

Abstract
T cells engineered to express chimeric antigen receptors (CARs) that are specific for tumour antigens have led to high complete response rates in patients with haematologic malignancies. Despite this early success, major challenges to the broad application of CAR-T cells as cancer therapies remain, including treatment-associated toxicities and cancer relapse with antigen-negative tumours. Targeting solid tumours with CAR-T cells poses additional obstacles because of the paucity of tumour-specific antigens and the immunosuppressive effects of the tumour microenvironment. To overcome these challenges, T cells can be programmed with genetic modules that increase their therapeutic potency and specificity. In this Review Article, we survey major advances in the engineering of next-generation CAR-T therapies for haematologic cancers and solid cancers, with particular emphasis on strategies for the control of CAR specificity and activity and on approaches for improving CAR-T-cell persistence and overcoming immunosuppression. We also lay out a roadmap for the development of off-the-shelf CAR-T cells.
Similar content being viewed by others

Main
The surface of virtually every cell in the body displays endogenous peptides, derived from intracellular proteins, as complexes with major histocompatibility proteins. This ensures that if a cell is infected with a pathogen or undergoes malignant genetic transformation, peptides derived from the pathogen or from the mutant proteins will be exposed to patrolling T cells. Each T cell has an exquisitely sensitive and specific T-cell receptor (TCR) that constantly surveys the organism in search of ‘non-self’ signals, triggering a cascading immune response when a foreign peptide is identified. T cells are therefore potent and precise killers of foreign entities, be these pathogenic invaders or malignant cells.
Tumours are frequently invaded by immune cells as part of the body’s response to malignant growth. Among these immune infiltrates, tumour-infiltrating lymphocytes (TILs) include T cells specific for mutated proteins that are present in cancer cells. These TILs can be isolated from a patient’s tumour tissues, expanded ex vivo to high numbers and adoptively transferred back into the patient to elicit a durable antitumour response1. However, this procedure carries several limitations: TILs need to be harvested during surgery, are not found in sufficient numbers for all cancers, and in many cases expand poorly2. A potentially broader therapeutic approach involves collecting polyclonal T cells from the blood and, through genetic engineering, inserting in them genes encoding a TCR that is specific for a known tumour antigen. For instance, T cells bearing an engineered TCR targeting the NY-ESO-1 antigen have yielded promising results in the treatment of patients with multiple myeloma and synovial cell sarcoma3,4.
Using tumour-specific TCRs enables the targeting of both membrane and cytoplasmic proteins; unfortunately, cancers can evade this endogenous recognition-and-response program by downregulating major histocompatibility complex (MHC) proteins or other factors required for antigen processing and presentation, rendering cancer cells invisible to patrolling T cells. To overcome this evasion strategy by the tumours, T-cell specificity can be directed to tumour-associated antigens (TAAs) in a MHC-independent manner. This can be achieved by employing chimeric antigen receptors (CARs).
CARs combine the specificity of a high-affinity recognition domain, most often derived from a monoclonal antibody (mAb), with the cytolytic properties of T cells. T cells modified using CARs (CAR-T cells) have shown unprecedented potency against haematologic malignancies, as exemplified by the several clinical trials of CD19-CAR-T-cell therapy that have reported complete response rates of 50–90% for refractory B-cell malignancies5,6,7,8,9,10,11,12,13. These results led to approval from the US Food and Drug Administration (FDA) in the second half of 2017 for the first CAR-T therapies14,15. Such early success has ignited a revolution in cancer immunotherapy, and intensified the focus on the development of strategies that extend the use of CAR-T cells to other haematological malignancies and, in particular, to solid tumours, which presents substantial challenges with regard to efficacy and safety. In this Review Article, we survey several next-generation approaches for the engineering of CAR-T cells that hold potential for overcoming these challenges. In what follows, we discuss the design of CARs for increased tumour specificity, and how functional modules and pathways can be implemented to regulate the activity of CARs for enhanced safety, including options available for enhancing the delivery of transgenes into T cells. We also discuss approaches for overcoming immunosuppression mechanisms that protect the tumour from the immune system, and describe how modulation of the tumour microenvironment can promote antitumour therapies.
The molecular architecture of CARs
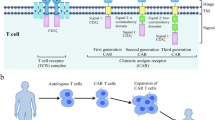
CARs transduce antigen recognition events into a signalling cascade that evokes T-cell effector functions, such as the secretion of cytotoxic factors and pro-inflammatory cytokines. The essential components of these synthetic receptors are an extracellular antigen-targeting moiety, such as a single-chain variable fragment (scFv), a transmembrane and hinge domain that anchors the receptor on the cell surface and projects the scFv out to the extracellular space, and intracellular signalling domains that are triggered on antigen engagement (Fig. 1a).

a, CARs comprise an extracellular antigen-targeting moiety, such as an scFv, a transmembrane and hinge domain that anchors the receptor to the cell surface and projects the scFv into the extracellular space, and intracellular signalling domains that activate the T cells following antigen engagement. First-generation CARs contain only a CD3ζ signalling domain. Second-generation and third-generation CARs incorporate one or more co-stimulatory domains (costim), respectively. b, The TCR complex is composed of variable α- and β-chains associated with CD3 invariant chains. Full T-cell activation requires signal 1 and 2 from the TCR complex, as well as co-stimulatory receptors. Signal 3, which is transmitted by cytokine receptors, is important for survival, differentiation and memory formation. TIM-3, T-cell immunoglobulin and mucin domain-containing protein-3; TIGIT, T-cell immunoreceptor with Ig and ITIM domains.
The antigen-binding domain
The antigen specificity of a CAR is most often conferred by the scFv module (the smallest functional domain of a mAb), which contains only the variable regions of the light and heavy chain fused via a flexible linker. The advantages of using scFvs as the CAR antigen-binding domain include their high specificity, the fact that they can be readily generated against most antigens of interest through well-established methods, and how easily they can be grafted into the CAR design16. Conversely, a disadvantage of using scFv-binding domains is their tendency for oligomerization, which can lead to tonic signalling and T-cell exhaustion17. Alternative binding domains have been used in preclinical studies, including receptors18, ligands19, cytokines20,21, DARPins (designed ankyrin repeat proteins)22, adnectins23, Fc receptor fragments24, nanobodies25, peptides26 and variable lymphocyte receptors27. The relative efficacy, safety and immunogenicity of these alternative binders have not yet been fully investigated and are the focus of substantial ongoing work.
The ability of a CAR to transduce a strong activating signal to the T cell is influenced by many factors, including the binding affinity to the antigen, the level of expression, antigen density on the target cell, and epitope proximity. The relationship between binding affinity and efficacy is more nuanced in the context of CARs as compared with mAbs, for which higher affinity is typically desirable. For example, on the basis of observations from preclinical studies, whereas a receptor tyrosine kinase-like orphan receptor 1 (ROR1)-CAR derived from a high-affinity scFv (with a dissociation constant of 0.56 nM) resulted in increased therapeutic index when compared with a lower-affinity variant28,29,30, other examples have reported that engineering the scFv for lower affinity improves the discrimination among cells with varying antigen density31,32,33, which could be useful for enhancing the therapeutic window for antigens differentially expressed on tumour versus normal tissues. However, this approach might increase the risk of immune evasion, owing to the emergence of low-antigen-expressing tumour cells.
Other factors such as the proximity of the target antigen-binding site to the cell surface can also have strong effects on lytic activity and cytokine production34,35, which in some cases supersedes the effects of affinity. In the context of CD22-CARs, a comparison between two scFvs targeting the same epitope but with varying affinities did not reveal a significant impact of affinity on CAR function, whereas an alternative CD22 binder (m971) with lower affinity proved most efficacious, possibly because of better accessibility of the targeted epitope34,35. Thus, for a given antigen density on a target cell, there is likely to be an optimal range of scFv affinities and CAR expression levels required for a specific and effective antitumor response36,37,38.
Co-stimulatory domains
First-generation CARs, which relied solely on the intracellular domain of CD3ζ for signalling, had limited efficacy in clinical trials owing to the weak expansion potential of the CAR-T cells in vivo39 and despite observations of significant antitumor responses with CARs of this kind that targeted the GD2 protein in neuroblastoma40,41. Incorporation of one or more co-stimulatory domains to provide additional activating signals (Fig. 1a) has resulted in enhanced clinical responses and persistence5,6,42,43,44. The choice of co-stimulatory domain influences the phenotype and metabolic signature of T cells. For example, CD28 co-stimulation yields a potent, yet short-lived, effector-like phenotype, with high levels of cytolytic capacity, interleukin-2 (IL-2) secretion and glycolysis45. By contrast, T cells modified with CARs bearing 4-1BB co-stimulatory domains tend to expand and persist longer in vivo, have increased oxidative metabolism, are less prone to exhaustion, and have an increased capacity to generate central memory T cells17,44,45,46,47,48. Whereas the vast amount of studies to date are based on CD28 or 4-1BB CARs, several other co-stimulatory domains are currently being evaluated (Table 1)48,49. Preclinical studies have reported that third-generation CARs — bearing both CD28 and 4-1BB domains — have superior efficacy when compared with second-generation constructs50,51; however, others reported opposite findings52,53. Expression of the 4-1BB ligand in CD28ζ-based CARs has also been shown to extend CAR-T-cell persistence, and is currently being tested in clinical trials47 (NCT03085173).
Hinge and transmembrane domains
The flexible hinge domain is a short peptide fragment that provides conformational freedom to facilitate binding to the target antigen on the tumour cell. It may be used alone or in conjunction with a spacer domain that projects the scFv away from the T-cell surface28,54. Previous studies have suggested that the optimal length of the spacer depends on the proximity of the binding epitope to the cell surface, with proximal epitopes requiring longer spacers and distal epitopes requiring shorter ones28,55.
