A inducible nickase; and the CCR genomics core for help with sequencing. K.W.C. is supported by Programme Grants from the UK Medical Research Council (MR/P010121/1), Cancer Research-UK (C6563/A7322), is an ERC Advanced Investigator (SIDSCA 694996) and received a Royal Society Wolfson Research Merit Award. The M.E.W. laboratory is supported by the NINDS Intramural Research Program, the Chan Zuckerberg Initiative, and the Packard ALS Center. S.E.H. received funding from the BrightFocus Foundation. The A.N. laboratory is supported by the Intramural Research Program of the NIH, an Ellison Medical Foundation Senior Scholar in Aging Award (AG-SS-2633-11), Department of Defense Awards (W81XWH-16-1-599 and W81XWH-19-1-0652), an Alex’s Lemonade Stand Foundation Award, and an NIH Intramural FLEX Award.
Author information
Authors and Affiliations
Contributions
W.W., S.E.H., W.J.N., J.P., K.W.C., M.E.W. and A.N. conceived, designed and analysed the experiments; W.W. designed bioinformatics pipelines, performed data analysis and designed the figures; S.E.H., E.C. and M.P. prepared i3Neurons for experiments; S.E.H. and E.C. performed gene knockdown, i3Neuron treatments, dissociation and collections in i3Neurons; J.P. and D.W. performed SAR-seq; W.J.N., J.P. and D.W. performed ChIP-seq; W.J.N. and N.V.W. performed S1 END-seq. S.E.H., E.C. and W.J.N. performed immunofluorescence imaging; A.C. developed SAR-seq in the A.N. laboratory; W.J.N. developed S1 END-seq; S.E.H. and M.E.W. selected guides for gene knockdown; K.S. performed ATAC-seq, did cloning for knock-down experiments and qRT–PCR; D.W. performed SEAL experiments; J.C.-M. prepared iMuscle cells; R.D.P. prepared primary rat neurons; S.E.H. treated and collected iMuscle and primary rat neurons; H.-Y.S. and S.C. conducted sequencing experiments in i3Neurons that were informative to the study; R.P. made MCF10A cells with Cas9-nickase; D.Z. and C.C. assisted with experiments; S.P. performed Hi-C, and S.K.J. and R.C. analysed the Hi-C data; H.H., P.J.M. and C.C. provided insights; K.W.C., M.E.W. and A.N. wrote the paper with input from all co-authors; M.E.W. and A.N. supervised the study.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Michael Weinfeld and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Discrete genomic loci in neurons are associated with ongoing DNA synthesis.
a, Schematic of SAR-seq (DNA synthesis associated with repair sequencing) methodology. Neurons grown in culture (1) are incubated with EdU to label sites of DNA repair synthesis (2). The incorporated genomic EdU is then conjugated to biotin via click chemistry (3), sheared by sonication to fragments of 150–200 bp and captured with streptavidin beads (4). Enriched DNA sequences are then PCR-amplified and subjected to next-generation sequencing (5). b, Genome browser screenshot displaying SAR-seq profiles as normalized read density (RPM) for i3Neurons. Three independent biological replicates are shown as well as input. Neurons were labelled with EdU for 18 h and collected on day 7 after induction of neuronal differentiation. All coordinates provided are from the hg19 reference genome for i3Neurons. c, Venn diagram showing the overlap of SAR-seq peaks in i3Neurons for three independent biological replicates. d, Scatter plots showing correlations of SAR-seq intensities (RPKM) between three replicates in i3Neurons. Pearson correlation coefficients and P values are indicated. e, Left, representative images of EdU–biotin staining (green) showing cell proliferation in iPS cells, but not in post-mitotic i3Neurons. i3Neurons were treated with EdU on day 3 or day 6 and fixed on day 7. iPS cells were treated with EdU for 24 h and fixed. Cells were counterstained with DAPI (blue). Different imaging conditions were used for iPS cells and i3Neurons in the representative images. Right, quantification of EdU-positive cells. Each dot represents the percentage of dividing cells in one image (iPS cell: n = 8 images, n = 397/410 cells EdU-positive; i3Neuron day 3: n = 8 images, n = 35/483 cells EdU-positive; i3Neuron day 7: n = 8 images, n = 0/523 cells EdU-positive). Data are mean ± s.e.m. and are representative of three independent experiments.
