

Abstract
Viperin is an interferon-induced cellular protein that is conserved in animals1. It has previously been shown to inhibit the replication of multiple viruses by producing the ribonucleotide 3′-deoxy-3′,4′-didehydro (ddh)-cytidine triphosphate (ddhCTP), which acts as a chain terminator for viral RNA polymerase2. Here we show that eukaryotic viperin originated from a clade of bacterial and archaeal proteins that protect against phage infection. Prokaryotic viperins produce a set of modified ribonucleotides that include ddhCTP, ddh-guanosine triphosphate (ddhGTP) and ddh-uridine triphosphate (ddhUTP). We further show that prokaryotic viperins protect against T7 phage infection by inhibiting viral polymerase-dependent transcription, suggesting that it has an antiviral mechanism of action similar to that of animal viperin. Our results reveal a class of potential natural antiviral compounds produced by bacterial immune systems.
Similar content being viewed by others


Main
Viperin is an antiviral protein that becomes highly expressed in cells stimulated by interferons1. In humans, this protein has broad antiviral activity against DNA and RNA viruses, including human cytomegalovirus, West Nile virus, dengue virus, hepatitis C virus and HIV1,3. Viperin was recently shown to catalyse the conversion of CTP to ddhCTP2. This modified nucleotide lacks a hydroxyl group at the 3′ carbon of the ribose, and when the viral polymerase incorporates it into the nascent chain of the viral RNA, it acts as a chain terminator that does not allow further polymerization of the RNA chain2. Thus, ddhCTP directly inhibits the RNA-dependent replication of RNA viruses such as Zika virus in vivo.
Some bacteria and archaea encode genes that have marked sequence similarity to vertebrate viperins, although their roles have remained unknown4,5,6. We set out to examine whether prokaryotic homologues of human viperin participate in defence against phages. We first performed a profile-based search for viperin homologues in a database of more than 38,000 bacterial and archaeal genomes. This search yielded 1,724 genes (1,112 non-redundant sequences) homologous to the human viperin gene (RSAD2), which we aggregated into 17 clusters on the basis of sequence similarity (Methods) (Supplementary Table 1).
Viperin is a member of the radical S-adenosyl-methionine (SAM) family of enzymes, and shows both sequence and structural homology to other members of that family, particularly the housekeeping molybdenum cofactor biosynthesis enzyme MoaA4. To differentiate between viperin homologues with housekeeping properties and homologues that may participate in defence against phages, we took advantage of the fact that, in prokaryotes, genes involved in antiviral activity tend to colocalize next to one another on the genome, forming ‘defence islands’7,8. Most clusters of viperin homologues did not show a tendency to colocalize with defence genes (Supplementary Table 1). However, in one of the clusters, 60% of the genes were found in the vicinity of CRISPR–cas systems, restriction-modification systems and other bacterial defence genes (Fig. 1a). This high propensity for colocalization with genes involved in defence systems is a strong predictor that the genes in the cluster have a role in phage resistance8,9. We denoted the genes in the defensive cluster pVips (for prokaryotic viperin homologues). As pVips are relatively rare in prokaryotic genomes (164 genes in the cluster), we performed an online homology search with additional genomes that were not included in our original database, retrieving 86 additional such genes, resulting in a total of 250 pVips (Supplementary Table 2).

a, Representative instances of pVip genes and their genomic neighbourhoods. Homologues of human viperin are shown in red, genes annotated as nucleotide kinase are brown, genes known to be involved in defence are yellow and genes with mobile genetic elements are dark grey. RM, restriction modification; TA, toxin–antitoxin; Gabija is a recently described defence system8. The bacterial species, and the accession of the relevant genomic scaffold in the Integrated Microbial Genomes (IMG) database20, are indicated on the left. b, Sizes of plaques caused by T7 phage infecting E. coli strains that express different viperins. Bacteria expressing pVips, negative controls (GFP or MoaA) or human viperin were grown on agar plates and phage lysate was added onto the plates. Graphs show mean of three replicates with individual data points overlaid. Asterisks indicate statistically significant differences compared with the GFP negative control. Two-sided t-test; *P < 0.0001 except for pVip46 (P = 0.0034)). c, Growth curves for E. coli strains expressing viperins that were infected with phage T7. Negative controls are GFP-expressing cells and are the same in all four graphs. Each growth curve represents the mean of 3 biological replicates, each with an average of 2 technical replicates, and the shaded area corresponds to the 95% confidence interval.
To determine whether pVips can defend against phages, we selected 59 genes that span the space of the pVip sequence diversity (Supplementary Table 2) and cloned them in Escherichia coli under the control of an inducible promoter. Similarly, we also cloned GFP and the MoaA gene from E. coli as negative controls. We then challenged the pVip-expressing bacteria with an array of phages spanning several major phage families (Myoviridae: P1; Siphoviridae: lambda, SECphi6, SECphi18 and SECphi27; Podoviridae: T7; Leviviridae: MS2 and Qβ) (Fig. 1b, c, Extended Data Fig. 1).
Around half of the tested pVips conferred clearly identifiable activity against phages. Most of these protected against T7, as evidenced by plaque assays (up to tenfold reduction in T7 plaque sizes) (Fig. 1b) and by a delay or absence of culture collapse in T7 infection assays in liquid culture (Fig. 1c, Extended Data Fig. 2a). Mutations in the cysteine residues predicted to coordinate the iron–sulfur cluster in the CxxxCxxC motif in pVips resulted in loss of defensive capacity against T7, suggesting that the catalytic activity of pVips is necessary for defence (Extended Data Fig. 2b). A subset of the pVips also protected against P1, lambda, SECphi6 and SECphi18 phages, reducing the observed number of plaques by between 10-fold and 10,000-fold (Extended Data Fig. 1). Of note, when the human viperin gene was cloned and expressed in E. coli under the same conditions, it protected against T7 infection in a similar manner to that observed for many pVips (Fig. 1, Extended Data Figs. 1, 2).
The pVips that we identified are present in phylogenetically distant organisms, suggesting an ancient evolutionary origin, frequent horizontal gene transfer or both. We found pVips in 176 species overall, belonging to 14 bacterial and archaeal phyla that include Proteobacteria, Firmicutes, Cyanobacteria, Actinobacteria, Bacteriodetes, Euryarchaeota and others (Supplementary Table 2). To better understand their diversity and phylogenetic distribution, we generated a phylogenetic tree of the viperin family, including pVips and eukaryotic viperins (Fig. 2, Supplementary Table 3). MoaA genes from bacteria and eukaryotes were added to the tree as an outgroup. We found that pVips are grouped into seven major clades that partially follow the phyletic grouping of the encoding microorganisms (Fig. 2). All eukaryotic viperins form a monophyletic clade within the tree, with the closest common ancestor predicted to localize to pVip clade 2, which is mostly composed of pVips from archaeal species. The clear monophyletic organization of the eukaryotic viperin clade and its position within the pVips tree suggest that the eukaryotic viperin was acquired from prokaryotes, via a single event in the ancient history of the eukaryotic lineage.