Besides promoting binding of the CAR to the target antigen, achieving an optimal distance between the T cell and the cancer cell may also help to sterically occlude large inhibitory molecules from the immunological synapse56. Long spacers typically include the CH2CH3 domain (~220 amino acids) of immunoglobulin G1 (IgG1) or IgG4 (refs 34,57), whereas the CH3 region can be used on its own to construct an intermediate spacer28 (~120 amino acids). Shorter spacers are derived from segments (<60 amino acids) of CD28 (refs 30,34,58), CD8α (refs 30,34,50,58,59), CD3 (ref. 60) or CD4 (ref. 61). Several CARs with long spacers comprised of the CH2CH3 domain were reported to have diminished efficacy and persistence in vivo despite potent effector function in vitro, a disparity that was shown to result from binding of Fc receptors expressed on macrophages, monocytes and natural killer (NK) cells. These off-target interactions resulted in increased activation-induced cell death of CAR-T cells and cross-activation and lysis of these innate immune cells62. Following these observations, deletion and mutation of CH2CH3 residues that resulted in abrogated Fc receptor binding led to a significant improvement of in vivo persistence and efficacy in murine models57,63.
The properties of the transmembrane domain have not been as meticulously studied, but they can potentially affect CAR expression and association with endogenous membrane proteins58,64,65.
Improving safety
Unlike traditional cancer drugs (such as small molecules and antibodies) that rely on passive targeting, T-cell-based therapeutic agents can actively home to disease sites, sense the microenvironment and integrate multiple inputs to mount a complex and dynamic immune response. These cells are also capable of self-amplification, renewal and differentiation into distinct effector subsets. Despite all these advantages, such complex behaviour can pose challenges with regard to safety. Recent clinical trials have reported CAR-T-associated toxicities, such as cytokine-release syndrome (CRS), neurotoxicity, off-tumour effects and acute respiratory distress syndrome, all of which are potentially fatal66,67,68. These adverse effects highlight the need for developing control programs to modulate the activity of CARs, and numerous approaches have been explored to program these genetically engineered systems with feedback control systems and safety modules, by leveraging recent advances in synthetic biology (Figs. 2 and 3).

a, The addition of a dimerizing drug activates iCasp9 signalling and leads to apoptosis69. b, T cells expressing HSV-TK convert the prodrug ganciclovir into a cytotoxic molecule that results in cell death244. c, A truncated form of EGFR acts as an epitope handle that can be used to target T cells with a mAb such as cetuximab71. T cells that are tagged with cetuximab are killed through antibody-dependent cell-mediated cytotoxicity or complement-dependent cytotoxicity. HSV-TK, herpes simplex virus thymidine kinase; GCV-MP, ganciclovir monophosphate; GCV-TP, ganciclovir triphosphate; tEGFR, truncated EGFR.

a, CAR domains can be split into separate non-functional receptors. Assembly of the CAR can be induced through the addition of a dimerizing drug76. b, sCARs are specific for a bi-orthogonal tag, such as a peptide neo-epitope (PNE). sCAR-T cells can be directed to a TAA through the addition of a Fab tagged with PNE78. c, CAR expression can be controlled by TRE3G-based expression systems. The addition of doxycycline induces a conformational change in TRE3G that enables it to turn on the expression of the CAR75. FKBP, FK506 binding protein; FRB, FKBP12-rapamycin binding domain; TRE3G, tet response element 3G.
Control switches
One strategy to control severe CAR-T-cell toxicity is to include a mechanism for rapid ablation of the transplanted cells through a suicide switch (Fig. 2). For example, cells expressing inducible caspase 9 (iCasp9) can be killed by the addition of a small-molecule drug that dimerizes and activates the protein to induce programmed cell death69. This system was used to eliminate 90% of T cells expressing iCasp9 within 30 minutes in patients receiving a hematopoietic stem cell transplant, and reversed graft-versus-host disease (GVHD)70. Alternative suicide genes include epitope tags that are recognized by FDA-approved mAb therapeutics such as cetuximab71,72 or rituximab73,74, which induce T-cell death through endogenous antibody-dependent cell-mediated cytotoxicity and complement-dependent cytotoxicity. These epitope tags can also serve as handles for purification of transduced T-cell populations11,12,13. Neither drug-induced nor mAb-mediated suicide switches have been tested clinically in the context of CAR-based therapeutics, and therefore it remains unclear how effective they may be in mitigating both on-target and off-target toxicity.
Suicide switches are effective at stopping the toxicity of the engineered cells, but result in the irreversible termination of the therapeutic treatment. Alternatively, non-cytotoxic reversible systems may be useful for controlling adverse toxicity without deleting the therapeutic cells (Fig. 3). For instance, the CAR gene can be placed under the control of inducible expression systems that toggle the transcription of the CAR between on and off states through small molecules such as doxycycline and tetracycline. These safety mechanisms may have limitations, such as a relatively slow induction or reduced receptor expression, and inherent leakiness of the system can lead to residual CAR expression even in the absence of the inducer75. Chemical inducers can also be used to control the assembly of the CAR signalling domains at the protein level through drug dimerization domains. In this context, a small molecule that brings together the ζ-chain and CAR to modulate CAR activation (Fig. 3a)76 and another similar model involving a MyD88/CD40 inducible co-stimulatory molecule49,77 have been reported.
Modular and switchable CAR (sCAR) designs in which the specificity is determined by a soluble antigen-binding fragment (Fab), administered in parallel to the sCAR-engineered cell, add important safety and specificity functionalities (Fig. 3b)78,79. Rather than recognizing an antigen, the sCAR binds to a bioorthogonal tag placed on a Fab derived from a mAb such as Herceptin. In this design, the Fab acts as an adapter that bridges the T cell and the TAA-expressing cell. Alternatively, CARs specific for Fc domains can be directed at TAAs through co-administration of commercially available or proprietary mAbs24,80. These approaches are only now being tested in the clinic, but preclinical results suggest that CAR activity and specificity can be tuned according to the type and dose of the bridging antigen-binding molecule administered, which may provide a means to both limit the activity of CARs for safety and target multiple antigens, thus hindering antigen escape by the tumour.
Combinatorial antigen sensing
An ideal tumour antigen is exclusively expressed by tumour cells and not by normal tissues, as in the case of epitopes derived from tumour-specific mutated or glycosylated proteins59,81. Although a great effort is being applied to discovering such antigens, the vast majority of candidate antigens are also expressed on healthy tissues, albeit at lower levels. Consequently, T-cell-based therapies targeting these antigens carry the potential for on-target, off-tumour toxicities82,83,84. One strategy to mitigate these effects and increase specificity is to require multiple TAAs to be engaged for full T-cell activation (AND gate, Fig. 4a). This strategy was used in a system that employed a synthetic notch receptor (synNotch) that releases a transcription factor on binding antigen ‘A’, which in turn drives expression of a CAR specific for antigen ‘B’85. These AND-gated T cells were able to clear tumours bearing both antigens in a xenograft murine tumour model while sparing cells expressing only one of the antigens. However, once the synNotch induced expression of the second CAR, that expression was sustained for some time; therefore, such cells could travel throughout the body and subsequently react against normal tissues expressing antigen B. Although this type of toxicity was not observed in mouse models, these were short-term experiments that do not fully replicate the effects of long-lived T cells in humans.

a, On binding antigen A, the synNotch receptor undergoes a conformational change that leads to the release of a transcription factor (TF), which subsequently translocates to the nucleus and turns on the expression of a CAR for antigen B85. b, iCARs dampen the T-cell response when a healthy antigen is encountered. T-cell activation only occurs in the absence of the healthy antigen86. c, Bispecific CARs can be activated in response to a cell expressing antigen A or antigen B.
CARs can also be made to discriminate between cancer and healthy cells through co-expression of an inhibitory CAR (iCAR) that dampens the T-cell response when a healthy antigen is present (AND–NOT gate, Fig. 4b)86. In iCARs, an antigen-binding domain against a ‘healthy tissue’ antigen is fused to inhibitory intracellular signalling domains, such as those in programmed cell death protein 1 (PD-1) or cytotoxic T-lymphocyte-associated protein 4 (CTLA-4). Other combinatorial antigen approaches are based on segregation of the CD3ζ domain and the co-stimulatory domain on two different CARs87,88; in this way, the ‘signal 1’ and ‘signal 2’ necessary for T-cell activation (Fig. 1b) are only combined in response to the engagement of both antigens. A potential disadvantage of this approach is that it assumes that individual CD3ζ or co-stimulatory CARs will not function alone; however, activity has been detected in CD3ζ-only CARs30,40, which could mediate deleterious effects if co-stimulatory signals from an inflammatory tumour microenvironment are provided.
The specificities of natural TCRs can provide additional input for CAR-T cell sensing. γδ T cells comprise a small fraction of circulating and tissue-resident lymphocytes that recognize antigens in a MHC-independent manner. A subset of these cells expressing the Vγ9Vδ2 TCR are thought to recognize transformed or metabolically dysfunctional cells by binding to specific phosphoantigens that act as ‘danger signals’ presented on the cell surface89. This property has been explored by inserting GD2-CARs into γδ T cells so that cytotoxicity towards GD2+ tumour cells was restricted only to those expressing these danger signals90. Furthermore, by employing a CAR that contained only a DAP10 co-stimulatory signalling domain, both the γδ TCR and CAR needed to be engaged for lytic activity — an approach that could potentially limit off-tumour effects.
Although targeting TAAs also expressed on normal tissues risks on-target, off-tumour toxicity, evidence from preclinical models suggests that CAR-T cells may not react to low-level antigen expression in healthy tissues33,36,37,38. Instances of human epidermal growth factor receptor 2 (HER2)-CAR off-tumour toxicity82 have been largely overshadowed by safe trials with a distinct HER2-CAR that demonstrated clinical efficacy in patients with sarcomas91,92. Combinatorial antigen-sensing programs have the potential to provide increased safety by adding an extra layer of specificity to avoid adverse toxicities; however, increasing the stringency of antigens required for CAR activation could facilitate cancer cell evasion via antigen escape.
Autonomous sensing and activation
CAR safety modules can also be designed to exert spatial control over targeting by activating the CAR-T cells only at the site of the tumour (Fig. 5). Masked CARs achieve this by including an inhibitory peptide, tethered extracellularly via a cleavable linker, that reversibly blocks the scFv and keeps the T cells in an off state until they reach the tumour microenvironment93. Once there, proteases specifically secreted locally cleave the inhibitory peptide and unmask the CAR, priming it for targeting tumour cells. One report showed that a masked CAR had comparable efficacy to non-masked CARs when targeting the epidermal growth-factor receptor (EGFR) in a murine xenograft model, but further studies are required to demonstrate that the strategy leads to decreased on-target, off-tumour effects given that the scFv used was derived from cetuximab, which has minimal cross-reactivity with mouse EGFR94. Another feature of the tumour microenvironment that can be explored for context-dependent activation of CARs is the hypoxic milieu generated by inadequate tumour vascularization95. This feature has been taken advantage of by the design of a CAR with oxygen-sensitive domains that is degraded in normoxic conditions but remains stabilized in hypoxic conditions96. Although the surface expression of this construct was low and the system had a limited dynamic range, it nevertheless demonstrates that the concept of engineering T cells with microenvironmental sensors can enable control over their activity.

