

Abstract
Neuromuscular disorders are often caused by heterogeneous mutations in large, structurally complex genes. Targeting compensatory modifier genes could be beneficial to improve disease phenotypes. Here we report a mutation-independent strategy to upregulate the expression of a disease-modifying gene associated with congenital muscular dystrophy type 1A (MDC1A) using the CRISPR activation system in mice. MDC1A is caused by mutations in LAMA2 that lead to nonfunctional laminin-α2, which compromises the stability of muscle fibres and the myelination of peripheral nerves. Transgenic overexpression of Lama1, which encodes a structurally similar protein called laminin-α1, ameliorates muscle wasting and paralysis in mouse models of MDC1A, demonstrating its importance as a compensatory modifier of the disease1. However, postnatal upregulation of Lama1 is hampered by its large size, which exceeds the packaging capacity of vehicles that are clinically relevant for gene therapy. We modulate expression of Lama1 in the dy2j/dy2j mouse model of MDC1A using an adeno-associated virus (AAV9) carrying a catalytically inactive Cas9 (dCas9), VP64 transactivators and single-guide RNAs that target the Lama1 promoter. When pre-symptomatic mice were treated, Lama1 was upregulated in skeletal muscles and peripheral nerves, which prevented muscle fibrosis and paralysis. However, for many disorders it is important to investigate the therapeutic window and reversibility of symptoms. In muscular dystrophies, it has been hypothesized that fibrotic changes in skeletal muscle are irreversible. However, we show that dystrophic features and disease progression were improved and reversed when the treatment was initiated in symptomatic dy2j/dy2j mice with apparent hindlimb paralysis and muscle fibrosis. Collectively, our data demonstrate the feasibility and therapeutic benefit of CRISPR–dCas9-mediated upregulation of Lama1, which may enable mutation-independent treatment for all patients with MDC1A. This approach has a broad applicability to a variety of disease-modifying genes and could serve as a therapeutic strategy for many inherited and acquired diseases.
Similar content being viewed by others


Main
MDC1A is caused by mutations in the LAMA2 gene—which encodes laminin-α2 chain. LAMA2 polymerizes with the β1 and γ1 chains to form the heterotrimer laminin-211, which is an extracellular matrix protein complex. Interactions between laminin-211, α-dystroglycan and α7β1 integrin in skeletal muscles and Schwann cells is important for the survival and stability of myotubes, proper neurite growth, axon myelination and migration of Schwann cells. Lack of laminin-211 in MDC1A causes degeneration of skeletal muscle and impaired differentiation of Schwann cells, resulting in a cascade of secondary events that include apoptosis or necrosis of muscle fibres, inflammation and fibrosis, which ultimately precipitate the disease. Despite considerable advances in our understanding of the pathophysiology that underlies MDC1A, there is currently no cure.
Owing to the genetic nature of the disease, the correction of LAMA2 mutations is a promising treatment option for MDC1A. We recently described the use of CRISPR–Cas9 technology to correct a mutation in Lama2 in vivo2. We focused on the dy2j/dy2j mouse model; dy2j/dy2j mice have a splice site mutation in Lama2 that causes spontaneous exon skipping and truncation of the N-terminal domain of the protein3. We developed an exon-inclusion strategy to correct the splice site mutation, leading to restoration of full-length Lama2, which established the direct correction of the primary genetic defect that underlies MDC1A in an in vivo model2. Nevertheless, there are over 350 pathogenic nonsense, missense, splice site and deletion mutations in LAMA2 reported to date. Given the number of MDC1A-causing genomic alterations, CRISPR–Cas9-mediated correction would require the design and thorough analysis of multiple single-guide RNAs (sgRNAs) that are specific to each mutation. Moreover, safety concerns regarding the potential mutagenic nature of the CRISPR–Cas9 system and the presence of off-target effects after gene editing remain, which together may prove to be challenging from a safety and regulatory point-of-view.
By contrast, the attenuation of disease pathogenicity by targeted modulation of the expression of disease-modifier genes would be a potentially safer alternative and beneficial to all individuals with MDC1A. One of the strongest reported disease modifiers for MDC1A is laminin-α1, which is structurally similar to laminin-α2 (Fig. 1a). However, laminin-α1 is not expressed in skeletal muscles or Schwann cells. Previous studies have demonstrated that transgenic Lama1 overexpression rescued myopathy and peripheral neuropathy in dy2j/dy2j mice4 and dy3K/dy3K mice1,4,5; the latter also had an increased lifespan. Although these studies established a compensatory function of laminin-α1 in MDC1A, the use of this modifier as postnatal gene therapy is hampered by the size of the Lama1 cDNA, which exceeds the packaging capacity of AAV vectors.

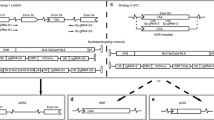
a, Protein alignment of Lama1 and Lama2. The total number of amino acids (aa) and percentage similarity between each domain are indicated. EGF, epidermal growth factor; G, globular; N, N terminus. b, Analyses of the proximal promoter of Lama1. Five sgRNAs (g1–g5) were designed to target the proximal promoter region of Lama1 immediately upstream of the transcription start site, as indicated by CAGE tags (red). Chromatin accessibility in skeletal muscle tissue and cells (retrieved using Digital DNase footprinting and assay for transposase-accessible chromatin using sequencing (ATAC-seq)) are shown in blue. Data were plotted according to positions from the UCSC Genome Browser. c, Positions of the five sgRNAs relative to Lama1 transcription start site. Arrowheads indicate the direction of each sgRNA. Sequences of each sgRNA (red) are immediately downstream (5′) of Sa protospacer adjacent motif (PAM) sequences (NNGRRT) (in blue). TSS, transcription start site. Lowercase and uppercase letters indicate intronic and exonic sequences, respectively. ATG indicates the translation start site. d, e, C2C12 myoblasts were transfected with a plasmid encoding SadCas9–2×VP64 and the corresponding sgRNA(s) targeting Lama1 and, 72 h after transfection, were analysed by reverse transcription PCR (RT–PCR) (d) and quantitative reverse transcription PCR (qRT–PCR) (e). d, A 100-bp ladder is shown. e, Data are mean ± s.d. from n = 3 biologically independent samples. P values are (from left to right): ****P < 0.0001, *P = 0.0136, ***P = 0.0006, **P = 0.0015, *P = 0.0224; one-way ANOVA. Basal Lama1 expression in GFP-transfected cells was set to 1. f, g, Single and a combination of the optimal sgRNAs were transfected into dy2j/dy2j myoblasts and expression of Lama1 was assessed by western blot (f); quantification is shown as fold change in expression (g). Flag expression serves as a transfection control and is used to normalize the Lama1 upregulation. NT, not transfected. ‘−’, no sgRNA. g, Data are mean ± s.d. from n = 3 biologically independent samples. P values are compared to ‘no sgRNA’: **P = 0.0023 (middle), **P = 0.0019 (right); one-way ANOVA. d–g, Experiments were repeated three times independently with similar results.
