

Abstract
Amyotrophic lateral sclerosis (ALS) is a complex neurodegenerative disorder, in which the clinical manifestations may be influenced by genetic and unknown environmental factors. Here we show that ALS-prone Sod1 transgenic (Sod1-Tg) mice have a pre-symptomatic, vivarium-dependent dysbiosis and altered metabolite configuration, coupled with an exacerbated disease under germ-free conditions or after treatment with broad-spectrum antibiotics. We correlate eleven distinct commensal bacteria at our vivarium with the severity of ALS in mice, and by their individual supplementation into antibiotic-treated Sod1-Tg mice we demonstrate that Akkermansia muciniphila (AM) ameliorates whereas Ruminococcus torques and Parabacteroides distasonis exacerbate the symptoms of ALS. Furthermore, Sod1-Tg mice that are administered AM are found to accumulate AM-associated nicotinamide in the central nervous system, and systemic supplementation of nicotinamide improves motor symptoms and gene expression patterns in the spinal cord of Sod1-Tg mice. In humans, we identify distinct microbiome and metabolite configurations—including reduced levels of nicotinamide systemically and in the cerebrospinal fluid—in a small preliminary study that compares patients with ALS with household controls. We suggest that environmentally driven microbiome–brain interactions may modulate ALS in mice, and we call for similar investigations in the human form of the disease.
Similar content being viewed by others

Main
ALS is a neurodegenerative disorder that is characterized by the premature death of motor neurons and has an average survival time of three to five years from diagnosis1. Extensive efforts are being made to develop ALS-specific drugs; however, so far only riluzole2 and edaravone3 have shown modest efficacy. Environmental factors have been postulated to modify the course of the disease4—these may include circulating low-molecular-mass metabolites that originate from the gastrointestinal tract and permeate the blood–brain barrier, after which they can modulate metabolic, transcriptional and epigenetic programs in neurons and in other resident cells5. However, the causative role of such environmental factors in ALS or in other pathologies of the central nervous system remains largely unknown6.
The gut microbiome is a source of these potentially disease-modifying bioactive metabolites, and has recently been suggested to contribute to the pathogenesis of neurological disorders7,8 by affecting neuronal transmission, synaptic plasticity, myelination and complex host behaviours9,10,11. Several observations suggest that the interface between the host and the gut microbiome may be altered in mouse models of ALS12,13, including that of an impaired gut barrier function and a dysbiotic microbiome configuration that was partially corrected by butyrate supplementation13. Analysis of 16S ribosomal DNA (rDNA) from stool samples of patients with ALS yielded conflicting results: one study noted dysbiosis in 6 patients with ALS compared with 5 healthy controls14, whereas another showed no differences between 25 patients with ALS and 32 healthy controls15. To our knowledge, there has been no direct investigation into the functional microbiome in an ALS setting. In this study, we used mice and preliminary human data to functionally assess the potential modulatory involvement of the microbiome in ALS.
Altered gut microbiome worsens ALS symptoms in mice
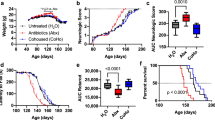
To assess the role of the gut microbiome in ALS, we used the Sod1G93A (herein Sod1-Tg) ALS mouse model. First, we depleted the microbiome of male and female Sod1-Tg mice or littermate controls by administrating broad-spectrum antibiotics at the age of 40 days (Extended Data Fig. 1a). Motor abilities were quantified using the rotarod locomotor test16, the hanging-wire grip test17 and neurological scoring18. Compared with Sod1-Tg mice that were given water only, we found that antibiotic treatment was associated with a significant exacerbation of motor abnormalities in the mice throughout the progression of ALS (pooled results, n = 14–20 mice per group, Fig. 1a–c; repeats, n = 4–8 mice in each group of each repeat, Extended Data Fig. 1b–j). To further assess time-, genotype- and treatment-dependency, we used a mixed linear model (methods) for each of the three motor readouts. Indeed, a poorer performance was noted in antibiotic-treated versus non-treated Sod1-Tg mice in the rotarod, grip score and neurological score (P value for genotype × treatment of 0.008, 0.058 and 5.6 × 10−36, and P value for time × genotype × treatment of 0.002, 0.002 and 1.1 × 10−36, respectively). Notably, at our vivarium we found that antibiotic treatment did not affect performances in the rotarod locomotor or the hanging-wire grip tests in wild-type littermate controls, when compared with wild-type mice that did not receive antibiotics (Fig. 1a–c, Extended Data Fig. 1b–j). A linear regression analysis further supported the negative effect of antibiotic treatment on these neuropathological measurements (Extended Data Fig. 1k–m).

a–c, Evaluation of motor symptoms in mice using the rotarod locomotor test (a), the hanging-wire grip test (b) and neurological scoring (c). *P = 0.0256 (a), ***P = 0.0006 (b) and ***P = 0.0005 (c), two-sided Mann–Whitney U-test. Results are pooled from three repeats. n = 20 Sod1-Tg water-treated mice; n = 18 Sod1-Tg antibiotic (Abx)-treated mice; n = 14 wild-type (WT) water-treated and n = 15 wild-type antibiotic-treated mice. Data are mean ± s.e.m. AUC, area under the curve. AU, arbitrary units. d, Survival of germ-free Sod1-Tg (GF; n = 15), specific-pathogen-free (SPF; n = 18) Sod1-Tg, germ-free wild-type (n = 9) and specific-pathogen-free wild-type (n = 22) mice. **P = 0.0062, two-sided log-rank test, and **P = 0.0011, Gehan–Breslow–Wilcoxon test. Results are pooled from two repeats.
In agreement with these findings, the histopathological analysis—using Luxol Fast Blue staining—of spinal cords from 140-day-old Sod1-Tg mice revealed a significant reduction in cell counts (suggestive of neurons) in antibiotic-treated compared with water-treated mice (Extended Data Fig. 1n, o). This suggests an increase in the death of motor-neuron cells after chronic exposure to antibiotics. Moreover, we performed T2-weighted magnetic resonance imaging of the mouse brain stem in areas that are known to degenerate in the Sod1-Tg model19 (Extended Data Fig. 1p), and found a prolonged T2 relaxation time in antibiotic-treated compared with water-treated Sod1-Tg mice (Extended Data Fig. 1q–x); this is indicative of higher levels of free water, enhanced brain atrophy and neurodegeneration20. An automated home-cage locomotion system revealed a significant reduction (P = 0.03) in the activity of antibiotic-treated Sod1-Tg mice at 100 days old, when compared with water-treated Sod1-Tg controls (Extended Data Fig. 1y). Antibiotic-induced aggravation of the motor function of Sod1-Tg mice was not associated with alterations in the main immune-cell sub-populations of the spinal cord, the small intestine or the colon lamina propria, compared with water-treated Sod1-Tg controls (Extended Data Fig. 1z–ae); this suggests that the antibiotic-associated phenotypic differences were not immune-mediated.
Notably, attempts at the rederivation of Sod1-Tg mice into the germ-free setting were associated with high mortality rates of Sod1-Tg mice but not of wild-type littermate controls; rederivation attempts failed with 30 pregnant Sod1-Tg dams over a period of 18 months. After rederivation succeeded, germ-free Sod1-Tg mice had significantly enhanced mortality when compared with germ-free wild-type littermates or with colonized Sod1-Tg mice (Fig. 1d, pooled results, n = 9–22 mice per group, Extended Data Fig. 1af, ag, two independent repeats, n = 3–16 per group); this persisted even when germ-free mice were spontaneously colonized at 115 days old, which suggests that microbial drivers affect the progression of ALS at an earlier stage of the disease. Collectively these results indicate that, in our vivarium, antibiotic-mediated microbiome alteration—or the absence of a microbiome in germ-free mice—could potentially exacerbate ALS in Sod1-Tg mice. This suggests that a locally dysbiotic gut microbiome configuration may modulate disease progression in this model.
Vivarium-dependent pre-clinical dysbiosis in mouse ALS
The suggested microbial-mediated effects on the neuropathology of ALS prompted us to identify locally prevalent commensal strains that potentially modulate the course of the disease. We used 16S rDNA sequencing to assess the composition of the faecal microbiome of Sod1-Tg mice and wild-type littermate controls at our vivarium. We found a significant difference between the two, which started early and persisted throughout the course of the disease as determined by a linear regression analysis (Fig. 2a–c, Extended Data Fig. 2a–m). Likewise, although limited by the numbers of mice used in these experiments (n = 6), a linear mixed model accounting for genotype and time (methods) yielded similar results (q = 2.01 × 10−29, 2.39 × 10−14, 2.46 × 10−3, 8.78 × 10−3, 0.016, 4.58 × 10−11, 4.9 × 10−4, 8.78 × 10−3, 0.028 and 8.16 × 10−4 for P. distasonis, Rikenellaceae, Prevotella, Lactobacillus, Sutterella, Allobaculum, Desulfovibrionaceae, Coprococcus, Oscillospira and Bifidobacterium, respectively). The total number of observed OTUs (alpha diversity) was higher in stool samples of Sod1-Tg mice at all time points (Extended Data Fig. 2n), although the total faecal bacterial load did not vary between Sod1-Tg mice and wild-type controls (Extended Data Fig. 2o). Additionally, at our vivarium we found that even the gut microbiome configurations of antibiotic-treated Sod1-Tg mice and their antibiotic-treated wild-type littermate controls were significantly different (Extended Data Fig. 2p–ab). Notably, the spontaneous colonization of germ-free Sod1-Tg mice and their germ-free wild-type littermates at our vivarium resulted in de novo dysbiosis (Extended Data Fig. 3a–i); however, dysbiosis was not observed in a second non-barrier (not specific-pathogen-free) vivarium in which Akkermansia, Parabacteroides, Erysipelotrichaceae and Helicobacteraceae were nearly absent (Extended Data Fig. 3j–m, Supplementary Table 1). Overall, these facility-dependent changes suggested that a combination of genetic susceptibility to ALS and a locally prevalent commensal signature drive early pre-clinical dysbiosis, which potentially contributes to the modulation of ALS.

a–c, Weighted UniFrac principal coordinate analysis at the ages of 40 days (pre-symptomatic; a), 100 days (disease onset; b) and 140 days (advanced disease; c). The experiment was repeated three times, n = 4 to 6 mice in each group. d, Species-level taxa summary obtained from gut microbiome metagenomic shotgun sequencing of stool samples from wild-type and Sod1-Tg mice during disease progression. e, Principal component analysis of entries in the KEGG database corresponding to the microbiomes of wild-type and Sod1-Tg mice. Spearman’s ρ = 1.57 × 10−14. n = 6 wild-type mice aged 40, 60 and 100 days, and Sod1-Tg mice aged 40 and 80 days; n = 5 wild-type mice aged 80 days and Sod1-Tg mice aged 60 and 100 days.
When measured by shotgun metagenomic sequencing, significant differences were noted in the faecal microbiome composition of Sod1-Tg mice compared with wild-type littermate controls by linear regression (Fig. 2d, Extended Data Fig. 4a–n). Similarly, although limited by the numbers of mice used in these experiments (n = 6), a linear mixed model (methods) accounting for genotype and time yielded similar results (q = 6.11 × 10−10, 1.08 × 10−15, 1.63 × 10−32, 2.37 × 10−50, 0.0007, 0.021, 5.06 × 10−74, 0.016, 4.97 × 10−13, 7.77 × 10-15, 7.71 × 10−7 and 5.24 × 10−5 for P. distasonis, Alistipes unclassified, Lactobacillus murinus, Eggerthella unclassified, Parabacteroides goldsteinii, Subdoligranulum unclassified, Helicobacter hepaticus, Lactobacillus johnsonii, Bacteroides vulgatus, Bifidobacterium pseudolongum, Lactobacillus reuteri and Desulfovibrio desulfuricans, respectively). Functionally, the faecal bacterial metagenomes of Sod1-Tg and wild-type mice clustered separately with respect to microbial genes (for PC1: 40 days old, P = 0.0002; 60 days old, P = 0.0002; 80 days old, P = 0.0005; 100 days old, P = 0.0005, Spearman correlation; KEGG orthology; Fig. 2e); in Sod1-Tg mice, compared with their wild-type counterparts, there was a marked reduction in the representation of genes encoding enzymes that participate in tryptophan metabolism (Extended Data Fig. 4o, p) and substantial alterations in those involved in nicotinamide (NAM) and nicotinate metabolism (Extended Data Fig. 4q). A detailed metabolic assessment of Sod1-Tg and wild-type mice at 60 days old (before the onset of clinical symptoms) found no significant differences in food and water intake, respiratory-exchange ratio, oxygen consumption, locomotion, heat production, and speed (Extended Data Fig. 4r–ac).