a, Masked CARs are kept in an off state through a decoy peptide that is tethered via a cleavable linker. Once the T cell reaches the tumour microenvironment (TME), specific proteases liberate the decoy peptide and unmask the CAR for targeting93. b, Oxygen-sensing CARs are selectively degraded under normoxia, yet stably expressed in hypoxic conditions. c, γδ TCRs can recognize transformed cells through danger signals expressed on the cell surface90.
If CAR therapy is to become widely applicable, it is without doubt that further developments of CAR-T technology need to address the potential for toxicity. The on-target, off-tumour effect observed with the CD19-CAR, which results in killing of healthy B cells, is generally well tolerated and can be managed with intravenous immunoglobulin therapy. However, these effects may be detrimental when targeting TAAs in solid tumours, as most of these are also expressed on essential tissues. Several cell-engineering strategies have provided possible means to improve specificity and control of basic CAR-T function in vivo. Future developments may include autonomous feedback loops and control mechanisms that enable engineered T cells to continuously sense key biochemical cues in the local microenvironment and to adjust their response accordingly. For instance, a CAR-T cell could potentially be programmed to secrete proteins that neutralize the effects of cytokines associated with CRS when inflammatory cytokines surpass a specific threshold. With many unresolved challenges left in the field, it is probable that CARs will continue to drive development and clinical testing of novel synthetic biology tools.
Improving efficacy and overcoming immune suppression
Improving the efficacy of CAR-T therapies to extend their application beyond blood cancers has met several formidable hurdles. After infusion into a patient, the T cells must traffic to the tumour site, infiltrate the tumour stroma and engage the specific TAA on malignant cells. Then, they must rapidly expand to high numbers in response to an antigen and mount the attack on cancer cells within a harsh tumour microenvironment that features multiple physical barriers97,98,99, a biochemical and metabolic milieu that is unfavourable to T-cell effector function100,101,102, and immunosuppression signals from an arsenal of inhibitory molecules displayed or secreted by cancer cells and supporting stromal cells alike103,104,105 (Fig. 6). Additionally, T-cell activation must be sustained to enable clearing of the tumour before the cells become dysfunctional or exhausted106,107. Ideally, the CAR-T cells should then persist after tumour elimination and form memory cells to provide long-lasting protection. Engineering T cells with additional functional modules, along with their use in combination therapies, can help circumvent these obstacles (Table 2).

T-cell homing and infiltration is impeded by mismatched chemokine receptors and by the lack of adhesion molecules in the dysregulated tumour vasculature. CAFs create a physical barrier to T-cell infiltration by maintaining a dense ECM that traps T cells at the periphery of the tumour. Tumour-associated cells such as Treg cells, MDSCs and TAMs support the tumour by suppressing immune responses. T cells can be inhibited through checkpoint receptors such as PD-1 and CTLA-4, which are engaged by cognate ligands expressed on tumour cells or tumour-associated cells. Soluble inhibitors in the form of suppressive cytokines (TGF-β, IL-10 and IL-6) disrupt T-cell effector functions. The metabolic milieu of the tumour microenvironment is unfavourable to effector T cells owing to a lack of nutrients, acidic conditions and toxic byproducts.
Homing and trafficking
To leave blood vessels and accumulate within solid tumours, circulating T cells must undergo extravasation, a highly coordinated process mediated by an array of factors such as adhesion molecules, chemokines and chemokine receptors108. The tumour vasculature, however, has a deregulated chemokine profile and downregulates key adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1) (ref. 109). In addition, natural T cells may not express the appropriate chemokine receptors to enable trafficking into specific tumour tissues, leading to poor homing and opening the door to tumour immunoevasion110. One strategy to overcome this deficit is to engineer T cells to express chemokine receptors such as CCR2 (refs 111,112), CCR4 (ref. 113) and CXCR1 (ref. 114). Tumours often secrete chemokines for these receptors to attract supporting stromal cells; thus, ectopic expression of these receptors or other factors that may enhance tumour trafficking in adoptively transferred T cells provides a potential means to preferentially direct their migration to the tumour site, which has been shown to improve antitumor efficacy in animal models.
Expansion and persistence
After trafficking to the tumour site and encountering their cognate antigen, T cells must undergo rapid expansion to attain the appropriate numbers relative to the tumour burden. The efficacy of CAR-T-cell treatment in clinical trials has been highly correlated with the T cells’ capacity for in vivo expansion and persistence5,44,115. Preconditioning treatments with chemotherapeutics that deplete native lymphocytes12,13 or small-molecule inhibitors such as ibrutinib have been shown to improve CAR-T-cell engraftment in haematologic malignancies115,116. For CAR-T cells to achieve optimal activation and proliferation, they require not only CD3ζ and co-stimulatory domain activity, which provide signals 1 and 2, but also a third signal provided by immunostimulatory cytokines. However, these activating cytokines are often downregulated in the microenvironment of solid tumours. To overcome this hurdle, several transgenic-based strategies have been developed to deliver those missing cytokines. Preclinical studies have shown that T cells engineered to secrete IL-12 (refs 117,118), IL-15 (ref. 119), IL-18 (ref. 120) and IL-21 (ref. 121) can help to promote the cells’ proliferation and reshape the tumour microenvironment. However, exogenous expression of such cytokines can ultimately hinder ex vivo expansion and result in increased toxicity in some cases122. In addition, several inflammatory cytokines may potentially counteract CAR-T-cell therapy by triggering negative feedback regulatory loops to dampen inflammation via indirect induction of immunosuppressive responses123,124,125.
Chimeric cytokine receptors — engineered chemokine receptors with modified functions — can be used to achieve selective expansion of T cells by an administered cytokine126. However, systemic administration of cytokines can result in significant toxicities127,128,129,130. It has been shown that an engineered, constitutively active IL-7 receptor (IL-7R) does not require extracellular IL-7 for the antitumor effects of GD2-CAR-T cells against metastatic neuroblastoma and orthotropic glioblastoma xenograft models131. The receptor, derived from a naturally occurring gain-of-function IL-7R mutant found in lymphoblastic leukaemias132,133, has a cysteine in the juxtamembrane domain that promotes dimerization of the receptor and initiates signalling independently of IL-7. In another example of this strategy, transgenic expression of cytokines tethered to their receptors can deliver stimulatory signals to the T cell while diminishing the risk of systemic toxicities associated with the administration of soluble cytokines134.
Vaccine-based approaches can also be used to enhance T-cell proliferation by using CAR-T cells that keep an identified native TCR, such as one that is specific for a viral antigen135,136,137,138. In this context, administration of an attenuated virus, such as vaccinia, or viral antigens present in host cells, provides stimulation through the native TCR and promotes T-cell activation. However, clinical trials evaluating virus-specific CARs have not shown a clear advantage compared with standard second-generation CAR-T therapy41,136,139. In fact, preclinical studies have shown that signalling through both the TCR and the CAR can accelerate T-cell exhaustion and result in diminished persistence46.
Targeting the tumour stroma and supporting cells
It has been well established that solid tumours coerce non-transformed stromal and immune cells, such as myeloid-derived suppressor cells (MDSCs), tumour-associated macrophages (TAMs), cancer-associated fibroblasts (CAFs), tumour endothelial cells (TECs) and regulatory T cells (Tregs), to support and maintain the tumour97. Importantly, these conditioned cells can directly suppress CAR-mediated antitumor effects in the tumour microenvironment53. In some cases, malignant cells can recruit these supporting cells to perform various functions, such as remodelling of the extracellular matrix (ECM), enhancement of angiogenesis and secretion of growth factors, and evasion of immune surveillance. Targeting supporting cells can thus help promote lymphocyte infiltration, and may weaken the immunosuppressive barriers of the tumour microenvironment. CAR-T cells have not only been administered concomitantly with agents that modulate the tumour microenvironment53, but have also been engineered to directly modulate it, for example through overexpression of heparanase for degradation of the ECM, which resulted in enhanced therapeutic efficacy in a neuroblastoma tumour model140. CAFs involved in maintaining the ECM can also be targeted using CARs specific for fibroblast activation protein141, and other cancer-associated cells such as TAMs, TECs and Tregs can be targeted using CARs directed against tumour-microenvironment-specific antigens118,142,143,144.
Immunosuppression in the tumour microenvironment
Tumour cells, together with tumour-associated stromal cells, suppress T-cell function through an array of immunosuppressive signals, such as transforming growth factor-β (TGF-β), IL-10 and the Fas ligand (refs 97,145). Tumour cells can also activate immune checkpoint receptors on T cells, such as the receptors PD-1 and CTLA-4, via expression of their ligands. Immune checkpoints are part of the physiological mechanisms for attenuating T-cell activation. Blocking the activation of immune checkpoint receptors by using mAbs, a strategy for reinvigorating T cells termed checkpoint blockade, has recently emerged as an effective immunotherapy for several cancers106. Preclinical studies have shown a synergistic effect when CAR-T therapy is combined with checkpoint blockade146,147, and recent clinical reports have suggested that anti-PD-1 agents enhance the efficacy of CD19-CAR-T therapy in patients with refractory diffuse large B-cell lymphoma and acute lymphoblastic leukaemia148,149. Despite these encouraging reports, a phase-I study that used a third-generation GD2-CAR for patients with neuroblastoma reported absence of improvements in efficacy when combined with anti-PD1 administration150. Therefore, the potential benefits of combining CAR-T-cell therapy with checkpoint blockade have to be tested further by taking into account the specific CAR used and the patient’s clinical history. Moreover, whereas combining checkpoint blockade with CAR-T cells may improve their potency, it could also increase toxicity, owing to the dampening of T-cell safety mechanisms.
