
Abstract
Purpose of Review
Muscular dystrophies (MDs) are a heterogeneous collection of inherited disorders which cause progressive muscle loss and weakness/hypotonia. Owing to the genetic root of MDs, CRISPR/Cas9 genome editing has been investigated as a possible therapy, with significant advancements having been made. This review aims to provide an overview of recent progress on the in vivo utilization of CRISPR/Cas9 in MD animal models.
Recent Findings
Three primary methods for correcting MD with CRISPR/Cas9 exist: restoration of the full-length protein, restoration of a truncated but partially functional protein, and modulation of gene expression. All these approaches have been (DMD) models with varying degrees of success. In congenital muscular dystrophy type 1A (MDC1A) mice, full-length protein restoration and disease modifier upregulation strategies significantly improved the phenotype. Lastly, efficient elimination of pathogenic CTG repeats via CRISPR/Cas9 was achieved in myotonic dystrophy type 1 (DM1) mice. Delivery of CRISPR machinery into MD animals was frequently accomplished with adeno-associated viruses (AAVs), which currently significantly outperform nanoparticle-based delivery. The targeting of satellite cells in vivo by AAVs has been evaluated by several groups in DMD mice, yielding conflicting results which require clarification.
Summary
Partial or nearly complete phenotypic rescue has been achieved in DMD, MDC1A, and DM1 animals with numerous CRISPR/Cas9 strategies. While considerable work will be necessary to advance CRISPR/Cas9 genome editing past preclinical stages, its therapeutic potential for MD is extremely promising and warrants the investment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Muscular Dystrophy
Muscular dystrophies (MDs) are a clinically and genetically heterogeneous group of diseases characterized by progressive weakness and loss of muscle mass [1]. The disorders differ by the affected muscles, age of onset, severity, and rate of progression [1, 2]. Medical interventions are currently restricted to symptom management or delaying disease progression [1, 3].
This review provides an overview of in vivo genome editing strategies in MD animal models that utilize clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein (Cas) 9 technology. Published studies applying CRISPR/Cas9 as an in vivo therapeutic strategy have been limited to Duchenne muscular dystrophy (DMD), congenital muscular dystrophy type 1A (MDC1A), and myotonic dystrophy type 1 (DM1). However, the techniques and approaches discussed here are potentially applicable to a broad range of MDs. Furthermore, we examine which delivery systems have been tested in vivo, critical long-term considerations, what challenges lay ahead for the field, and key items to be addressed for genome editing to become a viable therapy.
DMD is the most common pediatric MD, affecting 1 in 5000 boys due to its recessive X-linked inheritance [4]. It is a life-limiting disorder resulting from mutations in the DMD gene encoding dystrophin [1, 5]. Dystrophin is a sub-sarcolemmal protein integral to the dystrophin glycoprotein complex which protects muscle from contraction-induced injury [6, 7]. DMD mutations generate out-of-frame transcripts, abolishing dystrophin expression and culminating in muscle atrophy [4]. In contrast, Becker muscular dystrophy (BMD) patients harbor DMD mutations which maintain the reading frame, producing a truncated yet partially functional dystrophin that results in a milder disease course [8, 9]. A major clinical goal is converting DMD mutations into BMD-like mutations to restore the dystrophin open reading frame (ORF) and improve the disease phenotype [10]. As the bulk of published in vivo MD genome editing studies are on DMD, this review will primarily summarize recent progress for DMD.
MDC1A is an autosomal, recessive neuromuscular disorder, characterized by neonatal onset of hypotonia, muscle weakness, and muscle wasting [1, 11]. Mutations in the LAMA2 gene, which encodes laminin-α2, cause MDC1A [11, 12]. Laminins are extracellular matrix proteins which form complexes and are essential basement membrane components [13].
DM1 is an autosomal dominant condition affecting 1 in 8000 individuals and is the most common adult-onset MD [2, 14]. Apart from muscle weakness and stiffness, patients can develop intellectual impairment, respiratory insufficiency, and cardiac conduction abnormalities [2, 14]. A cytosine, thymine, guanine (CTG) repeat expansion in the 3′UTR of the dystrophia myotonica protein kinase (DMPK) gene causes DM1 [2]. These transcripts form nuclear foci which cause deleterious splicing defects in numerous pre-mRNAs [14].
Genome Editing Strategies
Various targeted MD therapies are currently approved or in clinical trials. These include gene therapy, antisense oligonucleotides (AONs), and stop codon read-through compounds [15,16,17,18,19]. However, relatively poor performance has hindered their widespread clinical use. An effective therapy must target and, ideally, permanently correct the genetic source of MDs. CRISPR/Cas9-based interventions hold extreme promise due to their unparalleled utility and precision in performing targeted genome editing [5, 20,21,22].
CRISPR/Cas9 Technology as a Genetic Engineering Tool
Genome editors include zinc finger nucleases, TALENs, and meganucleases, but their use has waned in favor of the more practical CRISPR/Cas9 [23]. The CRISPR/Cas9 system was first discovered in bacteria and archaea as an antiviral defense mechanism and has been repurposed as a programmable genome editor [24•, 25]. The most utilized type II CRISPR system has two components: a single guide RNA (sgRNA) with a region complementary to a target sequence and a Cas9 endonuclease [26]. Once guided to its DNA target by an sgRNA, Cas9 generates a double-strand break (DSB) upstream of its protospacer adjacent motif (PAM) [24•]. The most popular Cas9 from Streptococcus pyogenes (SpCas9) uses an NGG or NAG PAM [24•]. The smaller Staphylococcus aureus Cas9 (SaCas9) recognizes NNGRR(T), while Campylobacter jejuni Cas9 (CjCas9) uses an extended NNNNRYAC PAM [27]. Numerous other Cas9s have been discovered or developed, ensuring the availability of a suitable system for nearly any application [27].
DSB Resolution by HDR and NHEJ
The power of CRISPR/Cas9 for genome editing comes from site-specific DSB generation. This DSB induces DNA repair pathways which can be utilized to yield desired genomic modifications. Depending on the proliferative status of the cell and the presence of an exogenous DNA template, the DSB will be repaired by homology directed repair (HDR) or non-homologous end joining (NHEJ) [24•, 28, 29].
HDR results in faithful resolution of the DSB but is typically restricted to S/G2 phases of proliferating cells [30]. HDR proceeds by homologous recombination, enabling knock-in of complete or partial wild-type genes [30]. Therefore, for CRISPR to initiate HDR, a DNA template with homology to the regions flanking the DSB must be provided alongside the Cas9 and sgRNA [24•]. Insertion of exogenous DNA is a powerful therapeutic strategy for MD. Unfortunately, an enormous barrier to applying HDR is its poor efficiency in post-mitotic muscle cells [29, 31].
In the absence of a DNA template, Cas9-induced DSBs are typically repaired via NHEJ. NHEJ is the primary DSB repair pathway, with the free ends being directly ligated. Imprecise repair can introduce random insertions and/or deletions (indels) [24•]. NHEJ-based strategies have been used to correct splicing and excise mutated sequences like duplications and out-of-frame exons [32•, 33,34,35,36].
