
Abstract
The study of polarized protein trafficking in live neurons is critical for understanding neuronal structure and function. Given the complex anatomy of neurons and the numerous trafficking pathways that are active in them, however, visualization of specific vesicle populations leaving the Golgi complex presents unique challenges. Indeed, several approaches used in non-polarized cells, and even in polarized epithelial cells, have been less successful in neurons. Here, we describe an adaptation of the recently developed Retention Using Selective Hooks (RUSH) system (Boncompain et al., Nat Methods 9:493–498, 2012), previously used in non-polarized cells, to analyze the polarized sorting of proteins from the Golgi complex to dendrites and axons in live neurons. The RUSH system involves the retention of a fluorescently tagged cargo protein fused to the streptavidin-binding peptide (SBP) in the endoplasmic reticulum (ER) through the expression of an ER-hook protein fused to streptavidin. Upon d-biotin addition, the cargo protein is released and its traffic to dendrites and axons can be analyzed in live neurons.
Access provided by CONRICYT – Journals CONACYT. Download protocol PDF
Similar content being viewed by others



Key words
- Hippocampal neurons
- Neuron dissection
- Neuronal culture
- Neuron transfection
- Live-cell imaging
- Polarized trafficking
- Axon initial segment
- RUSH system
- Trafficking from the Golgi complex
- Kymograph analysis
1 Introduction
Neurons are polarized cells with a highly asymmetric structure. They comprise axonal and somatodendritic domains with different compositions of proteins in both the cytoplasm and plasma membrane. Both domains also have distinct subdomains, including presynaptic terminals at axon tips and postsynaptic terminals in dendrites [1–3]. The asymmetry of protein localization across these domains reflects their specialized functions and allows the vectorial transmission of information from the presynaptic terminals of one neuron to the postsynaptic terminals of another [1–3]. Maintenance of polarity and dynamic response to developmental and environmental inputs are important to proper neuronal function [1–3]. Thus, the means by which polarized sorting occurs, as well as the native role of each protein in its intended domain, are of mechanistic interest both in vivo and in vitro.
A common method for studying neuronal structure and function is the primary culture of hippocampal neurons from embryonic rats or mice. When maintained in culture, neurons progress through a characteristic sequence of developmental stages [4]. Within 2 days of plating, cells begin to establish polarity, and the one neurite destined to become the axon grows to several times the length of the others. The first synapses begin to form after 3–4 days in culture [4]. Around day in vitro (DIV) 5, a key event in neuronal polarization occurs with the accumulation of proteins such as ankyrin G (AnkG) at the axon initial segment (AIS) [3]. Among other roles, the AIS proteins play a major part in neuronal polarity, serving as a barrier to plasma membrane diffusion between the axonal and somatodendritic domains [3]. Axonal growth continues beyond this stage, producing dense networks of polarized, interconnected cells as additional synapses form over the following days [4].
The ability to transport proteins to their correct intracellular locations is critical for achieving neuronal polarity, and cells employ a variety of mechanisms to maintain this composition [2, 5, 6]. As in non-neuronal cells, newly synthesized proteins transferred from the endoplasmic reticulum (ER) to the Golgi complex undergo biosynthetic sorting [1]. The Golgi in neurons takes on a unique structure consisting of a somatic Golgi stack, which is preferentially oriented toward the longest dendrite in cultured neurons, as well as Golgi outposts present in dendrites but excluded from the axon (Fig. 1) [7]. Upon arrival at the trans-most compartment of the Golgi complex, the trans-Golgi network (TGN), p roteins undergo sorting into specific transport carriers depending on their intended destinations, after which p olarized transport mechanisms move these carriers into either axonal or somatodendritic domains [1]. Various machineries responsible for this sorting have been characterized, chief among them the adaptor protein (AP) complexes and the coat protein clathrin [1, 6]. Following budding from the TGN, proteins and their transport carriers may be subject to additional sorting and fusion events. For example, proteins may undergo active endocytosis in one domain but retention in another [5]. Certain proteins reach their destinations via transcytosis and thus traffic to one domain before being endocytosed, moving once again through the soma, and entering the opposite domain [5]. Proteins may also be targeted to lysosomes for degradation in a domain-specific manner [8].

Golgi complex in hippocampal neurons. (a, b) Immunofluorescence of neurons on DIV 10 stained for the Golgi marker GM1 30 (a) or the trans-Golgi netwo rk (TGN) marker TGN38 (b), together with the AIS marker AnkG. GM130 and TGN38 staining is shown in negative grayscale and green in the merged images, and AnkG is shown in red in the merged images. Red arrows point to the AIS. Scale bar, 5 μm. Notice the localization of the Golgi complex to the soma, and dendrites as indicated by arrowheads
In studying the initial sorting and ultimate fate of transmembrane proteins, direct visualization of trafficking from the Golgi complex is a useful approach. It was in neurons that Camillo Golgi first observed and described the apparatus that bears his name [9], and the work of recent decades has established trafficking from the Golgi complex as a fundamental aspect of neuronal development and function [1, 6]. Knowledge of the mechanisms and dynamics of protein sorting is important in understanding normal cellular processes and neuropathological disorders [2]; however, the study of sorting in live neurons has been challenging because proteins may be simultaneously present in the biosynthetic, endosomal, and lysosomal pathways at steady state [5]. Photobleaching and photo-convertible reporter expression have been widely used to study plasma membrane insertion of transmembrane proteins in neurons [10–12], but these methods are less useful when imaging the sorting of proteins from the Golgi complex.
