

Abstract
Glycans have diverse physiological functions, ranging from energy storage and structural integrity to cell signalling and the regulation of intracellular processes1. Although biomass-derived carbohydrates (such as d-glucose, d-xylose and d-galactose) are extracted on commercial scales, and serve as renewable chemical feedstocks and building blocks2,3, there are hundreds of distinct monosaccharides that typically cannot be isolated from their natural sources and must instead be prepared through multistep chemical or enzymatic syntheses4,5. These ‘rare’ sugars feature prominently in bioactive natural products and pharmaceuticals, including antiviral, antibacterial, anticancer and cardiac drugs6,7. Here we report the preparation of rare sugar isomers directly from biomass carbohydrates through site-selective epimerization reactions. Mechanistic studies establish that these reactions proceed under kinetic control, through sequential steps of hydrogen-atom abstraction and hydrogen-atom donation mediated by two distinct catalysts. This synthetic strategy provides concise and potentially extensive access to this valuable class of natural compounds.
Similar content being viewed by others


Main
Simple structural and storage polymers including starch, cellulose, and hemicellulose are important sources of the monosaccharide feedstocks d-glucose, d-xylose, d-galactose, d-mannose and l-arabinose (Fig. 1a). Isomerization is a useful strategy for the synthesis of so-called ‘rare’ sugars (Fig. 1b) from biomass precursors; however, these processes remain challenging owing to the structural and stereochemical complexity of sugars. Chemical isomerization reactions (for example, the Lobry de Bruyn–Alberda van Ekenstein and Bilik reactions) are typically unselective, leading to complex thermodynamic distributions of products and often intractable separations (Extended Data Fig. 1a)8. In contrast, enzymatic methods offer an added level of precision and have emerged as a powerful synthetic alternative to chemical strategies9. Enzymatic isomerizations feature prominently in industrial sugar processing, including in the syntheses of d-fructose and d-ribose from d-glucose10,11. In principle, multistep enzymatic synthesis also provides synthetic access to rare hexose, pentose and tetrose isomers but low yields and prohibitive production costs nonetheless limit implementation of these strategies12,13. For example, d-allose has potential value as a low-glycemic sweetener and shows promising anti-inflammatory and immunosuppressive activity14. The enzymatic synthesis of d-allose can be achieved in overall 2.5% yield from d-glucose through sequential treatment with d-xylose isomerase (50% yield), d-tagatose 3-epimerase (20% yield), and l-rhamnose isomerase (25% yield allose + 8% yield altrose) (Extended Data Fig. 1b)15. Like chemical isomerizations, nearly all enzymatic isomerizations proceed through reversible polar enolization mechanisms under equilibrium control. Maximum product yields are therefore dictated by thermodynamic considerations under reaction conditions constrained by temperature-dependent enzyme activity. Reaction scope is also mechanistically restricted: for example, 2-deoxygenated sugars cannot be substrates under enolization-based isomerization conditions.

a, Only a handful of monosaccharides can be obtained from natural sources. b, Hundreds of rare sugars exist and feature prominently in glycosylated natural products. c, Radical SAM-dependent epimerization by NeoN of neomycin C involves sequential HAT from substrate to the 5′-deoxyadenoxyl radical, and from cysteine thiol (Cys-249–SH) to the substrate. (Met, methionine.) d, Proposed direct chemical epimerization of biomass sugars to the rare sugars described here.