Applications of a Nuclease Deficient Cas9
A groundbreaking application of CRISPR/Cas9 has been the manipulation of gene expression with a catalytically inactive or “dead” Cas9 (dCas9) [24•, 35]. While DNA binding is retained, dCas9 cannot generate DSBs. Gene repression is achievable by targeting dCas9 to regulatory elements, sterically hindering transcription machinery [37]. Expression can alternatively be activated or enhanced by fusing transcriptional activators to dCas9 and localizing them to promoters [29].
Single nucleotide mutations can be corrected without DSBs through base editing [38]. A Cas9 capable of generating single-stranded DNA (ssDNA) breaks, called a nickase, is fused to a nucleobase deaminase which can chemically alter a base [38]. Current deaminase enzymes are restricted to C-to-T and A-to-G transitions, limiting which point mutations can be corrected [38, 39]. Unfortunately, base editors risk off-target editing when adjacent bases, identical to the target, are present [39]. Nevertheless, when correctly applied, base editing can restore full-length protein expression.
NHEJ-Mediated Restoration of a Truncated Dystrophin Protein
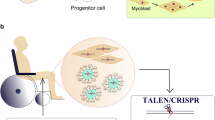
As previously mentioned, one promising strategy is converting DMD mutations into BMD-like variants, with the expectation that disease phenotype will improve. The most straightforward method to restore the reading frame is by Cas9-mediated deletion, skipping, or reframing of exons via NHEJ (Fig. 1a). It is important to note that while the following approaches are applicable to a wide range of DMD patients, converting DMD into BMD can only reduce disease severity, not cure it. Table 1 provides a comprehensive summary of the Cas9 variants, delivery vectors, routes of administration, and treatment regimens from these studies.

CRISPR/Cas9-mediated genome editing strategy for the treatment of MDs. On the left side are gene stretches shown with an exon 2 deletion mutation, a nonsense mutation (*), an exon 2 duplication or expanded CTG repeats in the 3′UTR region. a Non-homologous end joining (NHEJ)-meditated exon deletion, exon skipping, exon reframing, or duplication removal. b CRISPR/Cas9-induced gene correction via homology-directed repair (HDR)-mediated exon knock-in with an additional DNA template. c CRISPR/Cas9-mediated targeted elimination of expanded repeats. These (CTG)n repeats are located in 3′UTR of a gene between the stop codon (stop) and the polyadenylation signal (pA). d A-to-G base transition carried out by CRISPR/nCas9 attached with an adenine deaminase. e CRISPR/dCas9-mediated gene regulation. Gene expression of a disease modifier gene can be modulated by CRISPR/dCas9 fused with a transcriptional modulator
CRISPR, clustered regularly interspaced short palindromic repeats; AAV, adeno-associated virus; PAM, protospacer adjacent motif; nCas9, Cas9 nickase; dCas9, catalytically deficient Cas9
Exon Deletion
The deletion of one or more exons with a pair of flanking sgRNAs has been demonstrated to effectively restore the DMD ORF. Single exon deletion has been accomplished in DMD mice [40,41,42, 52, 60, 61, 62•, 64] and, more recently, pigs [57••]. Moretti et al. induced robust dystrophin expression in a Δ52 DMD pig model following systemic administration of SpCas9 and a pair of sgRNAs to achieve exon 51 deletion [57••]. Only two other studies using CRISPR/Cas9 in large DMD animals have been conducted [54•, 58]. Data from these studies have provided compelling support for the clinical utility of gene editing in DMD patients. The deletion of multiple exons has also been demonstrated in DMD mice, a strategy which could be applied across a greater range of mutations [36, 43, 44, 45•, 47, 48, 63]. For additional information on this topic, we invite the reader to consult additional reviews [5, 68,69,70].
Exon Skipping and Reframing with a Single sgRNA
Simultaneous DSBs produced by sgRNA pairs increase the risk of unintended insertions, deletions, and other complex rearrangements [71, 72]. A single sgRNA targeting the splice acceptor or donor site of a frameshifted exon can restore the ORF while minimizing undesirable mutations. NHEJ indels will disrupt the splice site, causing omission of the frameshifted exon from the mature mRNA [73]. Another option is to target an sgRNA near a premature stop codon. Indels could remove the stop codon and reframe the transcript. Exon skipping and reframing using CRISPR/Cas9 has been accomplished in DMD mice [50, 52, 55, 56, 59, 74] and dogs [54•].
CRISPR-Mediated Restoration of a Full-Length Protein
To truly correct MD, restoration of the full-length protein is required. To achieve this, CRISPR/Cas9 editing typically requires HDR (Fig. 1b) or base editing (Fig. 1d) or, in unique circumstances, NHEJ. Most strategies are mutation specific and are therefore not broadly applicable, which may provide barriers for rapid regulatory approval.
Gene Correction Via HDR
Unfortunately, HDR is highly inefficient in muscle, and as such only three studies have applied CRISPR/Cas9-mediated HDR in vivo [45•, 49, 58]. Bengtsson et al. achieved an HDR rate of 0.18% following intramuscular injection of SpCas9 and a repair template into the mdx4c DMD mouse model [45•]. Interestingly, HDR occurred in a fraction of myogenic cells, but not satellite cells due to promoter choice. Lee et al. achieved a 0.8% HDR frequency in mdx mice, demonstrating HDR worked, albeit inefficiently, in muscle [49]. In the third study, a splice site mutation was corrected in GRMD dogs using HDR, which yielded dystrophin recoveries between 2 and 16% [58].
To date, HDR-based strategies have yet to restore clinically relevant levels of dystrophin, which precludes their consideration as an MD therapy. These studies highlight the limitations of HDR and that novel approaches to enhance its efficiency in muscle, such as by upregulation of HDR factors, are necessary.
NHEJ-Mediated Intronic Deletion to Restore Correct Splicing
The dy2J/dy2J MDC1A mouse model has a LAMA2 donor splice site mutation in intron 2 which results in aberrant exon 2 skipping, yielding a truncated and unstable protein [75]. Kemaladewi et al. used two sgRNAs to excise a genomic region that included the defective splice site and harnessed NHEJ repair to restore a functional donor site [32•]. Systemic administration of SaCas9 and sgRNAs into neonatal pups resulted in robust restoration of full-length laminin-α2 in muscle and sciatic nerve, improving motility and paralysis. Given the diversity of Cas9 enzymes with different PAM specificities, NHEJ splice site restoration may be applicable to a variety of pathogenic splice site mutations [27].
Therapeutic Base Editing to Correct Point Mutations
The correction of a specific mutation via the CRISPR/Cas9 system can also be accomplished with base editing. In the context of DMD, Ryu et al. utilized this method to correct a nonsense mutation and restore full-length dystrophin [53]. With an adenine base editor (ABE), the nonsense mutation was converted into glutamine via an A-to-G transition. Intramuscular injection of the Cas9-ABE led to dystrophin restoration in 17% of myofibers.