In contrast to neurons, in non-polarized cells and even in polarized epithelial cells, which contain apical and basolateral domains, several approaches have been successfully used to dissect and image the sorting of proteins from the Golgi complex. For example, previous studies have used temperature reduction to 19 °C to retain proteins in the TGN [13, 14], the temperature-sensitive mutant of the Vesicular Stomatitis Virus glycoprotein (VSVG tsO45) to retain proteins in the ER [15–17], and pharmacologically controlled aggregation of proteins in the ER [18]. The most powerful and extensively used procedure for analyzing traffic in the biosynthetic pathway has been cargo synchronization using the VSVG transport assay, in which the mutant protein is misfolded and retained in the ER at 40 °C. Upon temperature shift to 32 °C, the protein folds and moves as a synchronous population to the Golgi complex before being transported to the plasma membrane [15–17]. However, this system has been of limited use in neurons because of their susceptibility to unphysiological conditions such as changes in temperature.
Recently, an alternative method for imaging synchronous transport through the secretory pathway, the Retention Using Selective Hooks (RUSH) system, was developed by Franck Perez and colleagues [19]. This system consists of the expression of two fusion proteins from a bicistronic expression vector: the “hook,” stably expressed in a donor compartment and fused to a core streptavidin, and the “reporter,” fused to the Streptavidin-Binding Peptide (SBP) , which reversibly interacts with the hook. Upon reversion of the interaction through addition of the vitamin d-biotin to the cell, the reporter can freely resume its traffic in the biosynthetic pathway [19].
We adapted the RUSH system to study the trafficking of transmembrane proteins from the Golgi complex to dendrites and axon in live neurons. Here we describe a detailed protocol for the preparation of hippocampal neurons, transfection of cDNAs including those of the RUSH system, staining of endogenous proteins to identify the somatodendritic and axonal domains, live-cell imaging using the RUSH system, and kymograph analysis for particle transport in dendrites and axons.
2 Materials
2.1 Neuron Preparation and Transfection
-
1.
Ultrapure water, cell cu lture grade.
-
2.
High-precision glass coverslips, 18-mm diameter, No. 1.5H (Marienfeld).
-
3.
70 % nitric acid.
-
4.
Borate buffer (0.1 M, pH 8.5, 1000 mL). Dissolve 38.14 g sodium tetraborate decahydrate ≥99.5 % in approximately 800 mL of ultrapure water, cell culture grade, using a magnetic stir plate and bar at high speed. After dissolution of sodium tetraborate, add 6.18 g boric acid ≥99.5 % and continue stirring until dissolved. Add 1 N HCl to pH = 8.5 and add water to final volume of 1000 mL. In a sterile laminar flow hood, filter the solution using two 500-mL filter units with 0.22-μm membranes. All subsequent solutions should be prepared in a sterile laminar flow hood and, when indicated, filtered using similar filter units of an appropriate size.
-
5.
Poly-l-lysine solution (1 mg/mL, 100 mL): Dissolve 100 mg of poly-l-lysine hydrobromide, molecular weight 30,000–70,000, in 100 mL of borate buffer and filter. Store at 4 °C when not in use. Poly-l-lysine solution can be reused many times; it should be replaced at the first sign that neurons are not properly adhered to the coverslips.
-
6.
Hank’s medium (1×, pH 7.3, 500 mL): Dilute 50 mL of 10× Hank’s Balanced Salt Solution (HBSS, no calcium, no magnesium, no phenol red) with 450 mL ultrapure water, cell culture grade, add 10 mL of 1 M HEPES (pH = 7.2–7.5), and filter the solution.
-
7.
Laminin solution (5 μg/mL, 100 mL): Dilute 1 mL of laminin from mouse Engelbreth-Holm-Swarm (EHS) sarcoma (Roche) with 100 mL of Hank’s medium and filter the solution. Store at 4 °C when not in use. Laminin can also be reused many times with the same precautions as for poly-l-lysine .
-
8.
Neuronal plating medium (1×, 500 mL): Add 50 mL horse serum (heat inactivated) and 5 mL penicillin–streptomycin (10,000 U/mL) to 450 mL of 1× Dulbecco’s Modified Eagle Medium (d-MEM, high glucose, HEPES, no phenol red) and filter. The medium can be stored at 4 °C for 1–2 weeks.
-
9.
Trypsin, 2.5 % (10×).
-
10.