Extended Data Fig. 2 Genomic characteristics of SAR-seq peaks.
a, Genome browser screenshot showing SAR-seq in i3Neurons treated with hydroxyurea (HU, n = 1) or polymerase-α inhibitor (POLαi, n = 2). NT, non-treated. b, Scatter plots showing SAR-seq intensities (RPKM) for HU- (left) and POLαi- (right) treated compared to non-treated (NT) samples. c, Flow cytometry cell cycle profile of pre-B cells treated or non-treated (NT) with POLαi. Cells were pulsed with EdU for 30 min before being collected for flow cytometry. Cells were counterstained with DAPI (n = 1). For an example of the gating strategy used for flow cytometry in c and in Extended Data Fig. 9b, see Supplementary Fig. 1. d, Histogram of individual SAR-seq peak widths, revealing a mean peak width of 901 bp. e, Genome browser screenshot showing SAR-seq in i3Neurons collected after 1, 2, 4, 8, or 18 h of EdU incubation (n = 2 for each). f, Fraction of EdU labelling in i3Neurons (relative to maximum labelling at 18 h) as a function of time, calculated from the top 2,000 peaks. Red points and error bars represent mean and s.d. of the relative levels of EdU measured from experimental data. Black line represents the theoretical model after fitting, with k being the rate of EdU labelling.
Extended Data Fig. 3 SAR-seq enrichment at neuronal intragenic regions.
a, Genome browser screenshot of chromosome 7 showing lack of localized DNA synthesis in two independent biological replicates of SAR-seq performed in iMuscle cells (n = 2) incubated with EdU for 18 h compared to SAR-seq in i3Neurons as well as input DNA. b, Genome browser screenshot displaying SAR-seq peak at a representative AsiSI restriction enzyme site (tick mark). AsiSI expression was induced for 18 h (+Dox, n = 1) compared with non-treated (−Dox, n = 1) conditions in G0-arrested, Abelson virus-transformed mouse pre-B cells as described19. c, Distribution of SAR-seq peaks with respect to different genomic features compared to genome-wide distribution of the hg19 human reference genome. Promoters are defined as 1 kb upstream of transcription start sites. Distal intergenic represents promoter-excluded intergenic regions. d, Fold enrichment of SAR-seq peaks in intergenic regions, intragenic regions and expressed genes compared to 1,000 sets of randomly shuffled regions of the same sizes and chromosome distribution (one-sided Fisher’s exact test, ****P < 2.2 × 10−16; NS, not significant). e, Left, scatterplot showing correlation of SAR-seq intensity (RPKM) with transcript level of genes containing SAR-seq peaks measured by RNA-seq (FPKM; n = 3) in i3Neurons. Seventy-one per cent of SAR-seq peaks are at expressed genes (FPKM ≥ 0.1; red dashed line, FPKM = 0.1). Right, correlation of SAR-seq intensity with transcript levels of linked genes determined by pcHiC in i3Neurons (red dashed line, FPKM = 0.1). Spearman correlation coefficients and P values are indicated. f, Genome browser screenshot comparing SAR-seq with strand-specific SAR-seq (n = 3), which discriminates which strand is labelled with EdU in i3Neurons. Both strands show labelling in three biological replicates. Strand-specific SAR-seq reads are separated into positive (black) and negative (grey) strands.
Extended Data Fig. 4 Motif discovery of SAR peaks and comparison with ATAC–seq peaks.
