

Abstract
Despite the efficacy of Hedgehog pathway inhibitors in the treatment of basal cell carcinoma (BCC)1, residual disease persists in some patients and may contribute to relapse when treatment is discontinued2. Here, to study the effect of the Smoothened inhibitor vismodegib on tumour clearance, we have used a Ptch1–Trp53 mouse model of BCC3 and found that mice treated with vismodegib harbour quiescent residual tumours that regrow upon cessation of treatment. Profiling experiments revealed that residual BCCs initiate a transcriptional program that closely resembles that of stem cells of the interfollicular epidermis and isthmus, whereas untreated BCCs are more similar to the hair follicle bulge. This cell identity switch was enabled by a mostly permissive chromatin state accompanied by rapid Wnt pathway activation and reprogramming of super enhancers to drive activation of key transcription factors involved in cellular identity. Accordingly, treatment of BCC with both vismodegib and a Wnt pathway inhibitor reduced the residual tumour burden and enhanced differentiation. Our study identifies a resistance mechanism in which tumour cells evade treatment by adopting an alternative identity that does not rely on the original oncogenic driver for survival.
Similar content being viewed by others


Main
Basal cell carcinoma is the most frequently occurring human cancer and results from aberrant activation of the Hedgehog (Hh) pathway4. Erivedge (vismodegib), a potent inhibitor of Hh signalling that acts at the level of Smoothened (SMO)5, has been approved for the treatment of locally advanced and metastatic BCC1,6,7, and is also effective for operable BCC2. However, the low rate of histological clearance observed raised concerns over whether residual BCCs can regrow once treatment is discontinued.
To determine how residual BCCs survive vismodegib treatment, we used a mouse model of BCC driven by inactivation of Ptch1 and Trp53 in skin basal cells3. Histological examination of skin from 8-week-old K14CreER;Ptch1fl/fl;Trp53fl/fl mice revealed superficial BCC, similar to the human condition (Fig. 1a, b). These tumours expressed high levels of the Hh target gene Gli1 and the BCC marker SOX98 (Fig. 1f, g). Ki67 staining revealed that roughly 50% of the tumour nests contained actively proliferating cells (Fig. 1h).

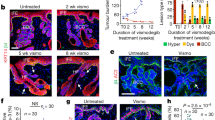
a–d, Representative images of haematoxylin and eosin (H&E)-stained skin sections showing response of mouse (n = 6 per group) and human BCC (n = 24, cohort 1 of ref. 2) to vismodegib (vismo) treatment. a, Skin section from an 8-week-old BCC mouse. b, Skin biopsy from a patient with BCC (pre-Rx, pre-treatment). c, Residual disease in a BCC mouse. White dashed line denotes boundary between epidermis and dermis. d, Residual disease (outlined in red) in the patient shown in b. e, Histology scores (mean ± s.d.; circles show individual scores) of skin samples from BCC mice treated with vehicle (black) or vismodegib (grey; n = 4 per group). f–k, Representative images of Gli1, SOX9 and Ki67 expression in mouse BCC and residual disease (n = 4 per condition). f, The Hh pathway is active in mouse BCC. g, Mouse BCCs stain positive for SOX9. h, Half of the tumours stain positive for Ki67. i, The Hh pathway is blocked in residual BCCs during vismodegib treatment. j, Residual disease identified by Sox9. k, BCCs stop proliferating within 1 day of vismodegib treatment. All experiments were replicated at least twice. Scale bars: b, d, 90 μm; other panels, 100 μm. DAPI nuclear stain is in blue (g, h, j, k); n represents the number of either mice or patients.
To test the effect of Hh pathway inhibition on established BCC, we treated 8 week-old mice with vismodegib for 28 days. Skin from treated mice frequently contained small residual tumours similar to the finger-like basaloid projections observed in patients2 (Fig. 1c, d). The absence of Gli1 expression in tumour nests indicated that vismodegib effectively blocked Hh signalling (Fig. 1i). Residual BCCs still expressed SOX9 (Fig. 1j) and stopped proliferating completely within 1 day of treatment initiation (Fig. 1k). Apoptosis was absent in untreated disease, but was observed in a small number of tumour cells early during treatment (Extended Data Fig. 1a–d). Residual tumours did not differentiate into normal epidermis (Extended Data Fig. 2) and remained present even after 56 days of drug treatment (Fig. 1e). Thus, residual BCCs in mice are quiescent and do not require active Hh signalling for their survival.
We next investigated whether residual lesions that survived drug treatment could reinitiate growth. Skin sections from BCC mice collected 0, 3, 6, or 12 days after the last dose revealed that superficial BCC returned quickly (Fig. 2a), with Gli1+ lesions resembling either residual tumours or newly formed tumour buds (Fig. 2b–d). Elevated Gli1 expression persisted as mice remained off drug for longer periods of time (Extended Data Fig. 1e, f). To distinguish growth of residual disease from de novo tumour formation due to inherent leakiness9 of the K14CreER, we labelled tumours with BrdU before treatment to mark residual disease (Fig. 2e, Extended Data Fig. 1g, h). Six days after cessation of vismodegib, labelled tumour nests stained positive for Ki67 and were diluting the incorporated BrdU, confirming that quiescent residual BCCs reinitiated tumour growth when drug treatment was discontinued (Fig. 2f, g).

a, Histology scores (mean ± s.d.) from BCC mice treated with vehicle or vismodegib for 28 days determined after the indicated number of days of recovery (rec) (n ≥ 3 per group). b–d, Representative images of Gli1 ISH on skin from BCC mice shown in a. b, Hh pathway is blocked in residual BCC immediately after treatment. c, d, A residual BCC (c, outlined) and a newly formed tumour bud (d, outlined), both with an active Hh pathway 3 days after treatment. e–g, Lineage tracing of residual BCCs and their subsequent growth after treatment. Representative images of residual BCCs stained for BrdU (green), Ki67 (red) and DAPI nuclear stain (blue) are shown (n = 4 per time point). e, BrdU labelling strategy of residual BCCs. d0, day 0 of recovery. f, BrdU+ residual tumour nests (outlined) are quiescent immediately after treatment. g, Residual BCCs have resumed growth 6 days after treatment. All experiments were replicated at least twice. Scale bars are 100 μm; n represents the number of mice.