Collectively, these results demonstrate that, at our vivarium, single-genotype-housed Sod1-Tg mice diverge from their wild-type littermates in terms of gut microbial composition and function, even before the appearance of motor-neuron dysfunction.
Commensal microbe contribution to ALS exacerbation
To determine whether these ALS-associated changes in the microbiome contribute to disease features, we tested 11 strains—Eggerthella lenta, Coprobacillus cateniformis, P. goldsteinii, L. murinus, P. distasonis, Lactobacillus gasseri, Prevotella melaninogenica, Eisenbergiella tayi (a member of the Lachnospiraceae family), Subdoligranulum variabile, R. torques and A. muciniphila—most of which were suggested by our composite 16S rDNA and shotgun metagenomic analyses to be correlated with the severity of ALS progression in the Sod1-Tg mouse model at our vivarium (Extended Data Figs. 2, 4). We mono-inoculated anaerobic cultures of each of the above strains (optical density = 0.4–0.7) into antibiotic pre-treated Sod1-Tg and wild-type mice, by repeated oral administration at 6-day intervals for a total of 15 treatments. Most of the stated bacteria did not affect ALS symptoms (Extended Data Fig. 5a–l). However, supplementation of antibiotic-treated Sod1-Tg mice with P. distasonis (Extended Data Fig. 5a–c) and R. torques (Extended Data Fig. 5m–p for pooled and Extended Data Fig. 5q–y for 3 independent repeats) exacerbated progression of the disease, whereas treatment with L. gasseri and P. melaninogenica showed disease-promoting effects in some—but not all—of the behavioural tests (Extended Data Fig. 5a–c). Of note, none of the 11 tested bacterial strains affected motor abilities in wild-type mice (Extended Data Fig. 5j–l for 9 tested bacterial strains, and Extended Data Fig. 5m–y for R. torques). Although treatment with R. torques did not affect the cellular number (suggestive of neurons) in the spinal cord (Extended Data Fig. 5z, aa), it significantly enhanced the T2 signal in the magnetic resonance images of multiple motor brain areas, which is indicative of neurodegeneration (Extended Data Fig. 5ab–ah). Notably, treatment with each of the strains resulted in a distinct microbiome composition in Sod1-Tg mice (Extended Data Fig. 5ai, aj). Taken together, these results suggest that several commensals might contribute to the degeneration of motor neurons in the Sod1-Tg mouse model of ALS.
AM colonization improves mouse ALS and survival
One of the species that was altered in Sod1-Tg mice compared with wild-type controls at our vivarium was A. muciniphila; both 16S rDNA sequencing (Fig. 3a, Extended Data Fig. 2c) and shotgun metagenomic sequencing (Extended Data Fig. 4b) showed a gradual reduction in the abundance of AM with disease progression in Sod1-Tg mice, whereas its levels remained relatively constant across time in wild-type littermates. The time-dependent reduction of AM in Sod1-Tg mice was confirmed using AM-specific quantitative PCR (Fig. 3b). In contrast to all of the other tested strains, treatment of antibiotic-pre-treated Sod1-Tg and wild-type mice with an anaerobically mono-cultured AM strain (BAA-835, optical density = 0.7), was associated with improved motor function in Sod1-Tg mice, as quantified by the rotarod locomotor and hanging-wire grip tests and by neurological scoring, assessed in pooled samples (n = 32–62 mice per group, Fig. 3c–e) or independently from 6 repeats (n = 5–26 mice in each group of each repeat, Extended Data Figs. 5a–c, 6). To further assess time-, genotype-, and treatment-dependency we used a mixed linear model (methods) for each of the three motor readouts (rotarod, grip score and neurological score), yielding a better performance of AM-supplemented versus non-supplemented Sod1-Tg mice in the majority of readouts (false discovery rate (FDR)-corrected P value for genotype × treatment of 0.0002, 0.0004 and 0.001, respectively, and a P value of time × genotype × treatment of 0.02, 0.43 and 0.01, respectively). This AM-mediated improvement in function was accompanied by increased cellular number (suggestive of neurons) in the spinal cords of AM-treated Sod1-Tg mice, as compared with those from vehicle-treated, antibiotic-pre-treated Sod1-Tg mice (Extended Data Fig. 7a, b, P < 0.05). Notably, treatment with AM significantly and substantially prolonged the lifespan of Sod1-Tg mice, as compared with vehicle-treated mice or with Sod1-Tg mice treated with other commensal microbiome species serving as bacterial controls (Fig. 3f). AM treatment reduced brain atrophy at 140 days old, as indicated by magnetic resonance imaging (Extended Data Fig. 7c–f). There were no significant differences in gut barrier function at 120 days old—as measured by the systemic influx of fluorescein isothiocyanate–dextran—between Sod1-Tg and wild-type mice treated with AM, PBS or other microbial species (Extended Data Fig. 7g). The microbiome metagenome of AM-treated Sod1-Tg mice clustered differently to that of PBS-treated Sod1-Tg control mice (Extended Data Fig. 7h). As expected, the relative abundance of AM was significantly increased in stool samples from AM-treated Sod1-Tg mice as compared with vehicle-treated Sod1-Tg mice (Extended Data Fig. 7i). By contrast, wild-type mice at our vivarium, which had high and stable indigenous levels of AM, showed competitive exclusion of exogenously administered AM, the levels of which increased only upon prolonged administration (Extended Data Fig. 7j). Moreover, AM was found to colonize more broadly and efficiently and in different regions of the gastrointestinal tract of Sod1-Tg mice compared to that of wild-type mice (Extended Data Fig. 7k, l). Consequently, AM supplementation after treatment with antibiotics altered the microbiome composition of both wild-type and Sod1-Tg mice in distinct ways (Extended Data Fig. 7m, n). Similarly, mono-colonization of antibiotic-pretreated Sod1-Tg mice and wild-type littermates with another strain of AM (ATCC 2869) induced a significant improvement in motor abilities (Extended Data Fig. 7o–q). Histopathological analysis of the distal colon mucus of AM- or PBS-treated 140-day-old Sod1-Tg mice showed an intact inner mucus layer in both AM-supplemented and PBS-treated Sod1-Tg mice (Extended Data Fig. 8a); however, penetrating bacteria were found in the inner mucus and rarely in the crypts only in AM-treated Sod1-Tg mice (Extended Data Fig. 8b, white arrows). A proteomic analysis did not find significant differences in the levels of mucus components in AM-supplemented Sod1-Tg mice compared with PBS-treated controls (Extended Data Fig. 8c–j). Collectively, the assessment of several differentially abundant gut commensals by their mono-inoculation into Sod1-Tg mice identified selected commensals that adversely (P. distasonis, R. torques, and potentially L. gasseri and P. melaninogenica) or favourably (AM) modulate the course and severity of ALS in mice.

a, Linear regression of the relative abundance of AM (assessed by 16S rDNA sequencing) in stool samples from Sod1-Tg and wild-type mice over time. FDR-corrected t-test for coefficient genotype × time P = 1.85 × 10−4. b, Analysis of AM 16S gene copies in faecal DNA extracts over time using quantitative PCR. n = 5 wild-type mice and n = 6 Sod1-Tg mice, two-sided Mann–Whitney U-test, **P = 0.0043. Data are mean ± s.e.m. c–e, Evaluation of motor symptoms in mice using the rotarod locomotor test (c), the hanging-wire grip test (d) and neurological scoring (e). n = 62 Sod1-Tg PBS-treated mice, n = 61 Sod1-Tg AM-treated mice, n = 34 wild-type PBS-treated mice and n = 32 wild-type AM-treated mice. **P = 0.0047 and ****P < 0.0001, two-sided Mann–Whitney U-test. Data are mean ± s.e.m. The experiment was repeated six times (n = 5–26 mice). f, Survival of PBS- (n = 8), AM- (n = 9), P. melaninogenica (PM, n = 4)- and L. gasseri (LG, n = 5)-treated mice ***P < 0.0003, two-sided log-rank test and **P = 0.0011, Gehan–Breslow–Wilcoxon test.
AM attenuates ALS in mice by increasing NAM levels
Because the gut microbiome is remote from the disease site of ALS, we proposed that a potential systemic influx of microbiome-regulated metabolites may affect the susceptibility of motor neurons in Sod1-Tg mice by translocating to the central nervous system7,21. We therefore used untargeted metabolomic profiling to identify candidate microbiome-associated molecules that were differentially abundant in the sera of AM-supplemented and vehicle controls during the early stage of ALS (at 100 days old). Out of 711 serum metabolites identified in Sod1-Tg mice, 84 metabolites were found to be significantly altered by AM supplementation, of which 51 were increased (Fig. 4a, Extended Data Fig. 9a–c). Of these, the biosynthetic genes (nucleotide sequences, KEGG database) of only six metabolites were aligned to our metagenomic index, with two metabolites—NAM and phenol sulfate—having the highest metagenomic probabilities to be synthesized by the wild-type microbiome and not the Sod1-Tg microbiome at our vivarium (Extended Data Fig. 9d). The administration of phenol sulfate, using subcutaneously implanted slow-release mini osmotic pumps, did not affect the symptoms of ALS in Sod1-Tg mice (Extended Data Fig. 9e–g).

a, Significantly increased serum metabolites in Sod1-Tg mice treated with AM (upper-right quadrant, n = 7 mice) compared with PBS-treated mice. Two-sided FDR corrected Mann–Whitney U-test. b, Serum levels of metabolites of the NAM pathway in Sod1-Tg and wild-type mice treated with AM or PBS. The heat map was generated using r log (Deseq). Two-sided FDR-corrected Mann–Whitney U-test. c, NAM levels in bacterial cultures. n = 4–5 cultures in each group, **P < 0.0035, ***P < 0.001, two-tailed t-test. AM vs M9 medium, P < 0.0001; AM vs AM HK, P = 0.0041, Kruskal–Wallis and Dunn’s test. Data are mean ± s.e.m. AM HK, heat-killed AM; M9 med., M9 minimal medium; RT, R. torques. d, NAM levels in the CSF of Sod1-Tg and wild-type mice treated with AM or PBS at 100 days old. **P = 0.0041 for AM- vs PBS-treated wild type and **P = 0.0059 for AM- vs PBS-treated Sod1-Tg. Two-sided Mann–Whitney U-test. Data are mean ± s.e.m. n = 7 for Sod1-Tg PBS, Sod1-Tg AM and WT PBS and n = 8 WT AM. e, NAM levels in the CSF of NAM-treated and vehicle-treated Sod1-Tg mice. n = 10 mice, *P = 0.0232, two-sided Mann–Whitney U-test. Data are mean ± s.e.m. f–h, Motor performance of Sod1-Tg mice, treated with NAM or with vehicle using subcutaneous osmotic pumps, as indicated by the rotarod locomotor test (f), the hanging-wire grip test (g) and neurological scoring (h). ****P < 0.0001 two-sided Mann–Whitney U-test. Data are mean ± s.e.m. The experiment was repeated 3 times, n = 30 mice for Sod1-Tg PBS and n = 29 for Sod1-Tg NAM.