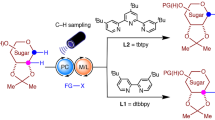
We envisioned that rare sugar isomers could be obtained directly from biomass-derived carbohydrates through site-selective radical epimerization reactions. A kinetically controlled epimerization process would require C–H bond cleavage and C–H bond formation steps to proceed through distinct mechanisms but could in principle afford product yields and selectivities exceeding those observed under canonical equilibrium-controlled sugar isomerization conditions. Our approach draws inspiration from recent reports of enzymatic radical epimerizations mediated by the cofactor S-adenosyl methionine (SAM)16,17. For example, in the biosynthesis of neomycin B from neomycin C, a high-energy 5′-deoxyadenosyl radical abstracts the C5 hydrogen atom from the terminal saccharide of neomycin C (Fig. 1c). Subsequent re-delivery of a hydrogen atom to the opposite face is achieved via a pendant cysteine thiol. Within the enzyme active site, diastereoselective hydrogen-atom transfer (HAT) steps are thus promoted through exquisite spatial control over the hydrogen-atom donor/acceptor pairs. The architecture of the enzyme further mitigates any chemical incompatibility of the 5′-deoxyadenosyl radical and cysteine thiol. Outside the physical context of an enzyme, however, other interactions would be necessary to achieve kinetic control and prevent reagent donor/acceptor quenching. Although several notable synthetic HAT-mediated epimerization reactions have been developed, these methods almost universally proceed through reversible HAT steps, affording equilibrium product distributions18. However, a deracemization reaction recently reported by Knowles has established the conceptual viability of contra-thermodynamic radical isomerization through sequential proton-coupled electron transfer and HAT steps19.
In addition to kinetic challenges posed by employing chemically incompatible reagents, the success of this transformation further requires numerous nearly identical C–H bonds to be distinguished within the context of a highly polar, densely functionalized, stereochemically complex unprotected carbohydrate substrate. Indeed, while site-selective functionalization of carbohydrate O–H bonds has been the subject of considerable recent attention20, examples of selective carbohydrate C–H oxidation and functionalization are extremely limited21. Precedents set by the Minnaard, Waymouth and Muramatsu laboratories have established the feasibility of site-selective oxidation of minimally-protected monosaccharides to keto-sugars using Pd(ii)/benzoquinone, Pd(ii)/O2, and Sn(iv)/Br2 catalyst systems, respectively22,23,24. Recent work by Minnaard has demonstrated that diastereoselective C–H alkylation of glucose derivatives can be achieved through site-selective hydrogen-atom abstraction by a quinuclidinium radical cation generated under photoredox conditions25. Taylor has further expanded the scope of this transformation by employing a borinic acid co-catalyst to promote stereoretentive C–H alkylation of cis-diol-containing monosaccharides26. Building on these findings, here we report a strategy for the synthesis of rare sugar isomers from biomass-derived precursors through the site-selective epimerization of unprotected sugars and glycans (Fig. 1d).
After extensive exploration of reaction conditions, the minimally protected model substrate, α-methylglucose, was found to react under photochemical conditions to afford α-methylallose as the sole reaction product in 92% yield within 3 h (Fig. 2). Optimal reaction conditions employ catalytic quantities of 1,2,3,5-tetrakis(carbazol-9-yl)-4,6-dicyanobenzene (4-CzIPN), quinuclidine, adamantane thiol and tetrabutylammonium p-chlorobenzoate in MeCN/DMSO at room temperature under blue light irradiation (see Supplementary Fig. 1). No epimerization was observed to occur in the absence of photocatalyst, thiol or light, and only trace product (<1%) was observed in the absence of quinuclidine. The reaction yield was considerably diminished in the absence of benzoate base (29% yield) or by employing tetrabutylammonium benzoate as the base (63% yield). Ir[(dF(CF3)ppy)2(dtbpy)]PF6 (IrF) is also an effective photoredox catalyst for this transformation, promoting the desired reaction at only 1 mol% loading. However, the observation of oxidation products, as well as considerable cost differences ($5 per mmol for 4-CzIPN versus $935 per mmol for IrF; dollar prices are in US$ in 2020) led us to select 4-CzIPN as the preferred reagent. Alkyl thiols were uniquely effective at promoting epimerization: no reaction was observed using thiophenols or thiobenzoic/thioacetic acid derivatives, nor when using any other hydrogen-atom donor surveyed (see Supplementary Information section 8).

Effect of changes to optimized reaction conditions. Yields determined by proton nuclear magnetic resonance (1H NMR) analysis using 4-fluoroanisole as internal standard. RSM, recovered starting material; MeCN, acetonitrile; DMSO, dimethylsulfoxide; RT, room temperature; LED, light-emitting diode; Me, methyl; DABCO, 1,4-diazabicyclo[2,2,2]octane; Ad-SH, adamantane thiol; Bz, benzoyl. aSee Supplementary Information sections 5 and 8 for full experimental details.