To uncover the mechanisms of BCC persistence, we performed RNA sequencing (RNA-seq) and compared the transcriptional profiles of untreated Ki67+ and Ki67− BCCs to those of residual tumours. Independent of proliferation status (Extended Data Fig. 3c), vismodegib affected the expression of known Hh targets (Fig. 3a) and a large number of other genes relative to untreated tumours (Extended Data Fig. 3a, b). Comparison with published skin compartment signatures (Extended Data Table 1) revealed that genes enriched in hair follicle bulge stem cells were downregulated, whereas genes enriched in basal cells from the interfollicular epidermis (IFE) or isthmus (ISTH) showed upregulation after treatment (Fig. 3b, Extended Data Fig. 3d–f). There was no significant overlap with hair germ, dermal papillae or outer root sheath (ORS) signatures (Fig. 3b, Extended Data Table 1). To confirm the shift from a hair follicle bulge to a mixed IFE/ISTH identity, we performed in situ hybridization (ISH) using key markers of these skin compartments. The transcription factor Lhx2, which has a key role in hair follicle bulge stem cells10 (Extended Data Fig. 3g), was abundant in untreated BCCs, but strongly depleted in residual disease (Fig. 3c, f, Extended Data Fig. 3l). On the other hand, IL-33 and Defb6, which are expressed in the normal IFE and ISTH, respectively (Extended Data Fig. 3h, i), were absent in established BCCs (Fig. 3d, Extended Data Fig. 3j), but induced in residual disease (Fig. 3g, Extended Data Fig. 3k), as was the ISTH marker MTS2411 (Extended Data Fig. 3l). Similar results for LHX2 and IL-33 were obtained in a lineage-tracing experiment with a Cre-inducible TdTomato reporter (Extended Data Fig. 4a–d), confirming that the cell identity shift occurs in residual tumour cells. Notably, the absence of IL-33 staining in untreated tumours and gradual appearance of this nuclear factor12 in residual disease (Extended Data Fig. 5) suggests that vismodegib induces a cell identity switch rather than selecting for pre-existing ISTH/IFE-like cells. The shift in cell identity was reversible upon cessation of drug treatment, as the proportion of BrdU+ residual tumour cells expressing LHX2 increased progressively in our BrdU labelling experiment (Extended Data Fig. 4e–i). Furthermore, GATA6 and KLF5 are normally restricted in skin to the ISTH and IFE, respectively13,14. Both transcription factors were undetectable in untreated BCCs, but were co-expressed in residual tumour cells (Fig. 3e, h), suggesting that an induced state of ‘lineage infidelity’14 may contribute to the development of residual disease.

a, Heat-map of Hh target gene expression in untreated and residual BCCs (n = 5 per group; data are represented as mean-centred normalized reads per kilobase of transcript per million mapped reads (nRPKM)). b, Quantitative set analysis for gene expression (QuSAGE) of indicated signatures in residual versus Ki67+ untreated BCC (n = 5 per group). Coloured violins depict differential expression of entire gene set; grey dots represent the log2[fold change] in expression of individual genes. BU, hair follicle bulge. c, d, f, g, Representative images of Lhx2 (hair follicle bulge) and Il33 (IFE) expression in untreated and residual BCCs (n = 4 per condition). c, Untreated BCC with high levels of Lhx2 mRNA. d, Untreated BCC (outlined) lacking IL-33 staining. Red arrowheads indicate nuclear localization of IL-33 in the epidermis. f, Residual BCCs downregulate Lhx2. g, Residual BCCs express IL-33 (red arrowheads). e, h, Representative images of KLF5 (IFE) and GATA6 (ISTH) antibody staining in untreated and residual BCC (n = 4 per condition). Untreated BCCs (e) lack expression of these lineage-specific transcription factors, whereas residual tumour cells (h, outlined) co-express KLF5 (green) and GATA6 (red). i, Regulatory potential exerted by chromatin regions (opening, green; closing, blue; unchanged, grey) on indicated gene sets (n = 3 per group). Box plots show median, two hinges (25th and 75th percentile), two whiskers (1.5 × inter-quartile range (IQR)), and all outlying points individually. Orange bars indicate the regulatory potential when using random genes. j, Graph depicting the association between differentially accessible enhancers with indicated transcription factor binding motifs and differentially expressed genes. A one-tailed Kolmogorov–Smirnov test was used to determine significance (n = 3 per group). k, Chromatin traces showing mean ATAC peaks at the Lhx2 and Klf5 loci. U, untreated; V, vismodegib-treated. l, Expression of Lhx2 and Klf5 in FACS-sorted BCC cells (n = 3 per group; data represented as mean ± s.d. log2 normalized read counts per million (CPM)). All experiments were replicated at least twice. Scale bars, 100 μm; n represents the number of mice.
To better define the mechanisms that drive this identity switch, we performed assay for transposase-accessible chromatin with high-throughput sequencing (ATAC–seq) coupled with RNA-seq on tumour cells isolated by fluorescence-activated cell sorting (FACS) before and after either 6 or 14 days of treatment (Extended Data Fig. 6a–f, i). Vismodegib had a relatively small effect on chromatin accessibility, as the majority of the open chromatin regions were shared between treatment groups (Extended Data Fig. 6g). Assessment of the regulatory potential15 exerted by differentially accessible chromatin on skin compartment signatures did not show enrichment for either opening or closing regions at these loci (Fig. 3i). Instead, these genes were more closely associated with constitutively open chromatin, indicating that in BCC, lineage-specific programs harbour a globally open chromatin structure that is poised for activation, similar to the small intestine16.
We next assessed whether chromatin accessibility correlated with general changes in gene expression. Closing regions were associated with vismodegib-downregulated genes, whereas opening chromatin was associated with upregulated genes (Extended Data Fig. 6j, k). Closing regions were enriched for GLI, the forkhead box (FOX) family, and LHX binding motifs (Extended Data Fig. 7a), consistent with inhibition of the Hh pathway and the BCC transcriptome resembling the hair follicle bulge compartment. Regions that opened during vismodegib treatment were enriched for binding motifs corresponding to SOX, NFAT, KLF and GATA transcription factors (Extended Data Fig. 7b), consistent with treated BCCs shifting towards an IFE and ISTH identity. Binding and expression target analysis (BETA) confirmed that peaks containing GLI, LHX2, SOX and KLF motifs were strongly associated with changes in gene expression (Fig. 3j, Extended Data Fig. 7c). Notably, TCF binding motifs were enriched in opening chromatin at the 6-day time point (Extended Data Fig. 7b), including the loci of the ISTH- and IFE-specific transcription factors Gata6 and Ahr13 (Extended Data Fig. 6h), suggesting that Wnt has an early role in mediating the identity switch.
Control of cell identity is thought to be regulated through binding and remodelling of chromatin regions termed super-enhancers, which control the expression of Lhx2 and Klf5 in hair follicle stem cells17. Strikingly, vismodegib-sensitive ATAC peaks were found in super-enhancers at both loci (Fig. 3k). The Lhx2 super-enhancer contains a GLI binding site that closed upon vismodegib treatment, whereas the Klf5 super-enhancer harbours two TCF binding sites that became more accessible. Accordingly, the expression of Lhx2 and Klf5 was modulated by vismodegib treatment (Fig. 3l). Collectively, our data support an escape mechanism of drug-induced cell identity switching in BCC that features a broadly open chromatin structure that remains sensitive to the activity of key transcription factors.
We next set out to identify environmental cues that drive cell identity switching in BCC and, on the basis of the above results, focused on Wnt. Consistent with published reports showing that Wnt is required during BCC formation18,19, initiating tumours expressed the universal Wnt target Axin220 (Fig. 4a). Unexpectedly, Axin2 was suppressed in established tumours, but reactivated in residual disease (Fig. 4a). Levels of Axin2 expression were inversely correlated with levels of Wif1, a potent secreted inhibitor of Wnt signalling21 (Fig. 4a, b), and were induced as early as 2 days after the initiation of treatment (Fig. 4b), well before significant tumour shrinkage occurred (Extended Data Fig. 8a, b). Expression of Wnt4 and Wnt10a, which have been implicated in the maintenance of normal basal epidermis22, followed the same pattern (Fig. 4b). Human tumours showed a similar pattern; AXIN2 mRNA was detectable in BCCs only after vismodegib treatment (Extended Data Fig. 8c–h). In addition to Lhx2, we also observed a return to pre-treatment levels of Axin2 and Wif1 in tumours that had been released from drug for 12 days (Extended Data Fig. 8i), again highlighting the plasticity of residual tumours.