While base editing can currently correct only a limited group of mutations, this study represents a milestone in demonstrating its feasibility in treating DMD without truncating dystrophin.
Modulation of Gene Expression
Gene editing strategies are applicable to MD patients harboring deletion, insertion, duplication, or point mutations. However, those lacking large genomic regions or possessing complex mutations require alternative therapeutic strategies. The application of CRISPR/Cas9 to modulate gene expression holds great promise for MD. It is primarily mutation independent, allowing for broad applicability. Typically, dCas9 is used, bypassing concerns for undesirable on- and off-target mutations resulting from DSBs [76]. Both the mutated gene and disease modulators can be targeted for modulation and multiplex regulation is possible (Fig. 1e) [76].
Activation and Upregulation of Disease Modifiers
In mdx mice, expression of utrophin, a homologue of dystrophin, can partially compensate for dystrophin deficiency [77]. Building on pioneering work by Wojtal et al., who achieved utrophin upregulation in DMD patient derived myoblasts [33], Liao et al. used a Cas9 activation system in mdx mice to upregulate utrophin [51]. For the first time in vivo, Cas9-mediated upregulation of the disease modifier utrophin was shown to improve the dystrophic phenotype.
Laminin-α1 protein is structurally similar to laminin-α2 and can compensate for its loss in MDC1A [78]. However, LAMA1, which encodes laminin-α1, is only expressed during embryogenesis [11]. Perrin et al. demonstrated that laminin-α1 expression can be induced by intramuscular delivery of dCas9-VP160 in mdx mice [46]. Recently, Kemaladewi et al. showed that systemic administration of dCas9-2xVP64 in neonatal dy2J/dy2J pups prevented muscle fibrosis and hindlimb paralysis [65••]. Furthermore, treatment of older, symptomatic mice resulted in drastic clinical improvements and effectively reversed disease progression.
Results from these studies demonstrate that upregulation of disease modifiers is a valuable therapeutic approach which could improve and possibly partially reverse some MDs. Additional studies are required to provide more insight into the long-term persistence and efficacy of transcriptional modulation.
Cas9 Interference (Deletion and Transcriptional Repression)
To treat DM1, the formation of nuclear foci from expanded DMPK transcripts must be prevented. This can be achieved by deletion or transcriptional repression of the expanded CTG repeats (Fig. 1c).
The DMSXL DM1 mouse model carries the human DMPK gene with > 1000 CTG repeats [79]. Lo Scrudato et al. decreased the number of pathological RNA foci within myonuclei by deleting the CTG repeats by intramuscular injection of SaCas9 and two sgRNAs. This provided compelling evidence that genome editing to remove a large trinucleotide expansion was a feasible strategy for treating DM1 afflicted muscle.
The transgenic HSALR mouse carries a fragment of the human skeletal actin (HSA) gene with 250 CTG repeats in the 3′UTR [79]. Pinto et al. treated these mice systemically with dCas9 targeted to CTG repeats, blocking their inclusion in HAS transcripts. They observed a notable decrease in repeat transcription and an improved phenotype, validating the therapeutic potential of dCas9 repression [66].
In Vivo CRISPR Delivery Systems
CRISPR/Cas9 gene editing strategies are composed of two elements: the editing machinery and the delivery system. Efficient treatment of MDs will require robust expression of CRISPR/Cas9 components throughout skeletal and cardiac muscle. As such, we will now cover the two predominant in vivo vectors for MD genome editing.
AAVs
The ssDNA adeno-associated viruses (AAVs) are strong candidates for use in CRISPR/Cas9 MD therapies. While lentiviruses and adenoviruses have been used as delivery vehicles of therapeutic components in the past, AAVs are becoming the frontrunners for efficient and safe systemic delivery to muscle. Unfortunately, even AAVs cannot be re-administered without significant intervention due to the production of neutralizing antibodies after treatment. For detailed information on AAV-based therapies, we refer to these excellent reviews [80••, 81,82,83].
At present, high AAV titers are required for MD therapies; thus, efforts have been made to reduce the effective dose, with some success. Moretti et al. coated AAV9s in polyamidoamine dendrimers which significantly increased skeletal and cardiac muscle transduction in DMD pigs [57••]. These dendrimers are suspected to enhance cellular uptake through electrostatic interactions with cell surfaces. Increasing the sgRNA-to-Cas9 ratio by encoding multiple sgRNA copies on a second AAV has enhanced corrective DMD exon skipping. Unexpectedly, Hakim et al. observed disproportionate depletion of sgRNA encoding AAVs following systemic administration [61]. Min et al. studied this phenomena in detail and concluded that optimization of the AAV ratio for dual-AAV strategies was necessary to minimize the impact of sgRNA loss on editing efficiency [55]. Zhang et al. circumvented this problem by packaging sgRNAs in self-complementary AAV (scAAVs), enabling efficient editing without sgRNA depletion [59••].
It is possible that encoding multiple, identical sgRNA sequences as ssDNA can cause vector loss. Upon reaching the nucleus, ssDNA of the AAV stabilizes by synthesizing its complementary strand, a step the dsDNA scAAVs skip [84]. AAVs also package plus and minus strands equally, allowing direct annealing and skipping second strands synthesis. We speculate that repetitive sgRNA sequences may result in mis-annealing between plus and minus strands, yielding unstable DNA species which are degraded. While this hypothesis is untested, it could explain why scAAVs were not disproportionally lost. Assessing if specific depletion of sgRNA encoding vectors occurs when only a single sgRNA cassette is present would shed light on this issue.
Nanoparticles
Nonviral nanoparticles are an attractive alternative to AAVs for CRISPR/Cas9 delivery. They can deliver DNA, RNA, or proteins for transient expression, are extremely diverse, and allow for re-treatment, giving them enormous potential [85, 86].
Gold nanoparticles (GNPs) are easily taken up by cells and bind both DNA and protein [49]. CRISPR-Gold consists of a GNP conjugated with DNA to allow hybridization with the HDR repair template [49]. A Cas9/sgRNA ribonucleoprotein then associates with the DNA. Lee et al. used CRISPR-Gold to restore full-length dystrophin via HDR in mdx mice [49]. Intramuscular injection of CRISPR-Gold corrected the mutated dystrophin gene to the wild-type sequence with an HDR frequency of 0.8%, which increased to 5.4% when cardiotoxin was used to induce muscle stem cell proliferation.
NanoMEDIC is a novel nanoparticle formulation consisting of vesicles purified from genetically modified packaging cells [60]. They contain SpCas9 protein and transcribed sgRNAs which are also produced by the packaging cells. NanoMEDIC was utilized by Gee et al. to correct NOG-mdx mice through exon skipping [60]. Analysis following intramuscular injection revealed an exon skipping efficiency of 1.6%.
Nanoparticles are extremely promising AAV alternatives. Unfortunately, the necessary editing efficiencies to restore fully protective dystrophin levels (estimated at ~ 20% of normal) have not been realized with these systems [87, 88]. Additionally, all MD studies assessing nanoparticles have been limited to local administration. The inability to perform systemic delivery to muscle is a major barrier to moving past the preclinical stage.