Although the basis for site-selectivity is not yet fully understood, the C3 selectivity observed here is congruent with the substrate-controlled selectivity previously noted by both Waymouth and Minnaard in oxidation reactions22,23,25. Nuclear magnetic resonance (NMR) titration experiments reveal an equilibrium interaction between 1a and tetrabutylammonium p-chlorobenzoate with alteration of substrate 1J(C–H) coupling constants, implicating the presence of hydrogen-bonding interactions between 1a and the base with concomitant weakening of α C–H bonds27. In contrast, no such interaction is observed between pentamethylated glucose and base, and importantly, no epimerization products were detected in reactions employing permethylated or peracetylated substrates.
A range of biomass-derived and abundant monosaccharides were evaluated as substrates under the optimized reaction conditions (Fig. 3). This strategy provides synthetic access to 4 of the 5 rare hexose isomers. In addition to d-allose products obtained from glucose-configured substrates, d-talose (2b), d-gulose (2c) and d-altrose (2d) products are obtained selectively from the reaction of β-methylgalactose, anhydrogalactose and anhydromannose, respectively. The biomass-derived pentose sugars d-α-methylxylose (1f) and l-β-methylarabinose (1e) afforded d- and l-ribose derivatives through C3 and C2 epimerization, respectively. Although d-ribose is accessible on a large scale through glucose fermentation, synthetic access to l-ribose remains extremely limited2,11. Despite the presence of an electron-rich acetamide substituent, the N-acetylglucosamine derivative 1g also undergoes productive epimerization, reacting to afford a 1.5:1 mixture of the C3- and C4-epimeric products, N-acetylallosamine, 2g, and N-acetylgalactosamine, 3g, respectively.

Scope of the reaction with respect to substrate. Reactions were conducted at 0.3-mmol scale in duplicate; the average 1H NMR yield is reported; duplicate reactions were combined, then isolated; isolated yields are in parentheses. a, Isolated in peracetylated form. b, 1 mol% Ir[(dF(CF3)ppy)2(dtbpy)]PF6 used instead of 4-CzIPN. c, Reaction was conducted using 5 mol% quinuclidine. d, Reaction was conducted using 2.5 mol% quinuclidine. See Supplementary Information Sections 5–7 for full experimental details and conditions.
Completely unprotected monosaccharides also undergo selective epimerization under these conditions: for example, 42% yield d-allose 2k is obtained from d-glucose (compared with a 2.5% total yield over 4 steps, using enzymatic methods), and 55% yield l-6-deoxytalose 2m is obtained from the reaction of l-fucose. The reaction of d-2-deoxyglucose affords d-2-deoxyallose in 61% yield: importantly, epimerization of 2-deoxygenated sugars cannot be realized using alternative, enolization-based isomerization methods.
More complex glycans were subsequently evaluated to assess further the selectivity and functional group compatibility of the reaction conditions. Allosucrose, 2o, can be obtained selectively from sucrose in 68% yield, and despite the presence of 14 stereogenic centres, the reaction of raffinose, 1p, affords a singly epimerized product, 2p, in 44% yield. Pyrimidine-containing pyranonucleoside, 1q, reacted to afford the C3-epimeric product, 2q. Finally, the C-glycoside SGLT2 inhibitor empagliflozin was also examined as a substrate, and was found to afford the C3-epimeric product, 2r, in 42% yield with no observation of epimerization at any other position in the molecule.
We performed a series of experiments to gather insight into the underlying mechanism of this transformation. Re-subjecting 6-deoxy-β-methyltalose, 2h, (formed in 70% yield from β-methylfucose, 1h) to standard reaction conditions did not result in the formation of any β-methylfucose starting material (Fig. 4a). A similar experiment was conducted using α-methylallose and adamantane thiol-d1 (where ‘d1’ indicates deuterium isotopic substitution). After 16 hours, 95% α-methylallose was recovered with 39% deuterium incorporation at the C3 position; no glucose products were detected. This experiment demonstrates that both α-methylglucose and α-methylallose can undergo hydrogen-atom abstraction but that both converge to the α-methylallose product (Fig. 4b). Taken in conjunction with established thermochemical data, these experiments provide preliminary evidence that these transformations do not proceed under simple equilibrium control28.