a, Representative images of Axin2 and Wif1 ISH on mouse skin during initiating (3-week-old), established (8-week-old) and residual stages of BCC (n = 4 per stage). b, Relative expression levels (mean ± s.e.m.) of Axin2, Wif1, Wnt4, and Wnt10a in untreated BCCs and tumours treated for 2 or 22 days (n = 3 per group). c, Representative images of H&E-stained skin sections showing the amount of residual BCC after indicated treatments (n = 7 per group). d, Average residual tumour counts per length of skin in BCC mice from c. A two-tailed unpaired t-test was used to determine significance between groups (*P = 0.0328). All experiments were performed at least twice. Scale bars, 100 μm; n represents the number of mice.
We next tested whether treatment of BCC with a combination of vismodegib and a Wnt pathway inhibitor would have an effect on residual disease. We used a function-blocking anti-LRP6 antibody23 (Extended Data Fig. 9a–c), as our RNA-seq analysis revealed that LRP6 was the predominant Wnt co-receptor in BCC. Treatment with vismodegib and anti-LRP6 led to a 33% decrease in the number of residual tumour nests compared to vismodegib monotherapy (Fig. 4c, d). Notably, the residual tumour burden correlated with the magnitude of Wnt pathway inhibition, as mice with fewer lesions experienced a greater decrease in Axin2 expression (Extended Data Fig. 9d). In addition, Oil Red O staining was enhanced within residual tumours after combination treatment (Extended Data Fig. 9e–h), consistent with attenuation of Wnt signalling being required for sebocyte differentiation24.
Finally, we investigated whether combined inhibition of Hh and Wnt signalling altered the regrowth of residual BCCs after treatment. Mice were dosed with BrdU to label tumour nests and were then treated for 28 days with either vismodegib alone or vismodegib and anti-LRP6. After discontinuation of treatment, tumour regrowth was delayed (but not abolished) in mice that were treated with combination therapy relative to those treated with vismodegib alone (Extended Data Fig. 9i–q), probably owing to incomplete Wnt inhibition (Extended Data Fig. 9d).
Together, our results show that despite the strong anti-tumour activity of vismodegib in BCC, residual tumour cells persist. Tumours display robust Hh pathway inhibition during treatment, indicating that the drug continues to block signalling. As such, residual tumour cells have not acquired drug resistance through de novo mutations, but have adopted an identity that no longer relies on Hh signalling. When treatment is discontinued, tumour cells reactivate the Hh pathway and resume growth. Although BCC patients experience clinical benefit from Hedgehog pathway inhibitors after a drug holiday2, this approach may lead to the development of de novo resistance if additional mutations in the Hh pathway are acquired25. It is therefore important to achieve complete elimination of all residual tumour cells. The Wnt pathway seems to be critical for cell identity switching in BCC through reprograming of super-enhancers that drive the expression of key transcription factors. However, tolerability of complete Wnt pathway inhibition remains a challenge26. The ability of residual tumour cells to adopt a state that allows them to survive while remaining fully quiescent may represent a more widespread mechanism of resistance to targeted therapies27. Strategies that block this process may provide an opportunity to increase the rate of complete responses.
Methods
BCC mice and dosing
All mouse experiments were performed according to the animal use guidelines of Genentech, a Member of the Roche Group, conforming to California state legal and ethical practices. BCC mice were generated using the following alleles28,29,30: K14CreER, Ptch1fl/fl (a generous gift from R. Toftgard and S. Teglund), and p53fl/fl. BCC mice were singly housed and monitored for signs of advanced tumour burden, such as a scruffy coat and ear thickening. Tumour formation occurred in the absence of tamoxifen, probably owing to inherent leakiness of the CreER recombinase9. Vismodegib was formulated as a suspension in 0.5% methyl-cellulose, 0.2% tween-80 (MCT). Minimal efficacious dosing of vismodegib by oral gavage was determined to be 75 mg/kg bodyweight twice a day, and this dosing schedule was used for all experiments. Anti-LRP6 bi-specific antibody23 was given once daily at 30 mg/kg bodyweight by intraperitoneal (IP) injection. For label-retaining experiments, mice were given BrdU injections (100 μl of 10mg/ml) for three consecutive days before the start of treatment to label all dividing cells in the skin. Subsequent treatment with vismodegib stopped proliferation and prevented the dilution of incorporated BrdU in tumour nests. Lineage tracing with the lox-stop-lox TdTomato reporter allele (Jackson strain code: 007914) was initiated in mice at weanling age with three consecutive daily doses of tamoxifen (100 μl of 20 mg/ml in sunflower oil).
BCC histology score
A pathologist determined the extent of BCC tumour burden using the following scoring system: 0, minimal basaloid nests involve <10% of linear extent of skin; 1, isolated basaloid nests are generally confined to superficial half of dermis (do not extend to base of hair bulb); 2, basaloid nests crowd superficial dermis and/or extend into deeper half of dermis, but do not expand to fill dermal space completely in areas of deeper penetration; 3, basal cells pack dermis in confluent masses in >50% of skin.
RNA sequencing
Hundreds of lesions were laser capture micro-dissected (LCM) from multiple skin dissections per animal. Adjacent skin sections were stained for Ki67 and counterstained with haematoxylin to determine the proliferation status of tumour nests from untreated BCC mice. Three groups, each consisting of five mice, were included: 1) Ki67+ tumours from untreated BCC mice; 2) Ki67– tumours from untreated BCC mice; and 3) residual tumours from BCC mice that were treated with vismodegib for 28 days. LCM samples were pooled on a per mouse basis and total RNA was extracted with the RNeasy kit (Qiagen). The concentration and integrity of the RNA were determined by NanoDrop 8000 (Thermo Scientific) and with a Bioanalyzer RNA 6000 Pico Kit (Agilent), respectively. cDNA was generated with the Ovation RNA-seq System V2 (Nugen) and sheared to 150–200 bp size using an LE220 focused ultrasonicator (Covaris). Fragment length was confirmed with the Bioanalyzer DNA 1000 Kit (Agilent) and samples were quantified with the Qubit dsDNA BR Assay (Life Technologies). A total of 1 μg of sheared cDNA and the TruSeq RNA Sample Preparation Kit v2 (Illumina) were used for the end repair step of each library. Library size was confirmed with High Sensitivity D1K screen tape on a 2200 TapeStation (Agilent Technologies), and concentration was determined by qPCR using the Library quantification kit (KAPA). Libraries were multiplexed and sequenced on a HiSeq 2500 (Illumina) to generate 30 million single-end 50-bp reads per library. Reads were first aligned to ribosomal RNA sequences to remove ribosomal reads. The remaining reads were aligned to the mouse reference genome (NCBI Build 38) using GSNAP31 version ‘2013-10-10’, allowing a maximum of two mismatches per 50 base pair sequence (parameters: ‘-M 2 -n 10 -B 2 -i 1 -N 1 -w 200000 -E 1–pairmax-rna = 200000–clip-overlap’). Transcript annotation was based on the RefSeq database (NCBI Annotation Release 104). To quantify gene expression, the number of reads mapped to the exons of each RefSeq gene was calculated. Read counts were scaled by library size, quantile normalized and precision weights calculated using the ‘voom’ R package32. Subsequently, differential expression analysis on the normalized count data was performed using the ‘limma’ R package33 by contrasting vismodegib-treated samples with untreated samples. Gene expression levels were considered significantly different across groups if we observed |log2[fold change]| ≥ 1 (estimated from the model coefficients) associated with a false discovery rate (FDR)-adjusted P ≤ 0.05. Gene expression was obtained in form of nRPKM as described previously34.