Satellite Cell Genome Editing
Satellite cells are quiescent muscle stem cells which are activated by signals triggered by muscle growth, turnover, or damage. Activated satellite cells generate myoblasts via asymmetric division which fuse together to yield new myotubes or repair existing ones.
Satellite cells are a critical target of MD genome editing strategies. If they remain uncorrected, fusion of their progenitor cells may render treatments ineffective due to dilution and loss of corrected nuclei within the myofiber [80••, 89]. While this may not be problematic for several years due to the slow rate of muscle turnover, it will be over the course of a patient’s life [80••]. Additionally, as already revealed with the role of dystrophin in satellite cells, other MD genes may also be essential for stem cell function and will require correction in these cells [90]. Therefore, future CRISPR/Cas9 MD strategies will likely need to target satellite cells to achieve long-lasting clinical benefits.
While in vitro correction of dystrophin in mdx satellite cells has been successful, in vivo results have been ambiguous. Arnett et al. reported rare transduction of satellite cells by AAV8 but none with AAV6 or AAV9 [91]. In contrast, Tabebordbar et al. achieved modest genome editing (~ 35%) of the satellite cell population with AAV9, whereas both Nance et al. and Goldstein et al. observed maximal genome modification rates of ~ 60% [42, 64, 92]. Due to significant differences in study design, the reasons behind these conflicting results are difficult to ascertain. Treatment age or injection routes might be factors as Goldstein et al. injected the AAVs systemically instead of intramuscularly. Transduction levels with Cre systems were notably higher than with CRISPR/Cas9, likely due to the superior editing efficiency of Cre. Despite the myriad of differentiating factors between these four studies, the most recent data is quite strong and suggests that AAVs, particularly AAV9, are capable of transducing satellite cells better than previously thought. While promising for AAV-based MD therapies, further validation is necessary.
Long-Term Considerations for the Use of In Vivo CRISPR Gene Editing
As the MD genome editing field embarks into new, uncharted territory, understanding the long-term impacts and effects is a necessity. A primary concern is the longevity of phenotypic rescue. Further research will be needed to answer this, though it is likely that without satellite cell correction, life-long benefits from a single treatment will be limited. This question is particularly relevant for MDs, as the optimal therapeutic window is early childhood, prior to severe disease progression. Rapid muscle growth at this age may swiftly render non-satellite cell targeting CRISPR therapies ineffective.
Important consideration must be given to the host’s immune system, owing to the introduction of several antigens by CRISPR/Cas9 treatments: the restored therapeutic protein, Cas9, and AAV capsid in the case of viral delivery. Restoring a protein which was absent during elimination of self-reactive lymphocytes can trigger immune responses. Anti-dystrophin antibodies and immune-system rejections have been noted in DMD animals and patients given corrective therapies [93,94,95,96,97]. Pre-existing Cas9 immunity has also been extensively reported; thus, caution should be taken towards Cas9 immunogenicity since AAV episomes can remain for at least a decade in human muscle, enabling prolonged expression of CRISPR machinery [98]. Xu et al. however demonstrated that at 19 months post-treatment, mdx mice developed a humoral response to the AAVs, not Cas9 [63]. Perhaps Cas9 immune responses are only transient in nature. Nevertheless, transient expression by nanoparticles is ideal, but they are far from ready for clinical use. Thus, rigorous safety monitoring will be essential during clinical trials to mitigate immune reactions in MD patients. Checking for Cas9 neutralization by the immune system should also be conducted to ascertain therapy effectiveness.
Recently, Nelson et al. revealed severe underestimations of AAV integration following Cas9 DNA cleavage [62•]. AAVs can integrate into the AAVS1 site of mammalian genomes at low frequencies [99, 100]. However, Nelson and colleagues demonstrated significant integration at on-target DSBs generated by Cas9 [62•]. This finding suggested that AAVs as delivery vehicles for genome editing applications are less benign than previously thought, though the authors still observed substantial dystrophin recovery in their mdx mice with no noted toxicity [62•]. On the other hand, 19 months post-treatment, Xu et al. found that the CRISPR-Cas9-induced DSBs were mainly repaired by the precise ligation of the two cut sites [63]. Further investigation into the impact of Cas9 on AAV integration is imperative to understanding the consequences of genomic incorporation of CRISPR systems.
Conclusions and Future Directions for the MD Gene Editing Field
Recently, pioneering advancements in CRISPR/Cas9 utilization have opened numerous new therapeutic opportunities for MD. There is palpable optimism in the field, but new challenges lie ahead, and the future of MD gene editing will be contingent on surmounting them. The ambiguity surrounding AAV transduction of satellite cells in vivo needs to be resolved, so focus can be appropriately directed towards optimizing vector targeting if necessary. It is straightforward to see the need for satellite cells correction, but it must be experimentally established if extensive muscle turnover will negatively impact CRISPR strategies.
The poor editing efficiency of HDR in muscle is another problematic area. Efficient integration of exogenous DNA would open a myriad of new CRISPR applications, even beyond MD. Either canonical HDR must be improved or alternative mechanisms will need to be developed. The post-mitotic nature of myotubes and dormant satellite cells suggests that the latter may be most successful. Three promising novel approaches are homology-independent targeted integration (HITI), homology-mediated end joining (HMEJ), and prime editing. HITI leverages NHEJ, achieving absolute knock-in rates of 3.4% and 10% in murine heart and quadriceps respectively after systemic AAV9 administration [101]. Unfortunately, relative knock-in rates for whole muscles were not reported. HMEJ is speculated to proceed through single-strand annealing by employing a modified HDR DNA template which incorporates sgRNA sites for excision from its delivery vector [102]. AAV9 administration into the cortex of mice yielded a ~ 50% knock-in rate within the transduced neuron population [102]. The post-mitotic state of neurons suggests that such a knock-in rate may be achievable in muscle. Prime editing is a recent development which fuses Cas9 nickase to reverse transcriptase [103]. Using a 3′-extended pegRNA encoding the desired template, knock-in rates similar to HDR have been achieved in vitro [103]. However, validation in muscle, miniaturization of the system, and greater editing efficiencies are necessary for in vivo MD applications.
An alternative method to restore full-length proteins is through duplication removal. Duplications are the second most common DMD mutation, and an elegant, single sgRNA approach has been demonstrated to restore dystrophin [33, 34]. At this time, CRISPR/Cas9-mediated duplication removal has only been performed in DMD patient cells and must now be evaluated in vivo [33, 34].
The first generations of MD CRISPR therapies will likely utilize AAVs due to their proven track record in in vivo studies and clinical trials [17, 80••]. AAVs may be the best available option currently, but future therapies will likely require an alternate delivery system. It may come in the form of nanoparticles as they can deliver a variety of transiently expressed cargo and are high modifiable. Their potential to avoid triggering immune responses will be particularly important if satellite cells cannot be corrected [104, 105]. Life-long rescue of MD would be possible through nanoparticle re-administration to mitigate the effects of muscle turnover. But, efficient in vivo editing with CRISPR via nanoparticle delivery has not been achieved in muscle. As this has not been a problem with AAVs, focus must be directed towards novel nanoparticle formulations rather than optimization of CRISPR/Cas9 systems. Systemic muscle delivery by nanoparticles remains a significant barrier as revealed by the lack of published studies. Local intramuscular injections are not feasible due to the amount of muscle in the human body and the need to target muscles within the thoracic cavity [106]. Development of an efficient, muscle-specific nanoparticle which can be delivered through circulation is paramount for future research.