Advances in CRISPR–Cas9 technologies have provided opportunities for regulating gene expression and creating epigenetic alterations without introducing DNA double-stranded breaks, which are known as the CRISPR transcriptional activation system. The strategy uses nuclease-deficient Cas9 (dCas9), which is unable to cleave DNA owing to mutations within the nuclease domains and retains the ability to specifically bind to DNA when guided by a sgRNA6,7. Using the previously described Streptococcus pyogenes (Sp)dCas9 fused to multiple copies of the VP16 transcriptional activator, our group and others have demonstrated the use of the CRISPR–dCas9 system to upregulate expression of modifier genes in vitro6,7,8. A major challenge for in vivo applications lies in the large size of SpdCas9 and its transcriptional activator fusion derivatives that exceed the genome-packaging capacity of AAVs. To accommodate this limitation, we adapted the transcriptional upregulation system and used a considerably smaller Cas9 protein, which is derived from Staphylococcus aureus (Sa)9, to upregulate Lama1. We hypothesized that CRISPR–dCas9-mediated transcriptional upregulation of Lama1 would be sufficient to compensate for the lack of Lama2 and ameliorate disease phenotypes in dy2j/dy2j mice.
First, we mutagenized the endonuclease catalytic residues (D10A, N580A) of SaCas9 to create SadCas9 and then fused it to transcriptional activators VP64 (four copies of VP16) on both N and C termini (Extended Data Fig. 1). We tested the ability of the system (hereafter SadCas9–2×VP64) to upregulate the expression of a minCMV-driven tdTomato gene in HEK293T cells. In the presence of SadCas9–2×VP64 and an sgRNA that targets the minCMV locus, we observed a high tdTomato fluorescence signal, which indicates the general applicability of this system to modulate expression of a gene of interest.
Subsequently, we tailored the system to upregulate Lama1 expression and designed five sgRNAs, denoted as g1 to g5, within the 500-nucleotide region immediately upstream of the Lama1 transcription start site (Fig. 1b, c). When co-expressed with SadCas9–2×VP64, three out of five sgRNAs (namely g1, g2 and g5) consistently induced a significant increase in Lama1 transcript expression in C2C12 mouse myoblasts (Fig. 1d, e), as well as protein expression in dy2j/dy2j-derived myoblasts (Fig. 1f, g), warranting further investigation in vivo.
We then treated three-week-old dy2j/dy2j mice with an AAV9 that contained Flag-tagged SadCas9–2×VP64 in the absence of sgRNA (no guide) or with g1 (single guide) or a combination of g1, g2 and g5 (three guides) (Fig. 2a). Owing to the packaging capacity of the AAV, dCas9–2×VP64 and the three guides were split into two vectors (Fig. 2a). Each mouse received a single intramuscular injection of 7.5 × 1011 viral genomes of AAV9—which was doubled to 1.5 × 1012 viral genomes in total for the three-guide cohort—in the right tibialis anterior muscle, and was euthanized 4 weeks after injection. Flag expression was detected by western blot in all SadCas9–2×VP64-injected right tibialis anterior muscles; however, only those injected with guide-containing constructs showed upregulation of Lama1 (Fig. 2b). Similarly, immunofluorescence staining revealed the expression of Lama1 in the sarcolemma (Fig. 2c), indicating proper protein localization. Analysis of haematoxylin and eosin staining showed that mice in the single- and three-guide treatment groups had considerably improved muscle architecture (Fig. 2c), compared to the no-guide controls.

a, Right tibialis anterior muscles of three-week-old dy2j/dy2j mice were injected with AAV9s containing no guide (n = 4; 7.5 × 1011 viral genomes), a single guide (n = 4; 7.5 × 1011 viral genomes) or three guides (n = 4; split into two vectors; total dose of 2 × 7.5 × 1011 viral genomes). i.m., intramuscular; ITR, inverted terminal repeats. CMV and U6 promoters are depicted as arrows. Left tibialis anterior muscles serve as control. b, Western blot analysis of Lama1, Flag-tagged SadCas9 and Gapdh expression. LTA, left tibialis anterior muscle; RTA, right tibialis anterior muscle. c, Immunofluorescence images of Lama1 (top) and haematoxylin and eosin (H&E) (top) of cross-sections of right tibialis anterior muscles from each treatment group. Scale bars, 50 µm. Samples from all mice (n = 4 mice per group) were analysed twice independently with similar results.
Next, we investigated whether upregulation of Lama1 could be achieved systemically in vivo and, if administered in presymptomatic neonatal dy2j/dy2j mice, whether it would prevent the manifestation of dystrophic pathophysiology and paralysis (Extended Data Fig. 2). AAV9 particles that contained either no guide or a combination of three guides were injected into the temporal vein of two-day-old (P2) dy2j/dy2j mice (Extended Data Fig. 2a). Then, seven weeks after injection, Lama1 expression was detected in the tibialis anterior (Extended Data Fig. 2b) and gastrocnemius (Extended Data Fig. 2c) muscles. Although AAV transduction efficiency varied slightly between individual muscles, overall upregulation of Lama1 led to a reduction in fibrosis (Extended Data Fig. 2d–h), larger fibre size (Extended Data Fig. 2g, i), as well as an absence of classical hindlimb contracture (Supplementary Videos 1, 2). Collectively, these data show successful prevention of the disease pathophysiology upon early intervention in dy2j/dy2j mice.
We subsequently examined the ability of Lama1 upregulation to reverse established muscular and peripheral nerve dysfunction by initiating the treatment at three weeks of age, when paralysis was already apparent10 (Fig. 3a). We first tested three different doses that ranged from 7.5 × 1010 to 3 × 1011 viral genomes per gram of mouse per AAV9, which was doubled for the three-guide cohorts owing to the use of two vectors, and found that the highest dose resulted in homogeneous Lama1-positive fibres (Extended Data Fig. 3a) and significant improvement of muscle function and mobility (Extended Data Fig. 3b, c). Longitudinal measurements of mouse mobility and stand-up activity in a non-invasive open field activity assay showed a significant difference between the no-guide- and three-guide-treated mice starting at five and six weeks old, respectively, which was sustained over time (Fig. 3b, c). Furthermore, specific tetanic force, which measures the aggregate torque produced by the dorsiflexor muscles, was also improved in the three-guide-treated mice (Fig. 3d). Consistent with this finding, we observed a significant increase in nerve conduction velocity, which is indicative of restoration of the myelination defect and contributes to neuromuscular functionality (Fig. 3e). The absence of paralysis in the hindlimbs and markedly improved movement of the mice were evident at the end of the treatment regimen (Supplementary Videos 3, 4).

