

Abstract
A high tumour mutational burden (hypermutation) is observed in some gliomas1,2,3,4,5; however, the mechanisms by which hypermutation develops and whether it predicts the response to immunotherapy are poorly understood. Here we comprehensively analyse the molecular determinants of mutational burden and signatures in 10,294 gliomas. We delineate two main pathways to hypermutation: a de novo pathway associated with constitutional defects in DNA polymerase and mismatch repair (MMR) genes, and a more common post-treatment pathway, associated with acquired resistance driven by MMR defects in chemotherapy-sensitive gliomas that recur after treatment with the chemotherapy drug temozolomide. Experimentally, the mutational signature of post-treatment hypermutated gliomas was recapitulated by temozolomide-induced damage in cells with MMR deficiency. MMR-deficient gliomas were characterized by a lack of prominent T cell infiltrates, extensive intratumoral heterogeneity, poor patient survival and a low rate of response to PD-1 blockade. Moreover, although bulk analyses did not detect microsatellite instability in MMR-deficient gliomas, single-cell whole-genome sequencing analysis of post-treatment hypermutated glioma cells identified microsatellite mutations. These results show that chemotherapy can drive the acquisition of hypermutated populations without promoting a response to PD-1 blockade and supports the diagnostic use of mutational burden and signatures in cancer.
Similar content being viewed by others

Main
Identifying genomic markers of response to immune checkpoint blockade (for example, PD-1 blockade) may benefit cancer patients by providing predictive biomarkers for patient stratification and identifying resistance mechanisms for therapeutic targeting. Gliomas typically have a low tumour mutational burden (TMB) and a highly immunosuppressive microenvironment—two features associated with immunotherapy resistance. Nevertheless, recent work has suggested that a subset of patients with high-TMB (hypermutated) gliomas might benefit from PD-1 blockade6. Although consistent with data from other cancers7,8,9, these initial observations were derived from unique disease contexts such as constitutional DNA mismatch-repair (MMR) deficiency syndrome6. Therefore, the extent to which glioma patients at large will benefit from this approach is unknown. While large amounts of genomic data on gliomas exist2,4,5,10,11,12, our understanding of the clinical landscape of hypermutation and the mechanisms that underlie its development remain unclear. Hypermutation is rare in newly-diagnosed gliomas (de novo hypermutation), but common in tumours that have recurred after the use of alkylating agents (post-treatment hypermutation)4,5,10,11. Given that gliomas exhibit substantial inter-patient and intra-tumoral genomic variation10,11,12, it remains to be determined whether molecular biomarkers (for example, IDH1 or IDH2 (hereafter IDH1/2) mutations) reliably predict the development of hypermutation or response to immunotherapy.
An association between hypermutation and MMR mutations has been observed in gliomas1,2,3,4,13, but most of the reported MMR mutations were not functionally characterized, and their role in causing hypermutation is unclear. Other studies have suggested that alkylating agents such as temozolomide are the direct cause of hypermutation3. This was supported by the discovery of a mutational signature (single base substitution (SBS) signature 11) characterized by the accumulation of G:C>A:T transitions at non-CpG sites in hypermutated gliomas after exposure to alkylating agents14. However, the fact that hypermutation is undetectable in most gliomas that recur after temozolomide treatment challenges this notion4,5. Furthermore, it remains unclear whether this mutational pattern enhances tumour immunogenicity and renders gliomas responsive to PD-1 blockade. Not all hypermutated cancers respond to such treatments7,8,9; a more accurate characterization of the phenotypic and molecular features of hypermutated gliomas therefore would help clinicians to manage such patients more effectively.
Mutational burden and signatures in gliomas
Previous studies included too few hypermutated gliomas to characterize the landscape of hypermutation in gliomas1,2,3,4,5. We therefore created a cohort of sufficient scale (n = 10,294) and subtype diversity by leveraging large datasets generated from clinical sequencing panels (DFCI-Profile, MSKCC-IMPACT and FMI)15,16,17. All samples from patients with a histopathological diagnosis of glioma were included and classified into molecular subgroups according to histopathology, mutational status of IDH1/2, and whole-arm co-deletion of chromosomes 1p and 19q (1p/19q co-deletion) (Extended Data Fig. 1, Supplementary Tables 1, 2). We quantified the TMB of all samples (median 2.6 mutations (mut.) per Mb (range 0.0–781.3)), established thresholds for hypermutation by examining the distribution of TMB (Extended Data Fig. 2)17, 18, and identified 558 (5.4%) hypermutated gliomas (median TMB 50.8 mut. per Mb (8.8–781.3)) for further analysis.
Using samples with detailed clinical annotation (DFCI-Profile), we found that the prevalence of hypermutation varied between and within subgroups (Fig. 1a, b, Extended Data Fig. 3a, b, Supplementary Table 3). Hypermutation was detected almost exclusively in diffuse gliomas (99.1% of hypermutated samples) with high-grade histology (95.6%) and was more prevalent in recurrent tumours (16.6% versus 2.0% in newly diagnosed tumours; Fisher’s exact test, P < 10−15) (Fig. 1b). In samples of recurrent tumours, hypermutation was associated with markers of response to alkylating agents, including IDH1/2 mutation (hypermutation in 1.4% of newly diagnosed versus 25.4% of post-treatment IDH1/2-mutant tumours, Fisher’s exact test, P = 2.0 × 10−13), 1p/19q co-deletion (0.0% versus 33.8%, P = 7.3 × 10−11), and MGMT promoter methylation (2.4% versus 24.2%, P = 9.0 × 10−12). The effect of IDH1/2 mutation was confirmed only in MGMT-methylated tumours (Extended Data Fig. 3c). These findings suggest that selective pressure from therapy may elicit progression towards hypermutation.

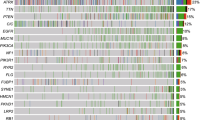
a, Integrated analysis of the DFCI-Profile dataset (n = 1,628 gliomas) depicting TMB, indels at homopolymer regions, and the single nucleotide variant (SNV) mutation spectrum in each tumour according to molecular status of IDH1/2, 1p/19q co-deletion, chromosome 7 gain and/or chromosome 10 deletion (7gain/10del), MGMT promoter methylation, histological grade, age at initial diagnosis, and prior treatment. Red line denotes high TMB (≥17.0 mut. per Mb). b, Prevalence of hypermutation in the DFCI-Profile dataset. Chi-squared test and two-sided Fisher’s exact test. NA, not available; TMZ, temozolomide; WT, wild-type; mut, mutant; codel, co-deleted.
The standard treatment for gliomas includes surgery, radiation and chemotherapy with alkylating agents19,20. To assess the role of each of these in the development of hypermutation, we analysed associations between TMB and detailed patterns of treatment in 356 recurrent gliomas. Hypermutation was associated with prior treatment with temozolomide (Fisher’s exact test, P < 10−15) in a dose-dependent manner (Fig. 1b, Extended Data Fig. 3d, e), but not with radiation (P = 0.88) or nitrosoureas (P = 0.78). Among recurrent tumours from patients who had received only one adjuvant treatment modality, TMB was increased only in temozolomide-treated samples (median 16.32 (interquartile range (IQR) 6.95–70.32) versus 6.08 (3.80–7.97) with surgery only, P = 4.0 × 10−7; Extended Data Fig. 3f). Of note, the prevalence of hypermutation in post-temozolomide samples correlated with the chemosensitivity of the primary, molecularly defined tumour type (1p/19q co-deleted oligodendrogliomas (59.5%) > IDH1/2-mutant astrocytomas (30.2%) > MGMT-methylated IDH1/2 wild-type glioblastomas (23.1%) > MGMT-unmethylated IDH1/2 wild-type glioblastomas (5.6%); P = 3.8 × 10−7; Fig. 1b). We observed a similar pattern in the FMI validation dataset (Extended Data Fig. 3g-i).
The systematic analysis of somatic mutation patterns by genome sequencing has identified a variety of mutation signatures in human cancer which are driven by known and unknown DNA damage and repair processes14. We examined the contributions of 30 previously reported signatures (COSMIC signatures v2) within our cohort to investigate the biological processes that cause hypermutation in gliomas. We first validated that mutational signatures can be predicted using large targeted panel sequencing in hypermutated samples (Extended Data Figs. 4, 5a–c). The majority of de novo hypermutated gliomas harboured mutational signatures associated with defects in the MMR pathway (COSMIC signatures 6, 15, 26 and 14) or the DNA polymerase POLE (10 and 14)14 (69% and 35% of samples, respectively; Extended Data Fig. 5d, e), implying that constitutional deficiency in MMR or POLE was likely to be the underlying genetic cause of hypermutation. By contrast, 98% of post-treatment hypermutated gliomas showed a mutational signature that has been previously associated with temozolomide exposure (signature 11). We also identified two distinct mutational signatures that were highly correlated with mutational signature 11 (Extended Data Fig. 5b, c) including a previously undescribed signature (S2) associated with 1p/19q co-deletion and lack of prior radiation therapy. Finally, half of the samples with a dominant signature 11 showed a co-existing minor MMR- or POLE-deficiency signature component (Extended Data Fig. 5e), suggesting that defective DNA repair and mutagen exposure cooperate to drive hypermutation in recurrent gliomas.
Molecular drivers of hypermutation
Only a subset of temozolomide-treated samples (58 of 225, 25.8%) showed evidence of hypermutation, suggesting that additional factors are required for its development. Although MMR defects have been consistently observed in hypermutated gliomas1,2,3,4,13, their co-occurrence with high TMB did not enable prior studies to determine the degree to which MMR mutations represent passenger versus hypermutation-causing driver events. We systematically characterized mutations and copy number variants (CNVs; Supplementary Figs. 1, 2) to identify hypermutation drivers using an unbiased approach that controlled for the increased incidence of passenger mutations associated with hypermutation21. In the merged DFCI-Profile/MSKCC-IMPACT dataset, 36 genes were significantly enriched (q value < 0.01) in hypermutated tumours (Fig. 2a). Collectively, MMR mutations stood out among the most enriched (91.2% versus 4.9% in non-hypermutated samples, q < 1.6 × 10−15), and mutations in MSH6 showed the highest enrichment (43.0% versus 1.2%, q = 3.3 × 10−7) (Extended Data Figs. 3j–l, 6a, b). MMR-variant allele frequencies (VAFs) and cancer cell fractions (CCFs) in gliomas were most similar to those in MMR-deficient colorectal (CRC) or endometrial cancers and were higher than in MMR-proficient hypermutated cancers (Extended Data Fig. 6c, d). Some MMR variants in post-treatment hypermutated samples matched the canonical signature 11 sequence context (Extended Data Fig. 5f), suggesting that a subset of these variants are likely to have been caused by temozolomide treatment.

a, Mutated genes and pathways enriched in hypermutated gliomas in the merged DFCI-Profile/MSKCC-IMPACT dataset (n = 2,173) using a permutation test to control for random mutation rate in the setting of hypermutability. b, Response to temozolomide across a panel (n = 30) of native spheroid glioma PDCLs (blue, MMR-proficient; red, MMR-deficient). Dose–response curves were calculated using mean surviving fractions from three independent assays. c, Response to temozolomide and CCNU in the glioblastoma PDCL BT145 following knockout of MSH2, MSH6, MLH1 or PMS2 by CRISPR–Cas9. Dose–response curves were calculated using mean surviving fractions from three independent assays (mean ± s.e.m.). d, Number of signature 11 variants after chronic temozolomide treatment of the PDCL DIPG13 with MSH2 or MSH6 knockout by CRISPR–Cas9. Mutational signatures could not be called in the vehicle-treated samples (too few variants). e, Tumour volume (n = 8 mice per group) during treatment with vehicle (blue) or temozolomide (red) in BT145 patient-derived xenografts (PDXs). f, Number of signature 11 variants found after chronic temozolomide exposure in BT145 PDXs. Mutational signatures could not be called in the vehicle-treated tumours (too few variants). g, Schematic representation of BT145 PDXs clonal evolution under temozolomide exposure. Two independent secondary resistant tumours (Resistant 1 and 2) and one vehicle-treated tumour are represented. Resistant tumours had four private variants that were not detected in the vehicle-treated tumour: an MSH6(T1219I) mutation (VAF 0.27 and 0.37 for resistant 1 and 2, respectively), and three non-coding variants of NF1 (VAF 1.0 and 0.99), RAC1 (VAF 0.86 and 0.86) and RAF1 (0.44 and 0.56). HGG, high-grade glioma; Chr, chromosome.
As most MMR variants lacked functional annotation, we next integrated sequencing data with immunohistochemistry for protein loss (Extended Data Fig. 6e). Overall, results from both assays were concordant, consistent with MMR mutations leading to loss of function. In rare samples that lacked MMR variants, signature analysis and MMR immunohistochemistry revealed evidence for MMR deficiency, suggesting that these samples harboured underlying MMR defects that could not be identified by sequencing (for example, promoter methylation). We identified several MMR mutational hotspots (Extended Data Fig. 6f, Supplementary Table 4), including a recurrent MSH6 mutation (p.T1219I, in 7.4% of hypermutated tumours) that has been previously identified in Lynch syndrome and shown to exert a dominant-negative effect without affecting protein expression22,23 (Extended Data Fig. 6g, h).
Immunohistochemistry on an independent cohort of 213 recurrent post-alkylator gliomas further validated these findings (Supplementary Table 2). MMR protein expression was lost in 22 post-treatment samples, and this loss was associated with IDH1/2 mutations (20% mutant versus 2% wild-type; Fisher’s exact test, P = 8.0 × 10−6) (Extended Data Fig. 7a, b). Sequencing of samples with MMR protein loss confirmed hypermutation, with MMR mutations in 18 of 19 (94.7%) of these samples. Subclonal loss of MMR proteins (that is, protein retained in more than 20% of tumour cells) was more common in post-treatment than de novo hypermutated gliomas (12 of 46 (26.1%) versus 0 of 16 (0.0%), P = 0.03) (Extended Data Fig. 7c–f).