Branches are coloured according to major clades. Bootstrap values (derived from the ultrafast bootstrap function in IQtree21) are indicated for major nodes. The presence of a nucleotide kinase in the genomic vicinity of the pVip is shown by a brown rectangle in the surrounding ring (or a dark grey rectangle, in cases in which the kinase is fused to the pVip gene). Triangles correspond to the type of ddh-nucleotide derivative produced by a specific pVip, as determined by mass spectrometry. The phylogenetic tree was generated using a set of 205 non-redundant pVip sequences.
In vertebrate genomes, the viperin gene is frequently located adjacent to a cytidylate kinase gene that is co-expressed with the viperin during the interferon response2,10. This kinase phosphorylates cytidine monophosphate to CTP, thus generating the substrate for viperin activity2. We found that 47 of the 250 pVips (19%) genes were adjacent to genes annotated as a nucleotide kinase in their genome of origin (Fig. 1a, Fig. 2) and that, in some cases, the kinase was fused to the pVip gene (Fig. 2, Supplementary Table 2). This further strengthens the hypothesis that the pVip substrate is a triphosphorylated nucleotide. Whereas some pVip-associated kinases were annotated as cytidylate kinases, as in vertebrates5, others were annotated as thymidylate or other kinases, suggesting that the substrates of some pVips might be triphosphorylated nucleotides other than CTP.
The animal viperin catalyses the production of ddhCTP2. We therefore investigated whether pVips produce ddhCTP and/or other modified nucleotides. We expressed pVips in E. coli and then extracted the fraction containing small molecules from the cell lysates, presuming that the product of the pVips would be in this fraction. We analysed these lysates using liquid chromatography followed by mass spectrometry (LC–MS) with an untargeted approach. As a positive control, we analysed cell lysates from cells expressing the human viperin protein. As expected, a compound consistent with the mass of ddhCTP was readily detected in lysates from cells expressing human viperin, but not in negative-control lysates derived from MoaA-expressing cells (Extended Data Figs. 3, 4). Other compounds found in the human viperin sample matched the masses of ddh-cytidine (ddhC) and ddh-cytidine monophosphate (ddhCMP), possibly derived from natural decay of ddhCTP, as reported to occur for CTP at neutral or acidic pH11. Analysis of fragment ions using tandem mass spectrometry (MS/MS) provided further evidence that the identified masses were probably derivatives of ddhCTP, which was additionally confirmed by analysing a ddhC standard using MS/MS (Extended Data Figs. 3a, 4a, 5). These results confirm that human viperin produces ddhCTP when expressed in E. coli, explaining its observed antiphage activity (Fig. 1).
We then analysed the small-molecule fraction from lysates of cells expressing 27 pVips that were found to have antiphage activity. Derivatives of ddhCTP (including ddhC, as verified by LC–MS with the synthesized ddhC chemical standard) were detected by LC–MS in the lysate of pVip50 (Fig. 3a, Extended Data Fig. 4), a protein derived from a methanogenic archaeon that is localized in clade 2 of the pVip tree, verifying that pVips are indeed functional homologues of human viperin. Derivatives of ddhCTP were detected in lysates of cells expressing several additional pVips, but for most pVips we could not detect ddhCTP or its derivatives in cell lysates expressing them. We therefore searched for other compounds that were markedly enriched in lysates of cells expressing pVips and absent from the negative-control lysate. For ten of the pVips we found masses that conform with ddhGTP and ddh-guanosine monophosphate (ddhGMP) (Fig. 3, Extended Data Fig. 4). In addition, for 15 pVips we found other molecules with masses matching ddhUTP and ddh-uridine monophosphate (ddhUMP) (Fig. 3, Extended Data Fig. 4). MS/MS analysis of fragment ions from the masses predicted as ddhGTP, ddhUTP and their monophosphorylated derivatives further supported that they most probably correspond to these molecules (Extended Data Fig. 5).

a, Top, structures of ddh derivatives. R represents H, monophosphate or triphosphate. Bottom, extracted-ion chromatograms for selected pVip lysates analysed by LC–MS. Chromatograms show singly charged masses with a precision of ±5 ppm corresponding to ddhC (m/z 226.0822, retention time (RT) of 2.2 min), ddhCMP (m/z 306.0486, RT 9.7 min), ddhCTP (m/z 465.9812, RT 10.7 min), ddhUMP (m/z 307.0326, RT 8.7 min), ddhUTP (m/z 466.9652, RT 9.9 min), ddhGMP (m/z 346.0547, RT 9.8 min) and ddhGTP (m/z 505.9874, RT 10.7 min). Normalization was performed on all pVips and MoaA (negative control) samples, with maximum values set to 1.0. Peaks assigned to ddh-nucleotides are in black. Peaks that appear in the negative controls and are not assigned to ddh-nucleotides are in grey. Results are representative of three replicates. b, Production of ddh-nucleotide derivatives by pVips. Coloured boxes depict compounds detected in the small-molecule fraction of lysates of E. coli expressing pVips. Coloured rectangles on the left and associated numbers represent the clade of pVips as described in Fig. 2. Euk, eukaryote. c, Chromatograms of ddh-nucleotides detected in reactions performed in vitro with purified pVips. The presence of a product corresponding to ddhCTP, ddhUTP and ddhGTP is observed in samples in which a pVip was incubated with SAM, dithionite and the respective nucleotide substrate. Results representative of two replicates.
To confirm that pVips convert the nucleotide substrates CTP, GTP and UTP to their ddh variants, we performed in vitro biochemical assays with purified pVip enzymes. Three isolated recombinant pVips, pVip6, pVip8 and pVip56 were incubated with the SAM cofactor and a nucleotide substrate, in the presence of an artificial electron donor (dithionite). LC–MS analysis confirmed the appearance of the expected ddh-ribonucleotide products (ddhCTP, ddhUTP and ddhGTP for pVip6, pVip8 and pVip56, respectively) in the reaction samples relative to control reactions without nucleotide substrate (Fig. 3c). Detailed analysis of the MS–MS fragmentation spectra of each compound further supported that these products correspond to ddh-ribonucleotides (Extended Data Fig. 6). Together, these results suggest that pVips produce antiviral ribonucleotides.
For most pVips, predicted derivatives of only one modified nucleotide were observed in the lysate (either ddhCTP, ddhGTP or ddhUTP). However, 11 pVips were found to produce derivatives of multiple ddh-ribonucleotides (Fig. 3). For five of the tested pVips, we did not detect any ddh-nucleotide or derivatives in the cell lysates, despite these pVips conferring antiviral activity to the bacteria that express them (Extended Data Fig. 1, Fig. 3). These pVips may produce a different antiviral molecule that could not be detected with our LC–MS protocol or could have evolved to confer defence by a mechanism that does not involve the production of antiviral molecules.
pVips produce ddh-ribonucleotides, but also protect against phages that have double-stranded DNA genomes (for example, T7) (Fig. 1). We therefore hypothesized that the products of these pVips affect phage-dependent transcription rather than DNA replication. In support of this hypothesis, it was previously shown that in mammalian cells, T7 RNA polymerase-dependent transcription of GFP was impaired if human viperin was co-expressed in the same cells12. We thus sought to examine whether T7 polymerase-dependent RNA synthesis is affected by pVips. We transformed E. coli BL21 DE3 with a plasmid that encodes a GFP reporter gene under the control of a T7 promoter (Fig. 4a). As expected, induction of the T7 RNA polymerase in these cells using isopropyl β-d-1-thiogalactopyranoside (IPTG) led to accumulation of a fluorescent GFP signal (Fig. 4b). When pVips or human viperin were co-expressed in the same cells, the GFP signal was repressed (Fig. 4c–f). Repression of GFP expression mediated by T7 RNA polymerase was observed upon expression of pVips producing ddhGTP (pVip60), ddhUTP and ddhCTP (pVip8 and pVip9), or ddhCTP (the human viperin) suggesting that T7 RNA polymerase is sensitive to multiple types of modified ribonucleotides.