Neuronal maintenance mediu m (50 mL): Prepare fresh medium the day of neuron isolation. Add 1 mL B-27® Supplement (50×), serum free (Gibco™), 500 μL GlutaMAX™ Supplement (100×) (Gibco™), and 500 μL penicillin–streptomycin (10,000 U/mL) to 50 mL of Neurobasal Medium (Gibco™) and filter.
-
11.
Opti-MEM® I Reduced-Serum Medium (1×) (Gibco™).
-
12.
Lipofectamine™ 2000 Transfection Reagent (Invitrogen™).
-
13.
Plasmids encoding RUSH system constructs. Several plasmids for use in the RUSH system are available in Addgene (http://www.addgene.org/) under the list of Franck Perez.
-
14.
Orbital shaker.
-
15.
Autoclave.
-
16.
Horizontal laminar flow hood.
-
17.
Dissecting microscope.
-
18.
Light guide.
-
19.
Dissecting tools (sterilized): microdissecting scissors (flat and curved), fine-tipped forceps (straight and curved).
-
20.
Syringe and needle, 19-gauge × 1.5″.
-
21.
Vertical laminar flow hood.
-
22.
Water bath at 37 °C.
-
23.
Hemocytometer for counting cells.
-
24.
Tissue culture incubator at 37 °C with humidified, 5 % CO2 atmosphere.
-
25.
Sterile glass Pasteur pipettes.
-
26.
Sterile filter units, 0.22-μm pore size.
-
27.
Sterile plasticware: 5-mL serological pipettes, 35- and 100-mm dishes, 12-well tissue culture plates, 15- and 50-mL conical centrifuge tubes, 1.5-mL microfuge tubes.
2.2 Endogenous Protein Labeling
-
1.
Anti-Pan-Neurofascin (external) antibody (clone A12/18, purified) (NeuroMab) or any primary antibodies against surface-anchored proteins (external epitope).
-
2.
Mix-n-Stain CF488, CF555, or CF640R antibody labeling kit (Biotium).
-
3.
Refrigerated centrifuge (10,000 × g at 4 °C).
-
4.
Vortex.
2.3 Live-Cell Imaging to Analyze Sorting of Proteins from Golgi Complex
-
1.
NeutrAvidin Protein (Thermo Scientific™). Prepare a stock of 10 mg/mL of NeutrAvidin using ultrapure water, cell culture grade.
-
2.
d-biotin : prepare a stock of 1-mM d-biotin using ultrapure water, cell culture grade.
-
3.
35-mm dish type magnetic chamber for 18-mm round coverslips (Quorum Technologies, Inc.).
-
4.
Spinning-disk confo cal microscope equipped with 63×, 1.4 NA or 100×, 1.4 NA objectives, EM-CCD camera for digital image acquisition, heating unit and temperature module control.
3 Methods
3.1 RUSH System Construct Design
-
1.
In Addgene, several plasmids deposited by Franck Perez (Curie Institute, Paris, France) for use with the RUSH system are available. A bicistronic expression plasmid (pIRESneo3) was used to generate the expression of two fusion proteins: The ER-hook (Ii, STIM1-NN, or KDEL retention signal) fused to a core streptavidin, and the reporter fused to SBP.
-
2.
The genes encoding the protein of interest (reporter) must be cloned and fused to SBP . A detailed protocol for subcloning into a RUSH plasmid has been described by Boncompain and Perez [20].
-
3.
The retention and release of the reporter can be fol lowed by live-cel l imagin g using any of the GFP, mCherry, or TagBFP fluorescent tags available in Addgene.
3.2 Preparation of Coverslips for Neurons
3.2.1 Cleaning and Sterilization of Coverslips
-
1.
At least 1 week prior to isolation of neurons, place 18-mm coverslips in 70 % nitric acid in a ceramic or glass rack inside a chemical fume hood and move gently with forceps to disperse any bubbles.
-
2.
After at least 2 days in nitric acid, remove coverslips to a dish with ultrapure water, cell culture grade. Rinse briefly, change to fresh water, then place dish on an orbital shaker to rinse for 20 min. Rinse five additional times for a total of 2 h.
-
3.
Use a pair of fine forceps to remove each coverslip, touch edges gently to a paper towel to remove excess water, and place in a glass dish.
-
4.
Autoclave dish and maintain in a sterile container or covering.
3.2.2 Coating of Coverslips
-
1.
At least 3 days prior to isolation of neurons, working in a sterile tissue culture hood, place one coverslip into each well of a 12-well plate and add 1 mL per well of poly-l-lysine solution.
-
2.
Leave plates to incubate at 37 °C for at least 2 days. Recover poly-l-lysine, wash each well with at least 0.5 mL ultrapure water, cell culture grade, three times for 10 min per wash, and place in an incubator at 37 °C between washes.
-
3.
Remove all water from last wash and add 1 mL laminin solution to each well. Return plates to 37 °C for 45 min.
-
4.
Recover laminin so lution and wash each well with water twice for 15 min per wash at 37 °C.
-
5.
Remove all water from las t wash and add 1.2 mL neuronal plating medium to each well.
-
6.