a, Three-week-old dy2j/dy2j mice were injected with AAV9s containing no guide (denoted as ‘−’; n = 9; 3 × 1011 viral genomes g−1 of mouse) or three guides (denoted as ‘+’; n = 8; split into two vectors; total dose of 2 × 3 × 1011 viral genomes g−1 of mouse) in the tail vein. Grey box represents the period before treatment. b, c, The open field activity assay was performed weekly, before (indicated as 3 weeks) and after treatment. NS, not significant. d, e, The mice were tested for muscle contractile properties (d) and nerve conduction velocity (e) at the end of the treatment regimen. b–e, Data are mean ± s.d.; two-tailed t-test. b, P values from left to right (including NS) are: P = 0.8230, P = 0.8467, **P = 0.0017, *P = 0.0201, *P = 0.0056. c, P values from left to right (including NS) are: P = 0.8026, P = 0.1725, P = 0.4061, **P = 0.0094, *P = 0.0008. d, **P = 0.0047. e, **P = 0.0059. Note that the number of mice analysed in the experiment shown in e were n = 8 (no guide) and n = 7 (three guides). f, g, Immunofluorescence (f) and western blot (g) analyses were carried out to analyse Lama1 expression. h–j, Haematoxylin and eosin staining (h) to quantify fibrosis (i; ****P < 0.0001, two-tailed t-test) and fibre size (j; P = 0.117, two-tailed t-test) was performed on gastrocnemius muscles. All mice (n = 9, no guide; n = 8, three guides) were analysed for experiments in f–j, which were repeated three times independently with similar results. k, Left, sciatic nerves were stained for expression of Lama1 (red) and neurofilament H (green). Nuclei were counterstained using 4′,6-diamidino-2-phenylindole (DAPI) (blue). Higher-magnification images from the dotted area are shown to the right. Middle, representative toluidine blue images are shown for each group. Asterisks indicate a region of amyelinated axon fibres. Right, electron micrographs are shown for each group. Three mice per group were analysed for the experiment in k, which was repeated three times independently with similar results. Scale bars, 100 µm (f), 50 µm (h) and 200 µm (k).
Molecular analysis of the treated mice revealed strong expression of Lama1 by immunostaining and western blot (Fig. 3f, g and Extended Data Fig. 4), which was accompanied by normalization of the expression of the α4 chain of the laminin subunit4 (Extended Data Figs. 5, 6), significant improvement in muscle histopathology (Fig. 3h) and approximately 50% reduction in the fibrotic area (Fig. 3i) compared to the no-guide control group. There was a trend towards larger fibres in the treated mice, although it did not reach statistical significance owing to the large variation between mice (Fig. 3j). In addition, upregulation of Lama1 was also observed in the endoneurium of sciatic nerves and resulted in restoration of the myelination defect (Fig. 3k and Extended Data Fig. 7), supporting the improvement of nerve conduction velocity and lack of paralysis in the mice (Fig. 3e and Supplementary Videos 3, 4). Quantification of the AAV genome copy number revealed accumulation of most of the viral genomes in the liver, which is expected from intravenous delivery. Nevertheless, approximately 1.11 ± 0.4 (mean ± s.e.m.) and 34.1 ± 4.3 copies per diploid genome of the viral genome were detected in the sciatic nerves and skeletal muscles, respectively (Extended Data Fig. 8). Even the relatively low transduction efficiency in sciatic nerves—compared to other tissues such as the skeletal muscle and liver—was sufficient to induce functionally significant expression of Lama1.
Finally, we investigated the genome-wide effects of CRISPR–dCas9-mediated upregulation of Lama1 by performing RNA sequencing on quadriceps muscles (Extended Data Figs. 9, 10 and Supplementary Tables 1–3). We observed a 3.6-fold upregulation (log2-transformed value) of Lama1 (defined by false-discovery rate-adjusted P < 0.05) when comparing the mice treated with AAV9 that contained no guide and three guides (Extended Data Fig. 9a). The transcriptional change was even higher (log2-transformed change in expression of 9-fold) when comparing untreated dy2j/dy2j with three-guide-treated dy2j/dy2j mice (Extended Data Fig. 9b, c). Hierarchical clustering between groups revealed clustering between wild-type mice and three-guide-treated mice, whereas the untreated cohort was grouped together with no-guide-treated mice (Extended Data Figs. 9d, 10). We also computationally predicted 704 potential off-target binding sites for the three sgRNAs that target the Lama1 promoter in the mouse genome, selected based on the presence of a six-base-pair (bp) proximal seed sequence of the protospacer adjacent motif and fewer than ten total mismatches to the cognate target sequence (Supplementary Tables 4, 5). None of the top 100 genes contained a predicted off-target site within 60 kilobases of the gene body and the mean distance from off-target site to the gene body was 2.2 megabases.
Taken together, our results provide strong evidence of the robustness and durability of CRISPR–dCas9-mediated upregulation of Lama1 for the treatment of MDC1A. Additionally, we show the therapeutic promise of the strategy for reversal of dystrophic features in skeletal muscle and peripheral neuropathy in the dy2j/dy2j mouse model of MDC1A, which ultimately halts progression of the disease, without any confounding off-target effects.
One challenge in developing a therapy for the treatment of MDC1A is that the heterogeneity of mutations often leads to variable disease severity and progression. Therefore, there is an urgent need to develop a universal, mutation-independent strategy that provides a treatment approach for all patients with MDC1A. Our study establishes a framework in which transcriptional upregulation of a disease-modifier gene, such as Lama1, ameliorates disease symptoms in vivo and has the potential to be applied to all patients with MDC1A, irrespective of the causative mutation. The ability to modify gene expression in postnatal mice also allowed us to begin to investigate the therapeutic window of intervention and address an important question in the development of therapeutics and clinical trials for the treatment of MDC1A11, such as the reversibility of symptoms caused by muscle fibrosis and nerve abnormalities. Here we show that upregulation of Lama1 leads to significant rescue of the phenotypes in three-week-old mice with already-apparent muscle fibrosis and hindlimb paralysis, indicating that postsymptomatic treatment provides a considerable benefit in the dy2j/dy2j mouse model.
Previous studies have demonstrated that the peripheral neuropathy found in the dy2j/dy2j mice involves impaired radial sorting, which originates prenatally owing to a lack of Lama2 in (immature) Schwann cells that prevents the cells from proliferating and differentiating properly. Many therapeutic interventions that start during the perinatal period (for example, at P2) influence the proliferation and differentiation of Schwann cells12,13. As our intervention starts at three weeks of age, it is unlikely that our strategy affects the proliferation and differentiation of Schwann cells and their progenitors. Nevertheless, the radial sorting process is not completely abolished in adult dy2j/dy2j Schwann cells14. In fact, dy2j/dy2j Schwann cells at P28 retain a limited capacity to initiate radial sorting that leads to partially impaired postnatal myelination, in which cooperation between Lama2 and its receptors—such as dystroglycan, α6β1 and α6β4 integrins—is crucial15. Therefore, we suggest that Lama1 upregulation compensated for the lack of Lama2 by establishing interactions with the integrins and/or dystroglycan in the basal lamina, which eventually restored the myelination defect. Examination of Lama1 interactomes would be required to investigate the cellular mechanism of the compensatory effect of postnatal Lama1 upregulation. Furthermore, previous studies14,15 have reported differences in the radial sorting defect between roots and proximal nerves of dy2j/dy2j mice, compared to their distal nerves. Additional studies to explore the effect of postnatal Lama1 upregulation on spatial differences in axonal radial sorting would be important both from disease mechanistic and translational perspectives.