We next assessed the relationship between MMR deficiency and acquired chemotherapy resistance. Because hypermutation and MMR defects were almost exclusively seen after temozolomide treatment, we hypothesized that nitrosoureas and temozolomide might not show complete cross-resistance. Analysis of temozolomide sensitivity in 30 cell lines derived from patients with glioma (patient-derived cell lines, PDCLs), including four derived from MMR-deficient gliomas (Extended Data Fig. 8a–c), showed that all native MMR-deficient PDCLs had striking temozolomide resistance compared to MMR-proficient PDCLs (6.46- and 1.35-fold increase in median area under the curve (AUC) versus MMR-proficient–MGMT-deficient and MMR-proficient–MGMT-proficient PDCLs, respectively) (Fig. 2b, Extended Data Fig. 8d–f). We next treated native and engineered isogenic MMR-knockout glioma models with temozolomide or the nitrosourea lomustine (CCNU), a chloroethylating alkylating agent that generates DNA interstrand crosslinks and double-strand breaks (Fig. 2c, Extended Data Fig. 8g–i). All MMR-deficient models were resistant to temozolomide and sensitive to CCNU, consistent with the lack of hypermutation in samples from nitrosourea-treated patients24 (Extended Data Fig. 3f).
Mismatch repair deficiency and signature 11
Our analyses indicated that MMR deficiency together with temozolomide exposure might cause signature 11, as opposed to it being a ‘pure’ temozolomide signature. To test this idea, we exposed isogenic models of MMR deficiency to temozolomide (Extended Data Fig. 9a, b). After treatment with temozolomide, MMR-deficient PDCLs developed hypermutation with signature 11, whereas MMR-proficient controls (expressing sgGFP) did not (Fig. 2d). We then chronically treated temozolomide-sensitive glioblastoma xenografts (PDXs) with temozolomide until resistance was acquired (Fig. 2e, Extended Data Fig. 9c, d). These tumours developed hypermutation with signature 11 (Fig. 2f, Extended Data Fig. 9e) and shared four unique variants; the dominant-negative MSH6 hotspot mutation (p.T1219I) and three non-coding variants (Fig. 2g), consistent with the theory that the MSH6 mutation drives both hypermutation and acquired temozolomide resistance (Extended Data Fig. 9f).
Collectively, these findings show that temozolomide exerts a previously underappreciated selective pressure in favour of MMR-deficient cells, which are resistant to temozolomide. Exposing MMR-deficient cells to temozolomide induces hypermutation with signature 11 by causing DNA damage in the absence of functional MMR. Therefore, hypermutation with signature 11 represents a potential biomarker for MMR deficiency and temozolomide resistance in gliomas (Extended Data Fig. 9g).
Characteristics of MMR-deficient gliomas
MMR deficiency recently emerged as an indicator of response to PD-1 blockade in patients with cancer8,25, leading to the first tissue-agnostic cancer-drug approval by the US Food and Drug Administration for use of the PD-1 blocker pembrolizumab in patients with MMR-deficient cancers. However, in CRCs and some other cancers, MMR inactivation occurs early in tumour progression, whereas in post-treatment gliomas it arises late. Gliomas might therefore differ from other cancers on which the approval was based and these differences might influence immune recognition of tumours and the response to immunotherapy.
To test this hypothesis, we first assessed the outcome of hypermutated gliomas. In CRC, MMR deficiency is associated with improved outcomes. By contrast, among patients with recurrent glioma, we observed worse survival in both hypermutated high-grade 1p/19q co-deleted oligodendrogliomas (median overall survival (OS) 96.5 months (95% confidence interval (CI) 20.8–NA (not applicable)) versus 137.2 months (95% CI 41.8–NA) in non-hypermutated tumours, P = 0.0009, two-sided log-rank test) and IDH1/2-mutant astrocytomas (median OS 15.7 months (95% CI 12.9–18.3) versus 21.5 months (95% CI 19.2–29.8), P = 0.0015) (Fig. 3a, Extended Data Fig. 10a–c). We observed a similar trend in IDH1/2 wild-type glioblastomas (P = 0.0809). The finding of poor survival in recurrent hypermutated gliomas remained significant in multivariable analysis (hazard ratio 2.16 (95% CI 1.38–3.38), P = 0.0008; Supplementary Table 5).

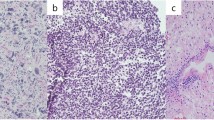
a, Survival of patients with recurrent high-grade glioma from the time of sample collection according to histomolecular group and TMB status (n = 333 recurrent samples; 238 from DFCI-Profile, 95 from MSKCC-IMPACT). Two-sided log-rank test. b, Quantification of tumour-infiltrating CD3-positive T-cells in CRC samples (n = 19). Left, representative low- and high-magnification images of CD3 immunolabelling (brown; intraepithelial lymphocytes, black arrowheads; stromal lymphocytes, black arrows) and nuclear counterstaining (blue). Dashed lines, border between tumour and stroma. Only intraepithelial lymphocytes were quantified. Scale bars; 100 µm (100×), 50 µm (200×). Right: boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range. Two-sided Wilcoxon rank-sum test. c, Quantification of tumour-infiltrating CD3-positive T-cells in gliomas according to their MMR status (n = 43). For each group, three areas with the maximal CD3 infiltration were selected for quantification (representative images, left). Scale bars: 500 µm (20×), 50 µm (200×). Right: boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range. Kruskal–Wallis test and Dunn’s multiple comparison test. d, TMB (top) and homopolymer indel burden (bottom) in hypermutated gliomas compared with other hypermutated cancers from the GENIE dataset. Tukey’s boxplots are shown. Two-sided Wilcoxon rank-sum test with Bonferroni correction. e, Pan-cancer analysis of cancer cell fractions in hypermutated gliomas (post-treatment) compared with other hypermutated cancers from the TCGA and ref. 4 exome datasets (n = 798). One hundred non-hypermutated samples from the TCGA were randomly selected as controls. Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range excluding outliers. Two-sided Wilcoxon rank-sum test with Bonferroni correction. f, Workflow for scWGS and bulk tumour DNA sequencing. g, Single-cell sequencing estimate of the number of G:C>A:T transitions at NCC and NCT trinucleotide contexts in 63 cells from a glioblastoma patient with post-temozolomide hypermutation using 1× scWGS sequencing. Error bars show 95% CI. The absolute computed purity was 0.66 for the primary tumour sample and 0.47 for the recurrent tumour sample in the bulk sequencing. h, Single-cell sequencing estimate of microsatellite mutation rate in eight cells from a patient with glioblastoma with post-temozolomide hypermutation. Eight cells were analysed for the presence of MSI using 10× scWGS sequencing. WGA, whole genome amplification; QC, quality control; nucl, nuclei; seq, sequencing.
The current hypothesis behind the response of MMR-deficient CRCs to PD-1 blockade is based on their increased neoantigen burden and immune infiltration. We therefore assessed the association between MMR deficiency and T-cell infiltration in gliomas (n = 43) and CRCs (n = 19). As expected, MMR-deficient CRCs exhibited significantly more infiltrating T-cells than their MMR-proficient counterparts (Fig. 3b). By contrast, both MMR-deficient and MMR-proficient glioma samples lacked significant T-cell infiltrates (Fig. 3c).
We next assessed whether the neoantigen burden was lower in MMR-deficient gliomas than in other hypermutated cancers using samples from the GENIE and TCGA datasets (n = 1,748 and 699 hypermutated cancers, respectively). As neoantigen prediction was not feasible using panel sequencing data, we used the nonsynonymous mutational burden as a surrogate measure. This showed that both de novo and post-treatment MMR-deficient gliomas had an increase in their nonsynonymous mutational burden, when compared to non-hypermutated gliomas, and the glioma nonsynonymous mutational burden was similar to other hypermutated cancers (Fig. 3d, Extended Data Fig. 11a, b, Supplementary Table 6). This finding suggested that the total number of neoantigens is unlikely to explain the differences in immune response between gliomas and other hypermutated cancers.
Recent data suggest that, among mutations associated with MMR deficiency, small insertions and deletions (indels) at homopolymers (microsatellites)—which accumulate in MMR-deficient cells and can cause frameshift mutations—are crucial for producing ‘high-quality’ neoantigens that are recognized by immune cells26,28. Unexpectedly, although the high TMB was associated with an increased homopolymer indel burden in MMR-deficient CRCs, this association was not found in MMR-deficient gliomas (de novo hypermutated gliomas showed a modest increase; Fig. 3d, Extended Data Fig. 11c). This was validated using testing for microsatellite instability (MSI), a clinical biomarker for MMR deficiency. Whereas MSI was identified in all MMR-deficient CRCs, all tested gliomas with MMR protein loss (n = 15) were microsatellite-stable (MSS) (Extended Data Figs. 7d–f, 11d).
We hypothesized that, in hypermutated gliomas, more of the homopolymer indels are subclonal and below the detection limits of bulk sequencing, relative to other MMR-deficient cancers. Indeed, analysis of CCFs indicated that hypermutated gliomas contained a greater burden of subclonal variants than did other hypermutated cancers (Fig. 3e, Extended Data Fig. 11e–h). We therefore performed single-cell whole-genome DNA sequencing (scWGS) of 28 cells from a hypermutated, post-temozolomide glioblastoma with an MSH6(T1219I) mutation, and compared these to 35 non-hypermutated cells from the matched pre-treatment sample (Fig. 3f, Extended Data Fig. 11i–k). In the post-temozolomide sample, 13 of 28 cells (46.4%) were hypermutated with signature 11 (Fig. 3g, Extended Data Fig. 11l). Strikingly, whereas this tumour harboured only a minor increase in its homopolymer indel burden at the bulk level (0.49 versus 0.0 per Mb), the scWGS analysis showed a ninefold increase in microsatellite mutations in all hypermutated cells (Fig. 3h). This suggested that glioma cells with an MSH6(T1219I) variant harbour a subtle MSI phenotype that is not revealed by standard bulk sequencing or clinical MSI assays (Extended Data Fig. 11m).
PD-1 blockade in MMR-deficient gliomas
As hypermutation in gliomas that acquire MMR deficiency tends to be subclonal and does not generate optimal antitumour T-cell responses, we hypothesized that these tumours might not have high response rates to PD-1 blockade. We performed a retrospective institutional review of patients treated with PD-1 pathway blockade for which the TMB at treatment initiation was available (n = 210). This identified 11 patients with MMR-deficient glioma (5 de novo, 6 post-treatment) who were treated with PD-1 blockade for a median of 42 days (range 13–145; Supplementary Table 7). Nine (81.8%) had disease progression as their best response (Fig. 4a), and the median progression-free survival (PFS) and OS were 1.38 months (95% CI 0.95–2.69) and 8.7 months (95% CI 2.79–15.08), which were not significantly different from the data for matched patients with non-hypermutated glioma (PFS 1.87 months (95% CI 1.28–2.92), OS 9.96 months (95% CI 7.56–15.08); Fig. 4b, Extended Data Fig. 10d).

a, b, Best radiological response (a, measured as the best change in the sum of the products of perpendicular diameters of target lesions), and overall survival (b) of 11 patients with hypermutated and MMR-deficient gliomas who were treated with PD-1 blockade. A cohort of patients with non-hypermutated gliomas who were treated with PD-1 blockade is depicted as control (n = 10, best matches according to diagnosis, primary versus recurrent status, and prior treatments). Two-sided log-rank test. c, Proposed model explaining differential response to PD-1 blockade in MMR-deficient CRCs and gliomas. In CRCs (top), MMR deficiency is acquired early in pre-cancerous cells, creating mutations and indels at homopolymer regions. Over time, clonal neoantigens of both types emerge and strong immune infiltrates are seen at diagnosis. Treatment with anti-PD-1 results in expansion of T cells that recognize these clonal neoantigens and substantial antitumour responses. In gliomas (bottom), few mutations are acquired early during tumorigenesis in the majority of tumours. Temozolomide drives the expansion of cells with MMR deficiency and late accumulation of random temozolomide-induced mutations. Ineffective antitumour responses may result from poor neoantigen quality (high burden of missense mutations versus frameshift-producing indels) and high subclonality associated with an immunosuppressive microenvironment. In some tumours, MMR-proficient subclones that have acquired therapy resistance through other pathways can co-exist with MMR-deficient subclones, giving rise to a mixed phenotype.
Because our prior analyses indicated that patients with hypermutated gliomas might have reduced survival, we used a second set of historical controls to compare the outcome of hypermutated gliomas treated with PD-1 blockade versus other systemic agents (Supplementary Table 7). Unexpectedly, we observed a longer median OS for patients treated with other systemic agents when compared to those treated with PD-1 blockade (16.10 months (95% CI 3.98–22.21) versus 8.07 (95% CI 2.79–15.08.21); P = 0.02, two-sided log-rank test; Extended Data Fig. 10e, f, Supplementary Table 8). In one patient with hypermutated glioma that showed rapid imaging changes, histopathologic analysis of samples taken before and after treatment with PD-1 blockade showed highly proliferative tumour in both samples, with no significant evidence of pathologic response or increase in immune infiltrates after PD-1 blockade (Extended Data Fig. 10g).