Immunosuppression in the tumour microenvironment can also be circumvented with engineered dominant-negative receptors, which are truncated receptors for inhibitory molecules (such as TGF-β) that lack intracellular signalling domains151. These decoy receptors compete with endogenous receptors by binding to and depleting the ligand, resulting in decreased signalling of inhibitory pathways in immune cells. Taking this approach to the next level, the immunosuppressive tumour microenvironment can also potentially be used against the tumour itself through switch receptors: using this approach, inhibitory signals can be reversed into stimulatory signals with fusion receptors composed of the extracellular domain of inhibitory receptors (such as PD-1) and the signalling domains of stimulatory molecules (such as CD28)152.
Selectively knocking out endogenous genes through targeted nucleases, a powerful technique that is revolutionizing T-cell engineering, can also be applied to prevent tumour-derived immunosuppression. Checkpoint molecules such as PD-1 (ref. 153), CTLA-4 (ref. 154) and lymphocyte activation gene 3 (LAG-3) (ref. 155) have been efficiently deleted by CRISPR–Cas9 (clustered regularly-interspaced short palindromic repeats–CRISPR-associated system 9) or TALENs (transcription activator-like effector nucleases), rendering T cells less prone to checkpoint inhibition and exhaustion, and resulting in improved antitumor efficacy and persistence in preclinical models. Advances in multiplexed assays have enabled targeting of multiple gene targets for simultaneous deletion154,156. However, caution must be exercised with such approaches as these inhibitory receptors are part of physiological processes involved in modulating T-cell activity, and permanently unleashing T cells with no feedback and attenuation mechanisms may increase the risk of toxicity owing to uncontrolled activation. Importantly, recent work demonstrated that PD-1 is a tumour suppressor gene, raising the possibility that deletion of PD-1 in engineered T cells could ultimately increase the risk of malignant transformation157.
Metabolism and suppression
Following activation, T cells must adapt their metabolism to meet the energetic demands associated with rapid proliferation and effector function, such as the production of cytokines and cytotoxic payloads. Consequently, the metabolic profile of T cells is intimately linked to the regulation of T-cell function and differentiation stage. The aberrant metabolic milieu in tumour microenvironments, as a result of the high metabolic activity of tumour cells and dysfunctional tumour vasculature, is both hypoxic and acidic, depleted of nutrients such as glucose and glutamine, and lacks key amino acids such as arginine and tryptophan due to the upregulation of the inhibitory enzymes indoleamine 2,3-dioxygenase-1 (IDO-1) and arginase by the tumour stroma100. This unfavourable environment inhibits T-cell effector functions and promotes a defective T-cell state. Metabolically reprogramming T cells by gene editing can potentially help them resist these conditions. Ectopic expression of PPAR-γ co-activator 1α (PCG1α) can rescue mitochondrial function and lead to increased cytokine production in T cells, resulting in enhanced antitumor efficacy158. Modifying cholesterol metabolism by knocking out acyl-CoA:cholesterol acyltransferase 1 (ACAT1), an enzyme involved in esterification of cholesterol, can also potentiate the effector functions of CD8+ T cells159, whereas resistance to reactive oxygen species can be conferred by expression of catalase, which relieves oxidative stress in the tumour microenvironment160.
The choice of CAR co-stimulatory domain can also drive T cells towards distinct metabolic profiles. Whereas co-stimulation through CD28 enhances aerobic glycolysis, 4-1BB drives mitochondrial biogenesis and oxidative metabolism, which may account for the phenotypic differences seen between CARs with these different domains17,45.
Avoiding antigen escape with bispecific CARs
Resistance to CAR-T therapy has been observed in the form of antigen escape, a process by which the tumour escapes CAR targeting by downregulating or mutating the CAR’s target antigen161 via selection of antigen-negative cells7 or via lineage switch162,163. Although clinical trials with the CD19-CAR in B-cell acute lymphoblastic leukaemia (B-ALL) have reported high complete response rates (70–90%), relapse with CD19-negative tumours occurred in >20% of responders5,7,11,164,165,166,167. In the context of solid tumours, antigen escape poses a potentially even greater challenge given their extensive heterogeneity in antigen expression. A recent trial of ten patients with glioblastoma treated with peripherally infused EGFR variant III (EGFRvIII)-specific CAR-T cells reported that five of seven patients who underwent tumour resection after treatment had reduced EGFRvIII expression in tumour tissue168. EGFRvIII antigen escape has also been implicated as a resistance mechanism for a peptide vaccine169. In a separate trial, a patient with glioblastoma treated with an IL-13 receptor α2 (IL13Rα2)-specific CAR had recurrent tumour with decreased antigen expresssion170. These clinical data, alongside preclinical studies demonstrating the emergence of antigen loss variants, underscore the need for targeting multiple antigens119,161,164,171,172,173.
Bispecific CARs have the potential to mitigate this escape mechanism by targeting two antigens simultaneously (OR gate, Fig. 4c)172. Tandem bispecific CARs can be constructed by linking two scFv domains in series174,175; these constructs may require optimization of the orientation of the heavy and light chains to achieve correct pairing and a favourable conformation for binding, and the linker length needed for targeting each antigen must be taken into consideration. For a bispecific CAR targeting CD19 and CD20, a CD19-proximal and CD20-distal scFv configuration was found to be ideal as these antigens require a short and long spacer, respectively174; in this format, the CD19 scFv effectively served as a linker to project the CD20 scFv away from the surface. In another example, a CD19–CD22 bispecific CAR required a loop configuration, with the CD19 variable heavy (VH) and light (VL) chains flanking the CD22 scFv (CD19VL–CD22VH–CD22VL–CD19VH); this CD19–CD22 bispecific CAR is currently being tested in clinical trials for both paediatric and adult B-ALL and lymphoma176 (NCT03241940 and NCT03233854).
Alternative formats of multi-antigen CAR therapies involve co-expressing two or more CARs of different specificities in the same cell using ribosomal skip sequences, or treatment with mixed CAR-T cell populations177,178,179. This combinatorial approach circumvents the need for engineering and optimizing a new CAR construct; yet it is important to note that dual expression of two full-length CARs substantially increases the genetic payload to be delivered to cells, whereas adding an additional scFv module has only a minimal impact on the payload. This is an important consideration when devising cell therapies: for viral-based gene delivery, for example, larger constructs are associated with decreased transduction efficiencies. In the only study that has directly compared multi-antigen CAR platforms to date, pooled infusions of monospecific CARs were inferior to the tandem CAR or to dual CAR expression on the same cells in a preclinical model of glioblastoma multiforme172.
The immune response induced by CAR-T-cell killing of tumour cells could also play an important part in preventing the rise of antigen-loss variants by inducing ‘epitope spreading’, which occurs when lysis of tumour cells in an inflammatory milieu leads to presentation of additional tumour antigens to lymphocytes, inducing parallel cell-mediated and humoral antitumour immune responses180,181. A preclinical study utilizing immunocompetent mice supported this model: mice whose EGFRvIII-positive tumours were cleared by treatment with EGFRvIII-CAR-T cells could reject EGFRvIII-negative tumour cells in subsequent re-challenge experiments, whereas tumour naive mice could not reject EGFRvIII-negative cells182. In clinical studies, patients receiving mesothelin-specific CAR-T cells developed elevated antibody responses against self-proteins, including mesothelin183. These data also raise the prospect of potentially enhancing the generation of physiological immune responses through combination therapies with checkpoint inhibitors that could potentially unleash tumour-reactive T cells and enable antitumour immunity beyond the specificity determined by the CAR. The contribution of epitope spreading to antitumor activity in patients undergoing CAR-T therapies remains unknown, as this phenomenon has not yet been thoroughly evaluated in clinical trials.
Other strategies for increasing efficacy
An alternative approach for cancer immunotherapy is based on the use of T cells as vehicles to deliver therapeutic agents to the tumour site. T cells engineered to secrete a soluble form of herpes virus entry mediator that inhibited B-cell lymphoma proliferation through the B and T lymphocyte attenuator signalling axis augmented the efficacy of CD19-CAR-T therapy184. A separate study reported a versatile system, termed ‘synNotch’, for the integration of signals from molecular stimuli in the local microenvironment into cellular responses185. Using this approach, T cells could be programmed to express antibodies, adjuvants, cytotoxic molecules or cytokines in response to binding to target cells.
MicroRNAs (miRs) are another attractive target for improving CAR-T therapy, as these non-coding RNAs are involved in the physiological regulation of T-cell development and effector function186,187,188,189. However, the immunosuppressive tumour microenvironment can downregulate key miRs such as miR-17-92 (ref. 190). Transgenic expression of miR-17-92 in CAR-T cells, for instance, led to enhanced proliferation, interferon-γ (IFN-γ) production and increased the durability of treatment in a murine glioma xenograft model191. miR engineering is a promising strategy to augment CAR-T-cell function, potentiated by its capacity for genetically encoded transcriptional control of endogenous or exogenous genes191,192.
Advances in biomaterials and drug delivery offer promising alternative avenues for increasing the efficacy of adoptively transferred cells. T cells can be outfitted with nanogel or liposome ‘backpacks’ loaded with therapeutic agents such as TGF-β inhibitors193,194 or IL-15 (ref. 195), endowing the treatment with time-limited release functionality. The release properties of these depot formulations can be fine-tuned by chemical means or even configured to unload during cytolysis through perforin-mediated rupture of the particles196. T cells can also be loaded inside biopolymers197, along with adjuvants, and implanted directly onto solid tumours198. In this approach, the combination of debris from dying cells and the adjuvant promotes activation of immune cells directly at the tumour site.
Towards an off-the-shelf T-cell product
The first two CAR-T therapies to be approved by the FDA, Kymria and Yescarta, are currently priced at US$475,000 and US$373,000, respectively199,200,201,202. Although some consider this a steep cost to pay, others deem it conservative given the robust potency, response rates and curative potential against otherwise refractory conditions. Nonetheless, providing personalized CAR-T treatment to the millions of patients with cancer worldwide may not be economically sustainable with current technologies. Furthermore, manufacturing autologous T cells ex vivo may be problematic when patients have low T-cell counts, as occurs with those who have been heavily pre-treated with chemotherapy regimens. Therefore, the development of universal, off-the-shelf T-cell products that are effective and persistent could dramatically reduce manufacturing costs and increase accessibility to this revolutionary cancer therapy.