We next focused on NAM, for the following reasons: the marked differences in the metagenomic NAM biosynthetic pathway between Sod1-Tg mice and wild-type controls (Extended Data Fig. 4q); the enrichment of NAM biosynthetic intermediates in serum upon supplementation with AM (Fig. 4b); the reduced abundance of genes from the gut-microbiome-derived tryptophan metabolizing pathway, which may be involved in the generation of NAM22 in naive Sod1-Tg mice (Extended Data Fig. 4o, p); and the alteration of metabolites in the tryptophan pathway upon treatment with antibiotics or upon AM supplementation (Extended Data Fig. 9h, i). Using targeted metabolomics, we first measured levels of NAM in anaerobically grown AM and control commensal isolates. We found significantly higher levels of NAM in the media of AM cultures, compared with the cultures of heat-killed AM or of other commensal isolates (Fig. 4c). In vivo, levels of NAM in the cerebrospinal fluid (CSF) were significantly higher in both AM-treated Sod1-Tg and AM-treated wild-type mice at 100 days old (early-stage disease, Fig. 4d). During advanced stages of the disease (140 days old), NAM levels in the CSF were significantly higher in AM-treated Sod1-Tg mice but not in AM-treated wild-type mice, as compared with untreated controls (Extended Data Fig. 9j); this could reflect differences in gut-colonization stability between wild-type and Sod1-Tg mice (Extended Data Fig. 7i–n). Notably, 8 out of the 10 genes that are related to the AM genome and encode enzymes participating in NAM metabolism were significantly enriched in AM-treated Sod1-Tg mice metagenome compared with vehicle-treated Sod1-Tg mice (Extended Data Fig. 9k, Supplementary Table 2). This indicates that AM supplementation in Sod1-Tg mice may directly modify the biosynthesis of functional NAM.
To causally link an increase in systemic NAM levels with the associated phenotypic effects seen upon AM supplementation, we continuously supplemented Sod1-Tg mice with NAM, administered subcutaneously through implanted mini osmotic pumps. We found that NAM levels were significantly increased in the sera (Extended Data Fig. 10a) and CSF (Fig. 4e) of NAM-treated Sod1-Tg mice compared with water-treated controls. Notably, in both behavioural and neurological motor tests, NAM-treated Sod1-Tg mice performed significantly better than vehicle-treated Sod1-Tg mice, as indicated by a pooled analysis (n = 29–30 mice per group, Fig. 4f–h) or independently in three repeats (n = 9–10 mice in each group of each repeat, Extended Data Fig. 10b–j). We further assessed time- and treatment-dependency in the rotarod, grip score and neurological score, using a mixed linear model (methods) for each of the three motor readouts, yielding a significantly improved motor performance in NAM-treated versus vehicle-treated Sod1-Tg mice (FDR-corrected P value for time × treatment of 3.14 × 10−7, 4.32 × 10−10 and 4.72 × 10-19, respectively) NAM treatment also resulted in a non-significant trend to improve survival (Extended Data Fig. 10k); this might reflect insufficient dosing or exposure time, or the necessity for the integration of other AM-mediated modulatory mechanisms in order to reach the survival benefit seen upon supplementation with AM (Fig. 3f). We further inoculated antibiotic-pretreated Sod1-Tg mice with either wild-type Escherichia coli, as a control, or with E. coli in which the nadA gene is deleted (∆nadA), which is compromised in NAM production (Extended Data Fig. 10l). It is notable that E. coli is considered to be a poor colonizer of the mouse gastrointestinal tract23. Whereas supplementation with ΔnadA E. coli did not affect performances in the rotarod locomotor test (Extended Data Fig. 10m for pooled and Extended Data Fig. 10p, s, v for individual repeats), it significantly worsened the hanging-wire grip test score and neurological score of Sod1-Tg mice compared with wild-type E. coli-treated mice (Extended Data Fig. 10n, o for pooled and Extended Data Fig. 10q, r, t, u, w, x for individual repeats). This suggests that NAM secreted from gut bacteria—even those with poor colonization capacity—may be able to affect some motor abilities in this mouse model of ALS.
Potential AM and NAM mechanisms of ALS modulation
To explore potential downstream mechanisms by which AM and NAM may support motor-neuron survival and ameliorate ALS progression in Sod1-Tg mice, we conducted bulk RNA-sequencing (RNA-seq) of spinal-cord samples collected from AM- and NAM-treated mice. After Sod1-Tg mice were treated with NAM, there was a significant change in the false discovery rate (FDR)-corrected expression of 213 genes (Extended Data Fig. 11a). Thirty-one of these genes also showed a significant change in expression pattern after treatment with AM (Extended Data Fig. 11b). Annotating the NAM-responsive genes to phenotype ontology resulted in a significant (21%) fit to four categories related to abnormal brain morphology, physiology and movement (Extended Data Fig. 11c). The gene-ontology pathways that were most significantly enriched after AM and NAM interventions (Extended Data Fig. 11d, e) were related to mitochondrial structure and function, nicotinamide adenine dinucleotide+ (NAD+) homeostasis and removal of superoxide radicals—canonical functions that are known to be disrupted in ALS. Notably, 28.6% of the promoters of the genes that showed significant changes in expression common to both AM and NAM treatments were found to share a binding site for the transcription factor nuclear respiratory factor-1 (NRF-1; Extended Data Fig. 12a), which is known to control mitochondrial biogenesis, electron transport chain activity and oxidative stress24,25,26,27,28. The potential contribution of NRF-1 to the modulatory effect of ALS merits further studies. Notably, treatment with NAM or AM did not alter the expression in the spinal cord of Slc1a2, a gene encoding the excitatory amino acid transporter 2 in astrocytes (Extended Data Fig. 12b–e); this suggests that the effect of these treatments may not arise from NAM-associated glutamate excitotoxicity.
Dysbiosis and impaired NAM levels in patients with ALS
Finally, we looked at preliminary links between the findings in Sod1-Tg mice at our vivarium and features of human ALS. To this aim, we performed a small-scale human observational study, which involved the collection of stool samples from 37 patients with ALS and 29 healthy BMI- and aged-matched family members as controls and sequencing their gut microbiome metagenomes. The microbiome composition of patients with ALS, as quantified by shotgun metagenomic sequencing, was significantly different to that of healthy control household members (Fig. 5a, for PC1: P = 1.39 × 10−5, Spearman correlation). We observed only a marginally significant difference in the abundances of specific bacterial species after FDR correction (Extended Data Fig. 13a), with five bacterial species reaching near-significance for the ALS microbiome (q < 0.1), out of which one showed significant correlation with serum NAM levels (Extended Data Fig. 13b, Supplementary Table 3). Functionally, ALS microbiomes showed a significant difference in the global bacterial gene content compared with healthy controls (Fig. 5b, for PC1: P = 2.55 × 10−10, Spearman correlation), accompanied by an FDR-corrected (adjusted for these pathways) decrease in several key genes participating in the metabolism of tryptophan and NAM (Extended Data Fig. 13c, Supplementary Table 4). Notably, several of the genes that showed significantly reduced abundance were mapped to the AM genome; this suggests a potential involvement of AM that merits larger studies in the future.

a, b, Principal component analysis of bacterial species composition (for PC1, Spearman’s ρ = 1.3 × 10−5) (a) and KEGG orthology annotated bacterial genes (for PC1, Spearman’s ρ = 2.55 × 10−10) (b) obtained by stool metagenomic sequencing from patients with ALS (n = 37) and healthy controls (family members, n = 29). c, d, Levels of NAM in the serum (c) and CSF (d) of patients with ALS (n = 60 for serum and 14 for CSF) and healthy controls (n = 33 for serum and 17 for CSF), ****P < 0.0001, ***P = 0.0001, Mann–Whitney U-test. Data are mean ± s.e.m.
Untargeted metabolomic profiling of sera from patients with ALS revealed significant changes in the levels of multiple metabolites, including increased levels of riluzole (an exogenously administrated treatment for ALS), creatine and 3-hydroxy-2-ethylpropionate, and reduced levels of methyl indole 3-acetate and triethanolamine (Extended Data Fig. 13d). Notably, there were significant alterations in the levels of key molecules of the tryptophan–nicotinamide metabolic pathway in sera of patients with ALS—among them indoleacetate, kynurenine, serotonin and circulating nicotinamide (Extended Data Fig. 13e)—which suggest aberrant metabolism of NAM in some of these cases of ALS in humans. Targeted serum metabolomics further validated the marked decrease in NAM in the sera of 60 patients with ALS compared with 33 healthy controls (Fig. 5c). Moreover, levels of NAM in the sera of patients with ALS mildly but significantly correlated both with better scores on the human ALS functional-rating scale (Extended Data Fig. 13f) and with the levels of the microbiome gene that encodes one of the rate-limiting reactions in the biosynthesis of NAM (Extended Data Fig. 13g). We further found that the levels of NAM in the CSF of 14 patients with ALS were significantly lower than those of 17 healthy controls; this was driven by some patients that had markedly low levels of NAM in the CSF (Fig. 5d). The clinical implications of these preliminary metabolomic findings and associations, and whether they apply to all or to subsets of patients with ALS, warrant further validation and exploration in human-focused studies.
Discussion
Our findings highlight the potential cooperative activity of genetic risk and locally varying environmental modulatory factors in affecting ALS, with the gut microbiome serving as a ‘hub’ that relays these environmental signals to the host; such an action has been noted in other multifactorial diseases such as inflammatory bowel disease29. We note that our study mainly focuses on one ALS mouse model, in the context of one vivarium, and mechanistically exemplifies one metabolite (NAM) out of several that are differentially expressed in ALS. Future studies may reveal complementary functions of other gut-derived commensals and metabolites in the central nervous system and of relevance to ALS. Importantly, we note that our human data are preliminary and observational, and are not aimed at nor are sufficient to constitute a treatment recommendation of any sort in this devastating disease. Larger, future prospective studies of human ALS—including those incorporating genetic disease variants—are needed to validate the suggested microbiome associations and assess the causal effects of potential microbiome modulators of human ALS. These limitations notwithstanding, our study suggests a potential modulatory involvement of the gut microbiome in ALS that may help to delineate some aspects of its pathophysiology, while providing an opportunity to identify modifiable environmental and microbial therapeutic targets.
Methods
Data reporting
No statistical methods were used to predetermine sample size. The experiments were not randomized, and the investigators were blinded to allocation during experiments and outcome assessment.
Mice
Animal experiments were approved by the Weizmann Institute Animal Care and Use Committee Following US National Institutes of Health, European Commission and the Israeli guidelines. mSod1G93A transgenic mice on a C57BL/6 background were provided by E. Hornstein (Weizmann Institute). In all experiments, age- and gender-matched mice were used and wild-type littermates were used as controls. Mice were 40 days of age at the beginning of experiments. All mice were kept on a strict 24-h reverse light–dark cycle, with lights being turned on from 22:00 to 10:00. For antibiotic treatment, mice were given a combination of vancomycin (0.5 g l−1), ampicillin (1 g l−1), neomycin (1 g l−1) and metronidazole (1 g l−1) in drinking water from 40 days of age as previously described30. For the A. muciniphila or R. torques colonization, 40-day-old mice were treated with antibiotics for two weeks, followed by 2 days of washout period, and were gavaged with 200 µl of PBS-suspended bacteria (OD = 0.7) weekly until the experimental end point. Food intake and other metabolic parameters were measured using the PhenoMaster system (TSE-Systems), which consists of a combination of sensitive feeding sensors for automated measurement, and a photobeam-based activity-monitoring system, which detects oxygen and carbon dioxide consumption and records ambulatory movements—including rearing and climbing—in each cage. All parameters were measured continuously and simultaneously. Mice were singly housed in identical cages before data acquisition. All experimental procedures were approved by the local Institutional Animal Care and Use Committee 02660418-3.