DISCUSSION
Collectively, these results support a model in which differences in the mutation landscape and antigen clonality of hypermutated gliomas relative to other hypermutated cancers markedly affect the response to immunotherapy (Fig. 4c) and may explain the lack of both recognition of MMR-deficient glioma cells by the host immune system and response to PD-1 blockade, compared to other MMR-deficient cancers8,25. A key difference is that MMR-deficient gliomas lack detectable MSI by standard assays, similar to data from patients with constitutional MMR deficiency syndromes30. Our scWGS analyses suggest that this discordance might be due to intratumour heterogeneity and a lack of sufficient evolutionary time to select clonal MSI populations. Mechanistically, selective pressure exerted by temozolomide drives the late evolution of MMR-deficient subclones, which further accumulate temozolomide-induced mutations in individual cells. In line with previous data, therapy-induced single nucleotide variant mutations might not elicit effective antitumour responses, possibly because of the quality (missense mutations versus frameshift-producing indels) or subclonal nature of their associated neoantigens8,27,28,29. However, future evaluation of longer treatment exposure or combinatorial strategies is warranted to determine whether checkpoint blockade can be effective in this or other selected populations (for example, individuals with newly diagnosed MMR- or POLE-deficient gliomas)6.
We have presented evidence that recurrent defects in the MMR pathway drive hypermutation and acquired temozolomide resistance in chemotherapy-sensitive gliomas. Although it is difficult to determine the origin of MMR deficiency by sequence context alone in individual post-treatment samples, our data suggests that some MMR variants are likely to be caused by temozolomide. However, as acquired MMR deficiency occurs in the most temozolomide-sensitive tumours, it is not clear whether the acquired MMR deficiency outweighs the positive effects of temozolomide in gliomas. Our finding that MMR-deficient cells retain sensitivity to CCNU supports the hypothesis that hypermutation reduces cellular fitness and tolerance to DNA-damaging agents other than temozolomide. These alternatives are of interest in light of recent evidence showing that the addition of CCNU to chemoradiation improves the outcome of patients with MGMT-methylated glioblastomas31. Future studies are warranted to address the possibility that upfront temozolomide with CCNU may attenuate the process of post-treatment hypermutation. Furthermore, mechanisms of resistance to temozolomide that are not associated with hypermutation will need to be addressed.
Finally, our data indicate that the absence of an immune response in gliomas is likely to result from several aspects of immunosuppression in the brain that require further characterization. Approaches that increase infiltration by cytotoxic lymphocytes into the glioma microenvironment will probably be required to improve immunotherapy response. Our data also suggest a change in practice whereby repeated biopsies and sequencing to identify progression and hypermutation could inform prognosis and guide therapeutic management.
Methods
Datasets
For the DFCI-Profile dataset, clinical data and tumour variant calls identified through targeted next-generation sequencing (NGS) panels of 1,628 gliomas sequenced between June 2013 and November 2018 as part of a large institutional prospective profiling program (DFCI-Profile) were included16 (Extended Data Fig. 1). The distinction between photon and proton radiotherapy was not systematically captured; the vast majority of patients underwent photon radiotherapy. For the MSKCC-IMPACT and FMI datasets, clinical data and tumour variant calls from a total of 545 and 8,121 samples, respectively, that could be assigned to a molecular subgroup (see below) were included15,17,32,33. For pan-cancer analyses in targeted panel sets, clinical data and tumour variant calls from the GENIE project (a repository of genomic data obtained during routine clinical care at international institutions) were downloaded from Synapse (public data, release v6.1)34. For pan-cancer analyses in whole-exome sequencing sets, clinical data and tumour variant calls from 17 hypermutated glioblastomas4 and from the pan-cancer TCGA dataset were downloaded from the NCI Genomic Data Commons35. In addition, 247 gliomas collected at one site between 2009 and 2017 were analysed for protein expression of four MMR proteins (MSH2, MSH6, MLH1, and PMS2) using immunohistochemistry. Written informed consent or IRB waiver of consent was obtained from all participants. Patients of the FMI dataset were not consented for release of raw sequencing data. The study, including the consent procedure, was approved by the institutional ethics committees (10-417/11-104/17-000; Western Institutional Review Board (WIRB), Puyallup, WA).
Tumour genotyping and diagnosis
For the majority of samples, genomic testing was ordered by the pathologist or treating physician as part of routine clinical care to identify relevant genomic alterations that could potentially inform diagnosis and treatment decisions. Patients who underwent DFCI-Profile testing signed a clinical consent form, permitting the return of results from clinical sequencing. In total, 1,628 gliomas were sequenced as part of a cohort of 21,992 tumours prospectively profiled between June 2013 and November 2018. Research tumour diagnoses were reviewed and annotated according to histopathology, mutational status of IDH1 and IDH2 genes, and whole-arm co-deletion of chromosomes 1p and 19q (1p/19q co-deletion), according to WHO 2016 criteria12. All samples were assigned to one of four main molecular subgroups: IDH1/2-mutant and 1p/19q co-deleted oligodendrogliomas (high- and low-grade), IDH1/2-mutant astrocytomas (high- and low-grade), IDH1/2 wild-type glioblastomas (high-grade only), and IDH1/2 wild-type gliomas of other histologies (high- and low-grade), the latter including grade I pilocytic astrocytomas, glioneuronal tumours and other unclassifiable gliomas. For simplification, IDH1/2 wild-type grade III anaplastic astrocytomas and grade IV diffuse intrinsic pontine gliomas were assigned to the group of IDH1/2 wild-type glioblastomas in all analyses. Samples for which the clinical diagnosis of glioma could not be confirmed (other histology or possible non-tumour sample) and five samples with missing minimal clinical annotation were excluded from all analyses. For the MSKCC-IMPACT and FMI datasets, patients also signed a consent form, and samples were classified using the same procedure. MGMT promoter methylation status was determined as part of routine clinical care using chemical (bisulfite) modification of unmethylated, but not methylated, cytosines to uracil and subsequent PCR using primers specific for either methylated or the modified unmethylated DNA in the CpG island of the MGMT gene (GenBank accession number AL355531 nt46931-47011).
Targeted panel next-generation sequencing (DFCI-Profile) was performed using the previously validated OncoPanel assay at the Center for Cancer Genome Discovery (Dana-Farber Cancer Institute) for 277 (POPv1), 302 (POPv2), or 447 (POPv3) cancer-associated genes16,36. In brief, between 50 and 200 ng tumour DNA was prepared as previously described16,37, hybridized to custom RNA bait sets (Agilent SureSelectTM, San Diego, CA) and sequenced using Illumina HiSeq 2500 with 2 × 100 paired-end reads. Sequence reads were aligned to reference sequence b37 edition from the Human Genome Reference Consortium using bwa, and further processed using Picard (version 1.90, http://broadinstitute.github.io/picard/) to remove duplicates and Genome Analysis Toolkit (GATK, version 1.6-5-g557da77) to perform localized realignment around indel sites38. Single-nucleotide variants were called using MuTect v1.1.439, insertions and deletions were called using GATK Indelocator, and variants were annotated using Oncotator40. Copy number variants and structural variants were called using the internally developed algorithms RobustCNV41 and BreaKmer42 followed by manual review. To filter out potential germline variants, the standard pipeline removes SNPs present at >0.1% in Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA (http://evs.gs.washington.edu/EVS/, accessed May 30, 2013), present in dbSNP, or present in an in-house panel of normal tissue, but rescues those also present in the COSMIC database43. For this study, variants were further filtered by removing variants present at >0.1% in the gnomAD v.2.1.1 database or annotated as benign or likely benign in the ClinVar database44,45. Arm-level copy number changes were generated using an in-house algorithm specific for panel copy number segment files followed by manual expert review. We set a copy number segment mean log2 ratio threshold at which we could accurately call arm amplification and deletion based on the average observed noise in copy number segments. Chromosome arms were classified as amplified or deleted if more than 70% of the arm was altered. A sample was considered co-deleted if more than 70% of both 1p and 19q were deleted.
Sequencing data from MSKCC-IMPACT were generated at the Memorial Sloan Kettering Cancer Center using a custom targeted panel capture to examine the exons of 341 (IMPACT341) or 398 (IMPACT410) cancer-associated genes as previously described17. The FMI dataset comprised specimens sequenced as a part of clinical care using a targeted next-generation sequencing assay as previously described (FoundationOne or FoundationOne CDx, Cambridge, MA)15,33. Germline variants without clinical significance were further filtered by applying an algorithm to determine somatic or germline status46. Results were analysed for genomic alterations, TMB, MSI and mutational signatures. TMB was assessed by counting all mutations and then excluding germline and known driver mutations33,43,47. The remaining count was divided by the total covered exonic regions15,33. MSI status was determined as previously described48. A log-ratio profile for each sample was obtained by normalizing the sequence coverage at all exons and ∼3,500 genome-wide SNPs against a process-matched normal control. This profile was corrected for GC-bias, segmented and interpreted using allele frequencies of sequenced SNPs to estimate tumour purity and copy number at each segment. Loss of heterozygosity (LOH) was called if local copy number was 1, or if local copy number was 2 with an estimated tumour minor allele frequency of 0%. To assess 1p/19q co-deletion, we calculated the percentage of each chromosome arm that was monoallelic (under LOH)46. A sample was considered 1p/19q co-deleted if both 1p and 19q were >50% monoallelic.
For the DFCI-Profile and FMI datasets, the appropriate cutoffs for hypermutation (17.0 and 8.7 mut/Mb, respectively) were determined by examining the distribution of TMB in all samples and further confirmed using segmented linear regression analysis (Extended Data Fig. 2). For the MSKCC-IMPACT datasets, a threshold previously validated in this dataset was used17. In all analyses, the homopolymer indel burden was calculated by computing the number of single base insertions or deletions in homopolymer regions of at least 4 bases in length and dividing the count by the total exonic coverage as previously established49. Somatic variants were annotated as previously described15,16,17,36,37. In addition, for the DFCI-Profile and MSKCC-IMPACT datasets, variants in a selected list of glioma- and DNA-repair associated genes (IDH1, IDH2, TERT, ATRX, CIC, H3F3A, HIST1H3B, EGFR, PDGFRA, FGFR1, FGFR2, FGFR3, MET, KRAS, NRAS, HRAS, BRAF, NF1, PTPN11, PTEN, PIK3CA, PIK3C2B, PIK3R1, CDKN2A, CDKN2B, CDKN2C, CDK4, CDK6, CCND2, RB1, TP53, MDM2, MDM4, TP53BP1, PPM1D, CHEK1, CHEK2, RAD51, BRCA1, BRCA2, ATM, ATR, MLH1, MLH3, PMS1, PMS2, MSH2, MSH6, EPCAM, SETD2, POLE, POLD1, MUTYH, WRN) were manually reviewed for oncogenicity using several clinical databases for variant annotation (OncoKB, ClinVar, COSMIC, ExAC, and ARUP).
Mutational signature analyses
All variants detected by the sequencing pipeline covered by at least 30× read depth were stringently filtered for germline origin using the gnomAD (population allele frequency greater than 0.1%), and ClinVar (benign or likely benign annotation) databases44,45, as well as manual review of VAF distributions and variants with VAFs consistent with possible germline origin (45–55% or over 95%). The mutational spectrum of variants filtered during these steps was similar to a previously published germline mutation spectrum50. Signature analysis was performed for hypermutated samples in a two-step approach starting with the SomaticSignatures package in R for de novo signature extraction within each group51. To account for the inherent heuristic quality of the NMF approach, the NMF clustering step was repeated 100 times and chosen result was selected based on identifying signatures with the strongest Pearson’s correlation coefficients when compared to the 30 well-established COSMIC signatures v2 (https://cancer.sanger.ac.uk/cosmic/signatures_v2)14 (Extended Data Fig. 5a–c). We then used the DeconstructSigs package in R to estimate the contribution of identified signatures using a regression model52. To account for the potential overfitting of a regression approach—owing to either lack of important signatures in the model, or inclusion of uninvolved signatures—we used only the signatures identified by the decomposition approach in step one, supplemented by any strong signature predictions identified through a first pass run of DeconstructSigs with the 30 COSMIC signatures to check for samples that may show strong correlation to an outlier signature. For the FMI dataset, mutational signatures were called as previously described17. All point mutations were included in the analysis except known oncogenic driver mutations and predicted germline mutations. A sample was deemed to have a dominant signature if a mutational signature had a score of 0.4 or greater.
To assess the ability of this method to detect hypermutation-associated signatures in targeted panel sequencing data, we compared the signature calls of exome-sequenced samples using all variants (previously published DeconstructSigs signature predictions52) versus using only variants that overlapped with the panel-targeted regions. Somatic variant calls for bladder cancer, colon adenocarcinoma, rectal adenocarcinoma, skin cutaneous melanoma, and lung adenocarcinoma (study abbreviations BLCA, COAD, READ, SKCM, LUAD) from the TCGA MC3 dataset were used53 to assess the detection of COSMIC mutational signatures associated with APOBEC (signatures 2 and 13), mismatch repair (signature 6), ultraviolet light (signature 7), POLE (signature 10), and tobacco (signature 4). Variant calls for 17 hypermutated and 12 non-hypermutated glioma exome-sequenced samples were used for assessing temozolomide (signature 11) detection4. There were two COAD samples with known POLE exonuclease domain oncogenic mutations and a POLE signature predicted by DeconstructSigs; these were used for assessing POLE signature detection. For a given threshold number of variants (X1), we considered how many samples had at least X1 variants, and what percentage of these samples could correctly predict the exome-based signature using panel-restricted variants (with a predicted signature fraction greater than 0.1–0.2). This analysis showed that panel-based signature calls for the APOBEC, mismatch repair, tobacco, and ultraviolet light signatures reached 90% sensitivity with at least 20 somatic variants. Owing to the low number of samples with POLE-associated and temozolomide-associated hypermutation, we did not assess the sensitivity of signature detection at each variant count threshold; we instead downsampled the number of variants in positive control samples to find the minimum number of variants necessary to reproducibly predict the known signature, which was also determined to be 20 somatic variants (Extended Data Fig. 4).