a, Motif analysis for sequences within 500 bp of the summit of the top 5,000 SAR-seq peaks in i3Neurons. Top, the best motif discovered by the MEME suite (1,384 out of 5,000 sites have this motif). Middle, TOMTOM motif tool used to compare SAR-seq motif shown above with databases of known motifs. The transcription factor ONECUT1 was identified as the most similar motif and its consensus sequence is shown. P value for motif comparison with ONECUT1 motif is indicated. Bottom, position distribution of the best motif (top) within 500 bp of the SAR-seq peak summit. The best motif is centred on the SAR-seq peak summit. b, Venn diagram illustrating the overlap between H3K4me1 ChIP–seq, ATAC–seq and SAR-seq peaks in i3Neurons. The statistical significance of the overlaps between SAR-seq, H3K4me1 ChIP–seq and ATAC–seq peaks was determined using randomly shuffled data sets (n = 1,000) by one-sided Fisher’s exact test (the P value for overlap between H3K4me1 ChIP–seq and SAR-seq peaks is P < 2.2 × 10−16, and for ATAC–seq/SAR-seq peaks is P < 2.2 × 10−16). Fraction of different overlapping groups are labelled in red for SAR-seq peaks, green for ATAC–seq peaks and blue for H3K4me1 ChIP–seq peaks. c, Heat maps of SAR-seq and ATAC–seq signals within 1 kb of SAR-seq peak summits in i3Neurons, ordered by SAR-seq intensity. d, Scatter plot comparing widths of ATAC–seq peaks and SAR-seq peaks for the top 10,000 overlapping peaks in i3Neurons. Pearson correlation coefficient and P value are indicated. e, Distribution of SAR-seq and ATAC–seq peaks with respect to different genomic features compared to genome-wide distribution on the hg19 human reference genome. Promoters are defined as 1 kb upstream of transcription start sites and distal intergenic represents promoter-excluded intergenic regions. f, Fold enrichment of SAR-seq and ATAC–seq peaks located at enhancers (black) and promoters (grey) compared to 1,000 sets of randomly shuffled regions of the same sizes and chromosome distributions, respectively (one-sided Fisher’s exact test, ****P < 2.2 × 10−16, NS: P = 0.0783, not significant). g, Heat map of RNA Pol II (n = 1) ChIP–seq and SAR-seq in i3Neurons for 1 kb on either side of the transcription start site (TSS) in i3Neurons, ordered by Pol II ChIP–seq intensity.
Extended Data Fig. 5 The correlation between SAR-seq and chromatin features.
a, Heat maps of SAR-seq and ChIP–seq data for enhancer markers (H3K4me1, H3K27ac and MLL4), other chromatin markers at accessible regions (H3K4me3, H3K36me3 and CTCF) and chromatin silencing markers (H3K27me3 and H3K9me3) for 1 kb on either side of the SAR-seq peak summit in i3Neurons, ordered by SAR-seq intensity. b, Scatter plots showing the correlation between SAR-seq intensity and ATAC–seq, H3K4me1, H3K27ac and MLL4 ChIP–seq intensities (RPKM) for 1 kb on either side of the SAR-seq peak summits in i3Neurons. Spearman correlation coefficients and P values are indicated. c, Fold enrichment of SAR-seq peaks at ATAC–seq peaks, ChIP–seq peaks of enhancer-related marks (H3K4me1, H3K27ac and MLL4), additional chromatin marks at accessible regions (H3K4me3, H3K36me3 and CTCF) (all n = 1) and chromatin silencing marks (H3K27me3 and H3K9me3) (both n = 1) in i3Neurons. n = 1,000 randomly shuffled data sets were generated to test the significance using one-sided Fisher’s exact test (P < 2.2 × 10−16 for H3K4me1, H3K27ac, ATAC–seq and MLL4; P = 1.85 × 10−316 for H3K4me3; P = 0.00116 for CTCF; **P < 0.001, ****P < 0.00001, NS, not significant). d, Heat maps of SAR-seq and H3K4me1 ChIP–seq signal for 1 kb on either side of the SAR-seq peak summit in primary rat cortical neurons, ordered by SAR-seq intensity. e, Venn diagram showing the overlap between H3K4me1 and SAR-seq peaks in rat primary neurons. n = 1,000 randomly shuffled data sets were generated to test the significance using one-sided Fisher’s exact test: P < 2.2 × 10−16. f, Scatter plot showing the correlation between SAR-seq and H3K4me1 ChIP–seq intensities (RPKM) for 1 kb on either side of the SAR-seq peak summits in rat primary neurons. Spearman correlation coefficient and P values are indicated.