Maintain plates in a CO2 incubator at 37 °C for up to several days before preparing neurons.
3.3 Isolation of Rat Hippocampal Neurons
The following protocol is a modification of that previously developed by Kaech and Banker [4]. Here we provide a detailed method for dissecting the brains of day-18 embryonic rats and isolating the hippocampus, as well as subsequent steps for dissociating and culturing hippocampal neurons. Coculture with a monolayer of glial feeder cells may be used if desired [4].
Following euthanasia, dissecti on steps are performed in a sterile horizonta l laminar flow cabinet. After hippocampus isolation, perform the remaining steps in a vertical laminar flow hood.
-
1.
Pre-warm an appropriate volume (approximately 15 mL) of neuronal plating medium to 37 °C.
-
2.
On embryonic day 18, sacrifice a pregnant female rat using an approved method of euthanasia and according to an approved animal use protocol. Remove the uterus and transfer to a sterile 100-mm tissue culture dish.
-
3.
Remove each fetus from the uterus and sacrifice by cutting just posterior to the forelimbs using flat scissors, depositing the head and upper torso in a dish of fresh Hank’s medium. Repeat for each embryo. Rinse one to two times with fresh Hank’s medium to remove any blood or debris.
-
4.
Secure the head using curved forceps and use small curved dissecting scissors to make a lateral incision at the base of the skull, just inferior to the cerebellum. The cerebral hemispheres and cerebellum should be visible beneath the surface (see Note 1 ). Make the incision wide enough to allow passage of the brain, then use closed scissors to gently push on the top of the skull in an anterior-to-posterior direction. Allow the brain to slide into a dish of fresh Hank’s medium. Repeat for each embryo. Rinse one to two times with fresh Hank’s medium and place dish on ice.
-
5.
Transfer a few brains to a sterile glass dish containing fresh Hank’s medium and place under a dissecting scope (Fig. 2a). Illumination may be used in order to better distinguish the regions of the brain.
Fig. 2 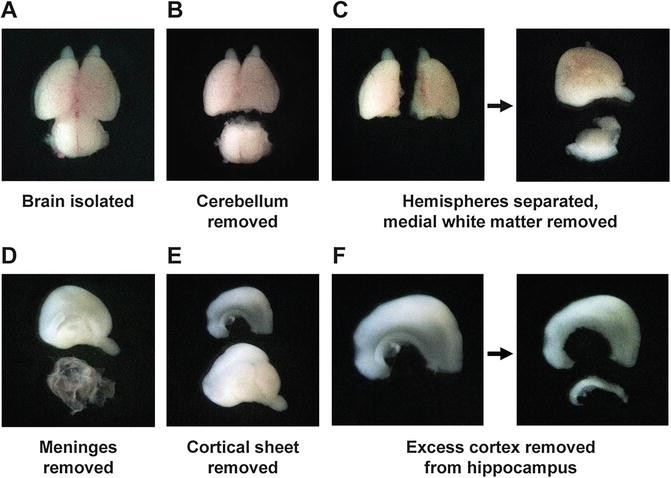
Hippocampal isolation. (a) Whole brain isolated from a day-18 rat embryo. (b) The cerebellum (lower) is removed by making a lateral cut just posterior to the cerebral hemispheres. (c) The hemispheres are separated along the midline, and the medially attached white matter (lower) is removed from each. (d) The meninges (lower) are peeled from the surface of the hemisphere. (e) A section of cortex (upper) containing the hippocampus is removed from the medial face of the hemisphere. The hippocampus can be seen as a more opaque band along the inner edge of this sheet. (f) The hippocampus (lower) is cut away from the attached cortical tissue using a needle to complete the isolation
-
6.
Using fine forceps, remove the cerebellum (Fig. 2b) and separate the cerebral hemispheres along the midline (Fig. 2c). For each hemisphere, remove the medial white matter not covered by meninges (Fig. 2c).
-
7.
Carefully remove the meninges (see Note 2 ) and locate the hippocampus (Fig. 2d). Look for a thin, curved structure on the medial aspect of the hemisphere that appears slightly more opaque than the surrounding cortex (Fig. 2d). Remove a relatively flat sheet of cortex from the medial face of the hemisphere hemisphere containing the hippocampus along its inferior edge (Fig. 2e).
-
8.
Using a 19-gauge × 1.5″ needle attached to a syringe, carefully cut the hippocampus away from this sheet of cortex (see Note 3 and Fig. 2f). Use one edge of the needle to cut the tissue against the surface of the glass plate.
-
9.
Repeat steps 5–8 for a few brains at a time, leaving the remaining brains in Hank’s medium on ice. Isolated hippocampi may be left in the glass dish until all brains have been dissected.
-
10.
Use forceps to transfer the isolated hippocampi to a sterile 35-mm tissue culture dish containing Hank’s medium. Use a pipet partially prefilled with fresh Hank’s medium to remove the hippocampi from the dish and transfer to a 15-mL conical tube. Add Hank’s medium to a volume of 4.5 mL and add 0.5 mL of 2.5 % trypsin. Swirl the tube twice to separate hippocampi. Incubate at 37 °C for a total period of 14–15 min, swirling the tube occasionally.