In addition to Lama1, a number of disease-modifying strategies are currently being explored in animal models of MDC1A, including miniaturized agrin (mini agrin)13,16, laminin-α1 LN-domain nidogen-1 (αLNNd) and a combination of both17,18. Although the efficacy of αLNNd and the combination of αLNNd and mini agrin have only been explored in transgenic mice thus far17, AAV-mediated delivery of mini agrin has been shown to normalize most histopathological parameters in skeletal muscle, and improve myelination and regeneration of Schwann cells of the peripheral nerves in the more severe dyW/dyW mouse model13. Despite the observed phenotypic improvements, the mini-agrin-treated dyW/dyW mice still have a lower survival rate than wild-type mice that expressed full-length agrin, suggesting a potential limitation of the strategy. It is important to note that many preclinical studies of MDC1A were carried out in the more-severe dyW/dyW and dy3K/dy3K mouse models. Therefore, it will be paramount to evaluate our approach in these more-severe mouse models to assess critical parameters such as survival, which is not possible in the dy2j/dy2j mice, as these mice have a near-normal lifespan.
Neuromuscular disorders have provided excellent examples to demonstrate the role of disease modifiers. In addition to MDC1A, several studies have demonstrated that upregulation of Lama1 stabilizes the sarcolemmal membrane in Duchenne muscular dystrophy. The most advanced approach is delivery of laminin-111 protein in dystrophin-deficient mice, although the need to produce a large amount of bioactive protein is challenging19. In addition, the use of the CRISPR–dCas9 system to upregulate Lama1 in dystrophin-deficient mice has been achieved locally using electroporation, which is not easily translatable into clinical settings20. Our strategy of using AAV-mediated infection of SadCas9 to upregulate Lama1 in vivo may be tested further as a potential therapy in Duchenne muscular dystrophy. Moreover, the CRISPR–dCas9-mediated transcriptional modulation may be used in conjunction with a mutation-correction approach for which the level of restoration of the defective gene is suboptimal, therefore necessitating further amplification to reach therapeutic efficacy2,21,22,23.
A recent study has described the use of the CRISPR transcriptional activation system to induce expression of target genes in skeletal muscle, kidney and liver tissues, resulting in phenotypic augmentation such as enhanced muscle mass and substantial improvement in disease pathophysiology24. However, it relied almost exclusively on a Cas9-expressing transgenic mouse model or on local intramuscular treatments and it is therefore difficult to extrapolate the efficacy of this strategy to disease-relevant models. By contrast, we successfully demonstrated robust upregulation of Lama1 after systemic delivery of therapeutic components in a relevant mouse model of disease that does not constitutively express Cas9.
Finally, the modular nature of the CRISPR–dCas9 system can be used not only to upregulate, but also to downregulate the expression of a target gene when coupled with the transcriptional repressor Kruppel-associated box (KRAB)25. An application as a combinatorial therapeutic approach, involving concurrent upregulation of protective disease-modifier genes and downregulation of detrimental genes, would represent a new paradigm for lessening disease phenotypes. In summary, our study establishes a framework to use CRISPR–dCas9 to modulate gene expression of disease modifiers that should be considered as a mutation-independent therapeutic strategy not only to treat MDC1A, but also for the treatment of various other inherited and acquired diseases.
Methods
Engineering of activation constructs
A fragment containing a catalytically inactive SadCas9 coupled to two flanking VP64 transactivator domains was synthesized by BioBasic Canada and cloned into pX601 (Addgene 61591) using AgeI and EcoRI directional cloning to generate 3×Flag-VP64-SadCas9(D10A/N580A)-NLS-VP64 plasmid (Fig. 2a and Supplementary Table 6). Each sgRNA (Supplementary Table 6) was subsequently introduced using BsaI directional cloning. To generate the three-guides-only construct (Fig. 2a), a fragment containing three repetitive regions of the U6 promoter and S. aureus guide scaffold was assembled, with short linkers in between each region (BioBasic Canada). The fragment was cloned into KpnI and NotI sites of a pX601-derivative plasmid.
Cell culture
Primary myoblasts were isolated from the extensor digitorum longus muscle of dy2J/dy2J mice and maintained in DMEM supplemented with 1% chicken embryo extract (GeminiBioscience), 10% horse serum, 1% penicillin–streptomycin and 1% l-glutamine (Gibco). HEK293 and C2C12 cells (ATCC) were maintained in DMEM supplemented with 10% FBS, 1% penicillin–streptomycin and 1% l-glutamine (Gibco). All cells were maintained at 37 °C with 5% CO2.
Transfection of HEK293 cells was performed using Lipofectamine 2000. C2C12 and dy2J/dy2J cells were transfected in 12-well-plates using the Neon Transfection System (Invitrogen). In brief, 400,000 cells were electroporated with 1.5 μg of DNA using optimization program 16 (pulse voltage, 1,400 V; pulse width, 20 ms; pulse number, 2). Cells were grown for 72 h, after which RNA or protein was subsequently collected for protein analysis and guide screening.
Mice, virus production and injections
dy2j/dy2j mice were purchased from the Jackson Laboratory and maintained in The Center for Phenogenomics. Both male and female mice were used in the analyses. The numbers of mice were based on the TREAT-NMD standard operating protocol for MDC1A or a previous study2. All mouse experiments were performed according to Animal Use Protocol number 20-0305 (The Center for Phenogenomics). SadCas9–2×VP64, single-guide and three-guide plasmids (Fig. 2a and Supplementary Table 6) were packaged into AAV9 vectors by Vigene Biosciences. For intramuscular and temporal vein injection into neonatal pups, the dose of 7.5 × 1011 viral genomes each was used. Owing to the limitation in packaging capacity, two AAVs were needed for the three-guide cohort (Fig. 2a), therefore the total virus injected was 1.5 × 1012 viral genomes per mouse. Injection volume was brought to 50 μl with 1× PBS (Gibco). For treatment initiated at neonatal age, no randomization was performed, for example, the entire litter of P2 newborn pups were injected with either no-guide- or three-guide vectors.
For the tail vein injection in young, 3-week-old mice, 3 different doses were initially tested: 7.5 × 1010, 1.5 × 1011 and 3 × 1011 viral genomes per gram of mouse (Extended Data Fig. 3). Randomization was performed among littermates. Similar to the intramuscular and temporal vein injections, two AAVs were needed for the three-guide cohort, therefore the total dose used in experiments described in Fig. 3 and Extended Data Figs. 4–10 was 6 × 1011 viral genomes per gram of mouse. Injection volume was brought to 100 μl with 1× PBS (Gibco). Detailed reporting of design, experimental procedures, mice and blinding are described in Supplementary Methods 1.