Administration of metabolites
For the in vivo administration of NAM and phenol sulfate, Alzet osmotic minipumps model 1004 (Charles River) were used (infusing the compound at a rate of 0.11 μl h−1 for 4 weeks). The pumps were filled with 100 μl 50 mg ml−1 nicotinamide (Cymit Quimica) or 33.33 mg ml−1 phenol sulfate sodium salt (TLC) diluted in sterile water (equivalent to 49.28 mg per kg per week of NAM and 30.8 mg per kg per week of phenol sulfate). Vehicle control pumps contained an equivalent volume of ultra-pure water. Six-week-old Sod1-Tg and wild-type littermate mice were anaesthetized by intraperitoneal (i.p.) injection of ketamine (100 mg kg−1) and xylazine (10 mg kg−1), the neck skin was shaved and sterilized with 70% ethanol, a 1-cm incision was made in the skin, and the osmotic minipumps were inserted after minimal blunt dissection and placed above the right hind flank. The cut was then closed with sterile surgical clips and the mice were carefully monitored for any signs of stress, bleeding, pain or abnormal behaviour. By replacing the pumps every 28 days—a total of 4 times between the ages of 40–152 days—we ensured the steady and continuous administration of metabolites to mice throughout the course of the disease.
Assessment of motor functions in mice
Throughout this assessment, key repeat experiments were independently scored by two researchers in a blinded fashion.
Rotarod locomotor test
To assess motor coordination and balance, each mouse was tested with a rotarod device (Panlab Le8500 Harvard Apparatus), in acceleration speed mode (increasing from 4 r.p.m. to 40 r.p.m. over 10 min), with a maximum test time of 5 min. The mice were habituated on the horizontal rotating rod and pre-trained for three trials before the formal tests. Each mouse was recorded three times at ages 60, 80, 100, 120 and 140 days. The apparatus automatically recorded the time that had elapsed when the mouse fell from the spindle.
Hanging-wire grip test
Mice were allowed to grip a 2-mm-thick horizontal metal wire (suspended 80 cm above the working surface) with their forepaws, and the latency to successfully raise their hind legs to grip the wire was recorded. The mice were observed for 30 s and scored as follows: 0, falls off within 10 s; 1, hangs onto bar by two forepaws; 2, attempts to climb onto bar; 3, hangs onto bar by two forepaws plus one or both hind paws; 4, hangs by all four paws plus tail wrapped around bar; 5, active escape to the end of bar.
Neurological scoring
Mice were neurologically scored by a system developed by the ALS Therapy Development Institute31: Score of 0: full extension of hind legs away from lateral midline when mouse is suspended by its tail, and mouse can hold this for two seconds, suspended two to three times. Score of 1: collapse or partial collapse of leg extension towards lateral midline (weakness) or trembling of hind legs during tail suspension. Score of 2: toes curl under at least twice when walking 12 inches, or any part of the foot is dragging along the bottom of the cage or table. Score of 3: rigid paralysis or minimal joint movement, foot not being used for generating forward motion. Score of 4: mouse cannot right itself within 30 s after being placed on either side.
Home-cage locomotion
The locomotion of mice was quantified over a period of 46 h in their home cage, by automated sensing of body heat using an InfraMot (TSE-Systems). The movements of individual mice were summed every 30 min.
Survival
From the age of 130 days, mice were monitored daily. The endpoint of the experiment was defined when a neurological score of 4 was reached and/or a reduction in body weight of greater than 15% was observed. The probability of survival was calculated using the Kaplan–Meier method, and statistical analysis was performed using a log-rank test.
Extraction of cerebrospinal fluid
Mice were anaesthetized by i.p. injection of ketamine (100 mg kg−1) and xylazine (10 mg kg−1). The skin of the neck was shaved, and the mouse was placed prone on the stereotaxic instrument. The head was secured with head adaptors. The surgical site was swabbed with 70% ethanol, and a sagittal incision of the skin was made inferior to the occiput. Under the dissection microscope, the subcutaneous tissue and muscles (m. biventer cervicis and m. rectus capitis dorsalis major) were separated by blunt dissection with forceps. A pair of micro-retractors was used to hold the muscles apart. The dura mater was blotted dry with a sterile cotton swab. CSF was collected using a capillary tube to penetrate into the cisterna magna (through the dura mater, lateral to the arteria dorsalis spinalis) and immediately frozen in liquid nitrogen and stored at −80 °C.
Magnetic resonance imaging
During magnetic resonance imaging (MRI) experiments, mice were anaesthetized with isoflurane (5% for induction, 1–2% for maintenance) mixed with oxygen (1 l min−1) and delivered through a nasal mask. Once anaesthetized, the mice were placed in a head-holder to assure reproducible positioning inside the magnet. The respiration rate was monitored and maintained throughout the experimental period at around 60–80 breaths per minute. MRI experiments were performed on a 9.4 T BioSpec Magnet 94/20 USR system (Bruker) equipped with a gradient coil system capable of producing a pulse gradient of up to 40 G cm−1 in each of the three directions. All magnetic resonance images were acquired with a receiver quadrature mouse brain surface coil and transmitter linear coil (Bruker). The T2 maps were acquired using the multi-slice spin-echo imaging sequence with the following parameters: a repetition delay (TR) of 3,000 ms, 16-time echo (TE) increments (linearly from 10 to 160 ms), a matrix dimension of 256 × 128 (interpolated to 256 × 256) and two averages, corresponding to an image acquisition time of 12 min 48 s. The T2 dataset consisted of 16 images per slice. Thirteen continuous slices with slice thickness of 1.00 mm were acquired with a field of view of 2.0 × 2.0 cm2.
Image analysis
A quantitative T2 map was produced from multi-echo T2-weighted images. The multi-echo signal was fitted to a monoexponential decay to extract the T2 value for each image pixel. All image analysis was performed using homemade scripts written in MATLAB R2013B. Inter- and intrasubject co-registration of the T2 maps was applied before voxel by voxel analysis. For optimal suitability to a mouse brain atlas (correction of head movements image artefacts), all images were subjected to atlas registration: reslicing, realignment and smoothing, using the SPM software (version 12, University College London). FDR correction was applied on the data as part of the SPM software processing. The results were reported as mean ± s.d. A t-test was used to compare the means of two groups, and a P value of less than 0.01 was considered statistically significant.
Histology
Sections from the spinal cord (C3-T6) were fixed in paraformaldehyde and embedded in paraffin for staining with Luxol Fast Blue and Cresyl Echt Violet. Subsequently, sections were examined in a blinded fashion and Cresyl-Echt-Violet-positive cells in the ventral horn were counted to estimate neuronal numbers. Colon tissues were fixed in dry methanolic-Carnoy and stained with the nuclear stain SYTOX Green and the Muc2 mucin with the anti-MUC2C3 antiserum and goat anti-rabbit-Alexa 555 (Thermo Fisher Scientific)32.
Measuring gut epithelial barrier permeability by FITC–dextran
On the day of the assay, 4 kDa FITC–dextran was dissolved in PBS to a concentration of 80 mg ml−1. Mice were fasted for 4 h before gavage with 150 μl dextran. Mice were anaesthetized 3 h after gavage and blood was collected and centrifuged at 1,000g for 12 min at 4 °C. Serum was collected and fluorescence was quantified at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Flow cytometry
Wild-type and Sod1-Tg mice treated with antibiotics from 40 days of age or with water as controls were used for small-intestinal, colonic and spinal cord cellularity analysis either at 140 days old (for small intestines and colons) or at 60 and 140 days old (for spinal cords). Small intestine and colonic samples were extensively washed to remove faecal matter followed by 2 mM EDTA dissociation in 37 °C for 30 min. Following extensive shaking, the epithelial fraction was discarded. Samples were then digested using DNaseI and collagenase for analysis of the lamina propria. Spinal-cord samples were collected from individual mice, homogenized and incubated with a HBSS solution containing 2% BSA (Sigma‐Aldrich), 1 mg ml−1 collagenase D (Roche), and 0.15 mg ml−1 DNaseI, filtered through a 70 μm mesh. Homogenized sections were resuspended in 40% Percoll, before density centrifugation (1,000g, 15 min at 20 °C with low acceleration and no brake). The isolated cells were washed with cold PBS and resuspended in PBS containing 1% BSA for direct cell-surface staining. Single‐cell suspensions were stained with antibodies for 45 min on ice against CD45, CD11b, CD11c, Ly6C, Ly6G, B220, CD3 and EpCAM (BioLegend). Stained cells were analysed on a BD‐LSRFortessa cytometer and were analysed with FlowJo software.
Mucus proteomic analysis
For the analysis of proteomes, isolated mucus samples were incubated overnight at 37 °C in reduction buffer (6 M guanidinium hydrochloride, 0.1 M Tris/HCl, pH 8.5, 5 mM EDTA, 0.1 M DTT (Merck)) and the soluble fraction was added on top of a spin-filter (10 kDa, PALL) for a filter-aided sample preparation following a previous protocol33; however, 6 M guanidinium hydrochloride was used here instead of urea. Proteins on the filters were alkylated and subsequently digested for 4 h with LysC (Wako) followed by an overnight digestion with trypsin (Promega). Heavy peptides (SpikeTides TQL, JPT Peptide Technologies) for the absolute quantification of Muc2 (10 peptides, 100 fmol each34) were added before trypsin digestion. Peptides released from the filter after centrifugation were cleaned with StageTip C18 columns35. Nano liquid chromatography coupled with tandem mass spectrometry (LC–MS/MS) was performed on an EASY-nLC 1000 system (Thermo Fisher Scientific) connected to a Q Exactive HF Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Fisher Scientific) through a nanoelectrospray ion source. Peptides were separated with an in-house-packed reverse-phase column (150 × 0.075 mm inner diameter, C18-AQ 3 µm) using a 30-min gradient from 10% to 45% of buffer B (A: 0.1% formic acid, B: 0.1% formic acid/80% acetonitrile) using a flow rate of 300 nl min−1. Full mass spectra were acquired from 350–1,600 m/z with a resolution of 60,000 (m/z 200). Up to the 15 most intense peaks (charge state ≥2) were fragmented and tandem mass spectra were acquired with a resolution of 15,000 and a dynamic exclusion of 20 s. For absolute quantification, a separate targeted mass spectrometry method was used in which only precursors and their fragments of the heavy and corresponding light peptides were scanned with a resolution of 30,000. Proteins were identified with the MaxQuant program (version 1.5.7.436) by searching against the mouse UniProt protein database (downloaded 11.07.2018) supplemented with an in-house database containing all the mouse mucin sequences (http://www.medkem.gu.se/mucinbiology/databases/). Searches were performed with full tryptic specificity, a maximum of 2 missed cleavages, precursor tolerance of 20 p.p.m. in the first search used for recalibration, followed by 7 p.p.m. for the main search and 0.5 Da for fragment ions. Carbamidomethylation of cysteine was set to fixed modification and methionine oxidation and protein N-terminal acetylation were set as variable modification. The required FDR was set to 1% for both peptide and protein levels and the minimum required peptide length was set to six amino acids. Proteins were quantified using the MaxQuant label-free quantification option, using a minimum of two peptides for quantification. Absolute quantification of Muc2 was performed with Skyline (version 4.2.037).
Bacterial cultures
A. muciniphila (ATCC BAA-835), A. muciniphila (ATCC BAA-2869), R. torques (ATCC 27756), L. gasseri (ATCC 33323), P. melaninogenica (ATCC 25845), C. cateniformis (DSM-15921), P. goldsteinii (DSM-19448), L. murinus (DSM-100194), P. distasonis (ATCC 8503), E. tayi (DSM-24404) and S. variabile (SDM-15176) were grown in chopped meat medium (BD 297307) under anaerobic conditions (Coy Laboratory Products, 75% N2, 20% CO2, 5% H2) in 37 °C without shaking. E. lenta (DSM-15644) was grown in chopped meat medium supplemented with 0.5% arginine. All strains were validated for purity by whole-gene 16S Sanger sequencing. Wild-type and ΔnadA E. coli were provided by S. Belkin (Institute of Life Sciences, the Hebrew University, Jerusalem, Israel), originally obtained from the ‘Keio collection’38 and were grown on LB media (wild-type) or LB supplemented with 30 μg ml−1 kanamycin (ΔnadA). To measure bacterial nicotinamide secretion in vitro, bacterial strains were grown in chopped meat medium until the stationary phase, centrifuged and washed twice with M9 minimal medium with trace elements and glucose (4 g l−1) and resuspended in M9 for 3 h under anaerobic conditions. After centrifugation, 50 μl of the supernatant was collected for targeted nicotinamide measurements, and protein was extracted from the pellet using the BCA method: in brief, bacterial pellets were homogenized in RIPA buffer containing protease inhibitors, incubated for 45 min at 4 °C and centrifuged for 20 min at 14,000 r.p.m. at 4 °C. Nicotinamide measurements in the media were then normalized to the total amount of protein in each sample.