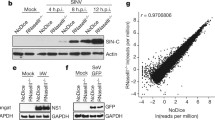
a, Schematic of the reporter system for T7 polymerase-dependent transcription. E. coli BL21 DE3 encodes a chromosomal T7 RNA polymerase (T7 RNAP) under the control of an IPTG-inducible promoter. A reporter plasmid encodes GFP under the control of a T7 promoter. Upon IPTG induction, the T7 RNA polymerase is expressed and drives the expression of GFP. The pVip (or MoaA control) is encoded on a second plasmid under the control of an arabinose (Ara) promoter. b–f, Application of the reporter assay for strains expressing MoaA (negative control), human viperin and pVips. Strains were first induced with arabinose for 45 min to express the pVip. At t = 0, IPTG was added to induce GFP expression. Curves show GFP fluorescence/optical density over time. Each curve shows the mean of two technical replicates and the shaded area shows the 95% confidence interval. Representative of two biological replicates. g, GFP expression measured by RNA-seq. GFP expression in reads per kilobase of transcript per million mapped reads (RPKM) in cells expressing viperins was compared to that in cells expressing the MoaA negative control. Data represent GFP expression as a percentage of GFP expression in cells expressing MoaA. The graph shows the average of two biological replicates, with individual data points overlaid.
Notably, when the above experiments were conducted with pVips mutated to inactivate their active sites, no repression of GFP expression was observed, implying that the catalytic activity of pVips and the production of the ddh-ribonucleotide products is required to inhibit T7 RNA polymerase-dependent expression (Extended Data Fig. 7a).
To more directly confirm that the effect of pVips on T7 polymerase-dependent GFP expression was caused by reduced RNA synthesis, we investigated GFP RNA levels using RNA sequencing (RNA-seq). RNA was extracted 1 h after T7-mediated induction of GFP expression in cells that co-express pVips or human viperin. We observed significant reductions in GFP RNA expression (as measured by RPKM) (Methods) when the pVips or human viperin were expressed in the cell, compared with control cells that expressed MoaA (Fig. 4g). The expression levels of genes driven by induced endogenous promoters (specifically, the T7 RNA polymerase gene itself) (Extended Data Fig. 7b) did not show marked changes during pVip expression, further supporting the idea that pVips specifically target transcription by the phage polymerase. Together, these results suggest that pVips defend against phage T7 by suppressing transcription by the viral RNA polymerase, presumably because their products form RNA chain terminators. Notably, expression of pVips does not appear to be toxic to E. coli (Extended Data Fig. 8), indicating that bacterial RNA polymerase may be less sensitive than T7 RNA polymerase to ddh-ribonucleotides. Similarly, ddhCTP produced by human viperin was previously shown to be non-toxic to human cells2.
Bacterial antiphage defence mechanisms frequently occur as multi-gene defence systems, with some genes in the system being responsible for identifying the invading phage and others functioning in mitigating the infection9,13,14. Most of the pVips that we examined formed part of a conserved cassette of genes (Extended Data Fig. 9). The most common configuration included two other genes in addition to pVip: a gene comprising an ankyrin repeat domain and a gene encoding a predicted HicA-like RNase (Extended Data Fig. 9). Ankyrin repeat domains are common biological recognition motifs involved in protein–protein interactions, and the ankyrin repeat domain may serve as a sensor for phage infection. It is possible that, following phage sensing, the protein containing the ankyrin repeats could activate expression of pVip in a manner conceptually similar to interferon-mediated activation of human viperin expression. The associated RNase may be responsible for degradation of prematurely terminated phage RNAs or some other auxiliary function.
Our data suggest that pVips protect against T7 infection by inhibiting transcription from the viral RNA polymerase. However, some pVips protect against lambda and P1 phages, which do not encode their own RNA polymerase and rely on the host polymerase for transcription15. It is possible that pVips inhibit phage lambda in a manner that is independent of the production of ddh-nucleotides, as suggested for viperin in the context of human viral infection16.
Small molecules of the anthracycline family, produced by Streptomyces species, have been shown to possess antiphage properties and efficiently inhibit phage replication, presumably through intercalation into phage DNA17. The identification of pVips reveals another strategy of chemical defence against phages and suggests that the use of small molecules for defence against phages may be a more common antiviral strategy than previously anticipated.
Many of the most potent antiviral drugs used in the clinic are synthetic nucleoside chain terminators. These include aciclovir, a drug commonly used against herpes viruses18, 3′-azido-3′-deoxythymidine (AZT), an anti-HIV drug used clinically since the 1980s18, and sofosbuvir, which is part of a successful treatment for hepatitis C19. The chain terminators described in our study could potentially be adopted for clinical treatment of human viruses. Moreover, if pVips turn out to be just one example of a chemical defence strategy widely used in bacteria, then bacteria may prove to host a repository of diverse antiviral molecules that could be adopted for clinical use. Thus environmental bacteria, which have long been used in the discovery of novel antibiotics, may once again serve in the battle against human pathogens.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
A search for viperin homologues in prokaryotic genomes
The human viperin protein sequence (NCBI accession NP_542388.2) was searched against the protein sequences of all genes in 38,167 bacterial and archaeal genomes downloaded from the IMG database20 in October 2017, using the ‘search’ option in the MMseqs2 package22 (release 6-f5a1c) with default parameters (3 iterations), as previously described9. Hits with an e-value higher than 10−5 were discarded. The resulting set of proteins was clustered using the ‘cluster’ option of MMseqs2 release v.6-f5a1c, with sensitivity parameter of ‘-s 7.5′, coverage parameter 60% and the remaining parameters being the default parameters (Supplementary Table 1). For each cluster, the fraction of genes associated with known defence genes was computed as previously described9. Additional candidate prokaryotic viperin homologues (pVips) were searched manually using the ‘top IMG homologs’ function in IMG for the identified genes in the cluster of pVips.
To generate the phylogentic tree, the protein sequences of prokaryotic viperins, eukaryotic viperins and MoaA sequences were aligned using mafft23 (v.7.402, default parameters). The sequences of the eukaryotic viperins and MoaA proteins used in the tree are provided in Supplementary Table 3. The tree was computed with IQ-TREE21 multicore v.1.6.5 (option –m TESTNEW in IQ-TREE). The phylogenetic model LG+R6 was used because it gave the lowest Bayesian information criterion among all models available for the tree. One thousand ultra-fast bootstraps were performed to evaluate node support (options –bb 1000 –wbtl in IQ-TREE). The online tool iTOL24 (v.5) was used for tree visualization.