The challenges ahead are arduous, but with the appropriate focus and investment of resources, solutions will arise, bringing therapeutic genome editing ever closer to the clinic. While this review was heavily centered on DMD, the discussed approaches and techniques can and undoubtedly will be applied to treating the plethora of other MDs.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Dowling JJ, Gonorazky HD, Cohn RD, Campbell C. Treating pediatric neuromuscular disorders: the future is now. Am J Med Genet A. 2018;176(4):804–41. https://doi.org/10.1002/ajmg.a.38418.
Shieh PB. Muscular dystrophies and other genetic myopathies. Neurol Clin. 2013;31(4):1009–29. https://doi.org/10.1016/j.ncl.2013.04.004.
Crispi V, Matsakas A. Duchenne muscular dystrophy: genome editing gives new hope for treatment. Postgrad Med J. 2018;94(1111):296–304. https://doi.org/10.1136/postgradmedj-2017-135377.
Bladen CL, Salgado D, Monges S, Foncuberta ME, Kekou K, Kosma K, et al. The TREAT-NMD DMD global database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36(4):395–402. https://doi.org/10.1002/humu.22758.
Wong TWY, Cohn RD. Therapeutic applications of CRISPR/Cas for Duchenne muscular dystrophy. Curr Gene Ther. 2017;17(4):301–8. https://doi.org/10.2174/1566523217666171121165046.
Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004;94(8):1023–31. https://doi.org/10.1161/01.RES.0000126574.61061.25.
Gumerson JD, Michele DE. The dystrophin-glycoprotein complex in the prevention of muscle damage. J Biomed Biotechnol. 2011;2011:210797–13. https://doi.org/10.1155/2011/210797.
Koenig M, Beggs AH, Moyer M, Scherpf S, Heindrich K, Bettecken T, et al. The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion. Am J Hum Genet. 1989;45(4):498–506.
Liechti-Gallati S, Koenig M, Kunkel LM, Frey D, Boltshauser E, Schneider V, et al. Molecular deletion patterns in Duchenne and Becker type muscular dystrophy. Hum Genet. 1989;81(4):343–8. https://doi.org/10.1007/bf00283688.
Verhaart IEC, ‘t Hoen PAC, Roos M, Vroom E, Workshop P. Meeting on data sharing for Duchenne 21-22 March 2019 Amsterdam, the Netherlands. Neuromuscul Disord. 2019;29(10):800–10. https://doi.org/10.1016/j.nmd.2019.08.010.
Gawlik KI, Durbeej M. Skeletal muscle laminin and MDC1A: pathogenesis and treatment strategies. Skelet Muscle. 2011;1(1):9. https://doi.org/10.1186/2044-5040-1-9.
Geranmayeh F, Clement E, Feng LH, Sewry C, Pagan J, Mein R, et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. 2010;20(4):241–50. https://doi.org/10.1016/j.nmd.2010.02.001.
Aumailley M. The laminin family. Cell Adhes Migr. 2013;7(1):48–55. https://doi.org/10.4161/cam.22826.
LoRusso S, Weiner B, Arnold WD. Myotonic dystrophies: targeting therapies for multisystem disease. Neurotherapeutics. 2018;15(4):872–84. https://doi.org/10.1007/s13311-018-00679-z.
Barthelemy F, Wein N. Personalized gene and cell therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2018;28(10):803–24. https://doi.org/10.1016/j.nmd.2018.06.009.
Robinson-Hamm JN, Gersbach CA. Gene therapies that restore dystrophin expression for the treatment of Duchenne muscular dystrophy. Hum Genet. 2016;135(9):1029–40. https://doi.org/10.1007/s00439-016-1725-z.
Duan D, Systemic AAV. Micro-dystrophin gene therapy for Duchenne muscular dystrophy. Mol Ther. 2018;26(10):2337–56. https://doi.org/10.1016/j.ymthe.2018.07.011.
Rodrigues M, Yokota T. An overview of recent advances and clinical applications of exon skipping and splice modulation for muscular dystrophy and various genetic diseases. Methods Mol Biol. 18282018:31–55.
McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, et al. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10101):1489–98. https://doi.org/10.1016/S0140-6736(17)31611-2.
Conboy I, Murthy N, Etienne J, Robinson Z. Making gene editing a therapeutic reality. F1000Res. 2018;7. https://doi.org/10.12688/f1000research.16106.1.
Sahel DK, Mittal A, Chitkara D. CRISPR/Cas system for genome editing: progress and prospects as a therapeutic tool. J Pharmacol Exp Ther. 2019;370(3):725–35. https://doi.org/10.1124/jpet.119.257287.
Doudna JA. The promise and challenge of therapeutic genome editing. Nature. 2020;578(7794):229–36. https://doi.org/10.1038/s41586-020-1978-5.
Khan SH. Genome-editing technologies: concept, pros, and cons of various genome-editing techniques and bioethical concerns for clinical application. Mol Ther Nucleic Acids. 2019;16:326–34. https://doi.org/10.1016/j.omtn.2019.02.027.
Wang H, La Russa M, Qi LS. CRISPR/Cas9 in genome editing and beyond. Annu Rev Biochem. 2016;85:227–64. https://doi.org/10.1146/annurev-biochem-060815-014607As this reference discusses the HDR and NHEJ repair systems in-depth, which are the fundamental mechanisms for Cas9 correction of MDs.
Wiedenheft B, Sternberg SH, Doudna JA. RNA-guided genetic silencing systems in bacteria and archaea. Nature. 2012;482(7385):331–8. https://doi.org/10.1038/nature10886.
Shmakov S, Smargon A, Scott D, Cox D, Pyzocha N, Yan W, et al. Diversity and evolution of class 2 CRISPR-Cas systems. Nat Rev Microbiol. 2017;15(3):169–82. https://doi.org/10.1038/nrmicro.2016.184.
Cebrian-Serrano A, Davies B. CRISPR-Cas orthologues and variants: optimizing the repertoire, specificity and delivery of genome engineering tools. Mamm Genome. 2017;28(7–8):247–61. https://doi.org/10.1007/s00335-017-9697-4.
Pawelczak KS, Gavande NS, VanderVere-Carozza PS, Turchi JJ. Modulating DNA repair pathways to improve precision genome engineering. ACS Chem Biol. 2018;13(2):389–96. https://doi.org/10.1021/acschembio.7b00777.
Hsu MN, Chang YH, Truong VA, Lai PL, Nguyen TKN, Hu YC. CRISPR technologies for stem cell engineering and regenerative medicine. Biotechnol Adv. 2019;37(8):107447. https://doi.org/10.1016/j.biotechadv.2019.107447.
Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20(11):698–714. https://doi.org/10.1038/s41580-019-0152-0.
Pini V, Morgan JE, Muntoni F, O'Neill HC. Genome editing and muscle stem cells as a therapeutic tool for muscular dystrophies. Curr Stem Cell Rep. 2017;3(2):137–48. https://doi.org/10.1007/s40778-017-0076-6.
Kemaladewi DU, Maino E, Hyatt E, Hou H, Ding M, Place KM, et al. Correction of a splicing defect in a mouse model of congenital muscular dystrophy type 1A using a homology-directed-repair-independent mechanism. Nat Med. 2017;23(8):984–9. https://doi.org/10.1038/nm.4367They utilized a unique strategy to correct an intronic donor splice site mutation through NHEJ.
Wojtal D, Kemaladewi DU, Malam Z, Abdullah S, Wong TW, Hyatt E, et al. Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am J Hum Genet. 2016;98(1):90–101. https://doi.org/10.1016/j.ajhg.2015.11.012.
Lattanzi A, Duguez S, Moiani A, Izmiryan A, Barbon E, Martin S, et al. Correction of the exon 2 duplication in DMD myoblasts by a single CRISPR/Cas9 system. Mol Ther Nucleic Acids. 2017;7:11–9. https://doi.org/10.1016/j.omtn.2017.02.004.
Chen M, Qi LS. Repurposing CRISPR system for transcriptional activation. Adv Exp Med Biol. 2017;983:147–57.
Duchene BL, Cherif K, Iyombe-Engembe JP, Guyon A, Rousseau J, Ouellet DL, et al. CRISPR-induced deletion with SaCas9 restores dystrophin expression in dystrophic models in vitro and in vivo. Mol Ther. 2018;26(11):2604–16. https://doi.org/10.1016/j.ymthe.2018.08.010.
Qi LS, Larson MH, Gilbert LA, Doudna JA, Weissman JS, Arkin AP, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–83. https://doi.org/10.1016/j.cell.2013.02.022.
Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet. 2018;19(12):770–88. https://doi.org/10.1038/s41576-018-0059-1.
Molla KA, Yang Y. CRISPR/Cas-mediated base editing: technical considerations and practical applications. Trends Biotechnol. 2019;37(10):1121–42. https://doi.org/10.1016/j.tibtech.2019.03.008.
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, Sanchez-Ortiz E, et al. Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science. 2016;351(6271):400–3. https://doi.org/10.1126/science.aad5725.
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, Castellanos Rivera RM, et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science. 2016;351(6271):403–7. https://doi.org/10.1126/science.aad5143.
Tabebordbar M, Zhu K, Cheng JKW, Chew WL, Widrick JJ, Yan WX, et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science. 2016;351(6271):407–11. https://doi.org/10.1126/science.aad5177.
Iyombe-Engembe JP, Ouellet DL, Barbeau X, Rousseau J, Chapdelaine P, Lague P, et al. Efficient restoration of the dystrophin gene reading frame and protein structure in DMD myoblasts using the CinDel method. Mol Ther Nucleic Acids. 2016;5:e283. https://doi.org/10.1038/mtna.2015.58.
Xu L, Park KH, Zhao L, Xu J, El Refaey M, Gao Y, et al. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol Ther. 2016;24(3):564–9. https://doi.org/10.1038/mt.2015.192.
Bengtsson NE, Hall JK, Odom GL, Phelps MP, Andrus CR, Hawkins RD, et al. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat Commun. 2017;8:14454. https://doi.org/10.1038/ncomms14454Because in this reference they used various strategies for genome editing and directly compared their differing efficiencies. Amongst others, they compared NHEJ with HDR, and SpCas9 with SaCas9.
Perrin A, Rousseau J, Tremblay JP. Increased expression of laminin subunit alpha 1 chain by dCas9-VP160. Mol Ther Nucleic Acids. 2017;6:68–79. https://doi.org/10.1016/j.omtn.2016.11.004.
Young CS, Mokhonova E, Quinonez M, Pyle AD, Spencer MJ. Creation of a novel humanized dystrophic mouse model of Duchenne muscular dystrophy and application of a CRISPR/Cas9 gene editing therapy. J Neuromuscul Dis. 2017;4(2):139–45. https://doi.org/10.3233/JND-170218.
El Refaey M, Xu L, Gao Y, Canan BD, Adesanya TMA, Warner SC, et al. In vivo genome editing restores dystrophin expression and cardiac function in dystrophic mice. Circ Res. 2017;121(8):923–9. https://doi.org/10.1161/CIRCRESAHA.117.310996.
Lee K, Conboy M, Park HM, Jiang F, Kim HJ, Dewitt MA, et al. Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat Biomed Eng. 2017;1:889–901. https://doi.org/10.1038/s41551-017-0137-2.
Amoasii L, Long C, Li H, Mireault AA, Shelton JM, Sanchez-Ortiz E, et al. Single-cut genome editing restores dystrophin expression in a new mouse model of muscular dystrophy. Sci Transl Med. 2017;9(418). https://doi.org/10.1126/scitranslmed.aan8081.
Liao HK, Hatanaka F, Araoka T, Reddy P, Wu MZ, Sui Y, et al. In vivo target gene activation via CRISPR/Cas9-mediated trans-epigenetic modulation. Cell. 2017;171(7):1495–507 e15. https://doi.org/10.1016/j.cell.2017.10.025.
Koo T, Lu-Nguyen NB, Malerba A, Kim E, Kim D, Cappellari O, et al. Functional rescue of dystrophin deficiency in mice caused by frameshift mutations using Campylobacter jejuni Cas9. Mol Ther. 2018;26(6):1529–38. https://doi.org/10.1016/j.ymthe.2018.03.018.
Ryu SM, Koo T, Kim K, Lim K, Baek G, Kim ST, et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat Biotechnol. 2018;36(6):536–9. https://doi.org/10.1038/nbt.4148.
Amoasii L, Hildyard JCW, Li H, Sanchez-Ortiz E, Mireault A, Caballero D, et al. Gene editing restores dystrophin expression in a canine model of Duchenne muscular dystrophy. Science. 2018;362(6410):86–91. https://doi.org/10.1126/science.aau1549This is the first study applying the CRISPR/Cas9 system in a large DMD animal model (dogs), intramuscularly as well as systemically.
Min YL, Li H, Rodriguez-Caycedo C, Mireault AA, Huang J, Shelton JM, et al. CRISPR-Cas9 corrects Duchenne muscular dystrophy exon 44 deletion mutations in mice and human cells. Sci Adv. 2019;5(3):eaav4324. https://doi.org/10.1126/sciadv.aav4324.
Amoasii L, Li H, Zhang Y, Min YL, Sanchez-Ortiz E, Shelton JM, et al. In vivo non-invasive monitoring of dystrophin correction in a new Duchenne muscular dystrophy reporter mouse. Nat Commun. 2019;10(1):4537. https://doi.org/10.1038/s41467-019-12335-x.