ATAC library preparation
ATAC–seq was performed as follows: 100,000 sorted cells were collected in 1 ml PBS + 3% FBS at 4 °C. Cells were centrifuged, then cell pellets were resuspended in 100 μl lysis buffer (Tris HCl 10 mM, NaCl 10 mM, MgCl2 3 mM, Igepal 0.1%) and centrifuged (500g) for 25 min at 4 °C. Supernatant was discarded and nuclei were resuspended in 50 μl reaction buffer (Tn5 transposase 2.5 μl, TD buffer 22.5 μl and 25 μl H2O – Nextera DNA sample preparation kit, Illumina). The transposase reaction was performed for 30 min at 37 °C and then blocked by addition of 5 μl clean up buffer (NaCl 900 mM, EDTA 300 mM). DNA was purified using the MinElute purification kit (QIAGEN). DNA libraries were PCR amplified (Nextera DNA Sample Preparation Kit, Illumina), and size selected for 200 to 800 bp (BluePippin, Sage Sciences) following the manufacturers’ protocols.
ATAC–seq
The libraries were sequenced on Illumina HiSeq 2500 sequencers. We obtained an average of 100 million paired-end reads (50 bp) per sample. Reads were aligned to the mouse reference genome (NCBI Build 38) using GSNAP31 version ‘2013-10-10’, allowing a maximum of two mismatches per read sequence (parameters: ‘-M 2 -n 10 -B 2 -i 1–pairmax-dna = 1000–terminal-threshold = 1000–gmap-mode = none–clip-overlap’). Reads aligning to locations in the mouse genome that contain substantial sequence homology to the MT chromosome or to blacklisted regions identified by the ENCODE consortium were omitted from downstream analyses. Properly paired reads derived from non-duplicate sequencing fragments were used to quantify chromatin accessibility according to the ENCODE pipeline standards with minor modifications as follows. Accessible genomic locations were identified by calling peaks with Macs235 using insertion-centred pseudo-fragments (73 bp; community standard) generated on the basis of the start positions of the mapped reads. Accessible peak locations were identified as described: in brief, we called peaks on a group-level pooled sample containing all pseudo-fragments observed in all samples within each group. Peaks in the pooled sample that were independently identified in two or more of the constituent biological replicates were retained for downstream analysis, using the union of all group-level reproducible peaks (https://www.encodeproject.org/atac-seq/#standards). We quantified the level of chromatin accessibility within each peak for each replicate as the number of pseudo-fragments that overlapped the peak in question and normalized these estimates using the TMM method36. To better understand the within- and between-group similarities in chromatin accessibility, we calculated the Pearson correlation coefficient for the top 5,000 most variable peaks and performed hierarchical clustering using the correlation measures.
We identified differentially accessible peaks between groups in the framework of a linear model implemented with the limma R package33 and incorporating precision weights calculated with the voom function in the limma R package32. Peaks that showed an increase in accessibility at either day 6 or day 14 of vismodegib treatment were called vismodegib peaks and control peaks were called untreated peaks. For subsequent analysis, peaks were divided into promoter regions (1 kb up- and 2 kb down-stream of transcription start sites) and enhancers (peaks outside of promoter regions).
We identified enriched transcription factor (TF) motifs using HOMER v4.737. To evaluate the significance of the TF enrichment we defined peaks as significantly differentially accessible based on a range of FDR adjusted P value thresholds between 1 and 0.01 and an |log2[fold change]| in accessibility ≥ 1 (estimated from the model coefficients). Given the strong enrichment of the top motifs across a wide range of P value cutoffs, we decided to consider peaks as different across groups for a |log2[fold change]| ≥ 1 and FDR adjusted P value ≤ 0.05 in subsequent analyses.
Chromatin accessibility was visualized as coverage tracks across genomic regions using the Integrated Genomics Viewer38 and as heatmaps using DeepTools v3.0.139.
Regulatory potential (BETA analysis)
We associated accessible chromatin regions with nearby genes using BETA15. The BETA minus mode was used to calculate the regulatory potential (determined through a distance-weighted measure) of specific sets of peaks within a certain distance to a target gene. The BETA basic mode allowed us to integrate differential expression with chromatin openness to evaluate whether the direct effect of changes in the chromatin landscape is promoting or repressing gene expression. In this mode all genes within 100 kb of a peak set are ranked (and listed along the x-axis) based on the regulatory potential using the ATAC–seq data. Subsequently, expression information is used to divide genes into downregulated by vismodegib (purple line), upregulated by vismodegib (red line) and transcriptionally unchanged (dashed line) genes. A one-tailed Kolmogorov–Smirnov test40 was used to determine whether the upregulated and downregulated groups differed significantly from the group of transcriptionally unchanged genes.
FACS sorting of tumour cells
Back skin was taken from BCC mice harbouring the Lgr5GFPDTR allele41 and processed into a single-cell suspension42. Cells were stained for CD34 and the live/dead marker SYTOX blue. GFP-only tumour cells were sorted on a BD Biosciences Influx ‘jet-in-air’ cell sorter equipped with a combination of 355-, 405-, 488-, 561-, and 640-nm lasers. Fluorescence emission was collected through 460/50 (Sytox Blue), 530/40 (GFP), and 670/30 (CD34-Alexa647) bandpass filters. Sorting was performed with an 86-μm nozzle, a pressure of 30 psi and a frequency of 48,610 Hz. Setup and alignment of the instrument were performed using 3 μm ultra rainbow polystyrene beads (URFP-30-20, Spherotech Inc.). See Extended Data Fig. 6 for gating strategies.
Skin epithelial signatures
We downloaded microarray studies that profiled gene expression across various skin epithelial compartments, including GSE1518543, GSE2156844, GSE4061219, GSE4170445, GSE2026946 and E-MTAB-160647. Data normalization was performed using either the affy or lumi R packages, and differential expression analysis was performed using the limma R package33. Genes were called significantly differentially expressed when |log2[fold change]| > 1 and adjusted P < 0.1. Genes were considered bulge-specific if they were significantly upregulated in either the Lgr5+ vs. Lgr6+ comparison (GSE20269) or the CD34+ vs. GFP– comparison (E-MTAB-1606). Genes were considered ISTH- or IFE-specific if they were significantly downregulated in the GSE20269 or E-MTAB-1606 studies, respectively. The ORS signature was obtained by downloading the gene count data from RNA-seq GEO study GSE9084748 and identifying genes that were significantly upregulated (log2[fold change] ≥ 1 and adjusted P < 0.01) in ORS as compared to hair follicle stem cells using the same voom and limma procedure as described above.
Gene set analysis
We performed QuSAGE49 to identify relevant biological processes associated with vismodegib treatment. For that purpose we compared vismodegib-treated samples with untreated samples. For each comparison we then calculated the gene set activity (the mean difference in log2 expression of the individual genes that comprise the set) for the four sets of bulge, isthmus, IFE and ORS marker genes identified from the above public microarray experiments. In addition, we assessed the up- or downregulation of bulge, isthmus, IFE and ORS genes using gene set enrichment analysis (GSEA)40. The significance of the enrichment (shown as FDR) was determined through 1,000 permutations of random gene sets.