Eukaryotic viperin sequences used in the phylogenetic tree were chosen as follows. A homology based-search was performed on the non-redundant eukaryotic proteins database of NCBI using HMMER 3.2.125 in the MPI bioinformatics toolkit26 with 205 non redundant pVips as a seed. This search yielded 4,915 hits that were used to build an initial phylogenetic tree. The sequences of pVips, MoaA and these hits were aligned using mafft23 (v.7.402, default parameters). The tree was computed with IQ-TREE21 multicore v.1.6.5 (option –m TESTNEW in IQ-TREE). On this tree, all the pVips were found in a monophyletic clade that also comprised 1,298 eukaryotic sequences (Extended Data Fig. 10). These 1,298 eukaryotic protein sequences were then used to build a second phylogenetic tree. Sequences of these 1,298 eukaryotic proteins, pVips and MoaA were aligned using mafft23 (v.7.402, default parameters). The tree was computed with IQ-TREE21 multicore v.1.6.5 (option –m TESTNEW in IQ-TREE). All the eukaryotic viperin sequences represented a monophyletic clade that is internal to the pVips clades. Representative eukaryotic sequences for the tree in Fig. 2 were then chosen to span the diversity of the eukaryotic viperin homologues, including sequences from mammals (human, dog and bat), animals such as mollusca and fungi (Extended Data Fig. 10).
Bacterial strains and growth conditions
E. coli strains (MG1655, Keio ΔiscR27, Keio ΔiscR-F+, DH5α, BL21 DE3, BL21 ΔiscR) were grown in LB or LB agar at 37 °C unless mentioned otherwise. Whenever applicable, media were supplemented with ampicillin (100 μg ml−1), kanamycin (50 μg ml−1), chloramphenicol (30 μg ml−1) or tetracycline (10 μg ml−1) to ensure the maintenance of plasmids.
Plasmids and strain construction
Primers used in this study are shown in Supplementary Table 4. pVip genes were codon-optimized for expression in E. coli and synthetized by Twist Bioscience (pVips 1–14) or by Genscript (pVips 15–63, MoaA control and human viperin). Synthetized pVip sequences are indicated in Supplementary Table 2. Each candidate sequence was cloned in plasmid pBad/His A (Thermofisher, Catalog number 43001). For pVips 1–14, flanking sequences were added to the synthesis for cloning purposes (upstream: GGATCCTACCTGACGCTTTTTATCGCAACTCTCTACTGTTTCTCCATACCCGTTTTTTGGGCTAACAGGAGGAATTAACC, downstream: TAAGAATTCCCAGGCATCAAATAAAACGAAAGGCTCAGTCGAAAGACTGGGCCTTTCGTTTTATCTGTTGTTTGTCGGTGAACGCTCTCCAGCTGGTACCATATGG) the vector fragment was generated by PCR (template pBad/His A, primer AB1 and AB2), and for each pVip, overhang sequences (matching primers AB3 and AB4) overlapping the plasmid backbone were added to allow Gibson assembly. pVips 1–14 were amplified by PCR (using the template of synthetized DNA, primers AB3 and AB4), and PCR fragments of pVip and plasmid backbone were joined using Gibson assembly. For pVips 15–63 and the MoaA, DNA synthesis and cloning into pBad/His A was performed by Genscript. The codon-optimized protein-coding sequence of the human viperin was synthesized and cloned into pBad/His A by Genscript. The sequence was then modified by PCR and Gibson assembly to remove the endoplasmic-reticulum-targeting sequence of the human viperin protein (residues 1–50, primers AB86, AB87, AB88 and AB89). All experiments involving human viperin were performed with this shortened version.
All of the pVip plasmids were initially cloned and propagated in E. coli DH5α and then purified and transformed into the Keio ΔiscR27 strain. Because pVips are iron–sulfur cluster proteins, they necessitate active production of iron–sulfur clusters for their enzymatic activity2. We therefore conducted the experiments in E. coli strains deleted for iscR, a repressor of iron–sulfur cluster production in E. coli28,29. For experiments involving phages Qβ and MS2, the F plasmid is necessary for infection. Thus, the Keio ΔiscR F+ strain was constructed by conjugation of strain Keio ΔiscR with strain top10 F+ and then later used as the relevant genetic background.
For protein purification, codon-optimized pVip genes were amplified by PCR (Supplementary Table 4) and cloned into the aTc-inducible expression vector pASG-IBA143 (IBA Lifesciences, pIBA143_vector_F, pIBA143_vector_R), for fusion of a Twin-Strep-tag to the C terminus of the pVips (pIBA143_VipX_F, pIBA143_VipX_R). To construct the suf operon expression vector pSuf, the complete suf operon (sufABCDSE) was amplified from E. coli MG1655 genomic DNA, and was cloned into the pACYC-184 (NEB) backbone (Suf_operon F, Suf_operon R) together with the arabinose expression system from pBad/His A vector (Thermofisher, Catalog number 43001). Tagged pVip8 and pVip56 were transformed in BL21 ΔiscR and tagged pVip6 was transformed in BL21 pSuf.
For experiments involving the GFP reporter assay, strains were constructed as follows. BL21 DE3 was knocked out for the iscR gene through P1 transduction, with P1-ΔiscR phages propagated from strain Keio ΔiscR27 followed by kanamycin selection. The final reporter plasmid, pAB151, was constructed to encode: (1) GFP under the control of T7 promoter; (2) a gene cassette encoding the T7 lysozyme to limit the activity of T7 RNA polymerase; (3) an insulator sequence between the chloramphenicol resistance gene and the GFP gene. pAB151 was constructed through three consecutive Gibson assemblies, each of which used two PCR fragments as described below. The first reaction used insert template pDR111 (provided by I. Kolodkin-Gal (Weizmann Institute of Science)) with primers OG630 and OG631, and vector template pACYC (Novagene), and primers OG629 and OG628, to generate plasmid pAB137. The second Gibson reaction used insert template pLysS (Novagene) with primers AB55 and AB56, and vector template pAB137 with primers AB53 and AB54, to generate pAB138. The third reaction used insert template pSG18 with primers AB121 and AB122, and vector template pAB138 with primers AB119 and AB120, to generate pAB151 (Supplementary Table 4).
For the design of the inactive mutants of human viperin, pVip8, pVip9 and pVip60, the conserved closely spaced cysteine residues in the CxxxCxxC motif, which coordinate the iron-sulfur cluster30, were mutated (Human viperin C32A, C36A, C39A; pVip8: C22A, C26A, C29A; pVip9: C17A, C21A, C24A; pVip60: C192A, C196A, C199A). Mutants were constructed using Q5 Site directed Mutagenesis kit (NEB) using primers presented in Supplementary Table 4 (AB156-AB163).
Plaque assays
Phages were propagated on E. coli MG1655 using the plate lysate method as previously described31. Lysate titre was determined using the small drop plaque assay method as previously described32. Phages used in this study are presented in Supplementary Table 5.
Plaque assays were performed as previously described32. Bacteria from overnight cultures were mixed with MMB agar (LB, 0.1 mM MnCl2, 5 mM MgCl2 and 0.5% agar) supplemented with arabinose (final concentration 0.004%) for induction of pVip expression. Serial dilutions of phage lysate in MMB were dropped on top of the agar plates. After the drops dried up, plates were incubated at 37 °C overnight.
Infection dynamics in liquid medium
Overnight cultures were diluted 1:100 in MMB medium and incubated at 37 °C with shaking at 250 rpm for 45 min, at which point arabinose was added to a final concentration of 0.2%. Cells were then incubated at 37 °C with shaking at 250 r.p.m. for 45 min. A 180-μl aliquot of the diluted cultures was transferred into wells in a 96-well plate containing 20 μl of phage lysate for a final multiplicity of infection (MOI) of 0.001. Infections were performed with technical duplicates and OD600 was monitored using a TECAN Infinite 200 plate reader with measurements every 5 min.