-
11.
Remove as much trypsin solution as possible without disturbing hippocampi at the bottom of the tube and add 10 mL of fresh Hank’s medium. Swirl the tube twice and incubate at room temperature for 6 min, swirling the tube every 2 min.
-
12.
Remove Hank’s medium and add 1.5 mL of neuronal plating medium pre-warmed to 37 °C. Use a 5-mL pipet to dissociate hippocampi by pipetting up and down approximately ten times. Next, pipet the hippocampi up and down approximately ten times using a glass Pasteur pipet whose tip has been narrowed to around 60 % of its original diameter. If chunks are still observed, use a glass Pasteur pipet whose tip has been narrowed to around 30 % and continue pipetting until a homogenous suspension has been achieved.
-
13.
Add 8.5 mL neuronal plating medium for a total volume of 10 mL.
-
14.
Measure cell concentration using a hemocytometer and determine the volume of suspension needed for the desired cell count. Typically, 60,000–80,000 cells per 18-mm coverslip are sufficient for live-cell experiments.
-
15.
Ensure that the cell suspension is uniform by pipetting up and down, then add the appropriate volume to a plate with coated coverslips in neuronal plating medium. Tap the plate on its sides many times to distribute the cells evenly across each well and place in a CO2 incubator at 37 °C.
-
16.
Pre-warm a sufficient volume of neuronal maintenance medium (at least 1 mL per coverslip) to 37 °C.
-
17.
After 3–4 h, check that most cells have attached to the coverslips, gently remove the neuronal plating medium and any unattached cells using a pipet, and replace with 1 mL per well of neuronal maintenance medium. Neurons may be maintained in this original medium in a CO2 incubator for several days before transfection.

3.4 Transfection of Rat Hippocampal Neurons (DIV 3-5)
-
1.
Pre-warm appropriate volumes of Neurobasal Medium (3 mL per well to be transfected) and Opti-MEM (200 μL per well) to 37 °C.
-
2.
In a laminar flow hood, prepare DNA mixtures for Lipofectamine 2000-based transfection according to the manufacturer’s instructions (100 μL Opti-MEM and approximately 1 μg DNA per well for a 12-well plate) (see Note 4 ).
-
3.
Prepare Lipofectamine 2000 mixture using 1.1–1.2 μL Lipofectamine and 100 μL Opti-MEM for each well to be transfected. Incubate for 5 min at room temperature.
-
4.
Combine Lipofectamine and DNA mixtures and incubate for 20 min at room temperature. While incubating, gently recover original culture medium, wash each well twice with 500 μL Neurobasal Medium per well, and add 1 mL of Neurobasal Medium for transfection (see Note 5 ).
-
5.
After incubation, add 200 μL of transfection mix to each well and return to a CO2 incubator at 37 °C for 1 h.
-
6.
During transfection, combine recovered culture medium with an additional 1/3 to ½ volume of fresh neuronal maintenance medium, filter using a 0.22-μm membrane, and return both filtered medium and Neurobasal Medium to 37 °C.
-
7.
After 1 h of transfection, gently wash each well twice with Neurobasal Medium and add 1 mL of filtered original medium plus fresh neuronal maintenance medium.
-
8.
Add NeutrAvidin (50 μL from a 10 mg/mL stock), a biotin-binding protein, if the RUSH s ystem plasmids have been transfected (see Note 6 ).
-
9.
Maintain plates in a CO2 incubator until ready for use. It is not necessary to change the culture medium during this time. Typical transfection efficiency is approximately 5 %.
3.5 Staining of the AIS for Live-Cell Imaging
This protocol may also be used for axonal and dendritic surface staining of proteins for live-cell imaging through selection of an antibody to the extracellular portion of a cell surface protein. Follow the manufacturer’s protocol for covalent coupling of each antibody to the Mix-n-Stain CF series dyes.
-
1.
Centrifuge 90 μL of Anti-Pan-Neurofascin (external) antibody (clone A12/18, purified) solution at 10,000 × g for 30 min.
-
2.
After centrifugation of the antibody, perform all subsequent steps in a laminar flow hood.
-
3.
Take the supernatant and incubate with the 10× Mix-n-Stain (10 μL) solution, pipetting up and down a few times.
-
4.
Transfer the entire solution to the vial containing the CF555, CF488, or CF640R dyes and vortex the vial for a few seconds.
-
5.
Incubate the mix for 30 min at room temperature in a dark room.
-
6.
Prepare 10-μL aliquots of the mixture and store at −20 °C.
-
7.
Prepare a 1:50 dilution of Anti-Pan-Neurofascin-CF with neuronal maintenance medium and pre-warm the solution to 37 °C. One aliquot is enough for one coverslip in a 12-well plate (10 μL of the Neurofacin-CF640R plus 500 μL of neuronal maintenance medium).
-
8.