Enrichment analysis
Mutation enrichment was statistically determined through a permutation test to control for confounders including variable mutability of different genes as well as sample mutation rates, which is of particular importance when assessing enrichment in hypermutated samples. First, we generated a list of every mutation in each of our samples. We calculated the difference in the mutation counts (Δ′) between the group of interest and the reference group. We then randomly permuted the mutations 100,000 times, preserving sample and gene mutation counts, and computed the Δ for each gene in each permutation. The P value for a given gene was determined by the fraction of permutations 1–n (in our case, n = 100,000) for which Δn ≥ Δ′. Storey q values were generated using the qvalue package in R to adjust for multiple comparisons. The analysis was first performed in the merged DFCI-Profile and MSKCC-IMPACT dataset, and further revalidated in the FMI dataset in an independent analysis.
Single-cell whole-genome sequencing
Frozen glioma samples were mechanically dissociated into pools of single nuclei as previously described54, following which single nuclei were isolated by flow cytometry, using a DAPI-based stain. Nuclei were subjected to whole-genome multiple displacement (MDA) amplification (Qiagen, REPLI-g) followed by next-generation sequencing library construction for Illumina Sequencing (Qiagen QIAseq FX DNA library kit). Libraries were sequenced on the Illumina HiSeq platform in paired end mode. Single cells were sequenced to 0.1–1× coverage. Bulk pooled nuclei were sequenced to 60× coverage while matched germline DNA (extracted from blood) was sequenced to 30× coverage.
Reads were aligned to hg38 using bwa mem, and variants were jointly called across bulk normal tissue, primary tumour single cells, and recurrent tumour single cells using the GATK best practices pipeline38 without variant quality score recalibration. Somatic mutations in single cells were called if they were monoallelic, had a homozygous reference genotype call but no alternate-allele support in bulk normal tissue, and had at least three supporting reads in a single cell. Germline heterozygous mutations (gHets) were called if they were monoallelic, were found in dbSNP (version 138, http://www.ncbi.nlm.nih.gov/snp), and had at least one supporting read and a heterozygous genotype call in bulk normal tissue. To assess sensitivity in each single cell, we computed the fraction of gHets detected with at least three supporting reads, analogous to our procedure for calling somatic mutations. To estimate the total number of somatic mutations present in each cell, we divided the total number of somatic mutations detected by sensitivity. To obtain 95% confidence intervals on the total mutational burden, we modelled the measurement of sensitivity using a beta distribution with Jeffrey’s prior, in which the beta parameters (α, β) are equal to the number of detected gHets + 0.5 and the number of undetected gHets + 0.5, respectively. We identified recurrent tumour single cells as hypermutated if their mutational burden was at least 1.5 times the highest mutational burden detected among primary tumour cells.
The method to detect microsatellite mutations was based on read-based phasing55,56 and was previously validated using scWGS data from neurons (I.C.-C. et al., manuscript in preparation). First, the human genome was scanned to define a reference set of microsatellite repeats that can be captured using short reads (that is, between 6 and 60 bp) as previously described57. Heterozygous SNPs were then detected in the bulk normal sample using the variant caller GATK38. Next, the reads in a given cell mapping to each heterozygous SNP allele detected in the bulk sample and their mates were extracted. If any of the microsatellites in the reference set were covered by these reads, the distribution of the allelic repeat lengths supported by the data was obtained by collecting the lengths of all intra-read microsatellite repeats mapped to the microsatellite locus under consideration. To discount truncated microsatellite repeats, we required 10-bp flanking sequences (both 5′ and 3′) of the intra-read microsatellite repeats to be identical to the reference genome. The same procedure was applied to the bulk sample. Finally, the distributions of microsatellite lengths from the single cell and the bulk sample were compared using the Kolmogorov–Smirnov test. The rates of microsatellite instability for each cell were computed as the number of sites mutated divided by the total number of microsatellites for which a call could be made. We applied FDR correction using 0.05 as a threshold for statistical significance, with a minimum of 8 single cell and 10 normal reads required to make a call. All the code is publicly available (https://github.com/parklab/MSIprofiler).
Immunohistochemistry
For the revalidation of MMR defects in an independent set, all prospectively collected surgical samples representing consecutive relapses of diffuse glioma following treatment with alkylating agents in adult patients (surgery between 2009 and 2015) were included. An expert neuropathologist reviewed histological samples from the IHC Pitié Salpêtrière cohort (Supplementary Table 2) in order to assess the WHO 2016 integrated diagnosis and to select the tumour areas for immunohistochemistry and for DNA extraction when molecular testing from formalin-fixed paraffin-embedded (FFPE) tissue material was required. Diffuse gliomas harbouring unambiguous positive IDH1(R132H) immunostaining were classified as IDH1/2-mutant. IDH1/2 status was tested by targeted sequencing in all diffuse gliomas harbouring negative or ambiguous IDH1(R132H) immunostaining. IDH1/2-mutant diffuse gliomas with loss of ATRX expression in tumour cells were classified as non 1p/19q co-deleted. 1p/19q co-deletion was tested in all IDH1/2-mutant diffuse gliomas with maintained ATRX expression. MGMT status was assessed in IDH1/2 wild-type gliomas. FFPE sections (3 μm thick) were deparaffinized and immunolabelled with a Ventana Benchmark XT stainer (Roche, Basel, Switzerland). The secondary antibodies were coupled to peroxidase with diaminobenzidine as brown chromogen. For immunohistochemistry performed at Pitié-Salpêtrière (PSL) Hospital, the following antibodies were used: mouse monoclonal anti-ATRX (Bio SB, clone BSB-108, BSB3296, 1:100), mouse monoclonal anti-IDH1(R132H) (Dianova, clone H09, DIA-H09, 1:100), rabbit monoclonal anti-CD3 (Roche, clone 2GV6, 790-4341, prediluted), rabbit polyclonal anti-IBA1 (Wako, W1W019-19741, 1:500), mouse monoclonal anti-MLH1 (Roche, clone M1, 790-4535, prediluted), mouse monoclonal anti-MSH2 (Roche, clone G219-1129, 760-4265, prediluted), mouse monoclonal anti-MSH6 (Roche, clone 44, 760-4455, prediluted), rabbit monoclonal anti-PMS2 (Roche, clone EPR3947, 760-4531, prediluted). For immunohistochemistry performed at BWH, the following antibodies were used: mouse monoclonal anti-MLH1 (Leica, clone ES05, MLH1-L-CE, 1:75), mouse monoclonal anti-MSH2 (Merck Millipore, clone Ab-2-FE11, NA27, 1:200), mouse monoclonal anti-MSH6 (Leica, clone PU29, MSH6-L-CE, 1:50), mouse monoclonal anti-PMS2 (Cell Marque, MRQ-28, 288M-14-ASR, 1:100). An expert neuropathologist blinded to the molecular status of MMR deficiency analysed the immunostaining. If loss of expression of one or several MMR proteins was observed in tumour cells, this result was confirmed in an independent laboratory by a second expert pathologist with separate stainer and reagents: FFPE sections were immunolabelled with a BOND stainer (Leica, Wetzlar, Germany). Primary antibodies were as follows: mouse monoclonal anti-MLH1 (clone G168-728, BD Pharmingen), mouse monoclonal anti-MSH2 (clone 25D12, Diagnostic BioSystems), mouse monoclonal anti-MSH6 (clone 44, Diagnostic BioSystems), mouse monoclonal anti-PMS2 (clone A16-4, BD Pharmingen). The loss of expression of MMR proteins was defined as the total absence of nuclear labelling in tumour cells associated with a maintained expression in normal cells (as a positive internal control in the same tissue area). The density of the immune infiltrate was studied after immunolabelling of T lymphocytes by CD3 and of macrophage/microglial cells by IBA1. The number of immunopositive cells was quantified by visual counting in the three areas (one square millimetre) of tumour tissue containing the highest density of immunopositive cells and a mean density was calculated.
Patient-derived cell lines
All PDCLs with a name starting with BT were established from tumours resected at Brigham and Women’s Hospital and Boston Children’s Hospital (Boston, MA) and were maintained in neurosphere growth conditions using the NeuroCult NS-A Proliferation Kit (StemCell Technologies) supplemented with 0.0002% heparin (StemCell Technologies), EGF (20 ng/ml), and FGF (10 ng/ml; Miltenyi) in a humidified atmosphere of 5% CO2 at 37 °C. The N16-1162 PDCL was established by the GlioTex team (Glioblastoma and Experimental Therapeutics) at the Institut du Cerveau et de la Moëlle épinière (ICM) laboratory and maintained as described above. SU-DIPG-XIII (DIPG13) cells were provided by Dr. Michelle Monje at Stanford University and were maintained in neurosphere growth conditions in a humidified atmosphere of 5% CO2 at 37 °C in tumour stem medium (TSM) consisting of Dulbecco’s modified Eagle’s medium: nutrient mixture F12 (DMEM/F12), neurobasal-A medium, HEPES buffer solution 1 M, sodium pyruvate solution 100 nM, non-essential amino acids solution 10 mM, Glutamax-I supplement and antibiotic-antimycotic solution (Thermo Fisher). The medium was supplemented with B-27 supplement minus vitamin A, (Thermo Fisher), 20 ng/ml human-EGF (Miltenyi), 20 ng/ml human-FGF-basic (Miltenyi), 20 ng/ml human-PDGF-AA, 20 ng/ml human-PDGF-BB (Shenandoah Biotech) and 2 μg/ml heparin solution (0.2%, Stem Cell Technologies). The identity of all cell lines established was confirmed by short tandem repeat assay or sequencing. All cell lines were tested for the absence of mycoplasma. Cell lines, xenografts, and model data available from the DFCI Center for Patient Derived Models.
Viability assays
For short-term viability assays, cells were plated in 96-well plates and treated the following day with temozolomide (Selleckchem) or CCNU (Selleckchem) for 7–9 days incubation. Fresh medium was added after four days of incubation. Cell viability was assessed using the luminescent CellTiter-Glo reagent (Promega) according to the manufacturer’s protocol. Luminescence was measured using the Modulus Microplate Reader (Promega). The surviving fraction (SF) for each [X] concentration was calculated as SF = mean viability in treated sample at concentration [X]/mean viability of untreated samples (vehicle). Dose–response curves and IC50 were generated using Prism 8 (GraphPad Software, San Diego, USA) after log transformation of the concentrations. Curves were extrapolated using nonlinear regression with four-parameter logistic regression fitting on triplicates from survival fractions of three independent replicates, following the model: y = Bottom + (Top – Bottom)/(1 + 10([logIC50 – x] × HillSlope)).
Generation of Isogenic MMR-deficient cell lines
Oligos of the form 5′-CACCG[N20] (where [N20] is the 20-nucleotide target sequence; sgGFP, GAGCTGGACGGCGACGTAAA; sgMSH2, ATTCTGTTCTTATCCATGAG; sgMSH6, TTATTGGAGTCAGTGAACTG; sgMLH1, ACTACCCAATGCCTCAACCG; sgPMS2, TCACTGCAGCAGCGAGTATG) and 5′-AAAC[rc20]C (where [rc20] is the reverse complement of [N20]) were purchased from Integrated DNA Technologies (IDT). For DIPG13 cells, oligos containing the sgRNA target sequence were annealed with their respective reverse complement and cloned into the lentiCRISPR all-in-one sgRNA/Cas9-delivery lentiviral expression vector (pXPR_BRD001; now available as pXPR_BRD023 lentiCRISPRv2) from the Broad Institute Genetics Perturbation Platform (GPP). For BT145 cells, oligos containing the sgRNA target sequence were annealed with their respective reverse complement and cloned into the pXPR_BRD051 CRISPRko all-in-one sgRNA/Cas9-delivery lentiviral expression vector (available from the Broad Institute Genetics Perturbation Platform, GPP). Successful cloning of each sgRNA target sequence was confirmed via Sanger Sequencing. To generate lentivirus from these vectors, HEK293T cells were transfected with 10 μg of each expression plasmid with packaging plasmids encoding PSPAX2 and VSVG using lipofectamine. Lentivirus-containing supernatant was collected 48 and 72 h after transfection. DIPG13 and BT145 cells were seeded in a 12-well plate at 1–3 × 106 cells/well in 3 ml medium and spin-infected (2,000 rpm for 2 h at 30 °C with no polybrene) with pLX311-Cas9 (DIPG13) or pXPR_BRD051 (BT145) lentiviral vectors and selected with blasticidin (10 μg/ml, DIPG13) or hygromycin (300 μg/ml, BT145) to generate Cas9-expressing or knockout cells. DIPG13-Cas9 cells underwent a subsequent lentiviral spin-infection with the lentiCRISPR sgGFP, sgMSH2, or sgMSH6 vectors described above. Puromycin selection (0.4 μg/ml for DIPG13 cells) commenced 48 h post-infection.
Chronic treatment and sequencing of isogenic MMR-deficient cell lines
DIPG13-sgGFP, -sgMSH2, and -sgMSH6 cells were seeded at 8 × 105 cells/well in 4 ml medium in a 6-well ULA plate. Each line was grown for 3 months under 3 conditions: no treatment, temozolomide (100 μM, Selleckchem), or DMSO vehicle. Cells were grown under these conditions in the absence of both blasticidin and puromycin. Cells were re-dosed with temozolomide or DMSO every 3–5 days, splitting over-confluent cells 1:2 or 1:4 as needed. After 3 months, genomic DNA was extracted using the QIAmp DNA Mini Kit. DNA was subjected to whole-exome Illumina sequencing. Reads were aligned to the Human Genome Reference Consortium build 38 (GRCh38). WES data were analysed using the Getz Lab CGA whole-exome sequencing characterization pipeline (https://docs.google.com/document/d/1VO2kX_fgfUd0x3mBS9NjLUWGZu794WbTepBel3cBg08/edit#heading=h.yby87l2ztbcj) developed at the Broad Institute which uses the following tools for quality control, calling, filtering and annotation of somatic mutations and copy number variation: PicardTools (http://broadinstitute.github.io/picard/) ContEst58, MuTect139, Strelka59, Orientation Bias Filter60, DeTiN61, AllelicCapSeg62, MAFPoNFilter63, RealignmentFilter, ABSOLUTE64, GATK38, Variant Effect Predictor65, and Oncotator40.