Nucleic acid extraction
DNA purification
Human genomic DNA was purified using a PowerMag Soil DNA isolation kit (Qiagen) optimized for the Tecan automated platform. Mouse faecal, luminal and mucosal genomic DNA was extracted with a Purelink Microbiome DNA purification kit (Invitrogen). For shotgun sequencing, Illumina libraries were prepared using a Nextera DNA Sample Prep kit (Illumina), according to the manufacturer’s protocol and sequenced using the Illumina NextSeq platform with a read length of 80 bp.
RNA purification
Spinal cord samples were collected from mice and snap-frozen in liquid nitrogen. Tissues were homogenized in Tri Reagent (Bio-Lab). RNA was purified using standard chloroform extraction. Two micrograms of total RNA was used to generate cDNA (High Capacity cDNA Reverse Transcription kit; Applied Biosystems).
Quantitative PCR (qPCR) was performed using Kapa Sybr qPCR kit (Kapa Biosystems) on a Viia7 instrument (Applied Biosystems). PCR conditions were 95 °C for 20 s, followed by 40 cycles of 95 °C for 3 s and 60 °C for 30 s. Data were analysed using the ∆∆Ct method with 16S serving as the reference housekeeping gene. 16S cycles were assured to be insensitive to the experimental conditions.
Nucleic acid processing and library preparation
16S qPCR protocol for quantification of bacterial DNA
DNA templates were diluted to 1 ng μl−1 before amplifications with the primer sets (indicated in Supplementary Table 5) using the Fast SYBR Green Master Mix (Thermo Fisher) in duplicate. Amplification conditions for A. muciniphila were as follows: denaturation at 95 °C for 3 min, followed by 40 cycles of denaturation at 95 °C for 3 s; annealing at 66 °C for 30 s followed by melting curve. Amplification conditions for total bacteria (16S rDNA) were: Denaturation at 95 °C for 3 min, followed by 40 cycles of denaturation at 95 °C for 3 s; annealing at 60 °C for 30 s followed by melting curve analysis. Duplicates with >2 cycle difference were excluded from analysis. The cycle threshold for any sample not amplified after 40 cycles was defined as 40.
16S rDNA sequencing
For 16S amplicon pyrosequencing, PCR amplification was performed spanning the V4 region using the primers 515F/806R of the 16S rDNA gene and subsequently sequenced using 2 × 250 bp paired-end sequencing (Illumina MiSeq). Custom primers were added to the Illumina MiSeq kit resulting in 253 bp fragment sequenced following paired-end joining to a depth of 110,998 ± 66,946 reads (mean ± s.d.). Read1: TATGGTAATTGTGTGCCAGCMGCCGCGGTAA; Read2: AGTCAGTCAGCCGGACTACHVGGGTWTCTAAT; Index sequence primer: ATTAGAWACCCBDGTAGTCCGGCTGACTGACTATTAGAA.
RNA sequencing
Ribosomal RNA was selectively depleted by RNaseH (New England Biolabs, M0297) according to a modified version of a published method39. Specifically, a pool of 50-bp DNA oligos (25 nM, IDT, indicated in Supplementary Table 6) that is complementary to mouse rRNA18S and 28S, was resuspended in 75 µl of 10 mM Tris pH 8.0. Total RNA (100–1,000 ng in 10 µl H2O) was mixed with an equal amount of rRNA oligo pool, diluted to 2 µl, and 3 µl 5x rRNA hybridization buffer (0.5 M Tris-HCl, 1 M NaCl, titrated with HCl to pH 7.4) was added. Samples were incubated at 95 °C for 2 min, then the temperature was slowly decreased (−0.1 °C s−1) to 37 °C. RNaseH enzyme mix (2 µl of 10U RNaseH, 2 μl 10 RNaseH buffer, 1 μl H2O, total 5 µl mix) was prepared 5 min before the end of the hybridization and preheated to 37 °C. The enzyme mix was added to the samples when they reached 37 °C and they were incubated at this temperature for 30 min. Samples were purified with 2.2x SPRI beads (AMPure XP, Beckmann Coulter) according to the manufacturer’s instructions. Residual oligos were removed with DNase treatment (Thermo Fisher Scientific, AM2238) by incubation with 5 µl DNase reaction mix (1 µl Turbo DNase, 1.25 µl Turbo DNase 10x buffer) that was incubated at 37 °C for 30 min. Samples were again purified with 2.2x SPRI beads and suspended in 3.6 µl priming mix (0.3 µl random primers from New England Biolabs, E7420, 3.3 µl H2O). Samples were subsequently primed at 65 °C for 5 min. Samples were then transferred to ice and 2 µl of the first strand mix was added for first strand cDNA synthesis (1 µl 5x first strand buffer, NEB E7420; 0.125 µl RNase inhibitor, NEB E7420; 0.25 µl ProtoScript II reverse transcriptase, NEB E7420; and 0.625 µl of 0.2 µg ml−1 actinomycin D, Sigma-Aldrich, A1410). The first strand synthesis and all subsequent library preparation steps were performed using NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England Biolabs, E7420) according to the manufacturer’s instructions (all reaction volumes reduced to a quarter) and sequenced using the Illumina NextSeq platform.
16S rDNA analysis
Overlapping paired-end FASTQ files were matched and processed in a data-curation pipeline implemented in Qiime2 version 2018.4.0 (Qiime2)40. Paired-end sequence data were demultiplexed according to sample-specific barcodes using Qiime2 demux-emp-paired. Trimming and amplicon sequence variant (ASV) picking were carried out with the use of DADA241. Alpha rarefaction curves were plotted using Qiime2 alpha-rarefaction and were used to set an appropriate subsampling depth for each comparison. Samples were rarefied using Qiime2 feature-table rarefy42. Samples with a read depth lower than the relevant subsampling depth were excluded from the analysis. ASVs were assigned with taxonomic annotations using a naive Bayes fitted classifier trained on August 2013, 97% identity Greengenes rRNA database43. Relative abundance tables were calculated using Qiime2 feature-table summarize-taxa. Ordination plots were calculated from unweighted and weighted UniFrac distance matrix using principal coordinate analysis (PCoA).
Metagenomic analysis
For metagenome analysis, metagenomic reads containing Illumina adapters and low-quality reads were filtered and low-quality read edges were trimmed. Host DNA was detected by mapping with GEM44 to the human or mouse genome (hg19 or mm10 respectively) with inclusive parameters, and host reads were removed. For Sod1-Tg versus wild-type naive mice metagenomes, 5 million reads were subsampled; for AM-treated mice, 1 million reads; and for humans, 7–10 million reads. Relative abundances from metagenomic sequencing were computed using MetaPhlAn245 with default parameters. MetaPhlAn relative abundances were capped at a level of 5 × 10−4. The relative frequency of each KEGG orthology term was obtained by mapping to KEGG46 bacterial genes database using DIAMOND47, considering only the first hit, and allowing the value of e to be <0.0001. The relative frequency of a KEGG orthology term was determined as the sum of all reads mapped to bacterial genes associated with that term, divided by the total number of mapped reads in a sample. For mouse samples, the relative frequency of each KEGG orthology term was obtained by first mapping to IGC—an integrated catalogue of reference genes in the human gut microbiome—and then summing the relative frequency of all the genes in each KEGG orthology term to obtain relative frequency. The relative frequencies of KEGG orthology terms were capped at a level of 2 × 10−5 for mice and 2 × 10−7 for humans. Taxa and KEGG orthology terms present in fewer than 10% of samples were discarded.
Metabolite selection
Using the top 12 significant serum metabolites altered by antibiotic treatment in wild-type and Sod1-Tg mice, we first downloaded all nucleotide sequences of KEGG genes with the potential to synthesize or degrade the 12 metabolites. Next, we built a bowtie index of KEGG genes and mapped to it Sod1-Tg and wild-type mouse metagenome samples. Finally, we obtained all mapped reads and, for every sample and KEGG gene, we report the number of reads mapped to the KEGG gene and its mean score. Scores are as defined by bowtie248 and range between 0 and −45, where 0 denotes a perfect match.
RNA sequencing analysis
Reads containing adapters and low quality reads were removed. Reads were aligned to the mm10 reference genome (UCSC) using STAR. Alignments were binned to genes using htseq-count. Transcript integrity number (TIN) medians were calculated using RSeQC. (tin.py.bed file: mm10 RefSeq.bed.gz downloaded from https://source-forge.net/projects/rseqc/files/BED/Mouse_Mus_musculus/). For each comparison, genes with read count ≥10−4 out of total reads and expressed in at least fifth of a group in each comparison were included in the analysis. Differentially expressed genes were found using DESeq2, DESeq2 models were fitted for each comparison separately with design formula counts ~ group + TIN. Genes with padj <= 0.05 were considered as differentially expressed. Heat maps were created using the regularized log transform (rlog) function of the Deseq2 package. Gene set enrichment analysis we done using the bulk.gsea function of the liger package and the mouse pathways gmt file found in http://ge-lab.org/gskb/2-MousePath/MousePath_GO_gmt.gmt as our universe model. The gene scores supplied to the bulk.gsea function were calculated as −log(Padj) * sign(log2FoldChange) for each gene.
Non-targeted metabolomics
Sera and caecal samples were collected, immediately frozen in liquid nitrogen and stored at −80 °C. Sample preparation and analysis were performed by Metabolon. Samples were prepared using the automated MicroLab STAR system (Hamilton). To remove protein, dissociated small molecules bound to protein or trapped in the precipitated protein matrix, and to recover chemically diverse metabolites, proteins were precipitated with methanol. The resulted extract was divided into five fractions: one for analysis by ultra performance liquid chromatography coupled with tandem mass spectrometry (UPLC–MS/MS) with negative ion mode electrospray ionization, one for analysis by UPLC–MS/MS with positive ion mode electrospray ionization, one for LC polar platform, one for analysis by gas chromatography coupled with mass spectrometry (GC–MS), and one sample was reserved as a backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. For LC, the samples were stored overnight under nitrogen before preparation for analysis. For GC, each sample was dried under vacuum overnight before preparation for analysis.
Data extraction and compound identification
Raw data were extracted, peak-identified and quality-control processed using hardware and software from Metabolon. Compounds were identified by comparison to library entries of purified standards or recurrent unknown entities.
Metabolite quantification and data normalization
Peaks were quantified using the AUC. For studies that spanned multiple days, a data normalization step was performed to correct for variation resulting from inter-day differences in instrument tuning. For metabolites, heatmaps r log normalisation (Deseq 2) was used where indicated.
Targeted metabolomics
d4-nicotinamide (50 ng ml−1; Cambridge Isotope Laboratories) was added to all samples as an internal standard. The samples (in 50% methanol) were dried in a speed vac to remove the methanol before drying was completed in a lyophilizer. All samples were re-dissolved in 100 μl of 0.1% formic acid.