Colony-forming unit (CFU) counts were measured using the same experimental setup and time points as above. Ten microlitres of cells were taken right after dilution (time 0), before induction (45 min), and 45 and 90 min after induction of pVip expression, serially diluted and plated on selective agar plates. CFUs were counted after overnight incubation at 37 °C.
Cell lysates preparation
Overnight cultures of E. coli Keio ΔiscR encoding pVips, human viperin, and MoaA and GFP negative controls were diluted 1:100 in 100 ml LB medium and grown at 37 °C (250 rpm) for 1 h 45 min. The expression of viperin or MoaA was induced by the addition of arabinose (final concentration 0.2%) and cells were further incubated at 37 °C (250 rpm) for 1 h. Cells were then centrifuged at 3,900 rpm for 10 min at 4 °C and samples kept on ice throughout the cell lysate preparation. Pellets were resuspended in 600 μl PBS buffer containing 100 mM sodium phosphate (pH 7.4). The resuspended pellet was supplemented with 1 μl of hen egg lysozyme (Merck) (final hen egg lysozyme concentration of 10 μg ml−1). The resuspended cells were then mixed with Lysing matrix B (MP Biomedicals) and beads and cells were disrupted mechanically using a FastPrep-24 bead-beater device (MP Biomedicals) (2 cycles of 40 s, 6 m s−1, at 4 °C). Cell lysates were then centrifuged at 12,000g for 10 min at 4 °C and the supernatant was loaded onto a 3-kDa filter Amicon Ultra-0.5 centrifugal filter unit (Merck) and centrifuged at 14,000g for 30 min at 4 °C. The resulting flow-through, containing substances smaller than 3 kDa, was used as the lysate sample for evaluating the presence of ddh-nucleotides by LC–MS.
Detection of ddh-nucleotides in cell lysates
Sample analysis was carried out by MS-Omics (Vedbæk) as follows. Samples were diluted 1:1 in 10% ultra-pure water and 90% acetonitrile containing 10 mM ammonium acetate at pH 9 then filtered through a Costar Spin-X centrifuge tube filter 0.22-μm nylon membrane. The analysis was carried out using a UHPLC system (Vanquish, Thermo Fisher Scientific) coupled with a high-resolution quadrupole-orbitrap mass spectrometer (Q Exactive HF Hybrid Quadrupole-Orbitrap, Thermo Fisher Scientific) at a resolution of 120,000 (at 200 m/z). An electrospray-ionization interface was used as ionization source. Analysis was performed in positive ionization mode from 200 to 1,000 m/z at a scan rate of 3 Hz. The UPLC was performed using a slightly modified version of the previously described protocol33. Peak areas were extracted using Compound Discoverer 3.0 (Thermo Fisher Scientific).
MS/MS of ddh-nucleotides was acquired using the same instrument with an inclusion list of the different ddh-nucleotide and ddh-nucleoside masses at a resolution of 30,000. Fragmentation was done through a higher-energy collisional dissociation cell using a normalized collision energy of 20, 40 and 60 eV in which the spectrum is the sum of each collision energy. Intensity threshold was set to 2 × 104, isolation window of 0.4 m/z and injection time of 100 ms. Analysis of ddhCTP and ddhGTP derivatives was performed in positive ionization mode, and for ddhUTP derivatives in negative ionization mode.
Raw data files were processed by Compound Discoverer 3.0 software. Unknown compounds were detected with a 3 ppm mass tolerance, signal to noise ratio of 3, 30% of relative intensity tolerance for isotope search, and minimum peak intensity of 5 × 105. The compounds were grouped with a 5 ppm mass and 0.2 min retention time tolerances. Blank samples were used to remove background noise, and annotated peaks that were 5 times higher than the blanks were kept. Metabolites identified were searched against ChemSpider chemical structure database with 3 ppm mass tolerance, mzCloud spectral library with a precursor and fragment mass tolerance of 3 and 5 ppm respectively, and an internal MS/MS library through mzVault with the same tolerance as mzCloud. Two data sources were searched in the ChemSpider database: Human Metabolome Database (HMDB) and E. coli Metabolome Database (ECMDB).
The raw data files for the MS and MS/MS data in this section, as well as additional technical details, are available for download on the Metabolights repository under study number MTBLS1750.
Quantification of ddhC
The ddhC molecule was synthesized by Jena Bioscience at a purity of 97.5% and was used as a standard for ddhC quantification in cell lysates using LC–MS. Sample analysis was carried out by MS-Omics as follows. Samples were diluted 1:1 in 10 mM ammonium formate and 0.1% formic acid in ultrapure water. The analysis was carried out using the LC–MS setup described above. An electrospray ionization interface was used as ionization source performed in positive ionization mode. The UHPLC method is based on Waters Application note 2011, 720004042en (Waters). Peak areas of ddhC were extracted using Trace Finder v.4.1 (Thermo Fisher Scientific) and quantified using an external calibration with the standard.
pVips purification and in vitro enzymatic assays
Overnight cultures of BL21-ΔiscR (pVip8 and pVip56) or BL21 pSuf (pVip6) cells freshly transformed with plasmids encoding the tagged pVip, were seeded at an initial OD600 of ~0.06 in 1–2 l of selective LB medium. pSuf expression was induced at OD600 0.2–0.3 (0.2% arabinose, 100 μM FeCl3, 100 μM l-cysteine). pVip expression was induced at OD600 0.6–0.8 (50 ng ml−1 anhydrotetracycline (aTc)) and incubated at 37 °C with shaking for 3–4 h. Pellets were then collected by centrifugation and stored at −20 °C.
Frozen cell pellets were resuspended in cold lysis buffer (50 mM Tris·HCl, 500 mM NaCl and 5 mM dithiothreitol (DTT), 0.5 M arginine, and 20% glycerol), and sonicated with a Branson Sonifier (15 s on, 45 s off, 10 min total on, 30% amplitude) on ice. Lysates were subjected to centrifugation for 30 min at 17,000g and 4 °C. The lysate was loaded onto a StrepTactin Superflow High Capacity (IBA Lifesciences) column, previously equilibrated with 20 column volumes of buffer W (100 mM Tris-HCl pH 8, 300 mM NaCl, 5 mM DTT, 10% glycerol). The column was washed twice with 10 column volumes of buffer W and eluted with buffer E (50 mM Tris-HCl pH 8, 300 mM NaCl, 5 mM DTT, 2.5 mM desthiobiotin, 20% glycerol). The presence of the pVip proteins in the resulting fractions was confirmed by SDS–PAGE. Purified proteins were frozen in liquid nitrogen and stored at – 80 °C.