Subcutaneous xenografts and drug treatment
BT145 cells (2 × 106) were resuspended in equal parts Hank’s buffered salt solution (Life Technologies) and Matrigel (BD Biosciences) and then injected into both flanks of eight-week-old NU/NU male mice (Charles River). Tumour-bearing mice (n = 8) were randomly assigned to the treatment or vehicle arm when tumours measured a volume of 100 mm3. Animals received 12 mg/kg/day temozolomide or vehicle (Ora-Plus oral suspension solution, Perrigo, Balcatta, Australia) by oral gavage for 5 consecutive days per 28-day cycle. An additional 4 weeks resting period without treatment was observed before the second cycle. Tumour volumes were calculated using the formula: 0.5 × length × width2. Body weights were monitored twice weekly. The investigators were not blinded to allocation during experiments and outcome assessment. Mice were euthanized when they showed signs of tumour-related illness or before reaching the maximum tumour burden. Tumours were subsequently removed, and a subset were submitted to Oncopanel sequencing for analyses of exonic mutations (POPv3, 447 genes) and mutational signature as defined above. To separate human and mouse sequenced reads in the DNA sequencing data generated for the PDX models, the ‘raw’ data were mapped to both the hg19 human and mm10 mouse reference genomes using BWA-MEM-0.7.17. The output of the alignment was name sorted by Samtools-1.7. We then used the software package Disambiguate (ngs_disambiguate-1.0) to assign each read to the human or mouse genome and to produce final alignment files in BAM format. Final hg19 BAM files were coordinate sorted by Samtools-1.7. Duplicate reads were marked and removed from the BAM files using Picard-2.0.1. GATK4.1.0.0 base recalibration was performed using BaseRecalibration and Applying Recalibration followed by CollectF1R2Counts and LearnReadOrientationModel to create a model for read orientation bias. Variant calling was performed using GATK-4.1.0.0/Mutect2 pipeline with the default parameters and filters except for the following modifications: (i) ‘af-of-alleles-not-in-resource’ was set to 0; (ii) ‘MateOnSameContigOrNoMappedMateReadFilter’ was disabled; (iii) the output of Step8 was used for fitting the read orientation model; and (iv) a germline resource from the gnomAD database was included (https://console.cloud.google.com/storage/browser/_details/gatk-best-practices/somatic-b37/af-only-gnomad.raw.sites.vcf). The capture targets intervals used for Mutect2 were POPv3. The generated variant calls were further filtered using the FilterMutectCalls module of GATK4.1.0.0 and the final output in VCF format was annotated with Ensemble Variant Effect Predictor (ensembl-vep-96.0) using vcf2maf-1.6.16. The calls were additionally annotated with the OncoKB dataset using oncokb-annotator and sorted as MAF files.
All in vivo studies were performed in accordance with Dana-Farber Cancer Institute animal facility regulations and policies under protocol number 09-016.
Immunoblotting
Proteins were extracted in lysis RIPA buffer (50 mM Tris, 150 mM NaCl, 5 mM EDTA, 0.5% sodium deoxycholic acid, 0.5% NP-40, 0.1% SDS) supplemented with protease inhibitor cocktail (Roche Molecular). Proteins were quantified using the PierceBCA Protein Assay Kit, according to the manufacturer’s protocol. Samples were then prepared with 1× NuPAGE (Invitrogen) LDS sample buffer, and NuPAGE (Invitrogen) sample reducing agent followed by heating to 95 °C for 5 min. The samples were then loaded onto NuPAGE 4–12% Bis-Tris Gel (Invitrogen) with NuPAGE MOPS SDS (Invitrogen) buffer and run through electrophoresis. The transfer onto membrane was then done at 40 V overnight at 4 °C in NuPAGE transfer buffer (Invitrogen) with 10% methanol. Membranes were blocked with 5% skim milk in TBST for 1 h, then incubated with the following primary antibodies added to 5% BSA and incubated overnight at 4 °C on a shaker: mouse monoclonal anti-MGMT (Millipore, MT3.1, MAB16200, 1:500), mouse monoclonal anti-MSH2 (Calbiochem, FE11, NA27, 1:1,000), mouse monoclonal anti-MSH6 (Biosciences, 44, 610918, 1:500), mouse monoclonal anti-MLH1 (Cell Signaling, 4C9C7, 3515, 1:500), mouse monoclonal anti-PMS2 (BD Biosciences, A16-4, 556415, 1:1,000), mouse monoclonal anti-beta-actin (Sigma, AC-74, A2228, 1:10,000). After several cycles of washing and incubation with secondary goat anti-mouse antibody (Invitrogen 31430, 1:10,000), membranes were imaged by chemiluminescence using the Biorad ChemidocTM MP imaging system.
Microsatellite instability analysis
PCR amplification of the five mononucleotide markers (BAT25, BAT26, NR21, NR24, MONO27) was performed with the MSI Analysis System kit (Version 1.2, Promega). PCR products were analysed by an electrophoretic separation on the polymer POP7 50cm in an Applied Biosystems 3130XL sequencer and using Genemapper Software 5.
Outcome of patients treated with PD-1 blockade
For comparison of PFS and OS in patients treated with PD-1 pathway blockade according to TMB and MMR statuses, we retrospectively identified patients with glioma who had been treated with PD-1 blockade (alone or in combination with bevacizumab) for recurrent disease at our institutions. Patients for whom sequencing was not performed at the time of recurrence were excluded. Magnetic resonance imaging (MRI) tumour assessments were reviewed using the Response Assessment in Neuro-Oncology (RANO) criteria by three independent reviewers (M.J.L.-F., S.A., and R.Y.H.) who were blinded to the groups. PFS and OS duration were calculated from cycle 1 day 1 of PD-1 blockade therapy.
Statistical analyses
Data were summarized as frequencies and proportions for categorical variables and as median and range for continuous variables. Continuous variables were compared using Mann–Whitney or Kruskal–Wallis tests; categorical variables were compared using Fisher’s exact or Chi-squared tests. Survival and PFS were estimated using the Kaplan–Meier method, and differences in survival or PFS between groups were evaluated by the log-rank test. Survival for subjects who were alive or lost to follow-up at the time of last contact on or before data cut-off was censored at the date of the last contact. Patient matching in a k-to-k fashion was conducted using coarsened exact matching according to diagnosis, primary versus recurrent status, and prior treatments. For evaluation of response to PD-1 blockade, patients with glioma from the DFCI-Profile cohort who were treated with anti-PD(L)-1 antibodies or other treatments (total n = 210) as part of their management were included in the analysis. For multivariable analysis, Cox proportional hazard regression was used to investigate the variables that affect survival. P values were considered statistically significant when <0.05. Statistical analyses were performed using STATA (v14.2, StataCorp LLC, College Station, USA), Prism 8 (GraphPad Software, San Diego, USA), and MedCalc Statistical Software, version 19.0.3 (MedCalc Software bvba, Ostend, Belgium). For enrichment analyses, mutated genes were considered significant when Q < 0.01. Where applicable, the means of population averages from multiple independent experiments (± s.d. or s.e.m.) are indicated. No statistical methods were used to predetermine sample size.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
Clinical and sequencing data from 1,495 samples from the DFCI-Profile and 545 samples from the MSKCC-IMPACT datasets are publicly available (GENIE v.6.1: https://genie.cbioportal.org or https://www.synapse.org/). All data for samples from the GENIE v.6.1 and TCGA pan-cancer datasets are publicly available. Data for samples from the FMI dataset are not publicly available, but de-identified, aggregated data can be accessed on request. dbGaP Study Accession: phs001967.v1.p1. All other data are available on request.
Code Availability
The code for the detection of microsatellite mutations in single-cell DNA sequencing is publicly available (https://github.com/parklab/MSIprofiler).
References
Hunter, C. et al. A hypermutation phenotype and somatic MSH6 mutations in recurrent human malignant gliomas after alkylator chemotherapy. Cancer Res. 66, 3987–3991 (2006).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455, 1061–1068 (2008).
Johnson, B. E. et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343, 189–193 (2014).
Wang, J. et al. Clonal evolution of glioblastoma under therapy. Nat. Genet. 48, 768–776 (2016).
Barthel, F. P. et al. Longitudinal molecular trajectories of diffuse glioma in adults. Nature 576, 112–120 (2019).
Bouffet, E. et al. Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J. Clin. Oncol. 34, 2206–2211 (2016).
Rizvi, N. A. et al. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128 (2015).
Le, D. T. et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 372, 2509–2520 (2015).
McGranahan, N. et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469 (2016).
Brennan, C. W. et al. The somatic genomic landscape of glioblastoma. Cell 155, 462–477 (2013).
Brat, D. J. et al. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. N. Engl. J. Med. 372, 2481–2498 (2015).
Louis, D. N. et al. World Health Organization Histological Classification of Tumours of the Central Nervous System (ed. 2) (International Agency for Research on Cancer, 2016).
Cahill, D. P. et al. Loss of the mismatch repair protein MSH6 in human glioblastomas is associated with tumor progression during temozolomide treatment. Clin. Cancer Res. 13, 2038–2045 (2007).
Alexandrov, L. B. et al. Signatures of mutational processes in human cancer. Nature 500, 415–421 (2013).
Frampton, G. M. et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat. Biotechnol. 31, 1023–1031 (2013).
Sholl, L. M. et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016).
Zehir, A. et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 23, 703–713 (2017).
Campbell, B. B. et al. Comprehensive analysis of hypermutation in human cancer. Cell 171, 1042–1056.e10 (2017).
Stupp, R. et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 352, 987–996 (2005).
van den Bent, M. J. et al. Adjuvant procarbazine, lomustine, and vincristine chemotherapy in newly diagnosed anaplastic oligodendroglioma: long-term follow-up of EORTC brain tumor group study 26951. J. Clin. Oncol. 31, 344–350 (2013).
Kim, J. et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat. Genet. 48, 600–606 (2016).
Berends, M. J. et al. Molecular and clinical characteristics of MSH6 variants: an analysis of 25 index carriers of a germline variant. Am. J. Hum. Genet. 70, 26–37 (2002).
Yang, G. et al. Dominant effects of an Msh6 missense mutation on DNA repair and cancer susceptibility. Cancer Cell 6, 139–150 (2004).
Ollier, E. et al. Analysis of temozolomide resistance in low-grade gliomas using a mechanistic mathematical model. Fundam. Clin. Pharmacol. 31, 347–358 (2017).
Marabelle, A. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J. Clin. Oncol. 38, 1–10 (2020).
Germano, G. et al. Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120 (2017).
Mandal, R. et al. Genetic diversity of tumors with mismatch repair deficiency influences anti-PD-1 immunotherapy response. Science 364, 485–491 (2019).
Rosenthal, R. et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 567, 479–485 (2019).
Gejman, R. S. et al. Rejection of immunogenic tumor clones is limited by clonal fraction. eLife 7, e41090 (2018).
Gylling, A. H. et al. Differential cancer predisposition in Lynch syndrome: insights from molecular analysis of brain and urinary tract tumors. Carcinogenesis 29, 1351–1359 (2008).
Herrlinger, U. et al. Lomustine-temozolomide combination therapy versus standard temozolomide therapy in patients with newly diagnosed glioblastoma with methylated MGMT promoter (CeTeG/NOA-09): a randomised, open-label, phase 3 trial. Lancet 393, 678–688 (2019).
Jonsson, P. et al. Genomic correlates of disease progression and treatment response in prospectively characterized gliomas. Clin. Cancer Res. 25, 5537–5547 (2019).
Chalmers, Z. R. et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 9, 34 (2017).
Consortium, A. P. G.; AACR Project GENIE Consortium. AACR Project GENIE: powering precision medicine through an international consortium. Cancer Discov. 7, 818–831 (2017).
Cancer Genome Atlas Research Network et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 45, 1113–1120 (2013).
Garcia, E. P. et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med. 141, 751–758 (2017).
Ramkissoon, S. H. et al. Clinical targeted exome-based sequencing in combination with genome-wide copy number profiling: precision medicine analysis of 203 pediatric brain tumors. Neuro-oncol. 19, 986–996 (2017).
McKenna, A. et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303 (2010).
Cibulskis, K. et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 31, 213–219 (2013).
Ramos, A. H. et al. Oncotator: cancer variant annotation tool. Hum. Mutat. 36, E2423–E2429 (2015).
Bi, W. L. et al. Clinical identification of oncogenic drivers and copy-number alterations in pituitary tumors. Endocrinology 158, 2284–2291 (2016).
Abo, R. P. et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res. 43, e19 (2015).
Tate, J. G. et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res. 47, D941–D947 (2019).
Karczewski, K. J. et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. Preprint at https://www.biorxiv.org/content/10.1101/531210v2 (2019).
Landrum, M. J. et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46, D1062–D1067 (2018).
Sun, J. X. et al. A computational approach to distinguish somatic vs. germline origin of genomic alterations from deep sequencing of cancer specimens without a matched normal. PLOS Comput. Biol. 14, e1005965 (2018).
Garofalo, A. et al. The impact of tumor profiling approaches and genomic data strategies for cancer precision medicine. Genome Med. 8, 79 (2016).