Remove the medium from one coverslip with neurons on DIV 6-14 and add the 500 μL of antibody solution. Return the plate with neurons to a CO2 incubator at 37 °C for 30 min to 1 h (see Note 7 ).
-
9.
Recover the 500 μL of antibody solution and transfer to another coverslip for the next experiment. The antibody solution can be used many times the same day. Wash the coverslip twice with pre-warmed neuronal m aintenance medium and add 1 mL of the same medium to the coverslip.
-
10.
Maintain the plate in the CO2 incubator at 37 °C until preparing the coverslips for live-cell imaging.
3.6 Live-Cel l Imaging Using the RUSH System
-
1.
Turn on the spinning-disk confocal microscope, set the temperature to 37 °C using a temperature controller connected to a heating device, and wait until the temperature is stabilized to 37 °C. Pre-warm the oil used for objectives to 37 °C.
-
2.
In a vertical laminar flow hood, remove the coverslip from the plate using sterile fine forceps, place the coverslip in a 35-mm dish type magnetic chamber for incubating 18-mm round coverslips during live-cell imaging, and add 500 μL of the original medium.
-
3.
Clean the bottom surface of the coverslip in the magnetic chamber with 70 % ethanol before imaging.
-
4.
In the spinning-disk microscope, add a pre-warmed drop of oil to a 63× oil or 100× oil objective and place the magnetic chamber in the microscope.
-
5.
Focus and find neurons expressing constructs of interest. Choose optimal conditions of laser power and exposure for imaging those particular constructs.
-
6.
Neurites can be identified by expression of GFP, RFP, CFP, or similar soluble fluorescent proteins, tagged tubulin, or tagged plasma membrane proteins in the transfection mix, and/or endogenous plasma membrane proteins stained with th e CF-series dyes (Figs. 3 and 4).
Fig. 3 
Expression of neuronal markers and staining of endogenous proteins. (a–c) One-frame images corresponding to live neurons transfected on DIV 4 with plasmids encoding mCherry (mCh) (a), 13 amino acids of the N-terminal sequence of the myristoylated and palmitoylated Lyn kinase to target mRFP to the plasma membrane (PM-RFP) (b), and mCherry-tagged alpha-tubulin (mCh-Tub) (c), and stained for the AIS with CF640R-conjugated antibody to neurofascin (Anti-NF-CF640R) on DIV 7 (a–c). Transgenic proteins are shown in negative grayscale (left panels) and red in the merged images (right panels), and surface neurofascin in negative grayscale (middle panels) and blue in the merged images (right panels). Blue arrows point to the AIS. Scale bar, 10 μm. Notice the staining of the whole neuronal cytoplasm for mCh and mCh-Tub, and the whole plasma membrane for PM-RFP. Staining with anti-NF-CF640R, on the other hand, is restricted to the AIS
Fig. 4 
Expression of RUSH system constructs to analyze sorting of proteins from the Golgi complex. (a) Schematic representation of the procedure to analyze protein sorting from the Golgi complex to dendrites and axon using the RUSH system. (b, c) Neurons transfected on DIV 4 with a bicistronic expression plasmid encoding two fusion proteins: the KDEL ER-retention signal fused to a core streptavidin (“hook”) and the transferrin receptor (TfR) fused to SBP and GFP (TfR-SBP-GFP). NeutrAvidin was added to the culture medium after transfection to compete with the d-biotin present in the neuronal maintenance medium. Neurons on DIV 7 wer e surface-stained by incubation with CF555-conjugated antibody to neurofascin (NF-CF555) for 45 min at 37 °C and analyzed using a spinning-disk confocal m icroscope. Images show the expression of TfR-SBP-GFP (negative grayscale in left and middle panels, and green in right panels) after 15 min (b) and 20 min (c) of incubation with d-biotin . NF-CF555 staining is shown in grayscale (middle panels) and red in the merged images (right panels). Red arrows point to the AIS. Scale bar 10 μm. Notice the accumulation of TfR-SBP-GFP at the Golgi complex after 15 min of d-biotin incubation. After 20 min of d-biotin incubation, some carriers containing TfR-SBP-GFP can be observed in dendrites (arrowheads), consistent with the somatodendritic localization of the TfR. A similar approach can also be used for studying axonal proteins sorting from the Golgi complex
-
7.
Find transfected neurons expressing proteins of interest using the RUSH system (see Note 8 ).
-
8.
Add 40 μM d-biotin to neurons. Dilute 24 μL of d-biotin (1-mM stock) with neuronal maintenance medium (100 μL) and add to the chamber containing 500 μL of medium. If necessary, adjust the focus again in the microscope.
-
9.
If the cargo molecules have been retained by interaction with an ER resident protein, it will take around 10–15 min to release the cargo molecules from the ER, and strong staining of the Golgi complex will appear (Fig. 4a–c).
-
10.
After accumulation of the cargo molecules at the Golgi complex, start the live-cell imaging to analyze trafficking of transmembrane proteins to the axon and dendrites (Fig. 4a).