RNA isolation, guide screening and RT–PCR
RNA was isolated from cultured cells and mouse tissue sections, and cDNA synthesis was performed as previously described3. PCR amplification was used to assess the efficiency of each guide in upregulating Lama1 expression using a primer in Lama1 exon 55 (RDC 1919) and a second primer spanning the junction of exons 55 and 56 (RDC 1920). Sequences are listed in Supplementary Table 6.
qPCR using Fast SYBR green Master Mix (Qiagen) on a Step One Plus Real Time PCR (Applied Biosystems) was performed. Lama1 expression was analysed using a primer in Lama1 exon 55 (RDC 1919) and one spanning the junction of exons 55 and 56 (RDC 1920). Primers against endogenous Gapdh (RDC 345 and 346) were used as an internal control. ΔΔCt was analysed to assess fold change in expression between treated and untreated samples.
Protein isolation and western blot
Proteins were isolated from dy2j/dy2j myoblasts and C2C12 cells by adding 150 μl of a 1:1 part solution of RIPA homogenizing buffer (50 mM Tris HCl pH 7.4, 150 nM NaCl, 1 mM EDTA) and RIPA double-detergent buffer (2% deoxycholate, 2% NP-40, 2% Triton X-100 in RIPA homogenizing buffer) supplemented with protease inhibitor cocktail (Roche). Proteins from mouse tissue sections were homogenized as previously described3. Total protein concentrations were measured using a Pierce BCA protein assay kit according to the manufacturer’s protocol (Thermo Fisher Scientific). Western blots were performed as previously described3. Primary antibodies used were rabbit anti-LNα1 E3 (a gift from P. Yurchenco, 0.6 μg ml−1), mouse monoclonal M2 anti-Flag (Sigma-Aldrich F1804, 1:1,000) and rabbit polyclonal anti-GAPDH (Santa Cruz sc-25778, 1:5,000). Uncropped gels are provided as Supplementary Fig. 1.
Immunofluorescence and haematoxylin and eosin staining
Muscles and nerves were sectioned at 8-μm thickness and processed for immunofluorescence analyses according to standard procedures. Antibodies used for immunofluorescence staining were rat monoclonal antibodies against laminin-α1 (mAb200, laboratory of M.D., 1:20), α2 (4H8-2, Sigma-Aldrich, 1:500), γ1 chain (clone A5, Thermo Fisher Scientific), a rabbit polyclonal antibody against laminin-α4 chain (provided by T. Sasaki), a mouse monoclonal antibody against NF-H (Biolegend SMI 31, 1:1,000), a goat polyclonal anti-rat Alexa Fluor 555 antibody (Thermo Fisher Scientific, 1:250) and a goat polyclonal anti-mouse Alexa Fluor 488 antibody (Thermo Fisher Scientific, 1:250). Haematoxylin and eosin staining was performed as previously described2. Both immunofluorescence and haematoxylin and eosin-stained slides were scanned with the 3Dhistech Pannoramic 250 Flash II digital scanner and analysed with CaseViewer software, with the exception of images shown in Extended Data Figs. 5, 6, which were analysed with a Zeiss Axioplan fluorescence microscope (Carl Zeiss) and images were captured using an ORCA 1394 ER digital camera (Hamamatsu Photonics) and Open Laboratory software version 4 (Improvision).
Toluidine blue staining and transmission electron microscopy
Freshly isolated mouse sciatic nerves were halved and fixed in a solution of 2% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer until further use. For embedding, the specimens were rinsed with the 0.1-M sodium cacodylate buffer, post-fixed in 1% osmium tetroxide in the washing buffer, dehydrated in a graded ethanol series followed by incubation in propylene oxide, and embedded in Quetol-Spurr resin overnight at 65 °C. Sections of 90-nm thickness were cut on a Leica EM UC7 ultramicrotome, stained with uranyl acetate and lead citrate, and imaged on a FEI Tecnai 20 electron microscope at 4,400×, 10,000× and 44,000× magnifications. The same 90-nm thick sections were stained with toluidine blue and imaged on a Leica DM-2000. All reagents were purchased from Electron Microscopy Sciences. Quantification of myelin thickness was measured using ImageJ from at least 14 random myelinated axons per field (at least 5 fields per mouse)26. No discrimination between axon diameters (for example, small or larger calibre nerves) was made during quantification.
Open field and in vivo muscle force assays
Open field activity test and assessment of in vivo muscle force were performed on tail-vein-injected cohorts at the Centre for Modelling Human Disease Mouse Phenotyping Facility of the Lunenfeld-Tanenbaum Research Institute. For the open field test, mice were placed in the frontal centre of a transparent Plexiglas open field (41.25 cm × 41.25 cm × 31.25 cm) illuminated by 200 lx. A trained operator, who was blinded to the nature of the projects and treatments, performed experiments. The VersaMax Animal Activity Monitoring System recorded vertical activities and total distance travelled for 20 min per mouse.
In vivo muscle contraction tests were performed using a 1300A: 3-in-1 Whole Animal System and analysed using the dynamic muscle control/analysis high-throughput software suite (Aurora Scientific). The mice were anaesthetized with intraperitoneal injection of a ketamine–xylazine cocktail at 100 mg kg−1 and 10 mg kg−1 of body weight, respectively. Contractile output was measured using percutaneous electrodes that stimulate specific nerves that innervate the plantar flexors. Specific tetanic force (200 ms of 0.5-ms pulses at 125 Hz) was recorded and corrected for body weight.
Nerve conduction velocity
Mice were anaesthetized with urethane (1.2 mg g−1; intraperitoneal injection) and underwent a tracheotomy to maintain their airway but were not artificially ventilated. Core body temperature was maintained at 34–36.5 °C using a feedback-controlled heating pad (TR-200; Fine Science Tools). The sciatic nerve was exposed at two locations through incisions (around 15 mm) above the left knee (site 1) and along the sacral region of the vertebral column (site 2). After separating the nerve from adjacent tissue, a bipolar hook electrode was applied to the nerve at each site. Each electrode comprised two chloride silver wires (0.01-inch diameter, A-M systems) placed approximately 1 mm apart and bent at the tip to form hooks, and attached to a stimulator (Model DS3, Digitimer). Stimulating electrodes were insulated from the underlying tissue using a small piece of plastic paraffin film. Stimulus duration was fixed at 20 μs and the current intensity was varied. Stimulus timing was controlled by a computer using a Power1401 computer interface and Signal v.5 software (Cambridge Electronic Design). The compound muscle action potential (CMAP) was recorded using needle electrodes (~30G, BD PrecisionGlide), one inserted into the gastrocnemius muscle and the other (reference) electrode inserted into the Achilles tendon. The CMAP signal was amplified, low-pass-filtered at 10 Hz and high-pass-filtered at 1 kHz (DAM 80, World Precision Instruments), and digitized at 40 kHz using the Power1401 and Signal v.5 software (Cambridge Electronic Design). To calculate conduction velocity from CMAP responses, the sciatic nerve was stimulated at just-maximal intensity (beyond which there is no change in CMAP amplitude or latency) three times at site 1 and again three times at site 2. Nerve conduction velocity was calculated as the difference between mean CMAP latencies for each stimulation site divided by the length of nerve (7.5–10 mm) separating the sites.