Protein reconstitution
Purified protein solutions were thawed on ice and introduced into in an MBraun anaerobic chamber maintained at <0.1 ppm oxygen. All subsequent steps were performed in anaerobic conditions at 12 °C. Purified pVips were incubated for 1 h with 50 mM DTT with gentle shaking. Protein solutions were supplemented with eightfold molar excess Fe(NH4)2(SO4)2, incubated for 15 min with gentle shaking, followed by the addition eightfold molar excess of Na2S droplet by droplet. After incubation for 3–4 h to overnight with slow shaking, the reconstituted pVips were transferred to the Reaction Buffer (50 mM HEPES pH 7.5, 150 mM KCl, 5 mM DTT, 20% glycerol) using PD-10 desalting columns (GE Healthcare) and concentrated using an Amicon Ultra centrifugal 10-kDa filter to a final protein concentration of 20–50 μM. Proteins were then flash-frozen with liquid nitrogen and stored at –80 °C.
In vitro enzymatic assays
For pVip6 and pVip8, reactions were performed in a total volume of 100 μl containing: 20–50 μM reconstituted enzyme in reaction buffer (2 mM SAM, 1 mM of nucleotide substrate and 5 mM sodium dithionite). Reactions were carried out inside the anaerobic chamber maintained at <0.1 ppm oxygen. A 10-μl aliquot was removed from the reaction mixture (sample before reaction). Reactions were then initiated with sodium dithionite and incubated at 37 °C for 1–2 h. After incubation, samples were taken out of the anaerobic chamber and stored at −80 °C until analysis.
For pVip56, to obtain sufficient amounts of ddhGTP for MS/MS analysis, an enzymatic reaction in a total volume of 1ml was performed, containing 113 mM pVip56, 2 mM SAM, 2 mM GTP and 5 mM dithionite in reaction buffer. Reactions were carried out in anaerobic conditions as previously described and incubated at 37 °C for 3 h. To remove the protein, 10-kDa centrifugal filters were used. The flow through was diluted twofold into cold 10 mM ammonium bicarbonate buffer pH 9.0 (buffer A), then loaded onto Capto HiRes Q 5/50 (GE Healthcare) pre-equilibrated with buffer A. The column was washed with 25 ml buffer A and elution was performed using linear elution gradient (100 ml) of 200 mM to 800 mM ammonium bicarbonate, pH 9. The purified product was lyophilized and resuspended in water before LC–MS analysis.
LC–MS analysis of in vitro assays
LC–MS measurements were performed with a Thermo Scientific Q Exactive Orbitrap mass spectrometry system equipped with a Dionex Ultimate 3000 UHPLC system. The software Thermo Xcalibur (v.4.2) was used for instrument control and data processing. Before analysis, 10 μl of sample from enzymatic assays were mixed with 40 μl of acetonitrile:methanol organic mixture (5:3 v/v ratio). The mixtures were vortexed, centrifuged at 17,000g for 2 min and 3 μl of supernatant was injected onto an SeQuant ZIC-pHILIC 5-μm polymeric 100 × 2.1 mm HPLC column. The mobile phase was composed of 20 mM ammonium carbonate pH 9.5 (solvent A) and 100% acetonitrile (solvent B). Samples were separated using a constant flow rate of 0.2 ml min−1: 80% solvent B was held for 2 min, followed by a gradient from 80% to 20% of solvent B for 15 min, before immediately returning to 80% solvent B for equilibration for 9 min. Data analysis was performed using Thermo Scientific FreeStyle software (v.1.6).
T7 dependent GFP expression assay
Overnight cultures of BL21 DE3 ΔiscR cells containing pAB151 and pVip-encoding plasmids (or plasmids encoding MoaA or the human viperin) were diluted 1:100 in LB medium and incubated in a 96-well plate format at 37 °C with shaking at 250 rpm until OD600 reached 0.1. Arabinose was then added to a final concentration of 0.2%. After 45 min of incubation at 37 °C and 250 rpm, the expression of T7 RNA polymerase was induced by the addition of IPTG to a final concentration of 0.1 mM. Fluorescence levels (wavelength excitation 488 nm, emission 520 nm) and cell density (OD600) were monitored using TECAN Infinite 200 plate reader with measurements every 15 min.
Quantification of GFP transcripts using RNA-seq
BL21 DE3 ΔiscR cells containing pAB151 and pVip-encoding plasmids (or plasmids encoding MoaA or the human viperin) were diluted 1:100 in 5 ml LB medium supplemented with antibiotics (chloramphenicol, kanamycin and ampicillin). These cells were grown at 37 °C with shaking at 250 rpm to OD600 of 0.3 and expression of the viperin (or MoaA) protein was induced by the addition of arabinose (final concentration 0.2%). After 45 min of incubation at 37 °C and 250 rpm, the expression of T7 RNA polymerase was induced by the addition of IPTG to a final concentration of 0.1 mM. After 1 h, samples were centrifuged for 10 min at 4,000 rpm at 4 °C. The supernatant was discarded, and pellets were used for RNA extraction. Bacterial pellets were lysed using TRIzol and phenol-chloroform. Bacterial pellets were treated with 100 μl of 2 mg ml−1 lysozyme (in Tris 10mM EDTA 1mM pH 8.0) and incubated at 37 °C for 5 min. One millilitre of TRI-reagent was added, samples were then vortexed for 10 s before addition of 200 μl chloroform. Following another vortexing step, the samples were left at room temperature for 5 min to allow phase separation and then centrifuged at 12,000g, 4 °C for 15 min. The upper phase was added to 500 μl of isopropanol. Samples were then incubated overnight at −20 °C. Finally, following 30 min of centrifugation at 12,000g at 4 °C, samples were washed twice with ice cold 70% ethanol, and resuspended in 50 μl water. RNA levels were measured using Nanodrop. All RNA samples were treated with TURBO DNase (Life technologies, AM2238). Ribosomal RNA depletion and RNA-seq libraries were prepared as previously described34, except that all reaction volumes were reduced by a factor of 4.
RNA-seq libraries were sequenced using Illumina NextSeq platform, Reads were mapped as previously described34 to the reference genome of E. coli BL21 DE3 (NC_012892) as well as the plasmids present in the relevant strain (pAB151 and plasmids encoding pVip/MoaA/human viperin). RNA-seq-mapped reads were used to generate reads-per-gene and RPKM counts.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Data that support the findings of this study are available within the article and its Extended Data and Supplementary Tables. IMG accessions, protein sequences and nucleotide sequences when relevant to the pVips appear in the Methods and Supplementary Table 2. Mass spectrometry data are available on Metabolights under study number MTBLS1750.
References
Rivera-Serrano, E. E. et al. Viperin reveals its true function. Annu. Rev. Virol. 7, 1–26 (2020).
Gizzi, A. S. et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 558, 610–614 (2018).
Helbig, K. J. & Beard, M. R. The role of viperin in the innate antiviral response. J. Mol. Biol. 426, 1210–1219 (2014).
Fenwick, M. K., Li, Y., Cresswell, P., Modis, Y. & Ealick, S. E. Structural studies of viperin, an antiviral radical SAM enzyme. Proc. Natl Acad. Sci. USA 114, 6806–6811 (2017).
Fenwick, M. K., Su, D., Dong, M., Lin, H. & Ealick, S. E. Structural basis of the substrate selectivity of viperin. Biochemistry 59, 652–662 (2020).
Honarmand Ebrahimi, K., Rowbotham, J. S., McCullagh, J. & James, W. S. Mechanism of diol dehydration by a promiscuous radical-SAM enzyme homologue of the antiviral enzyme viperin (RSAD2). ChemBioChem 21, 1605–1612 (2020).