An alternative — and potentially less costly — approach is to use T cells from an allogeneic source46,203,204,205, but this strategy bears additional complications in the form of potential cellular rejection and GVHD206. In this context, disruption of key genes through gene-editing tools can offer solutions156: a TALEN-based editing approach was used to create an allogeneic CD19-CAR-T treatment for two infants with B-ALL (ref. 207), whereby native TCRs from the donor cells were depleted by disrupting the TCR-α constant locus (TRAC) to render them non-reactive to the recipient’s endogenous cells. The CD52 locus was also targeted for deletion so that the infused cells could be spared from lymphodepletion by the anti-CD52 antibody administered to deplete the host T cells. The infants achieved molecular remission after treatment but developed GVHD, possibly due to residual TCR-expressing cells present in the donor product207. This strategy is currently being investigated in clinical trials for CARs targeting CD19 (NCT02808442) and CD123 (NCT03190278 and NCT03203369). Another strategy to avoid GVHD is to express the CAR in haematopoietic stem cells or other T-cell precursors that can be rendered tolerant to host antigens through the natural process of T-cell selection and maturation in a functional thymus208,209,210. Alternatively, cells may be sourced from induced pluripotent stem cells by using cellular reprogramming techniques, providing a potentially unlimited supply of T-cell precursors that can be expanded indefinitely and differentiated in vitro into T-cell subsets expressing specific CARs211,212.
Knocking out the TCR effectively avoids GVHD, but the infused cells can still be rejected by the host immune system. A possible solution involves the disruption of the β-2 microglobulin gene, which can ablate human leukocyte antigen I (HLA-I) expression to help donor CAR-T cells evade host CD8 T cells154,156. However, the CAR-T cells could still be vulnerable to rejection via CD4 cytolytic cells or lysis by NK cells as part of the ‘missing-self’ response213,214. Inserting non-classical HLA-E molecules, which are much less polymorphic than HLA-I, may help to avoid NK-cell-mediated rejection by the missing-self response215,216.
An alternative approach to overcome the costs of ex vivo individualized cell generation is to transduce the T cells in vivo. In this regard, a recent study has provided proof-of-concept evidence of nanoparticle carriers loaded with DNA encoding the CAR and a transposase that facilitates transgene integration217. The modified cells showed significant expression levels and expression frequencies, and mediated antitumor effects in a preclinical model. T cells can be targeted in this approach by using anti-CD3ε antibodies decorated on the nanoparticle surface.
Delivery of CAR genes
Delivery of CAR-encoding genes to T cells ex vivo is most commonly achieved with viral vectors5,6,218. Gamma-retroviral- and lentiviral-based delivery result in high expression and transduction efficiencies, but these have a limited payload (~8 kilobases (kb)) and pose the theoretical risk of insertional oncogenesis219. The potential for this risk arises from the random nature of integration into the host genome, although no instances of this occurring in clinical trials using CAR-T cells engineered through these methods have been reported. Alternatively, transposon-based systems are capable of delivering a larger payload (~100 kb), cost effective, scalable and potentially less immunogenic than viral vectors220,221,222. This approach requires delivery of plasmid DNA encoding the transgene and transposase components, which is most commonly achieved through electroporation221, a method that can be toxic to the cells and is not as efficient as viral-based delivery. Hybrid systems that utilize non-integrating viral vectors to deliver the transposase and transgene are currently under investigation223,224,225.
Non-integrating transient expression systems such as mRNA electroporation avoid the potential for insertional mutagenesis, but expression of the CAR is short-lived and is diluted with each cell division183. The development of targeted nucleases such as CRISPR–Cas9226, TALENs207, zinc fingers227 and homing endonucleases228 enable the targeting of the CAR gene to specific sites in the genome. Recent work has also demonstrated that delivering the CD19-CAR gene into an endogenous TCR locus by CRISPR–Cas9 resulted in uniform CAR expression, reduced tonic signalling and TCR-like regulation of expression, and enhanced T-cell potency226. The ideal target locus for other CARs may differ depending on the level of expression needed for optimal function.
Clinical results
The majority of CAR-T cell clinical trials conducted thus far have targeted CD19 to treat B-cell malignancies. CD19 is an ideal target, owing to it being highly expressed in the B-cell lineage and not being expressed in other tissues. Moreover, the on-target, off-tumour toxicity that results in B-cell aplasia can be managed with intravenous immunoglobulin. Remarkable responses have been observed with CD19-CAR-T therapy in patients with refractory B-ALL, chronic lymphocytic leukaemia, non-Hodgkin lymphoma, follicular lymphoma and diffuse large B-cell lymphoma229. Additionally, the first results of a CAR targeting CD22 on B-cell malignancies, published within the past few years, showed impressive activity in CAR-naive patients and patients who relapsed with CD19-negative disease following CD19-CAR-T therapy230,231,232. Clinical trials of targeting other B-cell antigens, such as B-cell maturation antigen (refs 233,234), CD20 (refs 235,236), Igκ (refs 237,238) and ROR1 (refs 239,240), are underway.
Adverse effects
The primary adverse effects observed with CAR-T therapies have been CRS and neurotoxicity. Neurotoxicity occurs in approximately 40% of patients and is characterized by aphasia, seizures, ataxia, delirium and other disturbances of the nervous system. In the vast majority, the symptoms are fully reversible, but in a small number of patients it led to cerebral oedema and death. Although the pathophysiology of this syndrome has remained enigmatic, a recent study proposed a plausible mechanism based on clinical data from patients receiving CD19.BBz-CAR-T cells68: CAR-T cells can induce endothelial cell activation in the central nervous system, resulting in increased permeability and coagulopathy; a low ratio of angiopoietin 1 (ANG1) to ANG2 then results in dysregulated endothelial cell activation and a breakdown of the blood–brain barrier. These data raise the prospect that approaches for normalizing the ANG1/ANG2 ratio could prevent or reverse the syndrome, and that agents such as recombinant BowANG1 could potentially be tested for this indication in the context of a clinical trial241. Less potent CARs might induce cytotoxicity but secrete lower cytokine levels. One such CAR targeting CD19 with a low-affinity binder is already in clinical trials, and in a series of nine patients, none experienced grade 3 or above neurotoxicity242. In summary, neurotoxicity remains an important concern following CD19-CAR-T therapy, but the majority of occurrences are fully reversible, and mortality rates are below 5%, which is in line with other treatments for aggressive B-cell malignancies.
Tackling solid tumours
Preliminary clinical results using CAR-T cells to treat solid tumours have not been as striking as those in blood cancers, but are only the first steps towards antitumor efficacy for this more challenging subset of tumours. One trial reported that 3 out of 11 patients treated with a GD2-CAR achieved complete remission40,243. In another, 4 out of 17 patients with sarcomas achieved stable disease91 with a HER2-CAR and no lymphodepletion, and 1 out of 6 patients achieved a complete response92 with a lymphodepleting regimen. Additionally, a patient treated with IL13Rα2-CAR-T cells delivered intracranially to treat glioblastoma showed regression of disease for at least 7.5 months21. These results are just the beginning of what will probably be a long road ahead for CAR-based therapies against solid tumours. The challenges include toxicities and limited efficacy in the face of a suppressive tumour microenvironment, but the engineering of new safety mechanisms as well as efficacy-enhancing elements will pave the way to success in the treatment of these cancers.
Outlook
The immune system has a remarkable ability to identify and eradicate dysfunctional cells, but acquired and inherited mutations in transformed cells enable their evasion from immune recognition. The engineering of CAR-T cells that redirect immune function against malignant cells marks the beginning of a new era in cancer therapy, marked by the encouraging initial success against B-cell malignancies with the CD19-CAR, which provides a proof-of-principle system for extending this therapeutic paradigm to other cancers. The primary hurdles limiting the success of CAR-T therapy thus far are treatment-associated toxicities and relapse of antigen-negative tumours. Tackling solid tumours poses several additional challenges, owing to a paucity of tumour-restricted antigens, high levels of tumour heterogeneity, poor T-cell trafficking and persistence, and the immunosuppressive effects of the tumour microenvironment. The next generation of CAR technology is moving towards the development of multifaceted smart T-cell machines that can: (1) simultaneously target multiple antigens; (2) be regulated either via small molecules or via intrinsic sensors; (3) be modified by gene editing to augment potency and endow resistance to suppressive factors present in the tumour microenvironment; (4) be equipped with recognition programs to discriminate between cancer and healthy cells; and (5) have intrinsic fail-safes and/or suicide switches. Bridging principles of immunology with recent developments in synthetic biology and genetic engineering will drive the development of the next generation of CAR-T therapies.
References
Rosenberg, S. A. et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin. Cancer Res. 17, 4550–4557 (2011).
Bethune, M. T. & Joglekar, A. V. Personalized T cell-mediated cancer immunotherapy: progress and challenges. Curr. Opin. Biotechnol. 48, 142–152 (2017).
Rapoport, A. P. et al. NY-ESO-1–specific TCR–engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat. Med. 21, 914–921 (2015).
Robbins, P. F. et al. Tumor regression in patients with metastatic synovial cell sarcoma and melanoma using genetically engineered lymphocytes reactive with NY-ESO-1. J. Clin. Oncol. 29, 917–924 (2011).
Maude, S. L. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 371, 1507–1517 (2014).
Brentjens, R. J. et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci. Transl. Med. 5, 177ra38 (2013).
Grupp, S. A. et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 368, 1509–1518 (2013).
Kochenderfer, J. N. et al. Chemotherapy-refractory diffuse large B-cell lymphoma and indolent B-cell malignancies can be effectively treated with autologous T cells expressing an anti-CD19 chimeric antigen receptor. J. Clin. Oncol. 33, 540–549 (2015).
Schuster, S. J. et al. Sustained remissions following chimeric antigen receptor modified T cells directed against CD19 (CTL019) in patients with relapsed or refractory CD19+ lymphomas. Blood 126, 183 (2015).
Lee, D. W. et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet 385, 517–528 (2015).
Gardner, R. A. et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood 129, 3322–3331 (2017).
Turtle, C. J. et al. CD19 CAR–T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J. Clin. Invest. 1, 2123–2138 (2016).
Turtle, C. J. et al. Immunotherapy of non-Hodgkins lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci. Transl. Med. 8, 355ra116 (2016).
CAR T-cells: an exciting frontier in cancer therapy. Lancet 390, 1006 (2017).
FDA approves CAR-T cell therapy to treat adults with certain types of large B-cell lymphoma. US Food and Drug Administration go.nature.com/2jqgcKX (18 October 2017).