Quantification of vector genomes
Evaluation of AAV genome distribution was performed as previously published27 with a few modifications. Genomic DNA was extracted from tibialis anterior, sciatic nerve and liver of treated mice using the Qiagen Blood and Tissue Kit. Then, 90 ng of DNA was amplified using primers located in between the two inverted tandem repeats (RDC 1687, RDC 1679; Supplementary Table 7) using Fast SYBR green master mix (Qiagen) on a Step One Plus Real Time PCR (Applied Biosystems). The Ct value of each reaction was converted to viral genome copy number by interpolating against the copy number of a standard curve of a known plasmid containing the sgRNA cassette (RDC 362). The amount of DNA input (90 ng per tissue) was used as a conversion factor to diploid genome (7 pg DNA = 1 diploid genome).
RNA-sequencing analyses
Total RNA was isolated from quadriceps muscles using the RNeasy kit (Qiagen) and quantified using a Qubit RNA HS assay (Thermo Fisher Scientific). RNA sequencing was performed by the Centre for Applied Genetics in Toronto using the Illumina HiSeq 2500 system, producing 120-bp paired-end reads. Alignment and analyses were performed as previously described25. In brief, raw transcript reads were aligned to the GRCm38 mouse genome (mm10) using HISAT2. HTSeq was used to determine the absolute number of read counts for each gene. Only genes with at least one read per million in at least three replicates were kept for downstream analysis. Normalization and differential expression analysis were completed using the R packages limma v.3.36.3 (https://bioconductor.org/packages/release/bioc/html/limma.html) and edgeR v.3.22.3 (https://bioconductor.org/packages/release/bioc/html/edgeR.html). Library sizes were scaled using trimmed mean of M-value normalization applied using the edgeR package. Using limma, paired sample data were fitted to a linear model, then all pairwise comparisons were made. False discovery and multiple testing were controlled for and an adjusted P value was computed using the Benjamini–Hochberg method. Differentially expressed genes were defined as genes with a more than twofold change and adjusted P < 0.05. Off-target analysis was conducted using a list of 704 computationally predicted using Cas-OFFinder. Using the Bedtools suite ‘closest’ function, each of the top 100 differentially expressed genes was matched to the nearest off-target loci to determine the shortest distance from off-target loci to differentially expressed gene body.
Statistical analyses
Statistical analyses for every experiment are described accordingly in the figure legends. Unless specified, GraphPad Prism version 8 was used to perform most statistical analyses.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
The authors declare that the main data supporting the findings of this study are available within the paper and its Supplementary Information.
References
Gawlik, K., Miyagoe-Suzuki, Y., Ekblom, P., Takeda, S. & Durbeej, M. Laminin α1 chain reduces muscular dystrophy in laminin α2 chain deficient mice. Hum. Mol. Genet. 13, 1775–1784 (2004).
Kemaladewi, D. U. et al. Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat. Med. 23, 984–989 (2017).
Sunada, Y., Bernier, S. M., Utani, A., Yamada, Y. & Campbell, K. P. Identification of a novel mutant transcript of laminin α2 chain gene responsible for muscular dystrophy and dysmyelination in dy 2J mice. Hum. Mol. Genet. 4, 1055–1061 (1995).
Gawlik, K. I., Harandi, V. M., Cheong, R. Y., Petersén, Å. & Durbeej, M. Laminin α1 reduces muscular dystrophy in dy 2J mice. Matrix Biol. 70, 36–49 (2018).
Gawlik, K. I., Li, J. Y., Petersén, A. & Durbeej, M. Laminin α1 chain improves laminin α2 chain deficient peripheral neuropathy. Hum. Mol. Genet. 15, 2690–2700 (2006).
Maeder, M. L. et al. CRISPR RNA-guided activation of endogenous human genes. Nat. Methods 10, 977–979 (2013).
Perez-Pinera, P. et al. RNA-guided gene activation by CRISPR–Cas9-based transcription factors. Nat. Methods 10, 973–976 (2013).
Wojtal, D. et al. Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet. 98, 90–101 (2016).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Kemaladewi, D. U., Benjamin, J. S., Hyatt, E., Ivakine, E. A. & Cohn, R. D. Increased polyamines as protective disease modifiers in congenital muscular dystrophy. Hum. Mol. Genet. 27, 1905–1912 (2018).
Bönnemann, C. G. et al. Diagnostic approach to the congenital muscular dystrophies. Neuromuscul. Disord. 24, 289–311 (2014).
Homma, S., Beermann, M. L. & Miller, J. B. Peripheral nerve pathology, including aberrant Schwann cell differentiation, is ameliorated by doxycycline in a laminin-α2-deficient mouse model of congenital muscular dystrophy. Hum. Mol. Genet. 20, 2662–2672 (2011).
Qiao, C. et al. Amelioration of muscle and nerve pathology in LAMA2 muscular dystrophy by AAV9-mini-agrin. Mol. Ther. Methods Clin. Dev. 9, 47–56 (2018).
Patton, B. L., Wang, B., Tarumi, Y. S., Seburn, K. L. & Burgess, R. W. A single point mutation in the LN domain of LAMA2 causes muscular dystrophy and peripheral amyelination. J. Cell Sci. 121, 1593–1604 (2008).
Previtali, S. C. et al. Expression of laminin receptors in Schwann cell differentiation: evidence for distinct roles. J. Neurosci. 23, 5520–5530 (2003).
Bentzinger, C. F., Barzaghi, P., Lin, S. & Ruegg, M. A. Overexpression of mini-agrin in skeletal muscle increases muscle integrity and regenerative capacity in laminin-α2-deficient mice. FASEB J. 19, 934–942 (2005).
Reinhard, J. R. et al. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci. Transl. Med. 9, eaal4649 (2017).
McKee, K. K. et al. Chimeric protein repair of laminin polymerization ameliorates muscular dystrophy phenotype. J. Clin. Invest. 127, 1075–1089 (2017).
Rooney, J. E., Gurpur, P. B. & Burkin, D. J. Laminin-111 protein therapy prevents muscle disease in the mdx mouse model for Duchenne muscular dystrophy. Proc. Natl Acad. Sci. USA 106, 7991–7996 (2009).
Perrin, A., Rousseau, J. & Tremblay, J. P. Increased expression of laminin subunit alpha 1 chain by dCas9–VP160. Mol. Ther. Nucleic Acids 6, 68–79 (2017).
Yuan, J. et al. Genetic modulation of RNA splicing with a CRISPR-guided cytidine deaminase. Mol Cell 72, 380–394 (2018).
Villiger, L. et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med. 24, 1519–1525 (2018).
Bengtsson, N. E. et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 8, 14454 (2017).