a, No epimerization is observed when reaction products are re-subjected to the standard reaction conditions. b, Deuterium labelling studies indicate that the reaction product reacts under standard reaction conditions, but both epimers converge to a common product. c, Reaction in the absence of thiol donor affords no isomerization product, implicating irreversible hydrogen-atom abstraction by the quinuclinium radical cation. d, Plausible mechanistic pathway for selective isomerization reactions.
To explore further the individual elementary steps of this reaction, Stern–Volmer fluorescence quenching was examined under two different sets of conditions (see Supplementary Information section 9). Preliminary experiments reveal that quinuclidine efficiently quenches the photocatalyst excited state, whereas adamantane thiol does not quench under the conditions examined. In the presence of both quinuclidine and adamantane thiol, quenching kinetics are identical to the ‘quinuclidine only’ conditions. These findings support a mechanism in which the excited photocatalyst is quenched by quinuclidine to generate quinuclidinium radical cation29. When catalyst loading is increased to 3 mol% and the reaction is run in the absence of thiol, small quantities (3% yield) of 3-keto sugar are obtained (Fig. 4c). Importantly, no epimerization is observed under these conditions, nor under any conditions tested where alkyl thiol is not present in the reaction mixture (see Supplementary Information section 9). These experiments establish that photocatalyst and quinuclidine are sufficient for C–H cleavage to occur but insufficient for epimerization. Together, they support a mechanism in which the quinuclidinium radical cation mediates an irreversible hydrogen-atom abstraction step.
To probe the role of the thiol co-catalyst, an analogous set of fluorescence quenching experiments was carried out using 4-bromothiophenol in the place of adamantine thiol. Under these reaction conditions no epimerization or consumption of α-methylglucose is observed. As with adamantane thiol, minimal fluorescence quenching was observed using 4-bromothiophenol alone. However, in the presence of both 4-bromothiophenol and quinuclidine, a substantial increase in fluorescence quenching was observed relative to the case with quinuclidine alone (see Supplementary Information section 9). We postulated that this enhanced fluorescence quenching might be due to the oxidation of thiolate—generated in situ by deprotonation of thiol by quinuclidine, or through proton-coupled electron transfer from a quinuclidine/thiol hydrogen-bonded complex—to the corresponding thiyl radical. Indeed, sodium thiophenolate was also found to quench the photocatalyst at a tenfold-higher rate than quinuclidine alone, and NMR titration studies performed under comparable conditions identified a small equilibrium interaction (Keq = 42 M−1) between 4-bromothiophenol (pKa = 9.0 in DMSO) and quinuclidine (quinuclidinium conjugate acid, pKa = 9.8 in DMSO), supporting the formation of thiolate in solution.
These experiments indicate that thiol acidity is an important parameter distinguishing productive versus unproductive reaction conditions. In the presence of quinuclidine, acidic thiols can be deprotonated to form thiolate salts. Preferential thiolate quenching of the photocatalyst results in the formation of thiyl radicals. No epimerization was observed under these, or any other photo-oxidative, photo-reductive and thermal conditions that we explored for the in situ generation of thiyl radical (see Supplementary Information section 9). These findings suggest that thiyl radical is not competent for hydrogen-atom abstraction. Accordingly, the thiol co-catalyst is instead implicated in a subsequent, irreversible HAT to the incipient sugar radical.
Collectively, the mechanistic studies presented here support a non-equilibrium epimerization mechanism proceeding through two sequential and distinct HAT steps: hydrogen-atom abstraction by quinuclidinium radical cation (homolytic bond dissociation enthalpy = 100 kcal mol−1) from substrate, followed by HAT from thiol (87 kcal mol−1) to the incipient sugar radical (Fig. 4d). Although both substrate and product can undergo hydrogen-atom abstraction by quinuclidinium radical cation, mechanistic data are consistent with irreversible hydrogen-atom abstraction followed by diastereoselective HAT from thiol. Attendant to a kinetically controlled epimerization mechanism, the reaction yields and product selectivities presented here exceed nearly all other direct isomerization yields reported so far, which have reflected thermodynamic product distributions.
Data availability
All data supporting the findings of this paper are available within the Article and its supplementary information files.