Makarova, K. S., Wolf, Y. I., Snir, S. & Koonin, E. V. Defense islands in bacterial and archaeal genomes and prediction of novel defense systems. J. Bacteriol. 193, 6039–6056 (2011).
Doron, S. et al. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 359, eaar4120 (2018).
Cohen, D. et al. Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695 (2019).
Kambara, H. et al. Negative regulation of the interferon response by an interferon-induced long non-coding RNA. Nucleic Acids Res. 42, 10668–10680 (2014).
Thiem, J. & Stangier, P. Preparative-enzymatic formation of cytidine 5′-monophosphosialate by integrated cytidine 5′-triphosphate regeneration. Liebigs Ann. Chem.1101–1105 (1990).
Dukhovny, A., Shlomai, A. & Sklan, E. H. The antiviral protein Viperin suppresses T7 promoter dependent RNA synthesis—possible implications for its antiviral activity. Sci. Rep. 8, 8100 (2018).
Makarova, K. S. et al. An updated evolutionary classification of CRISPR–cas systems. Nat. Rev. Microbiol. 13, 722–736 (2015).
Tock, M. R. & Dryden, D. T. F. The biology of restriction and anti-restriction. Curr. Opin. Microbiol. 8, 466–472 (2005).
Calendar, R. & Abedon, S. T. The Bacteriophages (Oxford Univ. Press, 2005).
Ebrahimi, K. H. et al. Viperin, through its radical-SAM activity, depletes cellular nucleotide pools and interferes with mitochondrial metabolism to inhibit viral replication. FEBS Lett. 594, 1624–1630 (2020).
Kronheim, S. et al. A chemical defence against phage infection. Nature 564, 283–286 (2018).
De Clercq, E. & Neyts, J. Antiviral Strategies (eds H. –G. Kräusslich & R. Bartenschlager) 53–84 (2009).
Feld, J. J. et al. Sofosbuvir and velpatasvir for HCV genotype 1, 2, 4, 5, and 6 infection. N. Engl. J. Med. 373, 2599–2607 (2015).
Chen, I. A. et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677 (2019).
Nguyen, L. T., Schmidt, H. A., von Haeseler, A. & Minh, B. Q. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274 (2015).
Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35, 1026–1028 (2017).
Katoh, K., Misawa, K., Kuma, K. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44 (W1), W242–W245 (2016).
Eddy, S. R. Accelerated profile HMM searches. PLOS Comput. Biol. 7, e1002195 (2011).
Zimmermann, L. et al. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 430, 2237–2243 (2018).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 2006.0008 (2006).
Roche, B. et al. Reprint of: Iron/sulfur proteins biogenesis in prokaryotes: formation, regulation and diversity. Biochim. Biophys. Acta 1827, 923–937 (2013).
Schwartz, C. J. et al. IscR, an Fe-S cluster-containing transcription factor, represses expression of Escherichia coli genes encoding Fe–S cluster assembly proteins. Proc. Natl Acad. Sci. USA 98, 14895–14900 (2001).
Jiang, D. et al. Identification of three interferon-inducible cellular enzymes that inhibit the replication of hepatitis C virus. J. Virol. 82, 1665–1678 (2008).
Fortier, L.-C. & Moineau, S. in Bacteriophages: Methods and Protocols Volume 1: Isolation, Characterization, and Interactions (eds Clokie, M. R. J. & Kropinski, A.) 203–219 (Springer, 2009).
Kropinski, A. M., Mazzocco, A., Waddell, T. E., Lingohr, E. & Johnson, R. P. in Bacteriophages: Methods and Protocols, Volume 1: Isolation, Characterization, and Interactions (eds Clokie, M. R. J. & Kropinski, A.) 69–76 (Springer, 2009).
Hsiao, J. J., Potter, O. G., Chu, T. W. & Yin, H. Improved LC/MS methods for the analysis of metal-sensitive analytes using medronic acid as a mobile phase additive. Anal. Chem. 90, 9457–9464 (2018).
Dar, D. et al. Term-seq reveals abundant ribo-regulation of antibiotics resistance in bacteria. Science 352, aad9822 (2016).
Acknowledgements
We thank M. Danielsen and D. Malheiro from MS-omics for conducting the MS experiments and for the extensive help with the data analysis and the Sorek laboratory members for comments on earlier versions of this manuscript. A.B. is the recipient of a European Molecular Biology Organization Long-Term Fellowship (EMBO ALTF 186-2018). A.M. was supported by a fellowship from the Ariane de Rothschild Women Doctoral Program and, in part, by the Israeli Council for Higher Education via the Weizmann Data Science Research Center. G.O. was supported by the Weizmann Sustainability and Energy Research Initiative (SAERI) doctoral fellowship. R.S. was supported, in part, by the Israel Science Foundation (personal grant 1360/16), the European Research Council (grant ERC-CoG 681203), the Ernest and Bonnie Beutler Research Program of Excellence in Genomic Medicine, the Minerva Foundation with funding from the Federal German Ministry for Education and Research, the Ben B. and Joyce E. Eisenberg Foundation and the Knell Family Center for Microbiology.
Author information
Authors and Affiliations
Contributions
A.B. and R.S. led the study and A.B. performed all experiments unless otherwise indicated. A.M. and A.B performed the computational analyses that appear in Figs. 1, 2, Extended Data Fig 9. H.S., M.M.R. and N.T. designed and performed purification of pVips and in vitro enzymatic assays that appear in Fig. 3, Extended Data Fig 6. G.M., C.A. and S.M. assisted with the plaque assays that appear in Extended Data Fig. 1. C.A. assisted in the preparation of cell lysates that appear in Fig. 3, Extended Data Figs 3–5. G.O. and G.A. assisted in the design and analysis of GFP-reporter studies presented in Fig 4, Extended Data Fig 7. R.S. supervised the study. R.S and A.B. wrote the paper together with the team.
Corresponding author
Ethics declarations
Competing interests
R.S. is a scientific cofounder and advisor of BiomX, Pantheon Bioscience and Ecophage. A.B., A.M. and R.S. are inventors on patent application PCT/IL2020/050377 licensed to Pantheon Bioscience. H.S., M.R. and N.T. are employed by Pantheon Bioscience. The other authors have no competing interests.
Additional information
Peer review information Nature thanks Pieter Dorrestein, Pei-Yong Shi and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Fig. 1 pVips protect against phage infection.
Bacteria expressing pVips, GFP or MoaA (negative controls), or the human viperin gene were grown on agar plates and tenfold serial dilutions of the phage lysate were dropped on the plates. a–h, Efficiency of plating (EOP) data, representing plaque-forming units per millilitre; each bar graph represents average of three replicates, with individual data points overlaid.
Extended Data Fig. 2 T7 infection in liquid culture in the presence of pVips.
a, For each pVip, growth curves of liquid cultures infected by phage T7 (MOI 0.001) are shown. Light and dark grey are uninfected and infected controls (strain expressing GFP), respectively. Light and dark red are uninfected and infected strains expressing pVips, respectively. Two technical replicates are presented as individual curves; representative of three biological replicates. The negative controls (GFP uninfected, GFP infected) are the same for pVips 6, 7, 8, 10, 15, 27, 37, 39, 42, 50, 54, MoaA, and for pVip12, 19, 32, 44, 46, 47, 48, 57, 58, 60, 61, 62, 63. b, The catalytic activity of pVips is required for defence against T7 phage. For each pVip and its respective mutant (mutation of three cysteines in the active site), growth curves of liquid cultures infected by phage T7 (MOI 0.001) are presented. Light and dark grey are uninfected and infected controls (strain expressing MoaA), respectively. Light and dark red are uninfected and infected strains expressing viperins, respectively. Light and dark blue are uninfected and infected strains expressing catalytically inactive mutants. Two technical replicates are presented as individual curves; representative of three biological replicates.