Liao, H. K. et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell 171, 1495–1507 (2017).
Thakore, P. I. et al. Highly specific epigenome editing by CRISPR–Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149 (2015).
Zhang, Y. et al. Notch1 signaling plays a role in regulating precursor differentiation during CNS remyelination. Proc. Natl Acad. Sci. USA 106, 19162–19167 (2009).
Hakim, C. H. et al. A five-repeat micro-dystrophin gene ameliorated dystrophic phenotype in the severe DBA/2J-mdx model of Duchenne muscular dystrophy. Mol. Ther. Methods Clin. Dev. 6, 216–230 (2017).
Acknowledgements
We thank members of the Cohn laboratory for technical support and input in this study; P. Yurchenco for providing reagents used in this study, C. Rand, I. Vukobradovic and R. Smith for assistance with functional and behavioural studies, D. Holmyard for transmission electron microscopy analysis and S. Pereira for genomic data acquisition. This work was supported by AFM-Telethon, Cure CMD, Muscular Dystrophy Association (to D.U.K.), Rare Disease Foundation Microgrant (to P.S.B. and S.E.), SickKids Restracomp (to S.E.), Crafoord Foundation (to K.I.G.), CIHR Summer Studentship (to R.M.M.), Swedish Research Council, Anna and Edwin Berger Foundation, Greta and Johan Kock Foundation, Olle Engkvist Byggmastare Foundation and Osterlund Foundation (to M.D.), Natural Sciences and Engineering Research Council of Canada, Canadian Institute for Health Research, SickKids Foundation and R. S. McLaughlin Foundation Chair in Pediatrics (to R.D.C.).
Reviewer information
Nature thanks James M. Ervasti, Reid D. Landes, Daniel Michele and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
D.U.K., E.A.I. and R.D.C. conceived and supervised the study. P.S.B., K.L., R.K. and K.M.P. cloned and screened guides in vitro. D.U.K., P.S.B., K.L. and E.H. performed in vivo injections and contractile assays. D.U.K., P.S.B., K.L., K.M.P. and R.M.M. analysed muscle histology. K.I.G. and M.D. performed extended muscle imaging and provided reagents. D.U.K. performed longitudinal open field assay and vector genome quantification. D.U.K. and P.S.B. performed nerve imaging with the help from K.M.P. and R.M.M. D.U.K. and D.A.-B. performed nerve conduction velocity under supervision by S.A.P. S.E. analysed RNA-sequencing data and off-target prediction. D.U.K. wrote the manuscript with inputs from the other authors. All authors provided feedback and agreed on the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 SadCas9–2×VP64 enhances expression of minCMV-driven tdTomato in vitro.
a, b, HEK293T cells were transfected with a plasmid encoding minCMV-driven tdTomato only. c, d, HEK293T cells were transfected with a combination of a plasmid containing minCMV-driven tdTomato and one containing SadCas9–2×VP64 and a sgRNA targeting the minCMV promoter. a, c, Schematic. b, d, Cells were imaged for tdTomato expression by fluorescent microscopy. Bright-field and tdTomato images are shown. The experiment was repeated three times independently with similar results. Scale bars, 50 µm.
Extended Data Fig. 2 Early intervention to upregulate Lama1 prevents disease progression in dy2j/dy2j mice.
a, P2 neonatal dy2j/dy2j mice were injected with AAV9s carrying no guide (n = 6; 7.5 × 1011 viral genomes) or three guides (n = 4; split into two vectors; total dose of 2 × 7.5 × 1011 viral genomes) in the temporal vein and euthanized 7 weeks later. b, c, Immunofluorescence staining shows Lama1 expression in tibialis anterior (b) and gastrocnemius (c) muscles. Scale bars, 100 µm. d, e, General muscle histopathology was evaluated by haematoxylin and eosin staining on tibialis anterior (d) and gastrocnemius (e) muscles. Scale bars, 200 µm. All mice (n = 6, no guide and n = 4, three guides) were analysed for experiments shown in b–e, which were repeated three times independently with similar results. f–i, Fibrosis (f, h) and minimum Feret diameter (g, i) were quantified for all mice from both treatment groups (n = 6, no guide and n = 4, three guides) by two independent experimenters with similar results. Data are mean ± s.d. **P = 0.0025 (f), *P = 0.0103 (g), *P = 0.0142 (h), P = 0.0711 (i); two-tailed Student’s t-test.
Extended Data Fig. 3 Upregulation of Lama1 corresponds to improvement of muscle functions.
Three-week-old dy2j/dy2j mice were injected systemically in the tail vein with AAV9s carrying no guide (n = 9) at the dose of 3 × 1011 viral genomes (vg) per gram of mouse, or three guides at different doses of 7.5 × 1010 (n = 4), 1.5 × 1011 (n = 3) or 3 × 1011 (n = 8) viral genome per gram of mouse. Two AAVs were needed for the three-guide cohorts, therefore the total doses of virus injected were 1.5 × 1011, 3 × 1011 and 6 × 1011 viral genomes per gram of mouse. a, Tibialis anterior muscles isolated that were isolated four weeks later were stained for Lama1 expression. Asterisks indicate Lama1-positive fibres in the low-dose cohort. The experiment was repeated three times independently with similar results. Scale bars, 100 µm. b, c, In vivo contractile force assays (b) and open field tests (c) were performed at the end of the treatment regimen. Data are mean ± s.d. b, P values from left to right are: P = 0.9849, P = 0.9082, **P = 0.0044; one-way ANOVA with Dunnett’s multiple comparisons test. c, P values from left to right are: P = 0.9640, P = 0.9646, **P = 0.0085; one-way ANOVA with Dunnett’s multiple comparisons test.
Extended Data Fig. 4 Representative images of Lama1-positive muscle sections.
Three-week-old mice were injected systemically with three guides (n = 5) at the dose of 3 × 1011 viral genomes per gram of mouse (split into two vectors, thus total dose was 2 × 3× 1011 viral genomes per gram of mouse) in the tail vein, and euthanized at the age of 11–12 weeks old. Muscles were stained for Lama1 expression (red). Scale bars, 500 µm. The experiment was repeated three times independently with similar results.
Extended Data Fig. 5 Expression of laminin subunits in tibialis anterior muscles.
Expression of laminin (LM) subunits in tibialis anterior muscles of wild-type, dy2j/dy2j (intravenously injected with AAVs carrying no guide at 3 × 1011 viral genomes per gram of mouse or three guides at a total dose of 2 × 3× 1011 viral genomes per gram of mouse) and dy2j/dy2j LMα1 (with transgenic overexpression of LMα1) mice (n = 3 for each group). Expression of the LMα1 chain in the tibialis anterior muscle is comparable between dy2j/dy2j mice treated with three guides and transgenic mice. No major differences in expression of LMα2 and LMγ1 were detected between the groups. The LMα4 chain is upregulated in muscles of dy2j/dy2j mice treated with the no-guide AAV and the expression of the LMα4 chain is partially normalized in muscle of dy2j/dy2j mice treated with the three-guide AAV. Scale bar, 50 μm. The experiment was repeated three times independently with similar results.