Extended Data Fig. 6 SAR-seq enrichment at neuronal enhancers.
a, Box plot showing Hi-C contacts (left, n = 4) and H3K27ac (right, n = 1) levels at enhancers with (red) or without (grey) SAR-seq peaks. Contacts were defined as Hi-C interactions between an H3K27ac+ enhancer (with and without SAR) and its closest promoter within the TAD domain. For comparative purposes, observed contacts were normalized to expected contacts. Centre line, median; box limits, 25th and 75th percentiles; whisker indicates 1.5× interquartile range. Statistical significance was determined using the one-sided Wilcoxon rank-sum test. P = 1.64 × 10−225 for HiC contacts and P < 2.2 × 10−16 for H3K27ac intensity (****P < 0.00001). b, Fold enrichment of SAR-seq peaks at in vivo-validated enhancers from the VISTA Enhancer Browser database that overlap with H3K4me1 ChIP–seq peaks (left, P = 1.42 × 10−53) or at promoter-interacting regions determined by pcHiC (right, P = 1.574 × 10−09). n = 1,000 randomly shuffled datasets were generated to test significance using one-sided Fisher’s exact test (**P < 0.001, ****P < 0.00001). c, d, Genome browser screenshots showing SAR-seq, H3K4me1 and H3K27ac ChIP–seq, and pcHiC and Hi-C profiles at representative enhancers (highlighted in orange) interacting with the CDK5RAP3 promoter (c) and the DRD2 promoter (d). Both enhancers have been validated to promote transcription of their respective genes using CRIPSR techniques in i3Neurons10. The CDK5RAP3 enhancer also overlaps with in vivo-validated enhancers from the VISTA Enhancer Browser database. In the Hi-C contact matrix (c, bottom) the intensity of each pixel represents the normalized number of contacts between a pair of loci. The maximum intensity is indicated at the lower left corner. e, Pie chart showing distribution of i3Neuron SAR-seq peaks in iPS cell-specific, i3Neuron-specific and shared iPS cell and i3Neuron enhancers. Approximately 56 million and 49 million single end reads were sequenced for the H3K4me1 ChIP–seq in iPS cell and i3Neurons, respectively, with approximately 100,000 peaks called in both cell types. f, Top biological processes enriched for genes containing the 2,000 most intense SAR-seq peaks determined by GO analysis. The x-axis represents the enrichment value as the logarithm of FDR. G, H3K27ac signal at enhancers in i3Neurons ranked by H3K27ac ChIP–seq intensity. Red dashed line indicates the inflection point of the H3K27ac signal used to identify super-enhancers (cutoff 1,000). Accordingly, 1,385 enhancers were defined as super-enhancers. h, Bar graph showing the fraction of super-enhancers (left) and conventional enhancers (right) that overlap with SAR-seq peaks. The super-enhancers in the i3Neurons were defined by H3K27ac ChIP–seq intensity in g.
Extended Data Fig. 7 PARP and XRCC1 recruited to DNA repair sites.
a, Quantification of PAR levels with (n = 422 cells) and without (n = 541 cells) MMS treatment (red line, mean). Each dot represents one cell. Statistical significance was determined using two sided Mann–Whitney test (****P < 0.0001). Data are representative of three independent experiments. b, Anti-ADP-ribose ChIP–seq signal (n = 1) at an AsiSI restriction enzyme cut site (tick mark) in Abelson virus-transformed mouse pre-B cells. Cells were arrested in G0, and AsiSI double-strand breaks were induced for 18 h before ChIP. ADP-ribose is enriched at cleaved AsiSI sites and is increased by 20 min treatment with PARGi before fixation (AsiSI + PARGi), which is indicative of the presence of PAR. c, Genome browser screenshot illustrating the overlap between SAR-seq (n = 3), PAR (n = 1) and XRCC1 (n = 1) ChIP–seq signals in i3Neurons. Cells for PAR ChIP–seq were incubated with PARGi for 20 min before fixation.
Extended Data Fig. 8 Mapping regions of DNA damage and repair in neurons.
a, Genome browser example of SAR-seq profiles in non-treated (NT) or etoposide (ETO)-treated (18 h, 50 μM) i3Neurons. Data are from two biological replicates. b, Heat maps for SAR-seq in nontreated (NT) or etoposide- (ETO) treated (18 h, 50 μM) i3Neurons at −2 kb to +5 kb from the transcription start sites (TSS), ordered by ETO SAR-seq intensity. c, Immunofluorescence staining of the DSB markers γH2AX (red) and 53BP1 (green) in non-treated or ETO-treated (1 h) i3Neurons. Data are representative of three independent experiments. d, Genome browser showing SAR-seq and END-seq profiles in non-treated i3Neurons. END-seq, which detects DSBs specifically19, does not detect any enriched signal (that is, above background) at SAR-seq peaks. END-seq signals are separated into positive (black) and negative (grey) strands. END-seq data are representative of two independent experiments. e, Heat maps of SAR-seq and XRCC1 ChIP–seq (n = 1) for 1 kb on either side of SAR-seq peak summits in cultured rat primary neurons, ordered by SAR-seq intensity. f, g, Scatter plots showing the correlation between SAR-seq and XRCC1 ChIP–seq intensities (RPKM) for 1 kb before and after SAR-seq peak summits in i3Neurons (f) and rat primary neurons (g). Spearman correlation coefficients and P values are indicated.