Lerner, R. A. Combinatorial antibody libraries: new advances, new immunological insights. Nat. Rev. Immunol. 16, 498–508 (2016).
Long, A. H. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat. Med. 21, 581–590 (2015).
Lynch, A. et al. Adoptive transfer of murine T cells expressing a chimeric-PD1-Dap10 receptor as an immunotherapy for lymphoma. Immunology 152, 472–483 (2017).
Niederman, T. M. J. et al. Antitumor activity of cytotoxic T lymphocytes engineered to target vascular endothelial growth factor receptors. Proc. Natl Acad. Sci. USA 99, 7009–7014 (2002).
Kahlon, K. S. et al. Specific recognition and killing of glioblastoma multiforme by interleukin 13-zetakine redirected cytolytic T cells. Cancer Res. 64, 9160–9166 (2004).
Brown, C. E. et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 375, 2561–2569 (2016).
Hammill, J. A. et al. Designed ankyrin repeat proteins are effective targeting elements for chimeric antigen receptors. J. Immunother. Cancer 3, 55 (2015).
Han, X. et al. Adnectin-based design of chimeric antigen receptor for T cell engineering. Mol. Ther. 25, 2466–2476 (2017).
Kudo, K. et al. T lymphocytes expressing a CD16 signaling receptor exert antibody-dependent cancer cell killing. Cancer Res. 74, 93–102 (2014).
Jamnani, F. R. et al. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: towards tumor-directed oligoclonal T cell therapy. Biochim. Biophys. Acta 1840, 378–386 (2014).
Thayaparan, T. et al. CAR T-cell immunotherapy of MET-expressing malignant mesothelioma. OncoImmunology 6, e1363137 (2017).
Moot, R. et al. Genetic engineering of chimeric antigen receptors using lamprey derived variable lymphocyte receptors. Mol. Ther. Oncolytics 3, 16026 (2016).
Hudecek, M. et al. Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 19, 3153–3164 (2013).
Yang, J. et al. Therapeutic potential and challenges of targeting receptor tyrosine kinase ROR1 with monoclonal antibodies in B-cell malignancies. PLoS ONE 6, e21018 (2011).
Lynn, R. C. et al. Targeting of folate receptor β on acute myeloid leukemia blasts with chimeric antigen receptor-expressing T cells. Blood 125, 3466–3477 (2015).
Drent, E. et al. A rational strategy for reducing on-target off-tumor effects of CD38-chimeric antigen receptors by affinity optimization. Mol. Ther. 25, 1946–1958 (2017).
Liu, X. et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res. 75, 3596–3607 (2015).
Caruso, H. G. et al. Tuning sensitivity of CAR to EGFR density limits recognition of normal tissue while maintaining potent antitumor activity. Cancer Res. 75, 3505–3518 (2015).
Haso, W. et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood 121, 1165–1171 (2013).
James, S. E. et al. Antigen sensitivity of CD22-specific chimeric TCR is modulated by target epitope distance from the cell membrane. J. Immunol. 180, 7028–7038 (2008).
Walker, A. J. et al. Tumor antigen and receptor densities regulate efficacy of a chimeric antigen receptor targeting anaplastic lymphoma kinase. Mol. Ther. 25, 2189–2201 (2017).
Watanabe, K. et al. Target antigen density governs the efficacy of anti-CD20-CD28-CD3ζ chimeric antigen receptor-modified effector CD8+T cells. J. Immunol. 194, 911–920 (2015).
Arcangeli, S. et al. Balance of anti-CD123 chimeric antigen receptor binding affinity and density for the targeting of acute myeloid leukemia. Mol. Ther. 25, 1933–1945 (2017).
Savoldo, B. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J. Clin. Invest. 121, 1822–1826 (2011).
Pule, M. A. et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat. Med. 14, 1264–1270 (2008).
Louis, C. U. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118, 6050–6056 (2011).
Kochenderfer, J. N. et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010).
Porter, D. L., Levine, B. L., Kalos, M., Bagg, A. & June, C. H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725–733 (2011).
Kalos, M. et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci. Transl. Med. 3, 95ra73 (2011).
Kawalekar, O. U. et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity 44, 380–390 (2016).
Ghosh, A. et al. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat. Med. 23, 242–249 (2017).
Zhao, Z. et al. Structural design of engineered costimulation determines tumor rejection kinetics and persistence of CAR T Cells. Cancer Cell 28, 415–428 (2015).
van der Stegen, S. J. C., Hamieh, M. & Sadelain, M. The pharmacology of second-generation chimeric antigen receptors. Nat. Rev. Drug Discov. 14, 499–509 (2015).
Foster, A., Mahendravada, A., Shinners, N. & Chang, W. Regulated expansion and survival of chimeric antigen receptor-modified T cells using small molecule-dependent inducible MyD88/CD40. Mol. Ther. 25, 2176–2188 (2017).
Carpenito, C. et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc. Natl Acad. Sci. USA 106, 3360–3365 (2009).
Zhong, X.-S., Matsushita, M., Plotkin, J., Riviere, I. & Sadelain, M. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication.Mol. Ther. 18, 413–420 (2010).
Abate-Daga, D. et al. A novel chimeric antigen receptor against prostate stem cell antigen mediates tumor destruction in a humanized mouse model of pancreatic cancer. Hum. Gene Ther. 25, 1003–1012 (2014).
Long, A. H. et al. Reduction of MDSCs with all-trans retinoic acid improves CAR therapy efficacy for sarcomas. Cancer Immunol. Res. 4, 869–880 (2016).
Kunkele, A. et al. Functional tuning of CARs reveals signaling threshold above which CD8+ CTL antitumor potency is attenuated due to cell Fas-FasL-dependent AICD. Cancer Immunol. Res. 3, 368–379 (2015).
Guest, R. D. et al. The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J. Immunother. 28, 203–211 (2005).
Taylor, M. J., Husain, K., Gartner, Z. J., Mayor, S. & Vale, R. D. A DNA-based T cell receptor reveals a role for receptor clustering in ligand discrimination. Cell 169, 108–119 (2017).
Hudecek, M. et al. The nonsignaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer Immunol. Res. 3, 125–135 (2015).
Alabanza, L. et al. Function of novel anti-CD19 chimeric antigen receptors with human variable regions is affected by hinge and transmembrane domains. Mol. Ther. 25, 2452–2465 (2017).
Johnson, L. A. et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci. Transl. Med. 7, 275ra22 (2015).
Weijtens, M. E., Willemsen, R. A., Valerio, D., Stam, K. & Bolhuis, R. L. Single chain Ig/gamma gene-redirected human T lymphocytes produce cytokines, specifically lyse tumor cells, and recycle lytic capacity. J. Immunol. 157, 836–843 (1996).
Willemsen, R. A., Ronteltap, C., Chames, P., Debets, R. & Bolhuis, R. L. H. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J. Immunol. 174, 7853–7858 (2005).
Hombach, A., Hombach, A. A. & Abken, H. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc spacer domain in the extracellular moiety of chimeric antigen receptors avoids off-target activation and unintended initiation of an innate immune response. Gene Ther. 17, 1206–1213 (2010).
Jonnalagadda, M. et al. Chimeric antigen receptors with mutated IgG4 Fc spacer avoid Fc receptor binding and improve T cell persistence and antitumor efficacy. Mol. Ther. 23, 757–768 (2015).
Nolan, K. F. et al. Bypassing immunization: optimized design of ‘designer T cells’ against carcinoembryonic antigen (CEA)-expressing tumors, and lack of suppression by soluble CEA. Clin. Cancer Res. 5, 3928–3941 (1999).
Bridgeman, J. S. et al. The optimal antigen response of chimeric antigen receptors harboring the CD3 transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 184, 6938–6949 (2010).
Neelapu, S. S. et al. Chimeric antigen receptor T-cell therapy — assessment and management of toxicities. Nat. Rev. Clin. Oncol. 15, 47–62 (2017).
Lee, D. W. et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124, 188–195 (2014).
Gust, J. et al. Endothelial activation and blood–brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells. Cancer Discov. 7, 1404–1419 (2017).
Diaconu, I. et al. Inducible caspase-9 selectively modulates the toxicities of CD19-specific chimeric antigen receptor-modified T cells. Mol. Ther. 25, 580–592 (2017).
Di Stasi, A. et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N. Engl. J. Med. 365, 1673–1683 (2011).
Paszkiewicz, P. J. et al. Targeted antibody-mediated depletion of murine CD19 CAR T cells permanently reverses B cell aplasia. J. Clin. Invest. 126, 4262–4272 (2016).
Wang, X. et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263 (2011).
Tasian, S. K. et al. Optimized depletion of chimeric antigen receptor T-cells in murine xenograft models of human acute myeloid leukemia. Blood 129, 2395–2407 (2017).
Philip, B. et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood 124, 1277–1287 (2014).
Sakemura, R. et al. A Tet-On inducible system for controlling CD19-chimeric antigen receptor expression upon drug administration. Cancer Immunol. Res. 4, 658–668 (2016).
Wu, C.-Y., Roybal, K. T., Puchner, E. M., Onuffer, J. & Lim, W. A. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science 350, aab4077 (2015).
Mata, M. et al. Inducible activation of MyD88 and CD40 in CAR T-cells results in controllable and potent antitumor activity in preclinical solid tumor models. Cancer Discov. 7, 1306–1319 (2017).
Rodgers, D. T. et al. Switch-mediated activation and retargeting of CAR-T cells for B-cell malignancies. Proc. Natl Acad. Sci. USA 113, 459–468 (2016).
Cao, Y. et al. Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew. Chem. Int. Ed. 55, 7520–7524 (2016).
Huet, H. A. et al. Targeting CD20+ relapsed refractory B cell lymphoma with ACTR087, antibody-coupled T-cell receptor (ACTR) engineered autologous T cells, in combination with rituximab. Blood 128, 3512 (2016).
Posey, A. D. et al. Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane mucin MUC1 control adenocarcinoma. Immunity 44, 1444–1454 (2016).
Morgan, R. A. et al. Case report of a serious adverse event following the administration of T Cells transduced With a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010).
Linette, G. P. et al. Cardiovascular toxicity and titin cross-reactivity of affinity-enhanced T cells in myeloma and melanoma. Blood 122, 863–871 (2013).