Liquid chromatography
Liquid chromatography was performed on a Waters Acquity UPLC system. Metabolites were separated on an Acquity HSS T3 column (2.1 × 150 mm, 1.8 μm particle size; Waters) at 40 °C using a 10-min program. The mobile phase consisted of water (A) and acetonitrile (B), each containing 0.1% formic acid. Gradient conditions were: 0–1 min, 99.9% A 0.1% B; 1–6 min, 0.1%–10.0% B; 6–7 min, 10%–100% B; 7.0–7.2 min, 100% B; 7.2–10 min, 99.9% A 0.1% B. Injection volume was 1.0 μl, and flow rate was 0.3 ml min−1.
Mass spectrometry
LC–MS/MS analysis was performed on a Waters Xevo triple quadrupole equipped with a Zspray electrospray ionization source. Multiple reaction monitoring was performed in the positive-ion mode. Other parameters included: desolvation temperature, 600 °C; desolvation gas flow, 900 l h−1; cone gas flow, 150l h−1; nebulizer pressure, 7 Bar; capillary voltage, 2.53 kV. The MRM transitions used were: nicotinamide: 123 > 78 and 123 > 80, CE 19, 13 V respectively; d4-nicotinamide: 127 > 81 and 127 > 84, CE 19, 17 V respectively. Argon (0.10 mg min−1) was used as the collision gas. TargetLynx (Waters) was used for qualitative and quantitative analysis.
Human observational study
The human observational study was approved by the Hadassah Medical Center Institutional Review Board (IRB approval number HMO-16-0396) and Weizmann Institute of Science Bioethics and Embryonic Stem Cell Research oversight committee (IRB approval number 365-1). Written informed consent was obtained from all subjects.
Exclusion and inclusion criteria
All subjects (Supplementary Table 7) fulfilled the following inclusion criteria: males and females, aged 18–70, who are currently not following any interventional diet modification other than advice to avoid nutritional deficiencies and are able to provide informed consent. Exclusion criteria included: (i) pregnancy or fertility treatments; (ii) use of antibiotics or antifungals in the three months before participation; (iii) consumption of probiotics in any form in the month before participation, (iv) active inflammatory or neoplastic disease three years before enrolment; (v) chronic gastrointestinal disorder, including inflammatory bowel disease and coeliac disease; (vi) myocardial infarction or cerebrovascular accident in the six months before participation; (vii) coagulation disorders; (viii) chronic immunosuppressive medication usage; (ix) pre-diagnosed type I or type II diabetes mellitus or treatment with anti-diabetic medication. None of the enrolled patients and controls reported constipation or were taking constipation medications. One of the patients with ALS had a chronic diagnosis of irritable bowel syndrome, another was diagnosed with asymptomatic primary biliary cholangitis 8 years before the development of ALS, and was consuming ursodecholic acid (1,500 mg per day). Two patients were diagnosed with chronic dyspepsia and one with reflux oesophagitis. Two of these patients were chronically treated (for years) with proton-pump inhibitors. Adherence to inclusion and exclusion criteria was validated by medical doctors.
Statistical analysis
Mann–Whitney U-test
Data are expressed as mean ± s.e.m. P < 0.05 were considered significant (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). For pooled analysis of behavioural test results from different independent repeats, all mice from the same experimental group were pooled and a new statistical comparison was made for the entire pooled experiment, as performed for the individual repeats. No intermediate statistical strategies were applied. The Mann–Whitney U-test was used when the distribution was not known to be normal and only between two groups. In all the behavioural-test analyses the area under the curve (AUC) was calculated. In behavioural tests, Mann–Whitney was only calculated on Sod1-Tg mice with and without treatment. Otherwise, we corrected multiple comparisons P values using the FDR.
Linear mixed model (LMM) and ordinary least squares (OLS)
The effect of antibiotics and AM over time in control and Sod1-Tg mice and of NAM in Sod1-Tg mice on neuro-phenotypical measurements (rotarod, hanging-wire grip test score and neurological score), were modelled, where indicated, as a function of time and treatment in a time-dependent manner using LMM or OLS. LMM:
OLS: phenotype = time + treatment × time + genotype × time + treatment × genotype × time. For NAM pooled behavioural data, only Sod1-Tg mice were used, and NAM time-depended treatment was evaluated. LMM:
in which ‘i’ is a mouse indicator and mousei is a random effect per mouse, time is the age of the mice in days (60, 80, 100, 120 and 140), treatment (±antibiotic or AM) and genotype (wild-type or Sod1-Tg) are binary indicators. Significance of treatment is then inferred by the FDR corrected P value of the time × genotype × treatment coefficient.
Microbial abundance change over time was evaluated using LMM and OLS. LMM:
OLS:
The significance of genotype affecting the abundance of operational taxonomic units was inferred by the P value of the time × genotype predictor (using t-test) after FDR correction for multiple operational taxonomic units. For this analysis we used Python statsmodels.formula.api.mixedlm with Restricted Maximum Likelihood (REML), and statsmodels.api.ols.
Given the relatively small numbers of mice used in the bacterial abundance experiments, we considered the possibility that the linear mixed model in this setting may produce unstable results with the REML not converging. Given this limitation, we analyzed these experiments with both linear regression and linear mixed model, with the results in both methods being in agreement with each other.
To analyse KEGG orthology terms of the nicotinamide and tryptophan metabolic pathways, the frequencies of KEGG orthology terms between groups were compared using the Mann–Whitney U-test. For this analysis we used Python stats.HigherLevelRanksum.directed_mannwhitneyu.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The sequencing data has been deposited at the European Nucleotide Archive database with the accession number PRJEB32767.
References
Turner, M. R. et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 12, 310–322 (2013).
Bensimon, G., Lacomblez, L. & Meininger, V. A controlled trial of riluzole in amyotrophic lateral sclerosis. N. Engl. J. Med. 330, 585–591 (1994).
The Writing Group, Edaravone (MCI-186) ALS 19 Study Group. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 16, 505–512 (2017).
Su, F. C. et al. Association of environmental toxins with amyotrophic lateral sclerosis. JAMA Neurol. 73, 803–811 (2016).
Kundu, P., Blacher, E., Elinav, E. & Pettersson, S. Our gut microbiome: the evolving inner self. Cell 171, 1481–1493 (2017).
Al-Chalabi, A. & Hardiman, O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat. Rev. Neurol. 9, 617–628 (2013).
Sampson, T. R. et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167, 1469–1480.e12 (2016).
Sharon, G. et al. Human gut microbiota from autism spectrum disorder promote behavioral symptoms in mice. Cell 177, 1600–1618.e17 (2019).
Blacher, E., Levy, M., Tatirovsky, E. & Elinav, E. Microbiome-modulated metabolites at the interface of host immunity. J. Immunol. 198, 572–580 (2017).
Veiga-Fernandes, H. & Mucida, D. Neuro–immune interactions at barrier surfaces. Cell 165, 801–811 (2016).
Sharon, G., Sampson, T. R., Geschwind, D. H. & Mazmanian, S. K. The central nervous system and the gut microbiome. Cell 167, 915–932 (2016).
Wu, S., Yi, J., Zhang, Y. G., Zhou, J. & Sun, J. Leaky intestine and impaired microbiome in an amyotrophic lateral sclerosis mouse model. Physiol. Rep. 3, e12356 (2015).
Zhang, Y. G. et al. Target intestinal microbiota to alleviate disease progression in amyotrophic lateral sclerosis. Clin. Ther. 39, 322–336 (2017).
Fang, X. et al. Evaluation of the microbial diversity in amyotrophic lateral sclerosis using high-throughput sequencing. Front. Microbiol. 7, 1479 (2016).
Brenner, D. et al. The fecal microbiome of ALS patients. Neurobiol. Aging 61, 132–137 (2018).
Ralph, G. S. et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 11, 429–433 (2005).
Aartsma-Rus, A. & van Putten, M. Assessing functional performance in the mdx mouse model. J. Vis. Exp. 27, 51303 (2014).
Sugiyama, K. & Tanaka, K. Spinal cord-specific deletion of the glutamate transporter GLT1 causes motor neuron death in mice. Biochem. Biophys. Res. Commun. 497, 689–693 (2018).
Niessen, H. G. et al. In vivo quantification of spinal and bulbar motor neuron degeneration in the G93A-SOD1 transgenic mouse model of ALS by T2 relaxation time and apparent diffusion coefficient. Exp. Neurol. 201, 293–300 (2006).
Zang, D. W. et al. Magnetic resonance imaging reveals neuronal degeneration in the brainstem of the superoxide dismutase 1 transgenic mouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 20, 1745–1751 (2004).
Hsiao, E. Y. et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463 (2013).
Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 350, 1208–1213 (2015).
Sweeney, N. J., Laux, D. C. & Cohen, P. S. Escherichia coli F-18 and E. coli K-12 eda mutants do not colonize the streptomycin-treated mouse large intestine. Infect. Immun. 64, 3504–3511 (1996).
Golko-Perez, S., Amit, T., Bar-Am, O., Youdim, M. B. H. & Weinreb, O. A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1G93A ALS mice. Neurotox. Res. 31, 230–244 (2017).
Scarpulla, R. C., Vega, R. B. & Kelly, D. P. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol. Metab. 23, 459–466 (2012).
Bergeron, R. et al. Chronic activation of AMP kinase results in NRF-1 activation and mitochondrial biogenesis. Am. J. Physiol. Endocrinol. Metab. 281, E1340–E1346 (2001).
Evans, M. J. & Scarpulla, R. C. NRF-1: a trans-activator of nuclear-encoded respiratory genes in animal cells. Genes Dev. 4, 1023–1034 (1990).
Virbasius, C. A., Virbasius, J. V. & Scarpulla, R. C. NRF-1, an activator involved in nuclear-mitochondrial interactions, utilizes a new DNA-binding domain conserved in a family of developmental regulators. Genes Dev. 7, 2431–2445 (1993).
Robertson, S. J. et al. Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes 4, 222–231 (2013).
Levy, M. et al. Microbiota-modulated metabolites shape the intestinal microenvironment by regulating NLRP6 inflammasome signaling. Cell 163, 1428–1443 (2015).
Hatzipetros, T. et al. A quick phenotypic neurological scoring system for evaluating disease progression in the SOD1-G93A mouse model of ALS. J. Vis. Exp. 104, 1–6 (2015).
Hansson, G. C. & Johansson, M. E. V. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Gut Microbes 1, 51–54 (2010).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Schroeder, B. O. et al. Bifidobacteria or fiber protects against diet-induced microbiota-mediated colonic mucus deterioration. Cell Host Microbe 23, 27–40.e7 (2018).
Rappsilber, J., Mann, M. & Ishihama, Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat. Protoc. 2, 1896–1906 (2007).
Cox, J. & Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 (2008).
MacLean, B. et al. Skyline: an open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
Baba, T. et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Syst. Biol. 2, 0008 (2006).
Adiconis, X. et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 10, 623–629 (2013).
Caporaso, J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336 (2010).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Weiss, S. et al. Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5, 27 (2017).
McDonald, D. et al. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6, 610–618 (2012).
Marco-Sola, S., Sammeth, M., Guigó, R. & Ribeca, P. The GEM mapper: fast, accurate and versatile alignment by filtration. Nat. Methods 9, 1185–1188 (2012).
Loh, P. R. et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016).
Kanehisa, M. et al. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 34, D354–D357 (2006).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012).
Reimand, J., Kull, M., Peterson, H., Hansen, J. & Vilo, J. g:Profiler—a web-based toolset for functional profiling of gene lists from large-scale experiments. Nucleic Acids Res. 35, W193–W200 (2007).