Extended Data Fig. 9 S1 END-seq mapping of SSBs.
a, Top, genome browser screenshot showing profiles of SAR-seq and S1 END-seq. Agarose plugs were incubated with or without the restriction enzyme Nt.BspQI before S1 treatment (n = 1). Bottom, expanded views of Nt.BspQI sites (tick mark) show detection of S1 END-seq upon Nt.BspQI treatment. S1 END-seq reads are separated into positive (black) and negative (grey) strands. b, Flow cytometry profile of G1-arrested MCF10A cells pulsed with EdU. MCF10A cells were treated with palbociclib (1 μM) for 48 h to arrest cells in G1. Doxycycline was added in the last 24 h to induce nickase expression. Data are representative of three independent experiments. For an example of the gating strategy used for flow cytometry in b and Extended Data Fig. 2c, see Supplementary Fig. 1. c, Genome browser screenshot showing S1 END-seq profiles at three Cas9 nickase targeting sites (tick marks: sgRNAs 1–3) in G1-arrested MCF10A cells treated with doxycycline (+Dox) to induce Cas9 expression (n = 1). S1 END-seq signals are separated into positive (black) and negative (grey) strands. d, Heat maps of SAR-seq and S1 END-seq signals in i3Neurons with ddN or without incubation (NT) for 1 kb on either side of the SAR-seq peak summits, ordered by SAR-seq intensity. e, Venn diagram showing the overlap between S1 END-seq peaks incubated with ddN and SAR-seq peaks in i3Neurons. n = 1,000 randomly shuffled datasets were generated to test the significance using one-sided Fisher’s exact test: P < 2.2 × 10−16. f, Distribution of the size of the gaps between positive- and negative-strand S1 END-seq peak summits in i3Neurons incubated with ddN. The median gap size is 25 bp (red dashed line). The positive-strand peak represents the right end and the negative-strand peak represents the left end of a detected DSB. g, Aggregate plot showing the distribution of CG dinucleotides (black) for 1 kb on either side of SAR-seq peak summits overlaid with SAR-seq signal (red). h, Aggregate plot showing the distribution of CG dinucleotides (black) for 1 kb on either side of SAR-seq peak summits or summits of ATAC–seq peaks (green) that are H3K4me1 positive but do not overlap with SAR-seq.
Extended Data Fig. 10 PARP, XRCC1 or POLB deficiency increases SAR.
a, Quantitative RT–PCR analysis showing the PARP1 mRNA transcript level in i3Neurons after CRISPRi knockdown (sgControl, control non-targeting sgRNA; sgPARP1, an sgRNA targeting PARP1), cultured in parallel with samples used for SAR-seq. P = 7.72 × 10−7 by unpaired two-tailed Student’s t-test; ****P < 0.00001 (n = 3). b, Genome browser screenshot displaying SAR-seq profiles from two biological replicates of i3Neurons treated with the PARP inhibitors olaparib, talazoparib, or veliparib, or using CRISPRi-mediated knockdown with sgControl or sgPARP1, in duplicates. NT, not treated. c, Quantitative RT–PCR analysis showing XRCC1 mRNA transcript level in i3Neurons after CRISPRi knockdown, cultured in parallel with samples used for SAR-seq. P = 1.88 × 10−5 by unpaired two-tailed Student’s t-test; ****P < 0.00001 (n = 3). d, Genome browser screenshots of SAR-seq profiles in i3Neurons expressing sgControl or sgXRCC1, in duplicate. e, Venn diagram showing the overlap of SAR-seq peaks between i3Neurons expressing sgControl and those expressing sgXRCC1. n = 1,000 random datasets were generated to test the significance of overlap (one-sided Fisher’s exact test: P < 2.2 × 10−16). f, Quantitative RT–PCR analysis showing POLB mRNA transcript levels in i3Neurons after CRISPRi knockdown, cultured in parallel with samples used for SAR-seq. P = 6.98 × 10−5 for sgPOLB1 and 5.82 × 10−5 for sgPOLB2 by unpaired two-tailed Student’s t-test; ****P < 0.00001 (n = 3). g, Western blot showing POLB protein levels in i3Neurons after CRISPRi knockdown, cultured in parallel with samples used for SAR-seq (n = 1). For gel source data, see Supplementary Fig. 2. h, Genome browser screenshots of SAR-seq profiles from two biological replicates of i3Neurons expressing sgControl or sgPOLB, in duplicate. i, Genome browser screenshots of SAR-seq profiles from two biological replicates of i3Neurons expressing sgControl or sgPOLB. Cells were pre-treated or not treated (NT) with 50 μM Aph for 24 h, and then also during incubation with EdU.