Cameron, B. J. et al. Identification of a titin-derived HLA-A1-presented peptide as a cross-reactive target for engineered MAGE A3-directed T cells. Sci. Transl. Med. 5, 197ra103 (2013).
Roybal, K. T. et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell 164, 770–779 (2016).
Fedorov, V. D., Themeli, M. & Sadelain, M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 5, 215ra172 (2013).
Kloss, C. C., Condomines, M., Cartellieri, M., Bachmann, M. & Sadelain, M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat. Biotechnol. 31, 71–75 (2013).
Lanitis, E. et al. Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol. Res. 1, 43–53 (2013).
Vantourout, P. & Hayday, A. Six-of-the-best: unique contributions of γδ T cells to immunology. Nat. Rev. Immunol. 13, 88–100 (2013).
Fisher, J. et al. Avoidance of on-target off-tumor activation using a co-stimulation-only chimeric antigen receptor. Mol. Ther. 25, 1234–1247 (2017).
Ahmed, N. et al. Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor–modified T cells for the immunotherapy of HER2-positive sarcoma. J. Clin. Oncol. 33, 1688–1696 (2015).
Hegde, M. et al. Expansion of HER2-CAR T cells after lymphodepletion and clinical responses in patients with advanced sarcoma. J. Clin. Oncol. 35, 10508 (2017).
Han, X. et al. Masked chimeric antigen receptor for tumor-specific activation. Mol. Ther. 25, 274–284 (2017).
Mamot, C. et al. Tolerability, safety, pharmacokinetics, and efficacy of doxorubicin-loaded anti-EGFR immunoliposomes in advanced solid tumours: a phase 1 dose-escalation study. Lancet Oncol. 13, 1234–1241 (2012).
LaGory, E. L. & Giaccia, A. J. The ever-expanding role of HIF in tumour and stromal biology. Nat. Cell Biol. 18, 356–365 (2016).
Juillerat, A. et al. An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 7, 39833 (2017).
Turley, S. J., Cremasco, V. & Astarita, J. L. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat. Rev. Immunol. 15, 669–682 (2015).
Joyce, J. A. & Fearon, D. T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015).
Engels, B., Rowley, D. A. & Schreiber, H. Targeting stroma to treat cancers. Semin. Cancer Biol. 22, 41–49 (2012).
Irving, M., de Silly, R. V., Scholten, K., Dilek, N. & Coukos, G. Engineering chimeric antigen receptor T-cells for racing in solid tumors: don’t forget the fuel. Front. Immunol. 8, 267 (2017).
Pavlova, N. N. & Thompson, C. B. The emerging hallmarks of cancer metabolism. Cell Metab. 23, 27–47 (2016).
Biswas, S. K. Metabolic reprogramming of immune cells in cancer progression. Immunity 43, 435–449 (2015).
Becker, J. C., Andersen, M. H., Schrama, D. & Thor Straten, P. Immune-suppressive properties of the tumor microenvironment. Cancer Immunol. Immunother. 62, 1137–1148 (2013).
Marvel, D. & Gabrilovich, D. I. Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J. Clin. Invest. 125, 3356–3364 (2015).
Noy, R. & Pollard, J. W. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41, 49–61 (2014).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 12, 252–264 (2012).
Pauken, K. E. & Wherry, E. J. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 36, 265–276 (2015).
Vestweber, D. How leukocytes cross the vascular endothelium. Nat. Rev. Immunol. 15, 692–704 (2015).
Dirkx, A. E. M. et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer Res. 63, 2322–2329 (2003).
Sackstein, R., Schatton, T. & Barthel, S. R. T-lymphocyte homing: an underappreciated yet critical hurdle for successful cancer immunotherapy. Lab. Invest. 97, 669–697 (2017).
Moon, E. K. et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 17, 4719–4730 (2011).
Craddock, J. A. et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 33, 780–788 (2010).
Di Stasi, A. et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood 113, 6392–6402 (2009).
Siddiqui, I., Erreni, M., van Brakel, M., Debets, R. & Allavena, P. Enhanced recruitment of genetically modified CX3CR1-positive human T cells into fractalkine/CX3CL1 expressing tumors: importance of the chemokine gradient. J. Immunother. Cancer 4, 21 (2016).
Porter, D. L. et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 7, 303ra139 (2015).
Fraietta, J. A. et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 127, 1117–1127 (2016).
Pegram, H. J. et al. IL-12-secreting CD19-targeted cord blood-derived T cells for the immunotherapy of B-cell acute lymphoblastic leukemia. Leukemia 29, 415–422 (2015).
Chinnasamy, D. et al. Local delivery of interleukin-12 using T cells targeting vascular endothelial growth factor receptor-2 eradicates multiple vascularized tumors in mice. Clin. Cancer Res. 18, 1672–1683 (2012).
Krenciute, G. et al. Transgenic expression of IL15 improves antiglioma activity of IL13Rα2-CAR T cells but results in antigen loss variants. Cancer Immunol. Res. 5, 571–581 (2017).
Hu, B. et al. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 20, 3025–3033 (2017).
Markley, J. C. & Sadelain, M. IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 115, 3508–3519 (2010).
Zhang, L. et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin. Cancer Res. 21, 2278–2288 (2015).
Spolski, R., Kim, H.-P., Zhu, W., Levy, D. E. & Leonard, W. J. IL-21 mediates suppressive effects via its induction of IL-10. J. Immunol. 182, 2859–2867 (2009).
Spolski, R. & Leonard, W. J. IL-21 is an immune activator that also mediates suppression via IL-10. Crit. Rev. Immunol. 30, 559–570 (2010).
Ahmadzadeh, M. & Rosenberg, S. A. IL-2 administration increases CD4+CD25hi Foxp3+ regulatory T cells in cancer patients. Blood 107, 2409–2414 (2005).
Whilding, L. M. et al. Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol. Ther. 25, 259–273 (2017).
Atkins, M. B. et al. High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J. Clin. Oncol. 17, 2105–2116 (1999).
Fyfe, G. et al. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J. Clin. Oncol. 13, 688–696 (1995).
Sportès, C. et al. Phase I study of recombinant human interleukin-7 administration in subjects with refractory malignancy. Clin. Cancer Res. 16, 727–735 (2010).
Conlon, K. C. et al. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 33, 74–82 (2015).
Shum, T., Omer, B., Tashiro, H., Kruse, R. & Wagner, D. Constitutive signaling from an engineered IL-7 receptor promotes durable tumor elimination by tumor redirected T-cells. Cancer Discov. 7, 1238–1247 (2017).
Zenatti, P. P. et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 43, 932–939 (2011).
Zhang, J. et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481, 157–163 (2012).
Hurton, L. V. et al. Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc. Natl Acad. Sci. USA 113, 7788–7797 (2016).
Tanaka, M. et al. Vaccination targeting native receptors to enhance the function and proliferation of chimeric antigen receptor (CAR)-modified T cells. Clin. Cancer Res. 23, 3499–3509 (2017).
Cruz, C. R. Y. et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood 122, 2956–2973 (2013).
Slaney, C. Y. et al. Dual-specific chimeric antigen receptor T cells and an indirect vaccine eradicate a variety of large solid tumors in an immunocompetent, self-antigen setting. Clin. Cancer Res. 23, 2478–2490 (2017).
Rossig, C. et al. Vaccination to improve the persistence of CD19CAR gene-modified T cells in relapsed pediatric acute lymphoblastic leukemia. Leukemia 31, 1087–1095 (2017).
Ahmed, N. et al. Autologous HER2 CMV bispecific CAR T cells are safe and demonstrate clinical benefit for glioblastoma in a Phase I trial. J. Immunother. Cancer 3, O11 (2015).
Caruana, I. et al. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 21, 524–529 (2015).
Wang, L.-C. S. et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2, 154–166 (2014).
Ruella, M. et al. Overcoming the immunosuppressive tumor microenvironment of Hodgkin lymphoma using chimeric antigen receptor T Cells. Cancer Discov. 7, 1554–1167 (2017).
Chinnasamy, D. et al. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Invest. 120, 3953–3968 (2010).
Perera, L. P. et al. Chimeric antigen receptor modified T cells that target chemokine receptor CCR4 as a therapeutic modality for T-cell malignancies. Am. J. Hematol. 92, 892–901 (2017).
Motz, G. T. et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 20, 607–615 (2014).
John, L. B. et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin. Cancer Res. 19, 5636–5646 (2013).
Gargett, T. et al. GD2-specific CAR T cells undergo potent activation and deletion following antigen encounter but can be protected from activation-induced cell death by PD-1 blockade. Mol. Ther. 24, 1135–1149 (2016).
Chong, E. A. et al. PD-1 blockade modulates chimeric antigen receptor (CAR)-modified T cells: refueling the CAR. Blood 129, 1039–1041 (2017).
Maude, S. L. et al. The effect of pembrolizumab in combination with CD19-targeted chimeric antigen receptor (CAR) T cells in relapsed acute lymphoblastic leukemia (ALL). J. Clin. Oncol. 35, 103–103 (2017).
Heczey, A. et al. CAR T cells administered in combination with lymphodepletion and PD-1 inhibition to patients with neuroblastoma. Mol. Ther. 25, 2214–2224 (2017).
Foster, A. E. et al. Antitumor activity of EBV-specific T lymphocytes transduced with a dominant negative TGF-β receptor. J. Immunother. 31, 500–505 (2008).
Liu, X. et al. A chimeric switch-receptor targeting PD1 augments the efficacy of second-generation CAR T cells in advanced solid tumors. Cancer Res. 76, 1578–1590 (2016).
Menger, L. et al. TALEN-mediated inactivation of PD-1 in tumor-reactive lymphocytes promotes intratumoral T-cell persistence and rejection of established tumors. Cancer Res. 76, 2087–2093 (2016).
Ren, J. et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget 8, 17002–17011 (2017).
Zhang, Y. et al. CRISPR-Cas9 mediated LAG-3 disruption in CAR-T cells. Front. Med. 11, 554–562 (2017).
Ren, J. et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin. Cancer Res. 23, 2255–2266 (2017).
Wartewig, T. et al. PD-1 is a haploinsufficient suppressor of T cell lymphomagenesis. Nature 552, 121–125 (2017).
Scharping, N. E. et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388 (2016).
Yang, W. et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 531, 651–655 (2016).