Moretti A, Fonteyne L, Giesert F, Hoppmann P, Meier AB, Bozoglu T, et al. Somatic gene editing ameliorates skeletal and cardiac muscle failure in pig and human models of Duchenne muscular dystrophy. Nat Med. 2020;26(2):207–14. https://doi.org/10.1038/s41591-019-0738-2As this reference showed correction of DMD via systemic AAV Cas9 treatment in a DMD pig model via exon deletion and that AAV9 could be more efficiently transduced by muscle using an easy to apply dendrimer coating.
Mata Lopez S, Balog-Alvarez C, Vitha S, Bettis AK, Canessa EH, Kornegay JN, et al. Challenges associated with homologous directed repair using CRISPR-Cas9 and TALEN to edit the DMD genetic mutation in canine Duchenne muscular dystrophy. PLoS One. 2020;15(1):e0228072. https://doi.org/10.1371/journal.pone.0228072.
Zhang Y, Li H, Min YL, Sanchez-Ortiz E, Huang J, Mireault AA, et al. Enhanced CRISPR-Cas9 correction of Duchenne muscular dystrophy in mice by a self-complementary AAV delivery system. Sci Adv. 2020;6(8):eaay6812. https://doi.org/10.1126/sciadv.aay6812For demonstrating that scAAVs are capable of negatingspecific loss of sgRNA AAVs, solving a problem which would necessitate significantly higher doses for dual AAV MD approaches with Cas9 (which many will need to be).
Gee P, Lung MSY, Okuzaki Y, Sasakawa N, Iguchi T, Makita Y, et al. Extracellular nanovesicles for packaging of CRISPR-Cas9 protein and sgRNA to induce therapeutic exon skipping. Nat Commun. 2020;11(1):1334. https://doi.org/10.1038/s41467-020-14957-y.
Hakim CH, Wasala NB, Nelson CE, Wasala LP, Yue Y, Louderman JA, et al. AAV CRISPR editing rescues cardiac and muscle function for 18 months in dystrophic mice. JCI Insight. 2018;3(23). https://doi.org/10.1172/jci.insight.124297.
Nelson CE, Wu Y, Gemberling MP, Oliver ML, Waller MA, Bohning JD, et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat Med. 2019;25(3):427–32. https://doi.org/10.1038/s41591-019-0344-3As this reference sheds light on the potentially serious concern of AAV integration at Cas9 DSBs, a phenomena which was not known or given much attention beforehand.
Xu L, Lau YS, Gao Y, Li H, Han R. Life-long AAV-mediated CRISPR genome editing in dystrophic heart improves cardiomyopathy without causing serious lesions in mdx mice. Mol Ther. 2019;27(8):1407–14. https://doi.org/10.1016/j.ymthe.2019.05.001.
Nance ME, Shi R, Hakim CH, Wasala NB, Yue Y, Pan X, et al. AAV9 edits muscle stem cells in normal and dystrophic adult mice. Mol Ther. 2019;27(9):1568–85. https://doi.org/10.1016/j.ymthe.2019.06.012.
Kemaladewi DU, Bassi PS, Erwood S, Al-Basha D, Gawlik KI, Lindsay K, et al. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature. 2019;572(7767):125–30. https://doi.org/10.1038/s41586-019-1430-xAs this is the first study systemically overexpressing a disease modifier to improve and effectively reverse disease progression in an MD animal.
Pinto BS, Saxena T, Oliveira R, Mendez-Gomez HR, Cleary JD, Denes LT, et al. Impeding transcription of expanded microsatellite repeats by deactivated Cas9. Mol Cell. 2017;68(3):479–90 e5. https://doi.org/10.1016/j.molcel.2017.09.033.
Lo Scrudato M, Poulard K, Sourd C, Tome S, Klein AF, Corre G, et al. Genome editing of expanded CTG repeats within the human DMPK gene reduces nuclear RNA foci in the muscle of DM1 mice. Mol Ther. 2019;27(8):1372–88. https://doi.org/10.1016/j.ymthe.2019.05.021.
Kemaladewi DU, Cohn RD. Exon snipping in Duchenne muscular dystrophy. Trends Mol Med. 2016;22(3):187–9. https://doi.org/10.1016/j.molmed.2016.01.007.
Lim KRQ, Yoon C, Yokota T. Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. J Pers Med. 2018;8(4). https://doi.org/10.3390/jpm8040038.
Min YL, Bassel-Duby R, Olson EN. CRISPR correction of Duchenne muscular dystrophy. Annu Rev Med. 2019;70:239–55. https://doi.org/10.1146/annurev-med-081117-010451.
Boroviak K, Fu B, Yang F, Doe B, Bradley A. Revealing hidden complexities of genomic rearrangements generated with Cas9. Sci Rep. 2017;7(1):12867. https://doi.org/10.1038/s41598-017-12740-6.
Park CY, Kim DH, Son JS, Sung JJ, Lee J, Bae S, et al. Functional correction of large factor VIII gene chromosomal inversions in hemophilia A patient-derived iPSCs using CRISPR-Cas9. Cell Stem Cell. 2015;17(2):213–20. https://doi.org/10.1016/j.stem.2015.07.001.
Ifuku M, Iwabuchi KA, Tanaka M, Lung MSY, Hotta A. Restoration of dystrophin protein expression by exon skipping utilizing CRISPR-Cas9 in myoblasts derived from DMD patient iPS cells. Methods Mol Biol. 1828;2018:191–217. https://doi.org/10.1007/978-1-4939-8651-4_12.
Zhang Y, Long C, Li H, McAnally JR, Baskin KK, Shelton JM, et al. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci Adv. 2017;3(4):e1602814. https://doi.org/10.1126/sciadv.1602814.
Saunier M, Bonnemann CG, Durbeej M, Allamand V, Consortium CMDAM. 212th ENMC international workshop: animal models of congenital muscular dystrophies, Naarden, the Netherlands, 29-31 May 2015. Neuromuscul Disord. 2016;26(3):252–9. https://doi.org/10.1016/j.nmd.2016.02.002.
Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, et al. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res. 2013;23(10):1163–71. https://doi.org/10.1038/cr.2013.122.
Tinsley JM, Fairclough RJ, Storer R, Wilkes FJ, Potter AC, Squire SE, et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One. 2011;6(5):e19189. https://doi.org/10.1371/journal.pone.0019189.
Gawlik K, Miyagoe-Suzuki Y, Ekblom P, Takeda S, Durbeej M. Laminin alpha1 chain reduces muscular dystrophy in laminin alpha2 chain deficient mice. Hum Mol Genet. 2004;13(16):1775–84. https://doi.org/10.1093/hmg/ddh190.
Gomes-Pereira M, Cooper TA, Gourdon G. Myotonic dystrophy mouse models: towards rational therapy development. Trends Mol Med. 2011;17(9):506–17. https://doi.org/10.1016/j.molmed.2011.05.004.
Crudele JM, Chamberlain JS. AAV-based gene therapies for the muscular dystrophies. Hum Mol Genet. 2019;28(R1):R102–R7. https://doi.org/10.1093/hmg/ddz128As this reference is an excellent, detail-oriented review for AAV-based gene therapies for MDs.