Immunohistochemistry and Immunofluorescence staining
Primary antibodies and their dilutions used in this study: rabbit Sox9 1:300 (Millipore, AB5535), rabbit Keratin1 1:1,000 (Covance, PRB-165 [AF109]), chicken Keratin5 1:2,000 (Biolegend, 90501), rabbit Keratin10 1:1,000 (Biolegend, 90541), rabbit Loricrin 1:1,000 (Covance, PRB-145P), goat IL-33 1:500 (R&D Systems, AF3626), rabbit Ki67 1:500 (GeneTex, GTX16667 [SP6]), rat BrdU 1:400 (BioRad, MCA2060), rabbit CC3 1:400 (Cell Signaling, 9661), and human Ep-CAM undiluted (Ber-EP4; Ventana 760-4383). Dorsal skin was shaved, dissected, and fixed in either 4% paraformaldehyde (PFA) or 10% neutral buffered formalin overnight. Fixed skin was washed in PBS and 70% ethanol, processed and embedded into paraffin. Fresh frozen skin was cut into strips and embedded in OCT (Sakura). Skin sections were cut at 6 μm on either a Leica RM2255 microtome or a Leica CM 3050 S cryostat. Fresh frozen skin sections were fixed in 4% PFA for 10 min before staining. For IHC, slides were de-paraffinized in xylenes, re-hydrated, and boiled in Dako target retrieval buffer for 10 min. Samples were then blocked with Dako protein-free blocking solution for 10 min and primary antibodies were diluted in Dako antibody diluent and exposed to samples overnight at 4 °C. Secondary antibodies (Invitrogen, Molecular Probes) for immunofluorescence staining were also diluted in Dako antibody diluent. IHC was performed using a Dako Envision+ system-HRP polymer detection kit.
In situ hybridization
Traditional RNA ISH was performed on paraffin-embedded tissue sections using digoxigenin-labelled probes according to standard protocols50. Alkaline phosphatase activity was detected on tissue sections using BM Purple staining solution (Roche) after overnight incubation in alkaline phosphatase buffer.
Hybridizations using the RNAscope method were performed according to the manufacturer’s protocol (Advanced Cell Diagnostics) using the RNAscope 2.5 HD Reagent Kit-RED (322350). Probes used were MmLhx2 (485791), MmAxin2 (400331), HuAXIN2 (400241), MmDefB6 (430141), MmWif1 (412361), and MmGli1 (311001).
Taqman quantitative real-time PCR
Tumours were collected from fresh frozen skin sections by LCM. Total RNA was extracted using the RNeasy micro kit (Qiagen) and cDNA was prepared using the TaqMan RNA to Ct 1 Step kit (Applied Biosystems). TaqMan analysis was performed on an ABI7900HT (Applied Biosystems), data were analysed with SDS 2.3 software (Applied Biosystems) and normalized to Hprt transcript levels. The following Applied Biosystems Taqman assays were used: Axin2 (Mm00443610_m1), Wif1 (Mm00442355_m1), Wnt4 (Mm01194003_m1), Wnt10a (Mm00839783_m1), Lhx2 (Mm00839783_m1) and Plet1 (MTS24 antigen; Mm01170995_m1).
Quantification of Oil Red O+ and BrdU+ tumours
The number of residual tumours in a linear unit of backskin equal to four lengths of a 5× magnification field of view was counted. Oil Red O+ and BrdU+ residual tumours were expressed as a fraction of the total number of tumours. BCCs were considered Oil Red O+ when they contained at least one Oil Red O+ cell and were not associated with a hair follicle (to avoid contamination from sebaceous glands). BCCs were considered BrdU+ when they contained at least 20 intense BrdU+ nuclei and did not contain any cells with a more weak, diluted nuclear signal (suggesting proliferation).
Statistics and reproducibility
See individual Methods sections for specific statistical methods. Experiments were independently repeated at least twice leading to similar results. No statistical method was used to predetermine sample size, and no mice were excluded from the analysis. BCC mice were age matched and randomly assigned to control and treatment groups. The investigators were blinded during outcome assessment. The data meet the assumptions of the statistical tests used and are presented as mean ± s.e.m.; P ≤ 0.05 was considered statistically significant. For RNA-seq, differential expression analysis was performed using the ‘limma’ R package, which uses the moderated t-statistic for significance analysis. The adjusted P value was calculated using the Benjamini and Hochberg method to account for the false discovery rate.
Human subject data
The study (ClinicalTrials.gov identifier: NCT01201915) was conducted per FDA regulations, International Conference on Harmonization E6 Guideline for Good Clinical Practice, and applicable local, state, and federal laws. The protocol was approved by institutional review boards where applicable. Patients gave written informed consent. The study evaluated the activity of vismodegib in patients with smaller operable BCC by measuring the rate and durability of complete histologic clearance (CHC) of lesions. Patients with new, operable, nodular basal cell carcinoma received vismodegib (150 mg/d) followed by excision and Mohs micrographic surgery to ensure clear margins. Samples from cohort 1 were analysed for this study: patients received vismodegib for 12 weeks and target sites were immediately excised by standard means, followed by Mohs micrographic surgery to obtain clear margins. Biospecimen type: skin; anatomical site: scalp/head/neck or upper aspect of trunk, greater trunk; disease status of patients: new, nodular BCC, operable; clinical characteristics of patients: alive; clinical diagnosis of patients: nodular BCC by biopsy; pathology diagnosis: nodular BCC by biopsy; collection mechanism: excision, Mohs surgery; type of stabilization: formalin fixed; type of long-term preservation: formalin fixation; constitution of preservative: 10% neutral-buffered formalin.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
RNA-seq and ATAC–seq data supporting the findings of this study have been deposited in the Gene Expression Omnibus (GSE116966). All other data are available from the corresponding author upon reasonable request.
References
Sekulic, A. et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 366, 2171–2179 (2012).
Sofen, H. et al. A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol. 73, 99–105.e1 (2015).
Wang, G. Y., Wang, J., Mancianti, M. L. & Epstein, E. H. Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/– mice. Cancer Cell 19, 114–124 (2011).
Rubin, A. I., Chen, E. H. & Ratner, D. Basal-cell carcinoma. N. Engl. J. Med. 353, 2262–2269 (2005).
Robarge, K. D. et al. GDC-0449-a potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 19, 5576–5581 (2009).
Von Hoff, D. D. et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 361, 1164–1172 (2009).
Basset-Seguin, N. et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): a pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 16, 729–736 (2015).
Vidal, V. P., Ortonne, N. & Schedl, A. SOX9 expression is a general marker of basal cell carcinoma and adnexal-related neoplasms. J. Cutan. Pathol. 35, 373–379 (2008).
Feil, R., Wagner, J., Metzger, D. & Chambon, P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem. Biophys. Res. Commun. 237, 752–757 (1997).
Folgueras, A. R. et al. Architectural niche organization by LHX2 is linked to hair follicle stem cell function. Cell Stem Cell 13, 314–327 (2013).
Nijhof, J. G. et al. The cell-surface marker MTS24 identifies a novel population of follicular keratinocytes with characteristics of progenitor cells. Development 133, 3027–3037 (2006).
Pichery, M. et al. Endogenous IL-33 is highly expressed in mouse epithelial barrier tissues, lymphoid organs, brain, embryos, and inflamed tissues: in situ analysis using a novel Il-33-LacZ gene trap reporter strain. J. Immunol. 188, 3488–3495 (2012).