References
Varki, A. et al. (eds) Essentials of Glycobiology 3rd edn (Cold Spring Harbor Laboratory Press, 2017).
de Leder Kremer, R. M. & Gallo-Rodriquez, C. Naturally occurring monosaccharides: properties and synthesis. Adv. Carbohydr. Chem. Biochem. 59, 9–67 (2004).
Chheda, J. N., Huber, G. W. & Dumesic, J. A. Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew. Chem. Int. Ed. 46, 7164–7183 (2007).
Imperiali, B. Bacterial carbohydrate diversity—a brave new world. Curr. Opin. Chem. Biol. 53, 1–8 (2019).
Herget, S. et al. Statistical analysis of the Bacterial Carbohydrate Structure Data Base (BCSDB): characteristics and diversity of bacterial carbohydrates in comparison with mammalian glycans. BMC Struct. Biol. 8, 35 (2008).
Frihed, T. G., Bols, M. & Pedersen, C. M. Synthesis of l-hexoses. Chem. Rev. 115, 3615–3676 (2015).
Elshahawi, S. I., Shaaban, K. A., Kharel, M. K. & Thorson, J. S. A comprehensive review of glycosylated bacterial natural products. Chem. Soc. Rev. 44, 7591–7697 (2015).
Angyal, S. The Lobry de Bruyn–Albereda van Ekenstein transformation and related reactions. In Glycoscience: Epimerisation, Isomerisation and Rearrangement Reactions of Carbohydrates (ed. Stütz, A. E.) 1–14 (Springer, 2001).
Buchholz, K. & Seibel, J. Industrial carbohydrate biotransformations. Carb. Res. 343, 1966–1979 (2008).
Bhosale, S. H., Rao, M. B. & Deshpande, V. V. Molecular and industrial aspects of glucose isomerase. Microbiol. Rev. 60, 280–300 (1996).
Wulf, P. & Vandamme, E. Production of d-ribose by fermentation. Appl. Microbiol. Biotechnol. 48, 141–148 (1997).
Granström, T. B., Takata, G., Tokuda, M. & Izumori, K. Izumoring: a novel and complete strategy for the bioproduction of rare sugars. J. Biosci. Bioeng. 97, 89–94 (2004).
Zhang, W., Zhang, T., Jiang, B. & Mu, W. Enzymatic approaches to rare sugar production. Biotechnol. Adv. 35, 267–274 (2017).
Hossain, M. A. et al. Effect of the immunosuppressants FK506 and d-allose on allogenic orthotopic liver transplantation in rats. Transplant. Proc. 32, 2021–2023 (2000).
Menavuvu, B. T. et al. Efficient biosynthesis of d-allose from d-psicose by cross-linked recombinant l-rhamnose isomerase: separation of product by ethanol crystallization. J. Biosci. Bioeng. 101, 340–345 (2006).
Kudo, F., Hoshi, S., Kawashima, T., Kamach, T. & Eguchi, T. Characterization of a radical S-adenosyl-l-methionine epimerase, NeoN, in the last step of neomycin B biosynthesis. J. Am. Chem. Soc. 136, 13909–13915 (2014).
Benjdia, A., Guillot, A., Ruffie, P., Leprince, J. & Berteau, O. Post-translational modification of ribosomally synthesized peptides by a radical SAM epimerase in Bacillus subtilis. Nat. Chem. 9, 698–707 (2017).
Wang, Y. et al. Epimerization of tertiary carbon centers via reversible radical cleavage of unactivated C(sp 3)–H bonds. J. Am. Chem. Soc. 140, 9678–9684 (2018).
Shin, N. Y., Ryss, J. M., Zhang, X., Miller, S. J. & Knowles, R. R. Light-driven deracemization enabled by excited-state electron transfer. Science 366, 364–369 (2019).
Dimakos, V. & Taylor, M. S. Site-selective functionalization of hydroxyl groups in carbohydrate derivatives. Chem. Rev. 118, 11457–11517 (2018).
Frihed, T. G., Bols, M. & Pedersen, C. M. C–H functionalization on carbohydrates. Eur. J. Org. Chem. 2740–2756 (2016).
Jäger, M., Hartmann, M., de Vries, J. G. & Minnaard, A. J. Catalytic regioselective oxidation of glycosides. Angew. Chem. Int. Ed. 52, 7809–7812 (2013).