Extended Data Fig. 11 Localized SSB repair in neurons correlates with sites of oxidized 5-methylcytosine.
a, Genome browser screenshot of chromosome 7 showing SAR-seq profiles from two biological replicates and input DNA in i3Neurons without MMS treatment (NT, n = 2) or after treatment with 0.1 mg ml−1 MMS (n = 2) for the final 15 min of an 18-h incubation with EdU. After streptavidin pull-down and PCR amplification, total DNA was quantified (NT rep 1: 0.95 μg; NT rep 2: 1.7 μg; MMS rep 1: 3.8 μg; MMS rep 2: 4.5 μg). Stochastic DNA damage results in loss of DNA synthesis at recurrent sites. b, Quantitative RT–PCR analysis showing PNKP mRNA transcript level in i3Neurons after CRISPRi knockdown, cultured in parallel with samples used for SAR-seq. P = 0.00015 by unpaired two-tailed Student’s t-test; ***P < 0.0001 (n = 3). c, Bottom, heat maps of SAR-seq intensities for 1 kb on either side of SAR-seq peak summits for i3Neurons expressing sgControl (n = 2) or sgPNKP (n = 2). Top, aggregate plots of SAR-seq intensity. d, Bottom, heat map of SAR-seq intensity for 1 kb on either side of the transcription start site in i3Neurons, ordered by SAR-seq intensity. i3Neurons expressing sgControl or sgPNKP were either not treated (NT, n = 2) or treated with 25 μM camptothecin (CPT, n = 2) during incubation with EdU. Top, aggregate plots of SAR-seq intensity. e, Scatter plots showing correlations of intensities (RPKM) between SSBs (ddN S1 END-seq) and 5fC or 5hmC, respectively, for 1 kb on either side of SAR-seq peak summits for i3Neurons. Spearman correlation coefficients and P values are indicated. f, Scatter plots showing correlations of intensities (RPKM) between SAR-seq for i3Neurons expressing sgPOLB and ddN S1 END-seq, 5fC, or 5hmC for 1 kb on either side of SAR-seq peak summits. Spearman correlation coefficients and P values are indicated.
Supplementary information
Supplementary Information
This file contains Supplementary Figure 1 (Flow cytometry gating) and Supplementary Figure 2 (Source Data gels).
Rights and permissions
About this article

Cite this article
Wu, W., Hill, S.E., Nathan, W.J. et al. Neuronal enhancers are hotspots for DNA single-strand break repair. Nature 593, 440–444 (2021). https://doi.org/10.1038/s41586-021-03468-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-021-03468-5
- Springer Nature Limited
This article is cited by
-
SSBlazer: a genome-wide nucleotide-resolution model for predicting single-strand break sites
Genome Biology (2024)
-
Methods and applications of genome-wide profiling of DNA damage and rare mutations
Nature Reviews Genetics (2024)
-
HLTF disrupts Cas9-DNA post-cleavage complexes to allow DNA break processing
Nature Communications (2024)
-
Base excision repair and double strand break repair cooperate to modulate the formation of unrepaired double strand breaks in mouse brain
Nature Communications (2024)
-
Covalent PARylation of DNA base excision repair proteins regulates DNA demethylation
Nature Communications (2024)