Joost, S. et al. Single-cell transcriptomics reveals that differentiation and spatial signatures shape epidermal and hair follicle heterogeneity. Cell Syst. 3, 221–237.e9 (2016).
Ge, Y. et al. Stem cell lineage infidelity drives wound repair and cancer. Cell 169, 636–650.e14 (2017).
Wang, S. et al. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nat. Protocols 8, 2502–2515 (2013).
Kim, T. H. et al. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature 506, 511–515 (2014).
Adam, R. C. et al. Pioneer factors govern super-enhancer dynamics in stem cell plasticity and lineage choice. Nature 521, 366–370 (2015).
Yang, S. H. et al. Pathological responses to oncogenic Hedgehog signaling in skin are dependent on canonical Wnt/β3-catenin signaling. Nat. Genet. 40, 1130–1135 (2008).
Youssef, K. K. et al. Adult interfollicular tumour-initiating cells are reprogrammed into an embryonic hair follicle progenitor-like fate during basal cell carcinoma initiation. Nat. Cell Biol. 14, 1282–1294 (2012).
Jho, E. H. et al. Wnt/β-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol. Cell. Biol. 22, 1172–1183 (2002).
Hsieh, J. C. et al. A new secreted protein that binds to Wnt proteins and inhibits their activities. Nature 398, 431–436 (1999).
Lim, X. et al. Interfollicular epidermal stem cells self-renew via autocrine Wnt signaling. Science 342, 1226–1230 (2013).
Tian, H. et al. Opposing activities of Notch and Wnt signaling regulate intestinal stem cells and gut homeostasis. Cell Reports 11, 33–42 (2015).
Niemann, C. Differentiation of the sebaceous gland. Dermatoendocrinol 1, 64–67 (2009).
Yauch, R. L. et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326, 572–574 (2009).
Kahn, M. Can we safely target the WNT pathway? Nat. Rev. Drug Discov. 13, 513–532 (2014).
Oser, M. G., Niederst, M. J., Sequist, L. V. & Engelman, J. A. Transformation from non-small-cell lung cancer to small-cell lung cancer: molecular drivers and cells of origin. Lancet Oncol. 16, e165–e172 (2015).
Vasioukhin, V., Degenstein, L., Wise, B. & Fuchs, E. The magical touch: genome targeting in epidermal stem cells induced by tamoxifen application to mouse skin. Proc. Natl Acad. Sci. USA 96, 8551–8556 (1999).
Kasper, M. et al. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl Acad. Sci. USA 108, 4099–4104 (2011).
Marino, S., Vooijs, M., van Der Gulden, H., Jonkers, J. & Berns, A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 14, 994–1004 (2000).
Wu, T. D. & Nacu, S. Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881 (2010).
Law, C. W., Chen, Y., Shi, W. & Smyth, G. K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29 (2014).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Srinivasan, K. et al. Untangling the brain’s neuroinflammatory and neurodegenerative transcriptional responses. Nat. Commun. 7, 11295 (2016).
Zhang, Y. et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010).
Robinson, J. T. et al. Integrative genomics viewer. Nat. Biotechnol. 29, 24–26 (2011).
Ramírez, F., Dündar, F., Diehl, S., Grüning, B. A. & Manke, T. deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191 (2014).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci. USA 102, 15545–15550 (2005).
Tian, H. et al. A reserve stem cell population in small intestine renders Lgr5-positive cells dispensable. Nature 478, 255–259 (2011).
Jensen, K. B., Driskell, R. R. & Watt, F. M. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat. Protocols 5, 898–911 (2010).
Greco, V. et al. A two-step mechanism for stem cell activation during hair regeneration. Cell Stem Cell 4, 155–169 (2009).
Garza, L. A. et al. Bald scalp in men with androgenetic alopecia retains hair follicle stem cells but lacks CD200-rich and CD34-positive hair follicle progenitor cells. J. Clin. Invest. 121, 613–622 (2011).
Blanpain, C., Lowry, W. E., Geoghegan, A., Polak, L. & Fuchs, E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell 118, 635–648 (2004).
Snippert, H. J. et al. Lgr6 marks stem cells in the hair follicle that generate all cell lineages of the skin. Science 327, 1385–1389 (2010).
Page, M. E., Lombard, P., Ng, F., Göttgens, B. & Jensen, K. B. The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell 13, 471–482 (2013).
Yang, H., Adam, R. C., Ge, Y., Hua, Z. L. & Fuchs, E. Epithelial-mesenchymal micro-niches govern stem cell lineage choices. Cell 169, 483–496.e13 (2017).
Yaari, G., Bolen, C. R., Thakar, J. & Kleinstein, S. H. Quantitative set analysis for gene expression: a method to quantify gene set differential expression including gene-gene correlations. Nucleic Acids Res. 41, e170 (2013).
O’Neill, J. W. & Bier, E. Double-label in situ hybridization using biotin and digoxigenin-tagged RNA probes. Biotechniques 17, 870, 874–875 (1994).
Beer, T. W., Shepherd, P. & Theaker, J. M. Ber EP4 and epithelial membrane antigen aid distinction of basal cell, squamous cell and basosquamous carcinomas of the skin. Histopathology 37, 218–223 (2000).
Acknowledgements
We thank J. Diaz for mouse colony management; the IVP team for animal dosing; S. Flanagan and V. Nunez for necropsy support; the Pathology core for help with histology; S. Biswas for help with LCM; the NGS laboratory for RNA-seq and ATAC–seq; I. Caro for help with human samples; and J. Svärd, R. Toftgard, and S. Teglund for Ptch1fl/fl mice.
Reviewer information
Nature thanks R. Shivdasani and the other anonymous reviewer(s) for their contribution to the peer review of this work.
Author information
Authors and Affiliations
Contributions
B.B., G.J.P.D., S.E.G. and F.J.d.S. conceptualized the project, designed experiments and analysed data. B.B. performed IHC, immunofluorescence and ISH on skin sections, isolated tumour nests by LCM and tumour cells by FACS, and performed qRT–PCR. G.J.P.D. performed initial characterization of residual disease. R.P. performed bioinformatics analyses. B.A. monitored mice for tumour burden and coordinated drug dosing. S.B. analysed ATAC–seq data. F.P. performed histopathological evaluation of skin sections. B.B., G.J.P.D. and F.J.d.S. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
All authors are employees of Genentech and own shares of Roche.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data figures and tables
Extended Data Fig. 1 Limited apoptosis in tumour nests early during vismodegib treatment and recovery of residual BCCs.
Related to Figs. 1a–d, 2e–h. a–d, Time-course analysis of cell death in our K14CreER;Ptch1fl/fl;Trp53fl/fl mouse model of BCC during vismodegib treatment. Representative images of skin sections collected after indicated length of vismodegib treatment were stained for cleaved caspase 3 (CC3, red) and nuclei (DAPI, blue) (n = 3 per time point). The apoptosis marker CC3 is lacking in untreated BCC (a), but could be detected in rare tumour cells after just 1 day of treatment (b). CC3+ tumour cells were most abundant after 3 days of treatment (c) but were no longer detectable after 7 days of treatment (d), indicating that additional mechanism(s) contribute to the tumour de-bulking that occurs at later time points. e, f, Additional images of Gli1 ISH on skin sections from vismodegib-treated BCC mice shown in Fig. 2a. Note that the robust Gli1 expression endures when animals remain off drug for 6 (e) or 12 days (f). g, h, Representative images of skin sections stained for BrdU (red) and nuclei (DAPI, blue) from untreated BCC mice and mice treated with vismodegib for 7 days (n = 3 per group). g, BrdU dosing before vismodegib treatment labels proliferating cells in BCCs and in the epidermis (above dotted line). h, Label-retaining BrdU+ cells can be found only in residual tumour nests (below dotted line) after 7 days of vismodegib treatment, as epidermal cells continue to proliferate in the presence of drug and dilute out the label. All experiments were performed at least twice. Scale bars, 100 μm; n represents the number of mice.