Trabucco, S. E. et al. A novel next-generation sequencing approach to detecting microsatellite instability and pan-tumor characterization of 1000 microsatellite instability-high cases in 67,000 patient samples. J. Mol. Diagn. 21, 1053–1066 (2019).
Papke, D. J. Jr et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod. Pathol. 31, 1882–1890 (2018).
Rahbari, R. et al. Timing, rates and spectra of human germline mutation. Nat. Genet. 48, 126–133 (2016).
Gehring, J. S., Fischer, B., Lawrence, M. & Huber, W. SomaticSignatures: inferring mutational signatures from single-nucleotide variants. Bioinformatics 31, 3673–3675 (2015).
Rosenthal, R., McGranahan, N., Herrero, J., Taylor, B. S. & Swanton, C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome Biol. 17, 31 (2016).
Ellrott, K. et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst. 6, 271–281.e277 (2018).
Francis, J. M. et al. EGFR variant heterogeneity in glioblastoma resolved through single-nucleus sequencing. Cancer Discov. 4, 956–971 (2014).
Lodato, M. A. et al. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359, 555–559 (2018).
Bohrson, C. L. et al. Linked-read analysis identifies mutations in single-cell DNA-sequencing data. Nat. Genet. 51, 749–754 (2019).
Cortes-Ciriano, I., Lee, S., Park, W. Y., Kim, T. M. & Park, P. J. A molecular portrait of microsatellite instability across multiple cancers. Nat. Commun. 8, 15180 (2017).
Cibulskis, K. et al. ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602 (2011).
Saunders, C. T. et al. Strelka: accurate somatic small-variant calling from sequenced tumor-normal sample pairs. Bioinformatics 28, 1811–1817 (2012).
Costello, M. et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 41, e67 (2013).
Taylor-Weiner, A. et al. DeTiN: overcoming tumor-in-normal contamination. Nat. Methods 15, 531–534 (2018).
Landau, D. A. et al. Evolution and impact of subclonal mutations in chronic lymphocytic leukemia. Cell 152, 714–726 (2013).
Lawrence, M. S. et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501 (2014).
Carter, S. L. et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 30, 413–421 (2012).
McLaren, W. et al. The Ensembl variant effect predictor. Genome Biol. 17, 122 (2016).
Acknowledgements
We thank the patients and families who took part in the study, as well as the staff, research coordinators and investigators at each participating institution. M.T. is supported by Fondation pour la Recherche Médicale (FDM 41635), Fondation Monahan, The Arthur Sachs Foundation and The Philippe Foundation. C.L.B. was funded by a Bioinformatics and Integrative Genomics training grant from NHGRI (T32HG002295). S.S. is supported by the Ludwig Center at Harvard. M.S. is supported by Institut National du Cancer (INCa), the Ligue Nationale contre le Cancer (Equipe Labelisée), and Investissements d’avenir. R.B. is supported by NIH R01 CA188228, R01 CA215489, and R01 CA219943, The Dana-Farber/Novartis Drug Discovery Program, The Gray Matters Brain Cancer Foundation, Ian’s Friends Foundation, The Bridge Project of MIT and Dana- Farber/Harvard Cancer Center, The Pediatric Brain Tumor Foundation, the Fund for Innovation in Cancer Informatics, and The Sontag Foundation. P.B. is supported by NIH K99 CA201592, R00CA201592-03, the Dana-Farber Cancer Institute and Novartis Institute of Biomedical Research Drug Discovery and Translational Research Program, the Pediatric Brain Tumor Foundation and the St Baldrick’s Foundation. F. Bielle is supported by Fondation ARC pour la recherche sur le cancer (PJA 20151203562), INCa, a grant Émergence (Sorbonne Université) and ARTC (Association pour la recherche sur les tumeurs cérébrales). K.L.L. is supported by R01CA188288, P01 CA163205, P50 CA165962, Pediatric Brain Tumor Foundation, and the Ivy Foundation. This work was in part supported by a the SiRIC CURAMUS, which is funded by INCa, the French Ministry of Solidarity and Health and Inserm (INCA-DGOS-Inserm_12560). We acknowledge K. Bryan, S. Valentin, B. Bonneau, A. Matos and I. Detrait for preparation and processing of samples; W. Pisano and S. Block for help in data collection; E. F. Cohen for mouse xenograft sequencing analyses; D. X. Jin and J. Moore for assistance with FMI dataset creation and curation; the members of the BWH Center for Advanced Molecular Diagnostics; Y. Marie, J. Gueguan and the ICM Genotyping and Sequencing Core Facility (IGENSEQ) for sharing expertise related to analysis of copy array and sequencing data; C. Perry and the DFCI Oncology Data Retrieval System (OncDRS) for the aggregation, management, and delivery of the operational research data used in this project; the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, and members of the consortium for their commitment to data sharing; the cBioPortal for Cancer Genomics (https://www.cbioportal.org) and the Memorial Sloan Kettering Cancer Center for data sharing of the MSKCC-IMPACT dataset. We greatly appreciate feedback and support from M. L. Meyerson regarding bioinformatics and genomics analysis, I. K. Mellinghoff and T. J. Kaley for scientific advice, V. Rendo for scientific review of the manuscript, and M. Monje for providing the DIPG13 parental cell line. The content is solely the responsibility of the authors.
Author information
Authors and Affiliations
Contributions
M.T., R.B., P.B., F. Bielle and K.L.L. designed the study. M.T., Y.Y.L., R.B., P.B., F. Bielle and K.L.L. wrote the initial draft, with input from all authors. Y.Y.L. validated mutational signature analyses using TCGA data. M.T., Y.Y.L., L.F.S., R.S., D. Pavliak and L.A.A. performed TMB and mutational signature analyses of the DFCI-Profile, MSKCC-IMPACT and FMI datasets, and integrated TMB, signature and clinical data. L.F.S. developed the code for permutation tests. M.T., Y.Y.L., L.F.S. and R.S. performed and analysed the permutation tests. M.T., A.N.B., K.P., C. Bellamy, N.C., J.B., K.Q., P.H., S.M., L.T., R.B., P.B. and K.L.L. performed in vitro experiments in native and engineered models and analysed experimental data. M.T., K.P., J.B. and K.L.L. performed in vivo experiments in native models and analysed experimental data. M.T., F. Beuvon, K.M., S. Alexandrescu, D.M.M., S.S., F. Bielle, and K.L.L. reviewed histological and immunohistochemistry data on human samples. M.T. and J.B.I. performed survival analyses. M.T., Y.Y.L., C.L.B., I.C.-C., P.J.P., R.B., P.B. and K.L.L. performed single-cell sequencing experiments and analysed data. C.L.B., I.C.-C. and P.J.P. developed computational tools for the analysis of single-cell data. M.T., Y.Y.L., L.F.S., C.L.B., I.C.-C., S.H.R., F.D., A.S., R.S., D. Pavliak, L.A.A., E.G., G.M.F., F.C., A.D., A. Cherniack, P.J.P., R.B., P.B. and K.L.L. reviewed and analysed the bulk sequencing genomic data. M.T., J.B.I., C. Birzu, J.E.G., M.J.L.-F., R.J., N.Y., C. Baldini, E.G., S. Ammari, F. Beuvon, K.M., A.A., C.D., C.H., F.L.-D., D. Psimaras, E.Q.L., L.N., J.R.M.-F., A. Carpentier, P.C., L.C., B.M., J.S.B.-S., A. Chakravarti, W.L.B., E. A. Chiocca, K.P.F., S. Alexandrescu, S.C., D.H.-K., T.T.B., B.M.A., R.Y.H., A.H.L., F.C., J.-Y.D., K.H.-X., D.M.M., S.S., M.S., P.Y.W., D.A.R., A.M., A.I., R.B., P.B., F. Bielle, and K.L.L. abstracted and reviewed clinical and treatment response data. Y.Y.L., L.F.S., C.L.B., I.C.-C., R.S., L.A.A., G.M.F., A. Cherniack, and R.B. created bioinformatics tools and systems to support data analysis. R.B., P.B., F. Bielle, and K.L.L. acquired funding and supervised the study. All authors participated in data analysis and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
M.T. reports consulting or advisory role for Agios Pharmaceutical, Integragen, and Taiho Oncology, outside the submitted work; travel, accommodations, expenses from Merck Sharp & Dome, outside the submitted work. Y.Y.L. reports equity from g.Root Biomedical. S.H.R., R.S., D. Pavliak, L.A.A., G.M.F. and B.M.A. report employment with Foundation Medicine and stock interests from Roche. K.M. reports advisory board honoraria from Bristol-Meyers Squibb, outside the submitted work. F.L.-D. reports fees from Pharmtrace, outside the submitted work. E.Q.L. reports consulting or advisory role for Eli Lilly; royalties from UpToDate; honoraria from Prime Oncology. L.N. reports consulting or advisory role for Bristol-Meyers Squibb, outside the submitted work. T.T.B. reports honoraria from Champions Oncology, UpToDate, Imedex, NXDC, Merck, GenomiCare Biotechnology; consulting or advisory role for Merck, GenomiCare Biotechnology, NXDC, Amgen; travel, accommodations, expenses from Merck, Roche, Genentech, GenomiCare Biotechnology. A.H.L. reports leadership from Travera (I); stock and other ownership interests from Travera (I); consulting or advisory role for Travera (I). K.H.-X. reports advisory board honoraria from Bristol-Meyers Squibb, outside the submitted work. S.S. reports personal fees from Rarecyte, outside the submitted work. P.Y.W. reports honoraria from Merck; consulting or advisory role for AbbVie, Agios Pharmaceuticals, AstraZeneca, Blue Earth Diagnostics, Eli Lilly, Genentech, Roche, Immunomic Therapeutics, Kadmon Corporation, KIYATEC, Puma Biotechnology, Vascular Biogenics, Taiho Pharmaceutical, Deciphera Pharmaceuticals, VBI Vaccines; speakers’ bureau from Merck, prIme Oncology; research funding from Agios Pharmaceuticals (Inst), AstraZeneca (Inst), BeiGene (Inst), Eli Lilly (Inst), Roche (Inst), Genentech (Inst), Karyopharm Therapeutics (Inst), Kazia Therapeutics (Inst), MediciNova (Inst), Novartis (Inst), Oncoceutics (Inst), Sanofi (Inst), Aventis (Inst), VBI Vaccines (Inst); travel, accommodations, expenses from Merck. D.A.R. reports honoraria from AbbVie, Cavion, Genentech, Roche, Merck, Midatech Pharma, Momenta Pharmaceuticals, Novartis, Novocure, Regeneron Pharmaceuticals, Stemline Therapeutics, Celldex, OXiGENE, Monteris Medical, Bristol-Myers Squibb, Juno Therapeutics, Inovio Pharmaceuticals, Oncorus, Agenus, EMD Serono, Merck, Merck KGaA, Taiho Pharmaceutical, Advantagene; consulting or advisory role for Cavion, Genentech, Roche, Merck, Momenta Pharmaceuticals, Novartis, Novocure, Regeneron Pharmaceuticals, Stemline Therapeutics, Bristol-Myers Squibb, Inovio Pharmaceuticals, Juno Therapeutics, Celldex, OXiGENE, Monteris Medical, Midatech Pharma, Oncorus, AbbVie, Agenus, EMD Serono, Merck, Merck KGaA, Taiho Pharmaceutical; research funding from Celldex (Inst), Incyte (Inst), Midatech Pharma (Inst), Tragara Pharmaceuticals (Inst), Inovio Pharmaceuticals (Inst), Agenus (Inst), EMD Serono (Inst), Acerta Pharma (Inst), Omnivox. A.I. reports grants and other from Carthera (September 2019); research grants from Transgene; grants from Sanofi, and Air Liquide; and travel funding from Leo Pharma, outside the submitted work. R.B. reports consulting or advisory role for Novartis, Merck (I), Gilead Sciences (I), ViiV Healthcare (I); research funding from Novartis; patents, royalties, other intellectual property—Prognostic Marker for Endometrial Carcinoma (US patent application 13/911456, filed June 6, 2013), SF3B1 Suppression as a Therapy for Tumors Harboring SF3B1 Copy Loss (international application No. WO/2017/177191, PCT/US2017/026693, filed July 4, 2017), Compositions and Methods for Screening Pediatric Gliomas and Methods of Treatment Thereof (international application No. WO/2017/132574, PCT/US2017/015448, filed 1/27/2017). P.B. reports research grants from the Novartis Institute of Biomedical Research; patents, royalties, other intellectual property—Compositions and Methods for Screening Pediatric Gliomas and Methods of Treatment Thereof (international application No. WO/2017/132574, PCT/US2017/015448, filed 1/27/2017). F. Bielle reports employment from Celgene (I); stocks from Crossject (I); research grants from Sanofi and Abbvie; travel, accommodations, expenses from Bristol-Myers Squibb for travel expenses, outside the submitted work. K.L.L. reports grants and personal fees from BMS, grants from Amgen, personal fees and other from Travera LLC, personal fees from InteraGen, personal fees from Rarecyte, grants from Tragara, grants from Lilly, grants from Deciphera, grants from X4, all outside the submitted work; and has patent US20160032359A1 pending. Inst. denotes institutional funding; I denotes a competing interest involving a first degree relative of the author. The other authors report no competing interests.
Additional information
Peer review information Nature thanks Stefan Pfister and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Overview of the clinical characteristics of the patients in the study and analyses performed.
a, Clinical datasets analysed and main demographics including age, histomolecular subtype and disease stage. 1,628 glioma samples from adult and paediatric patients were sequenced as part of a large institutional prospective sequencing program of consented patients (DFCI-Profile) and subsequently clinically annotated. We identified 545 and 8,121 gliomas with sequencing from the MSKCC-IMPACT and FMI datasets, respectively, and used them as a replication set (total set of 10,294 sequenced samples). In addition, 314 tumours—including 247 consecutive recurrent gliomas—were analysed for protein expression of four MMR proteins (MSH2, MSH6, MLH1, and PMS2) using immunohistochemistry. b, Analyses performed and key clinical questions that were addressed in the study.