3.7 Analysis of Live-Cell Imaging Data
Trafficking of proteins to the axon and dendrites can be studied using kymograph analysis .
-
1.
Open images containing the frames in ImageJ (Wayne Rasband, NIH; http://imagej.nih.gov).
-
2.
Isolate axon and dendrites from a neuron. Select a line width between 20 and 40 pixels (Edit/Options/Line width). Then use the segmented line tool to select a region from the axon or dendrite (Fig. 5a, b). Straighten the selected line (Edit/Selection/Straighten) and select “Process Entire Stack” (Fig. 5c). Draw each line beginning in the soma and ending in the axon or dendrite in order to visualize anterograde transport from left to right and retrograde transport from right to left. Use the same parameters of line width and axon or dendrite length for all experiments for direct comparison of transport parameters.
Fig. 5 
Kymograph analysis of the transport of proteins to dendrites and axon. (a) Single-frame images corresponding to a live neuron on DIV 7 co-transfected on DIV 4 with GFP-tagged transferrin receptor (TfR-GFP) (negative grayscale in left panel, green in merged image in right panel) and mCherry-tubulin (mCh-Tub) (negative grayscale in middle panel, red in merged image in right panel). Note the somatodendritic distribution of TfR. Live-cell imaging was performed using a spin ning-disk confocal microscope (Intelligent Imaging Innovations). Digital images were acquired with an Evolve electron-multiplying charge-coupled device (EM-CCD)Fig. 5 (continued) camera (Photometrics). Images of TfR-GFP and mCh-Tub were sequentially acquired at 500-ms intervals for 120 s. Arrows point to the axon. Scale bar 10 μm. (b–f) An example of selection and isolation of dendrite and axon segments for kymograph analysis . (b) Lines 30 pixels wide and 35 μm long in a dendrite (magenta region) and axon (cyan region) were selected with orientation S → D and S → A from a 240-frame dual-color image. (c) Selected dendrite and axon lines from the 240-frame dual-color image were straightened. TfR-GFP and mCh-Tub images are shown separately and merged. (d) The straightened dendrite was re-sliced to produce a Z-projection in order to generate a kymograph. (e) Kymograph generated from a dendrite, where the X-axis is distance (35 μm) and Y-axis is time (120 s). Vertical lines correspond to stationary particles that did not move during the entire period of imaging. Lines with negative slopes and positive slopes correspond to anterograde and retrograde movement, respectively, of TfR-GFP in the dendrite during the 120 s of recording. (f) Representation of anterograde (magenta lines) and retrograde (blue lines) particles from the kymograph
-
3.
Save isolated axon and dendrite frames as a TIFF file.
-
4.
To quantify the transport of cargoes to axon and or dendrites, generate kymographs (Fig. 5d, e). Re-slice the isolated axon or dendrite stack (Image/Stacks/Reslice). Generate a Z-projection from the re-sliced stack (Image/Stacks/Z-projection) (Fig. 5e).
-
5.
Stationary particles are observed as vertical lines; anterograde and retrograde transport are observed as lines with negative and positive slopes, respectively (Fig. 5e).
-
6.
Anterograde and retrograde transport can be manually represented by removing the stationary particles from the kymograph (Fig. 5f).
-
7.
The number of stationary, anterograde, and retrograde particles can be determined through the quantification of the number of events from kymographs of the same duration and neurite length.

4 Notes
-
1.
If the cerebral hemispheres and cerebellum are not visible through the skull, or if the base of the skull is rigid and difficult to cut using scissors, the embryos obtained may be older than 18 days. Brains obtained from embryos obtained after day 18 may be more difficult to dissect, and it may be necessary to make an additional incision in the skull along the midline in order to remove the brain.
-
2.
It is typically easiest to peel away the meninges by creating a small tear at either the anterior or posterior end of the hemisphere, then using one pair of forceps to pull on the membranes while gently holding the hemisphere stationary with the other. If removed carefully, the meninges will peel away from the cortex as a single sheet.
-
3.
The hippocampus is most easily identified and separated when what was formerly the medial surface of the hemisphere is facing upward. There are various techniques for removing the hippocampus using the needle. Generally, it is easiest to use a pair of forceps in the nondominant hand to hold the cortical sheet stationary while the hippocampus is cut away. The forceps can either be placed directly on the cortex or in the growing incision between the hippocampus and remaining cortical tissue.
-
4.
If transfecting more than two 12-well plates, it may be necessary to wash each plate once prior to the 5-min Lipofectamine incubation step. After washing once, return plates to incubator and resume washing during the 20-min incubation of the Lipofectamine–DNA mixture.
-
5.
Primary neurons are fragile and susceptible to detachment of neurites if media are added too forcefully. Examine neurons after initial washes and again after transfection to determine whether cell survival is affected by either step. If cell density decreases noticeably after washing, be sure to wash gently by touching the pipet tip to the side of the well and adding the medium slowly.
-
6.