Extended Data Fig. 2 Vismodegib treatment does not cause BCCs to differentiate into normal epidermis.
Related to Fig. 1. Detailed time-course analysis of epidermal differentiation marker expression in BCC. Skin samples were collected from BCC mice after indicated length of vismodegib treatment. Representative images of skin sections are shown for n = 4 per time point. Residual tumour nests could be identified with the BCC marker SOX9 throughout treatment (column 1; red). By contrast, the supra-basal marker KRT1 (column 2; red) and the terminal differentiation marker loricrin (column 4; red) were at no point expressed in either untreated or residual BCCs. The supra-basal marker KRT10 was detected in both untreated and residual BCC (column 3; red). Its expression pattern within tumour nests gradually changed from a proximal/dermal location in untreated BCCs to the centre of residual BCCs, where it no longer overlapped with the basal marker KRT5 (green; outlined in 28 d vismo). Scale bars, 100 μm; n represents the number of mice. DAPI nuclear stain is blue.
Extended Data Fig. 3 Identification of differentially expressed genes in BCC after vismodegib treatment and their normal expression pattern in telogen skin.
Related to Fig. 3. a, b, Volcano plots depicting the number of differentially expressed genes that are either significantly upregulated (green dots) or downregulated (blue dots) in residual disease after 28 days of vismodegib treatment relative to either Ki67+ (a) or Ki67– (b) untreated BCC. Significance was tested with a moderated t-statistic (two-sided) and P values were adjusted for multiple testing with the Benjamini–Hochberg procedure (log2FC ≥ 1, adjusted P ≤ 0.05; n = 5 per group). c, Robust correlation between the two differential gene expression estimates from the comparisons made in a and b. d, QuSAGE of indicated signatures in vismodegib-treated versus Ki67– untreated BCC samples (n = 5 per group). Coloured violins depict the differential expression of the entire gene set quantified by a probability density function calculated using QuSAGE. Each grey dot represents the log2[fold change] in expression of a particular gene in the signature. e, f, GSEA ranking of indicated skin compartment-specific gene signatures using the differential gene expression estimates from the comparisons made in a, b. Changes in expression were determined for n = 5 per group. g–i, Expression pattern of select skin epithelial markers in wild-type telogen skin. Representative images are shown for n = 4 per condition. g, Lhx2 mRNA is particularly enriched in the bulge of hair follicles. h, Antibody stain revealing nuclear localization of IL-33 in the isthmus and interfollicular epidermis. i, DefB6 mRNA is particularly enriched in the isthmus region of hair follicles. j, k, Representative images of Defb6 ISH on skin from untreated BCC mice (j) and animals treated with vismodegib for 28 days (k; n = 4 per condition). Arrowheads indicate DefB6+ residual BCCs. l, Relative expression of Lhx2 (hair follicle bulge) and MTS24 (ISTH) in BCCs treated with vismodegib for the indicated length of time. Data are plotted as mean ± s.e.m.; n = 3 per condition. Scale bars, 100 μm; n represents the number of mice.
Extended Data Fig. 4 Residual BCCs reinitiate the hair follicle bulge program when vismodegib treatment is discontinued.
Related to Fig. 3. a–d, Lineage tracing in BCC mice with a Cre-inducible TdTomato reporter allele after 28 days of vismodegib treatment (n = 3 per group). Untreated TdTomato+ BCCs display robust LHX2 antibody staining (a), but completely lack IL-33 antibody staining (c). TdTomato+ residual BCCs lack LHX2 antibody staining (b), but stain positive for IL-33 protein (d). e–h, Return of LHX2 expression in residual BCCs after vismodegib treatment. Representative images of skin sections stained for BrdU (red), nuclei (DAPI, blue) and LHX2 (green) from BCC mice treated with vismodegib for 28 days that were allowed to recover for the indicated times (n = 3 per group). e, BrdU and LHX2 co-staining is virtually absent in residual BCC (outlined) immediately after treatment. f, LHX2 staining is weak in BrdU-labelled tumour nests after 3 days of recovery. g, LHX2 staining is more prominent in residual tumours after 6 days of recovery. h, LHX2 staining is the most robust and shows maximum overlap with the BrdU label after 12 days of recovery. i, Proportion of BrdU–LHX2 double-positive tumour cells in skin sections from e–h. Graph shows mean ± s.d. Each data point represents the proportion of BrdU-labelled cells that were LHX2-positive within a single tumour nest (n ≥ 12 tumours from 3 mice per time-point). Scale bars, 100 μm; n represents the number of mice except for i, where it represents the number of tumours.
Extended Data Fig. 5 Il-33 expression in BCCs during vismodegib treatment.
Related to Fig. 3. a, Expression of Il33 in LCM tumours from untreated BCC mice and mice treated with vismodegib for 28 days (n = 5 per group). RNA-seq expression data were quantile normalized and represented as nRPKM. Data plotted are mean ± s.e.m.; data points (dots, squares) indicate Il33 expression in tumours from individual mice. b–f, Detailed time-course analysis of IL-33 expression in BCCs during vismodegib treatment. Representative images of skin samples collected from both untreated and treated BCC mice are shown (n = 3 per treatment and time point). IL-33 gradually appears in residual BCCs over the course of treatment. This nuclear factor can readily be detected in the basal layer of the epidermis, but is lacking in untreated BCCs (b) and in tumours treated with vismodegib for just 1 (c) or 3 (d) days. Nuclear IL-33 begins to emerge in a fraction of the tumour nests after 7 days of treatment (e), and most residual BCCs express IL-33 by 14 days of treatment (f). Scale bars, 100 μm; n represents the number of mice.
Extended Data Fig. 6 ATAC–seq of FACS-sorted BCC cells and regulatory potential of accessible chromatin regions.