Acknowledgements
We thank the members of the Elinav and Segal laboratories, and members of the DKFZ cancer-microbiome division for discussions, and apologize to authors whose work was not included owing to space constraints. We thank R. Straussman, D. Nejman and E. Hornstein for suggestions and help with experimental methodologies; C. Bar-Nathan for assistance with animal work; S. Belkin for providing reagents and A. Godneva for computational assistance. We thank J. Suez and N. Zmora for extensive editing during production. E.B. is supported by the Weizmann Institute Dean of Faculty fellowship. D.R. received a Levi Eshkol PhD Scholarship for Personalized Medicine by the Israeli Ministry of Science. N.Z. is supported by the Gilead Sciences International Research Scholars Program in Liver Disease. C.K. is supported by an MD fellowship of the Boehringer Ingelheim Fonds. Y.K. is the incumbent of the Sarah and Rolando Uziel Research Associate Chair. H.S. is the Incumbent of the Vera Rosenberg Schwartz Research Fellow Chair. E.S. is supported by the Crown Human Genome Center; the Else Kroener Fresenius Foundation; Donald L. Schwarz, Sherman Oaks, CA; Jack N. Halpern, NeNY; Leesa Steinberg, Canada; and grants funded by the European Research Council and the Israel Science Foundation. E.E. is supported by Y. and R. Ungar; the Abisch Frenkel Foundation for the Promotion of Life Sciences; the Gurwin Family Fund for Scientific Research; the Leona M. and Harry B. Helmsley Charitable Trust; the Crown Endowment Fund for Immunological Research; the Else Kroener Fresenius Foundation; the estate of J. Gitlitz; the estate of L. Hershkovich; the Benoziyo Endowment Fund for the Advancement of Science; the Adelis Foundation; J. L. and V. Schwartz; A. and G. Markovitz; A. and C. Adelson; the French National Center for Scientific Research (CNRS); D. L. Schwarz; The V. R. Schwartz Research Fellow Chair; L. Steinberg; J. N. Halpern; A. Edelheit; and by grants funded by the European Research Council, the Israel Science Foundation and the Helmholtz Foundation. E.E. is the Sir Marc and Lady Tania Feldmann Professorial Chair in Immunology, Weizmann Institute of Science, Israel; is the Director of the Microbiome Research Division, DKFZ, Heidelberg, Germany; is a senior fellow of the Canadian Institute of Advanced Research (CIFAR) and is an international scholar of The Bill and Melinda Gates Foundation and Howard Hughes Medical Institute (HHMI).
Author information
Authors and Affiliations
Contributions
E.B., S.B. and H.S. designed, performed and interpreted the experiments, and wrote the manuscript; S.B. and D.R. performed and analysed all the microbiome sequencing and analysis. U.M., Y.C., N.B. and I.L. assisted in computational analysis. M.D.-B., C.K., C.M., Y.H., D.K., N.Z., N.A., I.B., Y.K., M.T. and L. Alfahel performed and supervised animal experiments. T.M. and A.B. performed the metabolomics experiments. M. Zur, M. Zabari and R.B.-Z.B. performed and supervised the human experimentation. A.H. oversaw animal experimentation including in germ-free mice. M.S. and A.I. provided help and insights on the project. L. Arike performed proteomic studies, M.E.V.J. performed the colon immunohistology staining and G.C.H. supervised these studies. M.G., E.S. and E.E. conceived the study, supervised the participants, interpreted the experiments and wrote the manuscript.
Corresponding authors
Ethics declarations
Competing interests
E.S. and E.E. are paid consultants at DayTwo and BiomX. None of this work is related to, shared with or licensed to these or any other commercial entity. None of the other authors have competing interests related to this work.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Peer review information Nature thanks Mani Arumugam, Premysl Bercik, Jeffrey Rothstein and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Extended data figures and tables
Extended Data Fig. 1 The effects of microbiome depletion on ALS symptoms in Sod1-Tg mice.
a, Experimental design. Sod1-Tg and wild-type littermate control mice were untreated or treated with broad-spectrum antibiotics in their drinking water from the age of 40 days until the experimental end point. Testing was performed at age 60, 80, 100, 120 and 140 days. b–j, Motor performance of the mice was assessed at age 60, 80, 100, 120 and 140 days by the rotarod locomotor test (b, e, h), the hanging-wire grip test (c, f, i) and neurological scoring (d, g, j). For b–d: n = 8 Sod1-Tg water-treated mice; n = 6 Sod1-Tg antibiotic-treated mice; n = 5 wild-type water-treated and n = 5 wild-type antibiotic-treated mice. For e–g: n = 5 mice in each group; for h–j: n = 7 Sod1-Tg water-treated mice, n = 7 Sod1-Tg antibiotic-treated mice, n = 4 wild-type water-treated mice and n = 5 wild-type antibiotic-treated mice. Data are mean ± s.e.m. k–m, Linear regression of motor functions over time in Sod1-Tg and wild-type treated mice indicated by the rotarod locomotor test (k), the hanging-wire grip test (l) and neurological scoring (m). n = 20 Sod1-Tg water-treated n = 18 antibiotic-treated mice, n = 14 wild-type water-treated mice and n = 15 wild-type antibiotic-treated mice. n, o, Histological images (n) and cell counts (suggestive of neurons) (o) in the spinal cord of 140-day-old water-treated and antibiotic-treated Sod1-Tg mice. n = 10 Sod1-Tg water-treated mice, n = 5 Sod1-Tg antibiotic-treated mice. The experiment was repeated twice. p, q, MRI of areas of the mouse brain affected by ALS (p) and an image of T2 maps (q). The experiment was repeated twice. r–x, Quantification of T2 relaxation time in water-treated and antibiotic-treated Sod1-Tg mice throughout the course of ALS. n = 9 mice in each group. y, Home-cage locomotion analysis over a period of 46 h, for mice aged 100–101 days (n = 5 mice). z–ae, Distributions of immune cell sub-populations in the small-intestine (z, aa), colon (ab, ac), spinal cord at 50 days old (ad) and 140 days old (ae) in water-treated and antibiotic-treated Sod1-Tg mice. n = 5 mice. af, ag, Survival of specific-pathogen-free and germ-free Sod1-Tg mice and wild-type mice (af) or Sod1-Tg mice (ag) that were spontaneously colonized at age 115 days. The experiment was performed twice, with the results of the different experiments shown in af and ag. For af: n = 13 specific-pathogen-free and 7 germ-free Sod1-Tg mice, 16 wild-type specific-pathogen-free and 6 wild-type germ-free mice; for ag: n = 5 specific-pathogen-free and 8 germ-free Sod1-Tg mice. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to a two-tailed Mann-Whitney U-test (b–j), two-tailed t-test (r–x), or Gehan-Breslow (af–ag). Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 2 Microbial compositional dynamics in the Sod1-Tg mouse model throughout the progression of ALS.
a, Taxa summary of bacterial phyla (a) and genera (b) from wild-type and Sod1-Tg mice, obtained by 16S rDNA sequencing of stool samples. c, Relative abundances of genera with significantly different representations in Sod1-Tg mice and wild-type mice. n = 6 mice in each group. d–m, FDR-corrected linear regression analysis of the change in relative bacterial abundance in stool samples of wild-type and Sod1-Tg mice during the progression of ALS. n = 6 Sod1-Tg mice and n = 4 wild-type mice. n, Alpha diversity of the microbiomes of Sod1-Tg mice and wild-type mice. n = 7 Sod1-Tg mice and n = 5 wild-type mice. The experiment was repeated 3 times. o, qPCR-based quantification of total 16S copy-number in 1 ng of DNA extracted from the stool samples of Sod1-Tg mice and wild-type mice. n = 6 Sod1-Tg and n = 5 wild-type mice. p, Taxa summary of bacterial phyla in individual antibiotic-treated wild-type and antibiotic-treated Sod1-Tg mice during the course of ALS. q–v, Weighted UniFrac PCoA on 47-day-old (pre-antibiotic; q) and 60–140-day-old (r–v) mice under a chronic antibiotic regime. n = 7 Sod1-Tg mice and n = 5 wild-type mice. w–ab, Volcano plots of significantly enriched bacterial genera in antibiotic-treated wild-type and antibiotic-treated Sod1-Tg mice during the course of murine ALS-like disease. n = 7 Sod1-Tg mice and n = 5 wild-type mice. ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to an FDR-corrected two-sided FDR-corrected two-tailed Mann–Whitney U-test, or or t-test for coefficient genotype × time (d–m). Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 3 Microbial spontaneous colonization in an ex-germ-free Sod1-Tg mouse model and between two vivaria throughout the progression of ALS.
a, Taxa summary of bacterial genera in individual ex-germ-free wild-type and Sod1-Tg mice undergoing spontaneous bacterial colonization during the course of murine ALS-like disease. b–e, Weighted UniFrac PCoA of ex-germ-free wild-type and Sod1-Tg mice on days 4, 5, 53 and 63 after spontaneous colonization. n = 6 mice in each group. f–i, Volcano plots of significantly enriched bacterial genera in ex-germ-free wild-type mice and ex-germ-free Sod1-Tg mice during the course of the disease on days 4, 5, 53 and 63 after spontaneous colonization. n = 6 mice in each group. j, k, Weighted UniFrac PCoA (j) and alpha diversity (k) of wild-type and Sod1-Tg mice housed in a different non-barrier vivarium (vivarium B, at Ben-Gurion University) at weeks 4, 6, 8 and 12 of age. n = 7–9 Sod1-Tg mice and n = 7–9 wild-type mice. l, m, Individual (l) and averaged (m) taxa summary of bacterial genera in 80-day-old wild-type mice at vivarium A (Weizmann Institute of Science) and vivarium B (Ben-Gurion University). The comparison was performed once, n = 5–8 mice in each group. Significance according to FDR-corrected two-tailed Mann-Whitney U-test. Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 4 Differences in metagenomic faecal microbiomes and a metabolic comparison between wild-type and Sod1-Tg mice.
a, b, PCoA plot of bacterial composition (a), and taxa summary representation at the species level (b) of the gut microbiome of wild-type and Sod1-Tg mice, obtained by metagenomic shotgun sequencing. The experiment was repeated twice. c–n, FDR-corrected linear regression analysis of the change in relative bacterial abundance in stool samples of wild-type and Sod1-Tg mice during the progression of murine ALS-like disease. o, p, Schematic representation of tryptophan metabolism. p, Heat map of the abundances of bacterial genes involved in tryptophan metabolism. q, Heat map of the abundances of bacterial genes involved in the nicotinamide and nicotinate biosynthetic pathway. r–ac, Representative recording (r, t, v, x, z, aa, ab) and quantification (s, u, w, y, ac) of food intake (r, s), water consumption (t, u), respiratory exchange ratio (v, w), O2 consumption (x, y), heat production (z), locomotion (aa) and speed (ab, ac) of 60-day-old wild-type and Sod1-Tg mice. Data are mean ± s.e.m. The experiment was repeated twice. ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to an FDR-corrected two-tailed Mann–Whitney U-test or t-test for coefficient genotype × time (c–n).
Extended Data Fig. 5 Mono-colonization of antibiotic pre-treated Sod1-Tg mice with selected microbiome strains that show a correlation with ALS severity.
a–c, Motor functions of antibiotic pre-treated Sod1-Tg mice treated with PBS (n = 8), E. lenta (EL, n = 7), C. cateniformis (CC, n = 6), P. goldsteinii (PG, n = 6), L. murinus (LM, n = 7), P. distasonis (PD, n = 5), L. gasseri (LG, n = 8), P. melaninogenica (PM, n = 4), or A. muciniphila (AM, ATCC 835, n = 6) as indicated by the rotarod locomotor test (a), the hanging-wire grip test (b) and neurological scoring (c). d–i, Motor functions of antibiotic-pre-treated Sod1-Tg mice treated with PBS or E. tayi (ET, n = 7) (d–f) or S. variabile (SV, n = 7) (g–i). j–k, Motor functions of antibiotic-pre-treated wild-type littermate controls treated with PBS, LM, PD, LG, PM or AM. m, Linear regression of the relative abundance of R. torques (16S rDNA sequencing) in stool samples of Sod1-Tg mice and wild-type mice. n–p, Motor functions of antibiotic-pre-treated wild-type and Sod1-Tg mice treated with PBS or RT, as assessed by the rotarod locomotor test (n), the hanging-wire grip test (o) and neurological scoring (p). n = 18 PBS-treated Sod1-Tg mice; n = 20 RT-treated Sod1-Tg mice; n = 23 PBS-treated and n = 23 PBS-treated or RT-treated wild-type mice. q–y, Assessment of the motor functions of antibiotic-pretreated Sod1-Tg mice and wild-type littermates after treatment with RT in three biological repeats, by the rotarod locomotor test (q, t, w), the hanging wire grip test (r, u, x) and neurological scoring (s, v, y). n = 5–10 mice. z, aa, Histological images (z) and cell counts (suggestive of neurons) (aa) in 140-day-old PBS-treated (n = 5) and RT-treated (n = 3) Sod1-Tg mice. The experiment was repeated 3 times. ab–ah, Brain areas (ab) and the quantification of their T2 relaxation times (ac–ah) of PBS-treated and RT-treated Sod1-Tg mice throughout the course of ALS. The experiment was repeated twice, n = 5 mice in each group. ai, aj, Averaged taxa summary of bacterial genera (ai) and unweighted UniFrac PCoA (aj) of antibiotic-pretreated Sod1-Tg mice treated with PBS (n = 6) or mono-colonized with the above-stated bacteria at 120 days old. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to a two-tailed Mann-Whitney U-test (a–l, n–y), two-tailed t-test (ac–ah), or t-test for coefficient genotype × time (m). Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 6 AM treatment attenuates ALS symptoms in Sod1-Tg mice.