B27 supplement contains d-biotin , which can release the cargo molecules from the donor compartment when using the RUSH system. This dissociation can be prevented by addition of NeutrAvidin to the medium on the day of transfection.
-
7.
AIS staining in young neurons (DIV 4–6) will be weak compared to old neurons (DIV 7–21) because Neurofascin accumulates during neuronal development. If staining on DIV 6 is desired, perform a longer incubation with the anti-Neurofascin-CF-series dyes (1 h). If staining for Neurofascin is perfo rmed on DIV 10 or later, 30 min of incubation will be sufficient to visualize the AIS.
-
8.
In transfected neurons, expression of a protein of interest using the RUSH system will appear weak if the protein has been “hooked” to an ER resident protein, but after adding d-biotin for 10–15 min, the protein will accumulate in the Golgi complex (Fig. 4b, c).
References
Lasiecka ZM, Yap CC, Vakulenko M, Winckler B (2009) Compartmentalizing the neuronal plasma membrane: from axon initial segments to synapses. Int Rev Cel Mol Biol 272:303–389
Tahirovic S, Bradke F (2009) Neuronal polarity. Cold Spring Harb Perspect Biol 1:a001644
Rasband MN (2010) The axon initial segment and the maintenance of neuronal polarity. Nat Rev Neurosci 11:552–562
Kaech S, Banker G (2006) Culturing hippocampal neurons. Nat Protoc 1:2406–2415
Lasiecka ZM, Winckler B (2011) Mechanisms of polarized membrane trafficking in neurons – focusing in on endosomes. Mol Cell Neurosci 48:278–287
Bonifacino JS (2014) Adaptor proteins involved in polarized sorting. J Cell Biol 204:7–17
Hanus C, Ehlers MD (2008) Secretory outposts for the local processing of membrane cargo in neuronal dendrites. Traffic 9:1437–1445
Nixon RA, Cataldo AM (1995) The endosomal-lysosomal system of neurons: new roles. Trends Neurosci 18:489–496
Golgi C (1898) Intorno alla struttura delle cellule nervose. Bollettino della Società Medico-Chirurgica di Pavia 13:3–16
Ashby MC, Ibaraki K, Henley JM (2004) It’s green outside: tracking cell surface proteins with pH-sensitive GFP. Trends Neurosci 27:257–261
González-González IM, Jaskolski F, Goldberg Y, Ashby MC, Henley JM (2012) Measuring membrane protein dynamics in neurons using fluorescence recovery after photobleach. Methods Enzymol 504:127–146
Hildick KL, González-González IM, Jaskolski F, Henley JM (2012) Lateral diffusion and exocytosis of membrane proteins in cultured neurons assessed using fluorescence recovery and fluorescence-loss photobleaching. J Vis Exp 60:e3747
Matlin KS, Simons K (1983) Reduced temperature prevents transfer of a membrane glycoprotein to the cell surface but does not prevent terminal glycosylation. Cell 34:233–243
Saraste J, Kuismanen E (1984) Pre- and post-Golgi vacuoles operate in the transport of Semliki Forest virus membrane. Cell 38:535–549
Bergmann JE (1989) Using temperature-sensitive mutants of VSV to study membrane protein biogenesis. Methods Cell Biol 32:85–110
Presley JF, Cole NB, Schroer TA, Hirschberg K, Zaal KJ, Lippincott-Schwartz J (1997) ER-to-Golgi transport visualized in living cells. Nature 389:81–85
Hirschberg K, Miller CM, Ellenberg J, Presley JF, Siggia ED, Phair RD, Lippincott-Schwartz J (1998) Kinetic analysis of secretory protein traffic and characterization of Golgi to plasma membrane transport intermediates in living cells. J Cell Biol 143:1485–1503
Rivera VM, Wang X, Wardwell S, Courage NL, Volchuk A, Keenan T, Holt DA, Gilman M, Orci L, Cerasoli F Jr, Rothman JE, Clackson T (2000) Regulation of protein secretion through controlled aggregation in the endoplasmic reticulum. Science 287:826–830
Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F (2012) Synchronization of secretory protein traffic in populations of cells. Nat Methods 9:493–498
Boncompain G, Perez F (2014) Synchronization of secretory cargos trafficking in populations of cells. Methods Mol Biol 1174:211–223
Acknowledgement
This work was funded by the Intramural Program of NICHD, NIH (ZIA HD001607).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2016 Springer Science+Business Media New York
About this protocol
Cite this protocol
Farías, G.G., Britt, D.J., Bonifacino, J.S. (2016). Imaging the Polarized Sorting of Proteins from the Golgi Complex in Live Neurons. In: Brown, W. (eds) The Golgi Complex. Methods in Molecular Biology, vol 1496. Humana Press, New York, NY. https://doi.org/10.1007/978-1-4939-6463-5_2
Download citation
DOI: https://doi.org/10.1007/978-1-4939-6463-5_2
Published:
Publisher Name: Humana Press, New York, NY
Print ISBN: 978-1-4939-6461-1
Online ISBN: 978-1-4939-6463-5
eBook Packages: Springer Protocols