Extended Data Fig. 3 Detection of ddhCTP and ddhCTP derivatives in cell lysates from an E. coli strain expressing the human viperin.
a, Extracted ion chromatogram of the ddhC standard. b–d, Extracted ion chromatogram for singly charged masses that are predicted to correspond to ddhC (m/z 226.0822, retention time (RT) of 2.2 min) (b), ddhCMP (m/z 306.0486, RT 9.7) (c) and ddhCTP (m/z 465.9812, RT 10.7) (d) in cell lysates from an E. coli strain expressing the human viperin. Representative of three replicates.
Extended Data Fig. 4 Detection of ddh-ribonucleotides in lysates of cells that express pVips.
a, Quantification of ddh-cytidine (ddhC) in lysates of cells expressing pVips. Detection and quantification of ddhC was performed using LC–MS with a synthesized chemical standard (Methods). For MoaA, the measurement was under the limit of detection (LOD 0.0003 uM). Bar graph represents average of three replicates, with individual data points overlaid. b–h, Relative abundance for singly charged masses that are predicted to correspond to ddhC (m/z 226.0822, retention time (RT) of 2.2 min) (b), ddhCMP (m/z 306.0486, RT 9.7) (c), ddhCTP (m/z 465.9812, RT 10.7) (d), ddhUMP (m/z 307.0326, RT 8.7) (e), ddhUTP (m/z 466.9652, RT 9.9) (f), ddhGMP (m/z 346.0547, RT 9.8) (g), and ddhGTP (m/z 505.9874, RT 10.7) (h). Average relative abundance is presented as bar graph, with individual data points from three biological replicates overlaid. Limit of detection (LOD) is indicated by a dashed grey line. A compound was defined as present, in Fig. 3, if all three replicates were above the LOD.
Extended Data Fig. 5 MS/MS fragmentation spectra for predicted compounds.
a–c, MS/MS data were acquired in positive ionization mode for a synthesized chemical standard ddhC (a) as well as for masses from the human viperin cell lysate predicted to correspond to ddhC (b), and ddhCMP (c). d, e, Similar data were obtained for masses from the pVip21 cell lysate predicted to correspond to ddhGMP (d), and ddhGTP (e). f, g, MS/MS data were acquired, in negative ionization mode, from the pVip47 cell lysate for masses predicted to correspond to ddhUMP (f), and ddhUTP (g). In all panels, assignment of hypothetical structures is indicated for informative fragment ions. The ddhC molecule is annotated to level 1, and all other molecules are annotated to level 2b, per the Metabolomics Standards Initiative nomenclature.
Extended Data Fig. 6 MS/MS fragmentation spectra for predicted compounds from in vitro reactions with purified pVips.
a, b, MS/MS data were acquired in positive ionization mode for the product detected in reaction samples using purified pVip6 or purified pVip56 and CTP and GTP as nucleotide substrates respectively; the resulting products are predicted to correspond to ddhCTP (a) and ddhGTP (b). c, MS/MS data were acquired in negative ionization mode for product detected in reaction samples using purified pVip8 UTP as substrate; the resulting product is predicted to correspond to ddhUTP.
Extended Data Fig. 7 Transcription during induction of WT and mutant pVips.
a, The catalytic activity of pVips is required for defence against T7 phage and repression of viral transcription. Application of the reporter assay (same as presented in Fig. 4a) for strains expressing the human viperin, pVips and their cognate catalytically inactive mutants. Strains are first induced with arabinose for 45 min to express the pVip. At t = 0, IPTG is added to express the GFP. Fluorescence/OD over time curves are presented for each strain. Dark and light red correspond to induced and non-induced wild type viperins, respectively; Dark and light blue correspond to induced and non-induced mutant viperins, respectively. Grey curve corresponds to negative control (WT viperin, no addition of IPTG). Two technical replicates are presented as individual curves. Representative of two biological replicates. b, T7 RNAP expression as measured by RNA-seq. The expression (RPKM) of T7 RNAP in cells expressing viperins was compared to that in cells expressing the MoaA negative control. Bar graphs represent average of two replicates, with individual data points overlaid.
Extended Data Fig. 8 Heterologous expression of pVips is not toxic is E. coli.
Expression of pVips, human viperin or negative controls (GFP, MoaA) was induced at 45 min by addition of arabinose (final concentration 0.2%). Colony forming units (CFU) were measured right after dilution from overnight culture (t = 0), before induction (t = 45), and 45 and 90 min after induction (t = 90, t = 135). Bar graphs represent average of three replicates, with individual data points overlaid.
Extended Data Fig. 9 Putative multi-gene defence systems that include pVips.
Representative instances of pVips and their genomic neighbourhood. Genes predicted to be part of the pVip-containing defence system are highlighted. Genes known to be involved in defence are in yellow. Genes of mobile genetic elements are in dark grey. RM, restriction-modification; TA, toxin-antitoxin. The name of bacterial species, and the accession of the relevant genomic scaffold in the IMG database20 are indicated on the left. a–d, Four common configurations of putative pVip-containing systems found in bacterial and archaeal genomes.
Extended Data Fig. 10 Phylogenetic tree of pVips and putative eukaryotic viperins.
MoaA sequences were used as an outgroup (grey). pVips are depicted in red and putative eukaryotic viperins selected for the phylogenetic tree presented in Fig. 2 are depicted in blue.
Supplementary information
Supplementary Table 1
Clusters of genes retrieved by the homology-based search of human viperin in prokaryotic genomes. Genes used to calculate defense scores were those present on DNA scaffolds of sufficient size with at least ten genes from each side of the viperin homolog.
Supplementary Table 2
A list of pVips. Gene and genome accessions in the IMG database are indicated.
Supplementary Table 3
MoaA and eukaryotic viperin sequences. MoaA genes (outgroup) and eukaryotic viperin sequences used in the phylogenetic tree in Figure 2.
Supplementary Table 4
A list of primers used in this study.
Supplementary Table 5
A list of phages used in this study.
Rights and permissions
About this article

Cite this article
Bernheim, A., Millman, A., Ofir, G. et al. Prokaryotic viperins produce diverse antiviral molecules. Nature 589, 120–124 (2021). https://doi.org/10.1038/s41586-020-2762-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2762-2
- Springer Nature Limited
This article is cited by
-
Unexpected regulatory functions of cyprinid Viperin on inflammation and metabolism
BMC Genomics (2024)
-
Evolutionary history of an immune protein conserved across all domains of life
Nature Ecology & Evolution (2024)
-
Conservation and similarity of bacterial and eukaryotic innate immunity
Nature Reviews Microbiology (2024)
-
Phages overcome bacterial immunity via diverse anti-defence proteins
Nature (2024)
-
Structure and rational engineering of the PglX methyltransferase and specificity factor for BREX phage defence
Nature Communications (2024)