Chung, K. & Waymouth, R. M. Selective catalytic oxidation of unprotected carbohydrates. ACS Catal. 6, 4653–4659 (2016).
Muramatsu, W. Catalytic and regioselective oxidation of carbohydrates to synthesize keto-sugars under mild conditions. Org. Lett. 16, 4846–4849 (2014).
Wan, I. C., Witte, M. D. & Minnaard, A. J. Site-selective carbon–carbon bond formation in unprotected monosaccharides using photoredox catalysis. Chem. Commun. 53, 4926–4929 (2017).
Dimakos, V., Su, H. Y., Garrett, G. E. & Taylor, M. S. Site-selective and stereoselective C–H alkylations of carbohydrates via combined diarylborinic acid and photoredox catalysis. J. Am. Chem. Soc. 141, 5149–5153 (2019).
Maiti, N. C., Zhu, Y., Carmichael, I., Serianni, A. S. & Anderson, A. E. 1JCH correlates with alcohol hydrogen bond strength. J. Org. Chem. 71, 2878–2880 (2006).
Goldberg, R. N. & Tewari, Y. B. Thermodynamic and transport properties of carbohydrates and their monophosphates: the pentoses and hexoses. J. Phys. Chem. Ref. Data 18, 809–880 (1989).
Jeffrey, J. L., Terrett, J. A. & Macmillan, D. W. C. O–H hydrogen bonding promotes H atom transfer from α C–H bonds for C–alkylation of alcohols. Science 349, 1532–1536 (2015).
Acknowledgements
We thank A. Seim for checking the reaction procedure and X. Gu for help with substrate synthesis. A.E.W. also thanks E. Kwan, A. Radosevich, D. Suess and Z. Wickens for discussions. Financial support for this work was provided by the Massachusetts Institute of Technology and the National Science Foundation (NSF) for funding through the National Science Foundation Graduate Research Fellowships Program (NSF-GRFP) to H.M.C.
Author information
Authors and Affiliations
Contributions
A.E.W. and Y.W. conceived the work. A.E.W., Y.W. and H.M.C. designed the experiments. Y.W. and H.M.C. conducted the experiments. A.E.W. directed the research and wrote the manuscript with input from all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature thanks Christian Marcus Pedersen and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Chemical and enzymatic isomerizations proceed through polar aldose-ketose mechanisms.
a, Chemical isomerization reactions of glucose lead to unselective and complex thermodynamic distribution of products. b, ‘Izumoring’ enzymatic synthesis of d-allose proceeds through reversible polar enolization mechanisms under equilibrium control; see ref. 12. d-XI is d-xylose isomerase; d-TE is d-tagatose 3-epimerase; and l-RhI is l-rhamnose isomerase.
Supplementary information
Supplementary Information
This file contains the following sections: 1. General Experimental Methods; 2. Materials and Reagents; 3. Instrumentation; 4. Synthesis of Catalysts (4CzIPN and 4-ClOBzBu4N) and Substrates; 5. General Reaction Procedure for Sugar Epimerization; 6. General Procedure for Purification of Epimeric Sugars; 7. Product Characterization; 8. Condition Optimization for Sugar Epimerization of 1a; 9. Mechanistic Studies; 10. References; and 11. NMR Spectra.
Rights and permissions
About this article

Cite this article
Wang, Y., Carder, H.M. & Wendlandt, A.E. Synthesis of rare sugar isomers through site-selective epimerization. Nature 578, 403–408 (2020). https://doi.org/10.1038/s41586-020-1937-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-020-1937-1
- Springer Nature Limited
This article is cited by
-
Electrocatalytic cyclic deracemization enabled by a chemically modified electrode
Nature Catalysis (2024)
-
Catalytic glycosylation for minimally protected donors and acceptors
Nature (2024)
-
Synthetic techniques for thermodynamically disfavoured substituted six-membered rings
Nature Reviews Chemistry (2024)
-
Characterization of a novel ribose-5-phosphate isomerase B from Curtobacterium flaccumfaciens ZXL1 for D-allose production
Food Science and Biotechnology (2024)
-
Functional-group translocation of cyano groups by reversible C–H sampling
Nature (2023)