Antibiotic-pretreated Sod1-Tg and wild-type littermate control mice were treated orally with AM (ATCC 835) or PBS as vehicle from 60 days of age until the experimental end point. a–o, At 60, 80, 100, 120 and 140 days old, motor performance of the mice was assessed by the rotarod locomotor test (a, d, g, j, m), the hanging-wire grip test (b, e, h, k, n) and neurological scoring (c, f, i, l, o). In g–i, Sod1-Tg mice were treated with PBS (n = 8), AM (n = 9) P. melaninogenica (n = 4) or L. gasseri (LG, n = 5). n = 4–26 mice. ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to a two-tailed Mann–Whitney U-test. Data are mean ± s.e.m.
Extended Data Fig. 7 The effects of AM treatment on the manifestation of ALS and on microbiome composition in Sod1-Tg mice.
a, b, Histological images (a) and motor-neuron quantification (b) of the spinal cords of 140-day-old PBS-treated and AM-treated Sod1-Tg mice. n = 7 mice in each group. The experiment was repeated twice. c–f, T2 relaxation times in PBS-treated and AM (ATCC 835)-treated and antibiotic-pretreated Sod1-Tg mice at 100 and 140 days old. n = 5 mice in each group. g, Systemic FITC–dextran measurement in 120-day-old wild-type and Sod1-Tg mice treated with PBS (n = 3 wild-type mice and n = 7 Sod1-Tg mice), AM (n = 3 wild-type mice and n = 9 Sod1-Tg mice), P. melaninogenica (PM, n = 5 wild-type mice and n = 4 Sod1-Tg mice) or L. gasseri (LG, n = 5 for both wild-type and Sod1-Tg mice). h, PCoA of bacterial species compositions in Sod1-Tg mice treated with PBS (n = 33) or AM (n = 31). i, j, Relative abundance of AM in Sod1-Tg mice (i) or wild-type mice (j) treated with PBS or AM. n = 8 mice. k, l, Individual (k) and averaged (l) fold-change of AM 16S copy number (analysed by qPCR) in mucosal and luminal samples across the gastrointestinal tract of 140-day-old AM-treated or PBS-treated wild-type and Sod1-Tg mice. n = 5 mice in each group. m, n, Summary of bacterial genera in Sod1-Tg mice (m) or wild-type mice (n) treated with PBS or AM. Antibiotic-pretreated Sod1-Tg and wild-type littermate control mice were treated orally with AM (ATCC 2869) or PBS as a vehicle from 60 days of age until the experimental end point. o–q, At 60, 80, 100, 120 and 140 days old, the motor performance of the mice was assessed by the rotarod locomotor test (o), the hanging-wire grip test (p) and neurological scoring (q). n = 6 PBS-treated Sod1-Tg mice, AM-2869-treated Sod1-Tg mice and AM-2869-treated wild-type mice, and n = 7 wild-type PBS-treated mice. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to a two-tailed t-test (c–f), or a two-tailed Mann-Whitney U-test (all others). Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 8 AM treatment alters mucus properties in Sod1-Tg mice.
a, b, Immunohistochemical assessment of distal colon mucosa of 140-day-old PBS-treated (a) and AM-treated (BAA-835) (b) antibiotic-pretreated wild-type and Sod1-Tg mice. DNA is stained with SYTOX Green (green) and the mucus is stained with an anti-MUC2C3 antiserum and goat anti-Ig (red). The non-stained area between the epithelium and the outer mucus or luminal bacteria is the inner mucus layer, enabling identification of the bacteria. The experiment was performed once. White arrows represent penetrating crypt bacteria. c, d, Heat-map representation of the total mucus proteomic landscape (c) and AM-related peptides (d). e–j, Quantification of key representative components of mucus. For c–j, n = 4 Sod1-Tg PBS-treated mice and n = 8 Sod1-Tg AM-treated mice, two-tailed Mann–Whitney U-test. p = NS. Data are mean ± s.e.m.
Extended Data Fig. 9 The serum metabolomic profile is affected by antibiotics or by AM treatment in ALS Sod1-Tg mice.
a–c, Heat-map representation of serum metabolites of 100-day-old naive Sod1-Tg and their wild-type littermates (a), water-treated or antibiotic-treated Sod1-Tg mice (b) and PBS-treated or AM-treated Sod1-Tg mice (c). d, Scoring of the top six serum metabolites that were significantly altered by antibiotic treatment in Sod1-Tg mice by their potential to originate from the gut microbiome. n = 8 mice in each group. e–g, Motor performances of Sod1-Tg mice that were treated with phenol sulfate (n = 8) or water (n = 7) administered using subcutaneous osmotic pumps, as indicated by the rotarod locomotor test (e), the hanging-wire grip test (f) and neurological scoring (g). h, i, Non-targeted metabolomics assessment of tryptophan metabolism of water- and antibiotic- treated (h) or PBS- and AM-treated (i) 100-day-old Sod1-Tg mice. n=5 for water- and antibiotic -treated Sod1-Tg mice and n = 7 for PBS- and AM-treated Sod1-Tg mice in each group. j, Nicotinamide levels in the CSF of Sod1-Tg and wild-type mice treated with AM or PBS at the age of 140 days. k, Schematic representation of the microbiome-derived nicotinamide-encoding genes in faecal samples from AM-treated Sod1-Tg mice.*P < 0.05, two-tailed Mann-Whitney U-test. Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 10 Nicotinamide ameliorates the progression of ALS in Sod1-Tg mice.
a, Serum NAM levels in NAM-treated and vehicle-treated Sod1-Tg mice (n = 10). b–j, Motor performances of Sod1-Tg mice that were treated with NAM or vehicle administered using subcutaneous osmotic pumps, as indicated by the rotarod locomotor test (b, e, h), the hanging-wire grip test (c, f, i) and neurological scoring (d, g, j). n = 9–10 mice in each group. k, Survival analysis of NAM-treated and vehicle-treated Sod1-Tg mice (n = 10 mice in each group). l, Nicotinamide levels in wild-type or ΔnadA E. coli cultures (n = 5). m–o, Motor performances of wild-type E. coli-inoculated or ΔnadA E. coli-inoculated antibiotic-pretreated Sod1-Tg mice as indicated by the rotarod locomotor test (m), the hanging-wire grip test (n) and neurological scoring (o). n = 18 wild-type E.coli-treated Sod1-Tg mice and n = 16 ΔnadA E. coli-treated Sod1-Tg mice. p–x, Motor performances of wild-type E. coli-inoculated or ΔnadA E. coli-inoculated antibiotic-pretreated Sod1-Tg mice indicated by the rotarod locomotor test (p, s, v), the hanging-wire grip test (q, t, w) and neurological scoring (r, u, x). n = 4–8 mice. ****P < 0.0001; ***P < 0.001; **P < 0.01; *P < 0.05. Significance according to a two-tailed Mann–Whitney U-test. Unless stated otherwise, data are mean ± s.e.m.
Extended Data Fig. 11 Uncovering potential downstream motor-neuron modulatory mechanisms of AM and NAM treatments.
a, Heat map of significantly (FDR-corrected two-tailed Mann-Whitney U-test) differentially expressed genes in the spinal cords of NAM-treated Sod1-Tg mice (n = 10 mice). b, Spearman correlation of the log2 fold change of spinal-cord transcripts comparing AM-treated and NAM-treated Sod1-Tg mice, n = 10 mice. c, Comparison of the significantly differentially expressed genes following NAM treatment with the COG database classified into 4 neuropathological groups. The blue shaded area represents the 95% confidence interval of the linear regression curve. d, e, Gene set enrichment distribution of spinal cord transcripts of NAM-treated (d) and AM-treated (e) Sod1-Tg mice sorted by biological process, molecular functions and cellular components. n = 10 mice.
Extended Data Fig. 12 NAM differentially expressed genes associated with NRF-1.
a, Representation of spinal-cord transcripts from Sod1-Tg mice that showed a similar change after treatment with AM and with NAM, and share the GCGCMTGCGCN binding site for the transcription factor NRF-1 (in green). The analysis was performed using the g:Profiler platform49. b, Spinal-cord global transcriptomic analysis in NAM-treated or water-treated Sod1-Tg mice. c, Individual representation of the expression of Slc1a2 (n = 10). d, Spinal-cord global transcriptomic analysis in AM-treated or PBS-treated Sod1-Tg mice. e, Individual representation of the expression of Slc1a2 (n = 7). FDR-corrected two-tailed Mann–Whitney U-test. Data are mean ± s.e.m.
Extended Data Fig. 13 Differences in gut microbiome composition in patients with ALS compared with healthy controls.
a, Relative absence of gut microbiome bacterial species in stool samples of patients with ALS (n = 37) and healthy family members (n = 29). b, Spearman correlation between the abundance of Bifidobacterium pseudocatenulatum and levels of NAM in the serum of patients with ALS (n = 37) and healthy controls (n = 29). c, Relative frequencies of KEGG orthology terms of microbiome-associated genes of the nicotinamide pathway in stool samples of patients with ALS (n = 36) and healthy controls (n = 28). d, Top 97 serum metabolites that are differentially represented between patients with ALS (n = 24) and healthy controls (n = 13). e, Levels of metabolites from the tryptophan and nicotinamide biosynthetic pathways in the serum of patients with ALS (n = 24) and healthy controls (n = 13). f, g, Correlation of NAM levels in serum with clinical scores on the functional rating scale (FRS, f) and bacterial l-aspartate oxidase copies (g) in patients with ALS (n = 60) and healthy controls (n = 33). ***P < 0.0001, **P < 0.01; *P < 0.05. Significance according to a two-tailed Mann–Whitney U-test.
Supplementary information
Supplementary Information
This file contains Supplementary Tables 1-7.
Rights and permissions
About this article

Cite this article
Blacher, E., Bashiardes, S., Shapiro, H. et al. Potential roles of gut microbiome and metabolites in modulating ALS in mice. Nature 572, 474–480 (2019). https://doi.org/10.1038/s41586-019-1443-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1443-5
- Springer Nature Limited
This article is cited by
-
Therapeutics for neurodegenerative diseases by targeting the gut microbiome: from bench to bedside
Translational Neurodegeneration (2024)
-
Peripheral extracellular vesicles in neurodegeneration: pathogenic influencers and therapeutic vehicles
Journal of Nanobiotechnology (2024)
-
Vitamin A carotenoids, but not retinoids, mediate the impact of a healthy diet on gut microbial diversity
BMC Medicine (2024)
-
Utilization of the microbiome in personalized medicine
Nature Reviews Microbiology (2024)
-
Disentangling direct vs indirect effects of microbiome manipulations in a habitat-forming marine holobiont
npj Biofilms and Microbiomes (2024)