Related to Fig. 3. a, b, We crossed our mouse model of BCC to the Lgr5DTRGFP allele41, to enable FACS isolation of both untreated (a) and residual (b) tumour cells. Representative images of skin sections stained for GFP (green) and nuclei (DAPI, blue) are shown for n = 3 per group. c, Gating for low side scatter enriched for epidermal cells after dissociation of back skin samples42. d, Final gating strategy used for the FACS isolation of BCC cells. Lgr5+ hair follicle bulge stem cells were excluded by staining for CD34. e, Heat-map of differentially accessible chromatin regions that either close (top) or open (bottom) in response to indicated length of vismodegib treatment. Data are shown as normalized peak counts per million genomic DNA fragments averaged from n = 3 per condition. f, Matrix of Pearson correlation coefficients showing the overlap in chromatin accessibility between samples based on the 5,000 most variable peak regions. Pearson’s ρ statistic was used to determine each correlation. g, Volcano plots depicting chromatin accessibility in sorted BCC cells after 6 (left) and 14 days (right) of vismodegib treatment relative to untreated (MCT) controls (n = 3 per group). Significance was tested with a moderated t-statistic (two-sided) and P values were adjusted for multiple testing with the Benjamini–Hochberg procedure. Each dot represents a peak (an open chromatin region). Common peaks are grey, and specific peaks are either blue (untreated) or green (6 and 14 days vismodegib). The table summarizes the number of open chromatin regions according to promoters and enhancers. h, Chromatin traces showing averaged ATAC peaks at the Gata6 and Ahr loci; n = 3 per group. Orange rectangles mark TCF binding elements in chromatin that are sensitive to vismodegib. i, Volcano plots depicting the number of differentially expressed genes that are either significantly upregulated (red dots; 150 at 6 d and 794 at 14 d) or downregulated (purple dots; 416 at 6 d and 1,129 at 14 d) in sorted BCC cells after 6 (left) and 14 days (right) of vismodegib treatment relative to untreated controls. Significance was tested with a moderated t-statistic (two-sided) and P values were adjusted for multiple testing with the Benjamini–Hochberg procedure (log2FC ≥ 1, adjusted P ≤ 0.05; n = 3 per group). j, k, BETA analysis graphs depicting the effect of differentially open enhancers (j) and promoter peaks (k) on gene expression in sorted BCC cells after 6 (top) and 14 days (bottom) of vismodegib treatment relative to untreated controls (n = 3 per group). Genes were ranked from high to low according to the regulatory potential of the corresponding chromatin peak. Purple lines represent vismodegib-downregulated genes, while red lines represent vismodegib-upregulated genes. A one-tailed Kolmogorov–Smirnov test40 was used to determine whether the up- and downregulated groups differed significantly (shown as P values in parentheses) from the static group of transcriptionally unchanged genes (dashed lines). Scale bars, 100 μm; n represents the number of mice.
Extended Data Fig. 7 Enrichment of transcription factor binding motifs in differentially regulated enhancers and their regulatory potential.
Related to Fig. 3. a, b, Enrichment of transcription factor binding sites in non-promoter peaks that are either differentially closing (a) or opening (b) after 6 (top) and 14 days (bottom) of vismodegib treatment relative to untreated BCC cells (n = 3 per group). A hypergeometric test implemented in HOMER was used to identify enriched motifs, which are ranked by P value (top to bottom) over increasingly stringent P value cutoffs for peak calling (left to right). c, d, BETA analysis graphs depicting the effect of differentially open enhancer peaks with indicated transcription factor binding motifs on gene expression in sorted BCC cells after 6 (c) and 14 days (d) of vismodegib treatment relative to untreated controls (n = 3 per group). Genes were ranked from high to low according to the regulatory potential of the corresponding chromatin peak. Purple lines represent vismodegib-downregulated genes, while red lines represent vismodegib-upregulated genes. A one-tailed Kolmogorov–Smirnov test40 was used to determine whether the up- and downregulated groups differed significantly (shown as P values in parentheses) from the static group of transcriptionally unchanged genes (dashed lines). n represents the number of mice.
Extended Data Fig. 8 AXIN2 is also induced in human residual BCC.
Related to Fig. 4. a, b, Representative images of H&E-stained skin sections from untreated BCC mice (a) and from animals treated with vismodegib for 2 days (b; n = 3 per group). Note that the relatively short treatment has no impact on the overall morphology and size of the tumour nests. c–h, Representative images of Ber-EP4 (brown) staining and AXIN2 ISH (red) on skin sections collected from a patient with BCC before and after 12 weeks of vismodegib treatment. The human BCC marker Ber-EP451 was used to identify nodular BCC in a screening biopsy (c) and residual BCC at the tumour site after treatment (f). d, e, g, h, Representative images of AXIN2 ISH on skin sections immediately adjacent to the ones shown in c and f. d, Untreated nodular BCC lacks AXIN2 mRNA. g, Residual BCC contains elevated AXIN2 mRNA levels. e, h, High-magnification views of boxed regions in d and g, respectively. i, Axin2, Wif1 and Lhx2 ISH on skin sections from BCC mice after 28 days of vismodegib treatment. Skin samples were collected 0 (top), 6 (middle) and 12 (bottom) days after the end of treatment, and representative images are shown for n = 3 per group. Note that Axin2 expression rapidly declines during the recovery period, whereas the expression of both Wif1 and Lhx2 progressively increase. All experiments were replicated at least twice. Scale bars: c, f, 90 μm; other panels, 100 μm.
Extended Data Fig. 9 Effects of the vismodegib and anti-LRP6 combination treatment on mouse BCC.
Related to Fig. 4. a–c, Representative images of Axin2 ISH on skin sections from wild-type telogen mice (a) and from BCC mice treated for 28 days with either vismodegib alone (b) or vismodegib and anti-LRP6 (c). Note that co-treatment with anti-LRP6 strongly reduces Axin2 mRNA levels in residual BCCs (n = 3 per group). d, Relative expression of Wnt target genes in the small intestine of BCC mice treated for 28 days with either vismodegib alone (circles) or vismodegib and anti-LRP6 (squares; n = 4 per group). Data are plotted as mean ± s.e.m. Three out of four BCC mice that received vismodegib and anti-LRP6 experienced at least an 80% reduction in expression of all four Wnt target genes that was accompanied by a low residual tumour burden (black squares; 6, 9 and 18 residual tumours per length of skin), while the one animal with poor Wnt pathway modulation had a much higher residual tumour burden (red square; 31 residual tumours per length of skin). e–g, Representative images of Oil Red O (orange)-stained skin sections from untreated BCC mice (e) and mice treated for 28 days with either vismodegib alone (f) or vismodegib and anti-LRP6 (g) (nuclear blue counter stain, haematoxylin; n = 4 per treatment). h, Proportion of Oil Red O+ tumours per length of skin in BCC mice from f, g. Average percentages ± s.d. are shown for n = 4 per group. i–p, BrdU labelling of residual BCCs and their subsequent growth after cessation of indicated treatment. Representative images of residual BCCs stained for BrdU (red), nuclei (DAPI, blue) and Ki67 (green) are shown (n ≥ 2 per treatment and time point) from BCC mice treated for 28 days with either vismodegib alone (i–l) or vismodegib and anti-LRP6 (m–p). Note that residual BCCs from both treatment groups are fully proliferating by 12 days of recovery and have diluted out the BrdU label. q, Proportion of BrdU+ tumours per length of skin from BCC mice from i–p. Average percentages ± s.d. are shown for n ≥ 2 per group. All experiments were replicated at least twice. Scale bars, 100 μm; n represents the number of mice.
Supplementary information
Supplementary Table
This file contains Supplementary Table 1
Rights and permissions
About this article

Cite this article
Biehs, B., Dijkgraaf, G.J.P., Piskol, R. et al. A cell identity switch allows residual BCC to survive Hedgehog pathway inhibition. Nature 562, 429–433 (2018). https://doi.org/10.1038/s41586-018-0596-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41586-018-0596-y
- Springer Nature Limited
Keywords
This article is cited by
-
Targeting therapy-persistent residual disease
Nature Cancer (2024)
-
Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance
Cancer and Metastasis Reviews (2024)
-
Modeling stress-induced responses: plasticity in continuous state space and gradual clonal evolution
Theory in Biosciences (2024)
-
Biomarkers in Cutaneous Keratinocyte Carcinomas
Dermatology and Therapy (2024)
-
Cancer drug-tolerant persister cells: from biological questions to clinical opportunities
Nature Reviews Cancer (2024)