Extended Data Fig. 2 Distributions of TMB, homopolymer indels, and SNV mutation spectra in the datasets used.
a, DFCI-Profile (de novo gliomas only); b, MSKCC-IMPACT; c, FMI (total n = 9,938). After examining the distribution of TMB in each dataset for breakpoints, thresholds for hypermutation were further confirmed using segmented linear regression analysis (analysis restricted to de novo gliomas for DFCI-Profile). This method showed the presence of a breakpoint at 17.0 and 8.7 mutations per Mb for the DFCI-Profile and FMI datasets, respectively. For the MSKCC-IMPACT dataset, the cutoff used for hypermutation (13.8 mutations per Mb) was previously determined17. The frequency of hypermutation was similar in the three datasets (85 (5.2%) in DFCI-Profile; 29 (5.3%) in MSKCC-IMPACT; 444 (5.5%) in FMI). The median tumour mutation burden (TMB) in the combined datasets was 2.6 mutations per Mb (range, 0.0–781.3). Compared with non-hypermutated gliomas, hypermutated tumours showed atypical patterns of SNVs, consistent with abnormal mutational processes operating in these samples. Bars represent median and interquartile range for each dataset (right). HPI, homopolymer indels.
Extended Data Fig. 3 Integrated analysis of tumour mutation burden in hypermutated gliomas in the DFCI-Profile, MSKCC-IMPACK and FMI datasets.
a, Distribution of TMB, homopolymer indels, MMR mutations, and SNV mutational spectrum according to molecular status of IDH1/2, 1p/19q co-deletion (1p/19q), gain of chromosome 7 and/or deletion of chromosome 10 (7gain/10del), and MGMT promoter methylation, histological grade, age at initial diagnosis, and history of prior treatment with alkylating agents or radiation therapy (the distinction between photon and proton therapy was not systematically captured) in the DFCI-Profile dataset (n = 84, data not shown for the single sample from other gliomas, IDH1/2-wt subgroup). b, Top, distribution of histomolecular groups in non-hypermutated and hypermutated gliomas from the combined sequencing dataset (n = 2,173). Bottom, distribution of molecular groups in de novo and post-treatment hypermutated gliomas from the DFCI-Profile dataset (n = 85) (annotation not available for the MSKCC-IMPACT set). c, Prevalence of hypermutation according to MGMT promoter methylation and IDH1/2 mutation status in post-temozolomide gliomas from the DFCI-Profile dataset (n = 150). Two-sided Fisher’s exact test. d, Number of temozolomide cycles according to IDH1/2 mutation status in post-temozolomide diffuse gliomas from the DFCI-Profile dataset (n = 211 gliomas). Patients who received combined chemoradiation but no adjuvant temozolomide were included. Two-sided Wilcoxon rank-sum test. e, Boxplots of TMB in post-treatment hypermutated gliomas according to the number of temozolomide cycles received before surgery. Kruskal–Wallis test and Dunn’s multiple comparison test. f, TMB in recurrent gliomas according to treatments received before surgery. Patients who received multiple treatment modalities were excluded. Kruskal–Wallis test and Dunn’s multiple comparison test. Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range (d–f). g, Integrated analysis of the FMI dataset (n = 8,121 gliomas) depicting tumour mutation burden, the number of indels at homopolymer regions, and the SNV mutation spectrum detected in each tumour according to molecular status of IDH1/2 and 1p/19q co-deletion (1p/19q), MSI status, and age at initial diagnosis. Dominant mutational signatures detected in hypermutated samples are depicted. The dotted line indicates the threshold for samples with a high mutation burden (8.7 mutations per Mb). h, Prevalence of hypermutation among molecularly defined subgroups in the FMI dataset (n = 8,121 gliomas). Chi-squared test. i, Dominant mutational signatures detected in hypermutated samples in the FMI dataset (n = 8,121 gliomas). Chi-squared test. j, Mutated genes and pathways enriched in hypermutated gliomas in the FMI dataset (n = 8,121). Enrichment was assessed using a permutation test to control for random effects of hypermutability in tumours with high TMB. k, l, Proportion of TMBhigh versus TMBlow samples with mutations in selected DNA repair genes and glioma drivers (e) and in the MMR pathway (MSH2, MSH6, MLH1 and PMS2; f). Permutation test; ****P < 0.0001, ***P < 0.001, **P < 0.01; ns, not significant.
Extended Data Fig. 4 Validation of known hypermutation-associated signatures using TCGA datasets.
Mutational signatures were predicted using exome-sequencing variants that overlapped with the panel-targeted regions, and then compared to previously published DeconstructSigs signature predictions based on all exome variants. The TCGA MC3 dataset was used to assess the detection of COSMIC mutational signatures associated with APOBEC (signatures 2 and 13), mismatch repair (signature 6), ultraviolet light (signature 7), POLE (signature 10), and tobacco (signature 4). Variant calls for 17 hypermutated and 12 non-hypermutated glioma exome-sequenced samples4 were used to assess temozolomide (signature 11) detection. a, Detection of APOBEC-associated mutational signature in TCGA BLCA samples (n = 129 out of 411 samples). b, Detection of ultraviolet-associated mutational signature in TCGA SKCM samples (n = 237 out of 466 samples). c, Detection of tobacco smoking-associated mutational signature in TCGA LUAD samples (n = 250 out of 513 samples). d, Detection of MMR-associated mutational signature in TCGA COAD (n = 188 out of 380 samples). e, Detection of POLE-associated mutational signature in TCGA COAD and READ samples (n = 277 out of 380 samples). f, Detection of temozolomide-associated mutational signature in ref. 4 (n = 29). g, Unsupervised clustering of hypermutated samples. A total of 865 hypermutated tumour samples from exomes (pan-TCGA and Wang et al.4) and targeted panels (DFCI-Profile and MSK-IMPACT) were analysed for known hypermutation signatures (tobacco, UV, MMRD, POLE, TMZ, APOBEC). Samples and signatures underwent 2D hierarchical clustering based on Euclidean distance. h, Performance of cancer panel versus other genesets in mutational signature calling. We analysed 622 hypermutated tumour exomes (pan-TCGA and Wang et al.4, black) for their mutational signature contributions when restricted to variants from i) DFCI-Profile OncoPanel cancer panel genes (red), or ii) 9 randomly selected gene sets (grey) of similar total capture size to the cancer panel. For each sample, we assessed known hypermutation signatures for cancer panels and gene sets for which at least 20 single base substitutions were retained in the sample after restriction. Samples and signatures underwent 2D hierarchical clustering based on Euclidean distance. i, The violin plots represent the number of variants (top) and the cosine similarity of signature contributions (bottom) when using all exonic variants versus restriction to cancer panel or the 49 random gene sets. Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range. Two-sided Welch’s t-test.
Extended Data Fig. 5 Mutational signature analysis of primary and secondary hypermutated cohort (n = 111).
a, Mutational signature analysis of newly diagnosed hypermutated gliomas in the DFCI-Profile dataset (n = 24). b, Mutational signature analysis of secondary hypermutated gliomas (samples in which hypermutation was detected in the recurrent tumour) in the DFCI-Profile dataset (n = 58). The novel COSMIC Signature 11-related signature (S2) was associated with 1p/19q co-deletion and lack of prior radiation therapy (66.7% of samples with high S2 versus 26.2% of samples with high S1 signature, Fisher P = 0.016). c, Mutational signature analysis of hypermutated gliomas from the MSKCC-IMPACT dataset (n = 29). d, Mutational signature analysis in de novo (hypermutated at first diagnosis, n = 26, left) and post-treatment hypermutated gliomas (hypermutation in a recurrent tumour, n = 59, right). Percentage of samples exhibiting the most common mutational signatures and their hypothesized causes are displayed. MMR, C6, C14, C15, C26; age-related, C1; POLE, C10, C14. Chi-squared test. e, Mutational signatures identified in individual de novo hypermutated gliomas (hypermutated at first diagnosis, n = 26, left) and post-treatment hypermutated gliomas (hypermutation in a recurrent tumour, n = 59, right). f, Mutational signature analysis of MMR variants in hypermutated gliomas from the DFCI-Profile and MSKCC-IMPACT datasets (n = 114). Ninety variants of the MMR genes MSH2, MSH6, MLH1, and PMS2 were merged into two groups (de novo, n = 18; post-treatment, n = 72) according to the type of sample in which they were found and analysed for mutational signatures using a regression model (Rosenthal et al.52). In each sample, only the MMR variant with the highest VAF was included, to limit the inclusion of possible passenger variants. For signature discovery in both cohorts (a–c), variants were analysed using the non-negative matrix factorization (NMF) method and correlated with known COSMIC mutational signatures14 using Pearson correlation.
Extended Data Fig. 6 Characteristics of MMR molecular variants in hypermutated gliomas.
a, b, Proportion of TMBhigh versus TMBlow samples with mutations in selected DNA repair genes and glioma drivers (a) and in the MMR pathway (MSH2, MSH6, MLH1 and PMS2) (b) in the merged DFCI-Profile/MSKCC-IMPACT dataset (n = 2,173). Permutation test; ****P < 10−5, **P < 10−2, *P < 0.05. c, CCFs of MMR gene mutations in post-treatment hypermutated gliomas versus other hypermutated cancers in the FMI dataset. Horizontal line, median. Two-sided Wilcoxon rank-sum test with Benjamini–Hochberg correction. d, VAF distribution of mutations in post-treatment hypermutated gliomas, non-glioma MMR-deficient cancers (diverse histologies) and other non-glioma hypermutated samples (diverse histologies) from the TCGA and MSKCC-IMPACT datasets. Each dot represents a mutation found in an individual sample (represented vertically). MMR mutations are depicted in red. Left, hypermutated samples from the pan-TCGA dataset; right, hypermutated samples from the MSKCC-IMPACT dataset. e, Integrated view of mutational signatures and MMR gene mutations and protein expression in hypermutated gliomas (n = 114). Tumours with the mutational hotspot MSH6(T1219I) (11.9% of post-treatment hypermutated gliomas) are highlighted. f, Mutation diagram of MSH2, MSH6, MLH1, and PMS2 mutations found in hypermutated gliomas from the DFCI-Profile and MSKCC-IMPACT datasets (n = 114). The hotspot MSH6 missense variant p.T1219I was found in nine samples. g, Hotspot MSH6 p.T1219I variant mapped to the bacterial MutS 3D structure (PDB 5YK4). h, Representative immunohistochemistry (IHC) images of the MMR proteins MSH2, MSH6, MLH1 and PMS2 in a hypermutated glioblastoma with MSH6(T1219I) mutation. Three independent samples were stained. Scale bar, 100 μm.
Extended Data Fig. 7 Results of MMR IHC screening in 213 consecutive recurrent gliomas and patterns of MMR protein expression loss in three de novo or post-treatment MMR-deficient gliomas.
a, Recurrent patterns of MMR protein loss identified by IHC in gliomas. Scale bar, 50 µm. b, Summary of MMR IHC screening results for 213 consecutive recurrent gliomas. All monocentric consecutive relapses of diffuse gliomas in adult patients following treatment with post-alkylating agents (surgery between 2009 and 2015) were included in the immunohistochemistry analysis. Further sequencing of samples in which MMR protein loss was identified showed hypermutation with MMR molecular defects in 18/19 (94.7%) samples. c, Percentage of tumour MMR protein loss in glioma samples with de novo (n = 16) or post-treatment (n = 46) MMR deficiency. Samples were scored by two pathologists in blinded fashion. Regional heterogeneity of MMR protein loss for the four MMR proteins MSH2, MSH6, MLH1, and PMS2 was scored as to the maximal percentage of protein loss among tumour cells for each sample (5% increments). Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range, excluding outliers. Two-sided Wilcoxon rank-sum test. d, Clonal MMR deficiency in a de novo high-grade glioma. Top left, low magnification of haematoxylin and eosin (H&E) staining of the large surgical tumour pieces obtained from surgical resection. Right, high magnification in three tumour areas (H&E staining, MLH1 and PMS2 immunostaining) showing a highly cellular tumour with an oligodendroglial phenotype and a loss of expression of MLH1 and PMS2 in all tumour cells (open arrowheads). Normal cells have a maintained MLH1 and PMS2 expression (solid arrowheads). Bottom left, microsatellite testing via PCR amplification of five mononucleotide markers (BAT25, BAT26, NR21, NR24, and MONO27) showed the tumour to be MSS. Array CGH showed a homozygous deletion of the entire coding region of MLH1. Scale bars; top left, 5 mm; right, 100 µm. e, Clonal MMR deficiency in a hypermutated post-treatment, IDH1-mutant glioblastoma. Top left, low-magnification image of H&E staining of tissue obtained from surgical resection, with three areas of tumour selected for images. Red dashed line delimits normal brain. Right, high-magnification images of H&E staining, showing highly cellular tumour and an astrocytic phenotype, and PMS2 IHC, showing loss of expression of PMS2 in all tumour cells (open arrowheads). Normal cells have maintained PMS2 expression (internal control, solid arrowheads). Bottom left, Microsatellite testing via PCR amplification of five mononucleotide markers (BAT25, BAT26, NR21, NR24, and MONO27) showed the tumour to be MSS. NGS showed a TMB of 120.1 per Mb and homopolymer indel burden of 3.8 per MB, with contributions from temozolomide (90%) and MMR-deficiency (10%) mutational signatures. A missense (p.P648L) hotspot MLH1 mutation known to be pathogenic from patients with Lynch syndrome with a VAF of 0.73 and loss of heterozygosity was present in this case. Scale bars, top left, 5 mm; right 100 µm. f, Subclonal MMR deficiency in a hypermutated post-treatment IDH1-mutant glioblastoma. Top left, low-magnification image of PMS2 immunostaining of the tumour pieces obtained from surgical resection. Right, high magnification images of three areas of PMS2 immunostaining showing heterogeneous PMS2 expression across the sample consistent with a subclonal tumour. Area 1 shows that PMS2 is retained in atypical tumour cells (arrow); area 2 is heterogeneous with loss (open arrowhead) in some but not all tumour cells; area 3 is an example of diffuse loss of expression in tumour cells (open arrowhead). Normal cells have a maintained PMS2 expression (solid arrowheads in all images). Bottom left, microsatellite analysis via PCR amplification of five mononucleotide markers (BAT25, BAT26, NR21, NR24, and MONO27) showed the tumour to be MSS. NGS showed a TMB of 236.5 per Mb and homopolymer indel burden of 2.3 per MB, with 95% contribution of temozolomide mutational signature. Scale bars, top left 5 mm; right 100 µm.