Wang D, Zhang F, Gao G. CRISPR-based therapeutic genome editing: strategies and in vivo delivery by AAV vectors. Cell. 2020;181(1):136–50. https://doi.org/10.1016/j.cell.2020.03.023.
Buning H, Schmidt M. Adeno-associated vector toxicity-to be or not to be? Mol Ther. 2015;23(11):1673–5. https://doi.org/10.1038/mt.2015.182.
Wang D, Zhong L, Nahid MA, Gao G. The potential of adeno-associated viral vectors for gene delivery to muscle tissue. Expert Opin Drug Deliv. 2014;11(3):345–64. https://doi.org/10.1517/17425247.2014.871258.
McCarty DM. Self-complementary AAV vectors; advances and applications. Mol Ther. 2008;16(10):1648–56. https://doi.org/10.1038/mt.2008.171.
Nance ME, Hakim CH, Yang NN, Duan D. Nanotherapy for Duchenne muscular dystrophy. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2018;10(2). https://doi.org/10.1002/wnan.1472.
Glass Z, Li Y, Xu Q. Nanoparticles for CRISPR-Cas9 delivery. Nat Biomed Eng. 2017;1(11):854–5. https://doi.org/10.1038/s41551-017-0158-x.
Wells DJ. What is the level of dystrophin expression required for effective therapy of Duchenne muscular dystrophy? J Muscle Res Cell Motil. 2019;40(2):141–50. https://doi.org/10.1007/s10974-019-09535-9.
Hoffman E. Variable dystrophin content within myofibers, between myofibers, and between regions of muscles: determining the level of dystrophin that makes a clinical impact in BMD and manifesting carriers of DMD.
Pawlikowski B, Pulliam C, Betta ND, Kardon G, Olwin BB. Pervasive satellite cell contribution to uninjured adult muscle fibers. Skelet Muscle. 2015;5:42. https://doi.org/10.1186/s13395-015-0067-1.
Dumont NA, Wang YX, von Maltzahn J, Pasut A, Bentzinger CF, Brun CE, et al. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat Med. 2015;21(12):1455–63. https://doi.org/10.1038/nm.3990.
Arnett AL, Konieczny P, Ramos JN, Hall J, Odom G, Yablonka-Reuveni Z, et al. Adeno-associated viral (AAV) vectors do not efficiently target muscle satellite cells. Mol Ther Methods Clin Dev. 2014;1:14038. https://doi.org/10.1038/mtm.2014.38.
Goldstein JM, Tabebordbar M, Zhu K, Wang LD, Messemer KA, Peacker B, et al. In situ modification of tissue stem and progenitor cell genomes. Cell Rep. 2019;27(4):1254–64 e7. https://doi.org/10.1016/j.celrep.2019.03.105.
Braun S, Thioudellet C, Rodriguez P, Ali-Hadji D, Perraud F, Accart N, et al. Immune rejection of human dystrophin following intramuscular injections of naked DNA in mdx mice. Gene Ther. 2000;7(17):1447–57. https://doi.org/10.1038/sj.gt.3301261.
Ferrer A, Wells KE, Wells DJ. Immune responses to dystropin: implications for gene therapy of Duchenne muscular dystrophy. Gene Ther. 2000;7(17):1439–46. https://doi.org/10.1038/sj.gt.3301259.
Lochmuller H, Petrof BJ, Pari G, Larochelle N, Dodelet V, Wang Q, et al. Transient immunosuppression by FK506 permits a sustained high-level dystrophin expression after adenovirus-mediated dystrophin minigene transfer to skeletal muscles of adult dystrophic (mdx) mice. Gene Ther. 1996;3(8):706–16.
Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363(15):1429–37. https://doi.org/10.1056/NEJMoa1000228.
Bowles DE, McPhee SW, Li C, Gray SJ, Samulski JJ, Camp AS, et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol Ther. 2012;20(2):443–55. https://doi.org/10.1038/mt.2011.237.
Buchlis G, Podsakoff GM, Radu A, Hawk SM, Flake AW, Mingozzi F, et al. Factor IX expression in skeletal muscle of a severe hemophilia B patient 10 years after AAV-mediated gene transfer. Blood. 2012;119(13):3038–41. https://doi.org/10.1182/blood-2011-09-382317.
Hamilton H, Gomos J, Berns KI, Falck-Pedersen E. Adeno-associated virus site-specific integration and AAVS1 disruption. J Virol. 2004;78(15):7874–82. https://doi.org/10.1128/JVI.78.15.7874-7882.2004.
Dutheil N, Yoon-Robarts M, Ward P, Henckaerts E, Skrabanek L, Berns KI, et al. Characterization of the mouse adeno-associated virus AAVS1 ortholog. J Virol. 2004;78(16):8917–21. https://doi.org/10.1128/JVI.78.16.8917-8921.2004.
Suzuki K, Tsunekawa Y, Hernandez-Benitez R, Wu J, Zhu J, Kim EJ, et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540(7631):144–9. https://doi.org/10.1038/nature20565.
Yao X, Wang X, Hu X, Liu Z, Liu J, Zhou H, et al. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27(6):801–14. https://doi.org/10.1038/cr.2017.76.
Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576(7785):149–57. https://doi.org/10.1038/s41586-019-1711-4.
Boraschi D, Italiani P, Palomba R, Decuzzi P, Duschl A, Fadeel B, et al. Nanoparticles and innate immunity: new perspectives on host defence. Semin Immunol. 2017;34:33–51. https://doi.org/10.1016/j.smim.2017.08.013.
Boraschi D, Costantino L, Italiani P. Interaction of nanoparticles with immunocompetent cells: nanosafety considerations. Nanomedicine (Lond). 2012;7(1):121–31. https://doi.org/10.2217/nnm.11.169.
Duan D. Duchenne muscular dystrophy gene therapy: lost in translation? Res Rep Biol. 2011;2011(2):31–42. https://doi.org/10.2147/RRB.S13463.
Acknowledgments
The Cohn lab members are gratefully acknowledged for their input in this study. We would specifically like to thank Teija Bily, Eleonora Maino, and Dwi Kemaladewi for their valuable feedback on this manuscript.
Funding
This work was funded by the Canadian Institutes of Health Research (R.D.C. and E.A.I.), the McArthur family (R.D.C.), Jesse’s Journey (R.D.C.), Duchenne UK (R.D.C.), Duchenne Research Fund (R.D.C.), and the Michael Hyatt Foundation (R.D.C.).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Monika Kustermann, Matthew J. Rok, Ronald D. Cohn, and Evgueni A. Ivakine declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of Topical Collection on Genome Editing
Rights and permissions
About this article

Cite this article
Kustermann, M., Rok, M.J., Cohn, R.D. et al. In Vivo Genome Engineering for the Treatment of Muscular Dystrophies. Curr Stem Cell Rep 6, 52–66 (2020). https://doi.org/10.1007/s40778-020-00173-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40778-020-00173-3