Extended Data Fig. 6 Expression of laminin subunits in gastrocnemius muscles.
Expression of laminin subunits in gastrocnemius muscles from wild-type, dy2j/dy2j (intravenously injected with AAVs carrying no guide at 3 × 1011 viral genomes per gram of mouse or three guides at a total dose of 2 × 3 × 1011 viral genomes per gram of mouse) and dy2j/dy2j LMα1 (with transgenic overexpression of LMα1) mice (n = 3 for each group). Expression of the LMα1 chain in the gastrocnemius muscles is comparable between dy2j/dy2j mice treated with three guides and transgenic mice. No major differences in expression of LMα2 and LMγ1 were detected between the groups. The LMα4 chain is upregulated in muscle of dy2j/dy2j mice treated with the no-guide AAV and the expression of the LMα4 chain is partially normalized in muscle of dy2j/dy2j AAV-mice treated with three guides. Scale bar, 50 μm. The experiment was repeated three times independently with similar results.
Extended Data Fig. 7 Myelination of sciatic nerves.
Transmission electron microscopy images of sciatic nerves isolated from dy2j/dy2j mice injected with AAV9s carrying no guide (3 × 1011 viral genomes per gram of mouse) or three guides (split into two vectors; total dose of 2 × 3 × 1011 viral genomes per gram of mouse). Three mice were analysed per group. Myelin thickness was measured as the difference between the distance of the myelin and axon (m+a, blue solid line) and axon only (a; red dotted line), quantified using ImageJ and presented as mean ± s.d. Statistical analysis was performed using a two-tailed t-test. *P = 0.0231. Scale bar, 5 μm. The experiment was repeated three times independently with similar results.
Extended Data Fig. 8 Quantitative evaluation of AAV genome distribution.
Genomic DNA isolated from the tibialis anterior muscle, sciatic nerve and liver of dy2j/dy2j mice (n = 8) injected with AAVs carrying three guides at the dose of 3 × 1011 viral genomes per gram of mouse (split into two vectors; total dose of 2 × 3 × 1011 viral genomes per gram of mouse) was amplified to analyse the presence of viral genomes by qPCR. Data are mean ± s.e.m. The experiment was repeated three times independently with similar results.
Extended Data Fig. 9 Genome-wide analysis of gene expression of SadCas9-2×VP64-treated dy2j/dy2j mice.
a–c, Differential expression analysis derived from RNA-sequencing results from the quadriceps of treated and untreated mice (n = 3 per group). a, b, No guide: dy2j/dy2j mice injected with AAV9 carrying only the SadCas9–2×VP64 at the dose of 3 × 1011 viral genomes per gram of mouse. a, c, Three guides: dy2j/dy2j mice injected with AAV9 carrying the SadCas9–2×VP64 and three guides at the dose of 3 × 1011 viral genomes per gram of mouse (total dose of 2 × 3 × 1011 viral genomes per gram of mouse). b, c, Untreated: dy2j/dy2j mice that had not been injected with AAV9. False discovery and multiple testing were controlled for and an adjusted P value was computed using the Benjamini–Hochberg method. Differentially expressed genes were defined as genes with a more than twofold change and adjusted P < 0.05. For each comparison, significantly differentially expressed genes (false-discovery rate-adjusted P < 0.05) are coloured. Red data points indicate a log-transformed fold change in expression greater than one; blue data points indicate a log-transformed fold change in expression less than one. d, Hierarchical clustering was performed on the normalized counts-per-million expression data.
Extended Data Fig. 10 Top 500 differentially expressed genes.
Heat map illustrating the log-normalized counts per million for the top 500 genes that were differentially expressed in quadriceps isolated from wild-type mice, or dy2j/dy2j mice injected with AAV9s carrying SadCas9–2×VP64 only (no guide, n = 3) or with three guides (three guides, n = 3), and compared to age-matched, untreated dy2j/dy2j mice. Red indicates higher expression; blue indicates lower expression.
Supplementary information
Supplemental Figure 1
Uncropped scans with size marker indications Boxes indicate area presented in the corresponding manuscript figures.
Supplemental Method 1
Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines Detailed reporting of design, experimental procedures, and experimental animals in this study.
Supplementary Tables
This file contains Supplementary Tables 1-6 (legends included at the top of each sheet).
Supplementary Video 1
Phenotype of control dy2j/dy2j mouse following early intervention. The dy2j/dy2j mouse was injected with AAV9 containing SadCas9-2xVP64 (no guide) at P2 (pre-symptomatic stage) via temporal vein and video was taken at the age of 7-week old. Hind limb paralysis, contracture and kyphosis resulting from lack of functional Lama2 and compensatory Lama1 are apparent.
Supplementary Video 2
Phenotype of treated dy2j/dy2j mouse following early intervention. The dy2j/dy2j mouse was injected with AAV9 containing SadCas9-2xVP64 and sgRNAs targeting Lama1 proximal promoter (three guides) at P2 (pre-symptomatic stage) via temporal vein and video was taken at the age of 7-week old. Upregulation of compensatory Lama1 expression ameliorates the hind limb paralysis, contracture and kyphosis.
Supplementary Video 3
Phenotype of control dy2j/dy2j mouse following intervention at symptomatic stage. The dy2j/dy2j mouse was injected with AAV9 containing SadCas9-2xVP64 (no guide) at 3-week old (pre-symptomatic stage) via tail vein. Video was taken at the age of 7-week old. Hind limb paralysis, contracture and kyphosis resulting from lack of functional Lama2 and compensatory Lama1 are apparent.
Supplementary Video 4
Phenotype of treated dy2j/dy2j mouse following intervention at symptomatic stage. The dy2j/dy2j mouse was injected with AAV9 containing SadCas9-2xVP64 and sgRNAs targeting Lama1 proximal promoter (three guides) at 3-week old (pre-symptomatic stage) via tail vein. Video was taken at the age of 7-week old. Dystrophic features and disease progression were significantly improved and partially reversed following upregulation of Lama1.
Rights and permissions
About this article

Cite this article
Kemaladewi, D.U., Bassi, P.S., Erwood, S. et al. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature 572, 125–130 (2019). https://doi.org/10.1038/s41586-019-1430-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-019-1430-x
- Springer Nature Limited
This article is cited by
-
Molecular mechanisms and therapeutic strategies for neuromuscular diseases
Cellular and Molecular Life Sciences (2024)
-
Comparison of DNA targeting CRISPR editors in human cells
Cell & Bioscience (2023)
-
Profiling the impact of the promoters on CRISPR-Cas12a system in human cells
Cellular & Molecular Biology Letters (2023)
-
Advances in CRISPR therapeutics
Nature Reviews Nephrology (2023)
-
Compact engineered human mechanosensitive transactivation modules enable potent and versatile synthetic transcriptional control
Nature Methods (2023)