Extended Data Fig. 8 Characterization of high-grade glioma PDCLs and their sensitivity to temozolomide and CCNU.
a, Clinico-molecular characteristics of four native newly diagnosed or recurrent glioma PDCL models harbouring hypermutation and MMR deficiency. b, Thirty glioma PDCLs, including four PDCLs derived from patients with de novo (BT1160, N16-1162, both established from patients with Lynch syndrome) or post-treatment (BT237, BT559) MMR deficiency were molecularly characterized using whole-exome sequencing. The panels show the tumour mutational burden (left) and homopolymer indel burden (right) in each model. Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range. Two-sided Wilcoxon rank-sum test. c, Mutational signature analysis was performed in the PDCL models of constitutional and post-treatment MMR deficiency using the R package DeconstructSigs to estimate the contributions of mutational signatures using a regression model (Rosenthal et al.52). For each PDCL, the contribution of the main COSMIC mutational signatures identified is expressed as decimal. d, Boxplots of temozolomide AUC in non-hypermutated versus hypermutated PDCLs. Boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range. Two-sided Wilcoxon rank-sum test. e, f, A panel of 12 glioma PDCL models representing the different MGMT and MMR classes was selected and assessed for sensitivity to temozolomide in a short-term viability assay (e; dots represent means). The temozolomide AUC was compared between the three groups using a Kruskal–Wallis test and Dunn’s multiple comparison test (f; mean ± s.d.). g, Western blot of the glioblastoma patient-derived cell line (BT145) in which the genes MSH2, MSH6, MLH1 or PMS2 have been knocked-out using the CRISPR–Cas9 system. h, i, A panel of 11 glioma PDCL models representing the different MGMT and MMR classes was selected and assessed for sensitivity to CCNU in a short-term viability assay (h; dots represent means). No CCNU data was available for the model BT172. The CCNU AUC was compared between the three groups using a Kruskal–Wallis test and Dunn’s multiple comparison test (i; mean ± s.d.).
Extended Data Fig. 9 MMR-deficient models of glioma, continued.
a, b, CRISPR–Cas9 MSH2 and MSH6 gene knockout in DIPG13 high-grade glioma cell line. a, Integrated genomics viewer (IGV) plots depicting MSH2 reads in between the guide RNAs in the MSH2 unedited line (sgGFP, left) and the MSH2 CRISPR knockout line (right) confirming knockout in the MSH2 edited line. b, IGV plots depicting MSH6 reads in between the guide RNAs in the MSH6 unedited line (sgGFP, left) and the MSH6 CRISPR knockout line (right) confirming knockout in the MSH6 edited line. c, Overview of in vivo temozolomide resistance study. Treatment of subcutaneous BT145 PDX-bearing animals was initiated at a volume of 100 mm3 and eight nude mice per group were randomized to 12 mg/kg/day temozolomide or vehicle for five consecutive days per 28-day cycle. Mice were treated until tumours reached a volume of 1,500 mm3, and tumours were sequenced to identify mutations and mutational signature. d, Survival of mice with BT145 xenografts (n = 8 mice per group) during treatment with vehicle (blue) or temozolomide (red). Two-sided log-rank test. e, Unique variants found in three sequenced BT145 tumours (two temozolomide-treated, and one vehicle-treated) were analysed for correlation with known mutational signatures. COSMIC Signature 11 was found in the two temozolomide-treated tumours. Mutational signatures could not be called in the vehicle-treated tumour (too few variants). After filtering of truncal variants common to all tumours, the two temozolomide-treated tumours shared only four variants, including an MSH6(T1219I) mutation and three noncoding variants. f, BT145 xenografts chronically treated with vehicle (n = 1) or temozolomide (n = 2) were removed, dissociated and cultured in serum-free medium to establish cell lines. After three passages in culture, sensitivity to temozolomide was assessed. The results of the short-term viability assays (mean ± s.e.m.) and temozolomide AUC of each cell line are depicted. g, Model of acquired hypermutation with mutational signature 11 in gliomas. Top, MMR-proficient cells repair TMZ damage and do not develop signature 11. Resistance in these cells is mediated by non-MMR pathways (for example, MGMT expression). Bottom, TMZ induces and/or selects resistant subclonal MMR-deficient cells. Further TMZ exposure produces accumulation of mutations at specific trinucleotide contexts, detected as hypermutation with signature 11.
Extended Data Fig. 10 Extended outcome data.
a–c, Survival of patients with recurrent high-grade glioma (WHO grade III or IV) from the time of initial diagnosis according to TMB status (solid curves, TMBlow; dotted curves, TMBhigh). The curves include 240 recurrent samples from DFCI-Profile with available survival data from initial diagnosis. Two-sided log-rank test. a, Survival of patients with recurrent high-grade 1p/19q co-deleted oligodendroglioma from the time of initial diagnosis. b, Survival of patients with recurrent high-grade IDH1/2-mutant astrocytoma from the time of initial diagnosis. c, Survival of patients with recurrent IDH1/2 wild-type glioblastoma from the time of initial diagnosis. d, PFS of 11 patients with hypermutated and MMR-deficient glioma who were treated with PD-1 blockade (single-agent or in combination with bevacizumab, red curve). A cohort of patients with non-hypermutated glioma who were treated with PD-1 blockade is depicted as control (n = 10, best matches according to diagnosis, primary versus recurrent status, and prior treatments, blue curve). A two-sided log-rank test is used. e, f, PFS (e) and OS (f) of 11 patients with hypermutated and MMR-deficient glioma who were treated with PD-1 blockade (red curves). A cohort of hypermutated patients treated with other systemic agents is depicted as control (best matches according to diagnosis, primary vs recurrent status, and prior treatments were selected from the cohort of sequenced gliomas, purple curves). Two-sided log-rank test. Clinical and histomolecular characteristics of patients from both cohorts are provided in Supplementary Table 7. g, Lack of immune response following PD1 blockade (pembrolizumab) in a patient with post-treatment hypermutated MMR-deficient glioblastoma. Top, timeline; middle, MRI images; bottom, H&E images and IHC for PMS2 expression and tumour infiltration with CD3-positive T cells and IBA1-positive macrophages in the primary (S1), recurrent pre-pembrolizumab (S3) and recurrent post-pembrolizumab (S4) tumours. The tumour acquired a focal PMS2 two-copy deletion, protein loss, and hypermutation in the post-temozolomide recurrent tumour (S3). Scale bar, 50 µm.
Extended Data Fig. 11 Molecular characteristics of hypermutated gliomas.
a, Pan-cancer analysis of TMB and homopolymer indel burden in the GENIE dataset (n = 44,389). Tumour samples from the GENIE dataset (v6.1) were analysed for mutational and homopolymer indel burden. Statistical comparisons between groups are provided in Supplementary Table 6. b, TMB in hypermutated gliomas (post-treatment) versus MMR-deficient cancers and other hypermutated cancers from the TCGA and Wang et al.4 exome datasets (n = 798). Two-sided Wilcoxon rank-sum test with Bonferroni correction. c, Pan-cancer analysis of the homopolymer indel burden in hypermutated gliomas (post-treatment) versus MMR-deficient cancers and other hypermutated cancers from the TCGA and Wang et al.4 exome datasets (n = 798). d, Results of MSI analysis using the standard pentaplex assay in glioma (n = 39) and CRC samples (n = 19) according to MMR status (MMR-d, MMR deficient; MMR-p, MMR-proficient). e, Pan-cancer analysis of cancer cell fractions in hypermutated gliomas (post-treatment) versus MMR-deficient cancers and other hypermutated cancers from the TCGA and Wang et al.4 exome datasets (n = 798). Two-sided Wilcoxon rank-sum test with Bonferroni correction. f, Weighted TMB in hypermutated gliomas (post-treatment) versus MMR-deficient cancers and other hypermutated cancers from the TCGA and Wang et al.4 exome datasets (n = 798). The weighted TMB was calculated by weighing each individual mutation to its cancer cell fraction. Two-sided Wilcoxon rank-sum test with Bonferroni correction. g, Distribution of VAFs (left) and mutation spectrum analysis of low-allelic frequency variants (<0.1, right) in TMBlow gliomas (n = 1,543, top), de novo hypermutated gliomas with MMR deficiency mutational signature (n = 12, middle), and post-treatment hypermutated gliomas (n = 59, bottom) from the DFCI-Profile dataset. h, Distribution of VAFs (left) and mutation signature analysis of low-allelic frequency variants (<0.1, right) in TMBlow CRCs (n = 1,265, top) and TMBhigh CRCs with MMR deficiency mutational signature (n = 110, bottom) from the GENIE dataset. i, Clinical timeline for the patient with hypermutated glioblastoma with an MSH6(T1219I) mutation in whom bulk and single-cell WGS was performed. j, Distribution of VAFs of mutations in the recurrent bulk sample. The median VAF in the recurrent sample was 0.11. The MSH6(T1219I) mutation had the 18th-highest VAF out of 4,350 coding mutations. k, Cancer cell fractions (CCFs) of mutations in the primary and recurrent tumour bulk samples. Each dot represents a coding mutation. The horizontal and vertical axes are estimated clonal frequency for each mutation in the primary and recurrent samples, respectively. Mutations of the four main MMR genes are depicted in red. l, Mutational spectra in 35 cells from the primary tumour (orange) and 28 from the recurrent tumour (green) submitted to scWGS sequencing (1×). Mutational signature analysis showed a strong contribution of mutational signature 11 in hypermutated cells from the recurrent tumour. m, Representative IGV plots (n = 2 distinct genomic segment for each sample) of microsatellite insertions in the normal (TMB low) and recurrent (TMB high) bulk samples and recurrent TMB low (n = 2) and TMB high (n = 2) single cells. Solid arrowheads represent microsatellite insertions phased with a flanking heterozygous SNP allele. Open arrowheads represent microsatellite insertions for which the reads do not reach the flanking heterozygous SNP allele. Both hypermutated single cells showed multiple phased microsatellite insertions consistent with a true somatic microsatellite mutation. In general, a few reads with similar microsatellite insertions correctly phased with the same flanking heterozygous SNP allele were found in the recurrent bulk, but not in the normal bulk or non-hypermutated cells. For a–c, e, f, biological subgroups were identified on the basis of mutational burden, dominant signature and histology. For b, c, e, f, 100 non-hypermutated samples were randomly selected as controls. For all box plots: boxes, quartiles; centre lines, median ratio for each group; whiskers, absolute range, excluding outliers. RT, radiation therapy; Cil, cilengitide; Cabo, cabozantinib; Bev, bevacizumab.
Supplementary information
Supplementary Figures
This file contains Supplementary Figures 1-2.
Supplementary Table 1
Clinical data for samples included in the sequencing dataset.
Supplementary Table 2
Clinical and histomolecular characteristics of the sample set.
Supplementary Table 3
Clinical, histologic and molecular characteristics of hypermutated gliomas from the Profile dataset (n=85).
Supplementary Table 4
Recurrent MMR gene alterations in hypermutated gliomas (n=558).
Supplementary Table 5
Multivariable proportional hazards for overall survival (OS) in recurrent gliomas (n=346).
Supplementary Table 6
Comparative analysis of TMB (left panel) and homopolymer indel burden (right panel) in the GENIE dataset.
Supplementary Table 7
Clinical and histomolecular characteristics of hypermutated glioma patients treated with PD-1 blockade (HM – PD-1i, n=11), as well as matched non-hypermutated glioma patients treated with PD-1i (non-HM –PD-1i, n=10), and matched hypermutated glioma patients treated with other therapies (HM – Other, n=10).
Supplementary Table 8
Multivariable analysis of overall survival (OS) in patients with recurrent high-grade glioma treated with anti-PD(L)-1 antibodies or other treatments (n=210).
Rights and permissions
About this article

Cite this article
Touat, M., Li, Y.Y., Boynton, A.N. et al. Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580, 517–523 (2020). https://doi.org/10.1038/s41586-020-2209-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-2209-9
- Springer Nature Limited
This article is cited by
-
From clinical management to personalized medicine: novel therapeutic approaches for ovarian clear cell cancer
Journal of Ovarian Research (2024)
-
A patient-derived cell model for malignant transformation in IDH-mutant glioma
Acta Neuropathologica Communications (2024)
-
Unveiling divergent treatment prognoses in IDHwt-GBM subtypes through multiomics clustering: a swift dual MRI-mRNA model for precise subtype prediction
Journal of Translational Medicine (2024)
-
Integration of 3D bioprinting and multi-algorithm machine learning identified glioma susceptibilities and microenvironment characteristics
Cell Discovery (2024)
-
Minimally-invasive implantable device enhances brain cancer suppression
EMBO Molecular Medicine (2024)