Ligtenberg, M. A. et al. Coexpressed catalase protects chimeric antigen receptor-redirected T cells as well as bystander cells from oxidative stress-induced loss of antitumor activity. J. Immunol. 196, 759–766 (2016).
Sotillo, E. et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 5, 1282–1295 (2015).
Jacoby, E. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat. Commun. 7, 12320 (2016).
Gardner, R. et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood 127, 2406–2410 (2016).
Yu, H. et al. Repeated loss of target surface antigen after immunotherapy in primary mediastinal large B cell lymphoma. Am. J. Hematol. 92, 11–13 (2017).
Curran, K. J. et al. Multi-center clinical trial of CAR T cells in pediatric/young adult patients with relapsed B-cell ALL. Blood 126, 2533 (2015).
Grupp, S. A. et al. Durable remissions in children with relapsed/refractory ALL treated with T cells engineered with a CD19-targeted chimeric antigen receptor (CTL019). Blood 126, 681 (2015).
Lee, D. W. et al. Safety and response of incorporating CD19 chimeric antigen receptor T cell therapy in typical salvage regimens for children and young adults with acute lymphoblastic leukemia. Blood 126, 684 (2015).
O’Rourke, D. M. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 9, eaaa0984 (2017).
Sampson, J. H. et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J. Clin. Oncol. 28, 4722–4729 (2010).
Brown, C. E. et al. Bioactivity and safety of IL13Rα2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 21, 4062–4072 (2015).
Hegde, M. et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol. Ther. 21, 2087–2101 (2013).
Hegde, M. et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J. Clin. Invest. 126, 3036–3052 (2016).
Yee, C. et al. Adoptive T-cell therapy using antigen-specific CD8+ T-cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T-cells. Proc. Natl Acad. Sci. USA 99, 16168–16173 (2002).
Zah, E., Lin, M.-Y., Silva-Benedict, A., Jensen, M. C. & Chen, Y. Y. T cells expressing CD19/CD20 bi-specific chimeric antigen receptors prevent antigen escape by malignant B cells. Cancer Immunol. Res. 4, 498–508 (2016).
Grada, Z. et al. TanCAR: a novel bispecific chimeric antigen receptor for cancer immunotherapy. Mol. Ther. Nucleic Acids 2, e105 (2013).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med 24, 20–28 (2018).
Ruella, M. et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J. Clin. Invest. 126, 3814–3826 (2016).
Anurathapan, U. et al. Kinetics of tumor destruction by chimeric antigen receptor-modified T cells. Mol. Ther. 22, 623–633 (2014).
Bielamowicz, K. et al. Trivalent CAR T-cells overcome interpatient antigenic variability in glioblastoma. Neuro. Oncol. 20, 506–518 (2017).
Beatty, G. L. Engineered chimeric antigen receptor-expressing T cells for the treatment of pancreatic ductal adenocarcinoma. Oncoimmunology 3, e28327 (2014).
Pilon, S. A., Kelly, C. & Wei, W.-Z. Broadening of epitope recognition during immune rejection of ErbB-2-positive tumor prevents growth of ErbB-2-negative tumor. J. Immunol. 170, 1202–1208 (2003).
Sampson, J. H. et al. EGFRvIII mCAR-modified T-cell therapy cures mice with established intracerebral glioma and generates host immunity against tumor-antigen loss. Clin. Cancer Res. 20, 972–984 (2014).
Beatty, G. L. et al. Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol. Res. 2, 112–120 (2014).
Boice, M. et al. Loss of the HVEM tumor suppressor in lymphoma and restoration by modified CAR-T cells. Cell 167, 405–418 (2016).
Roybal, K. T. et al. Engineering T cells with customized therapeutic response programs using synthetic Notch receptors. Cell 167, 419–432 (2016).
Rossi, R. L. et al. Distinct microRNA signatures in human lymphocyte subsets and enforcement of the naive state in CD4+ T cells by the microRNA miR-125b. Nat. Immunol. 12, 796–803 (2011).
Steiner, D. F. et al. MicroRNA-29 regulates T-box transcription factors and interferon-γ production in helper T cells. Immunity 35, 169–181 (2011).
Dooley, J., Linterman, M. A. & Liston, A. MicroRNA regulation of T-cell development. Immunol. Rev. 253, 53–64 (2013).
Okada, H., Kohanbash, G. & Lotze, M. T. MicroRNAs in immune regulation—opportunities for cancer immunotherapy. Int. J. Biochem. Cell Biol. 42, 1256–1261 (2010).
Sasaki, K. et al. miR-17-92 expression in differentiated T cells — implications for cancer immunotherapy. J. Transl. Med. 8, 17 (2010).
Ohno, M. et al. Expression of miR-17-92 enhances anti-tumor activity of T-cells transduced with the anti-EGFRvIII chimeric antigen receptor in mice bearing human GBM xenografts. J. Immunother. Cancer 1, 21 (2013).
Wong, R. S., Chen, Y. Y. & Smolke, C. D. Regulation of T cell proliferation with drug-responsive microRNA switches. Nucleic Acids Res. 46, 1541–1552 (2017).
Zheng, Y., Tang, L., Mabardi, L., Kumari, S. & Irvine, D. J. Enhancing adoptive cell therapy of cancer through targeted delivery of small-molecule immunomodulators to internalizing or noninternalizing receptors. ACS Nano 11, 3089–3100 (2017).
Park, J. et al. Combination delivery of TGF-β inhibitor and IL-2 by nanoscale liposomal polymeric gels enhances tumour immunotherapy. Nat. Mater. 11, 895–905 (2012).
Stephan, M. T., Moon, J. J., Um, S. H., Bershteyn, A. & Irvine, D. J. Therapeutic cell engineering with surface-conjugated synthetic nanoparticles. Nat. Med. 16, 1035–1041 (2010).
Jones, R. B. et al. Antigen recognition-triggered drug delivery mediated by nanocapsule-functionalized cytotoxic T-cells. Biomaterials 117, 44–53 (2017).
Stephan, S. B. et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat. Biotechnol. 33, 97–101 (2014).
Smith, T. T. et al. Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest. 127, 2176–2191 (2017).
Tindera, M. Incoming Novartis CEO on $475,000 cancer therapy: ‘no question that the list price raises eyebrows’. Forbes go.nature.com/2HMNuPa (30 November 2017).
Rosenbaum, L. Tragedy, perseverance, and chance—the story of CAR-T therapy.N. Engl. J. Med. 377, 1313–1315 (2017).
Kite’s Yescarta™ (axicabtagene ciloleucel) becomes first CAR T therapy approved by the FDA for the treatment of adult patients with relapsed or refractory large B-cell lymphoma after two or more lines of systemic therapy. Gilead go.nature.com/2wb51PE (18 October 2017).
With FDA approval for advanced lymphoma, second CAR T-cell therapy moves to the clinic. National Cancer Institute go.nature.com/2FDRTC5 (25 October 2017).
Brudno, J. N. et al. Allogeneic T cells that express an anti-CD19 chimeric antigen receptor induce remissions of B-cell malignancies that progress after allogeneic hematopoietic stem-cell transplantation without causing graft-versus-host disease. J. Clin. Oncol. 34, 1112–1121 (2016).
Cai, B. et al. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J. Hematol. Oncol. 9, 131 (2016).
Kochenderfer, J. N. et al. Donor-derived anti-CD19 chimeric-antigen-receptor-expressing T cells cause regression of malignancy persisting after allogeneic hematopoietic stem cell transplantation. Blood 122, 151 (2013).
Jacoby, E. et al. Murine allogeneic CD19 CAR T cells harbor potent antileukemic activity but have the potential to mediate lethal GVHD. Blood 127, 1361–1370 (2016).
Qasim, W. et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 9, eaaj2013 (2017).
Zakrzewski, J. L. et al. Tumor immunotherapy across MHC barriers using allogeneic T-cell precursors. Nat. Biotechnol. 26, 453–461 (2008).
Yang, L. & Baltimore, D. Long-term in vivo provision of antigen-specific T cell immunity by programming hematopoietic stem cells. Proc. Natl Acad. Sci. USA 102, 4518–4523 (2005).
Larson, S. M. et al. Pre-clinical development of gene modification of haematopoietic stem cells with chimeric antigen receptors for cancer immunotherapy. Hum. Vaccin. Immunother. 13, 1094–1104 (2017).
Themeli, M. et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat. Biotechnol. 31, 928–933 (2013).
Vizcardo, R. et al. Regeneration of human tumor antigen-specific T cells from iPSCs derived from mature CD8+ T cells. Cell Stem Cell 12, 31–36 (2013).
Liao, N. S., Bix, M., Zijlstra, M., Jaenisch, R. & Raulet, D. MHC class I deficiency: susceptibility to natural killer (NK) cells and impaired NK activity. Science 253, 199–202 (1991).
Bix, M. et al. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature 349, 329–331 (1991).
Torikai, H. et al. Toward eliminating HLA class I expression to generate universal cells from allogeneic donors. Blood 122, 1341–1349 (2013).
Gornalusse, G. G. et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat. Biotechnol. 35, 765–772 (2017).
Smith, T. T. et al. Regulation of T cell proliferation with drug-responsive microRNA switches regulation of T cell proliferation with drug-responsive microRNA switches. Nat. Nanotech. 46, 1541–1552 (2017).
Garfall, A. L. et al. Chimeric antigen receptor T cells against CD19 for multiple myeloma. N. Engl. J. Med. 373, 1040–1047 (2015).
Hacein-Bey-Abina, S. et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J. Clin. Invest. 118, 3132–3142 (2008).
Tsukahara, T. et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 22, 209–215 (2015).
Kebriaei, P. et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Invest. 126, 3363–3376 (2016).
Nakazawa, Y. et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol. Ther. 19, 2133–2143 (2011).
Soifer, H. et al. Stable integration of transgenes delivered by a retrotransposon-adenovirus hybrid vector. Hum. Gene Ther. 12, 1417–1428 (2001).
Staunstrup, N. H. et al. Hybrid lentivirus-transposon vectors with a random integration profile in human cells. Mol. Ther. 17, 1205–1214 (2009).
Yant, S. R. et al. Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nat. Biotechnol. 20, 999–1005 (2002).
Eyquem, J. et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117 (2017).
Wang, J. et al. Highly efficient homology-driven genome editing in human T cells by combining zinc-finger nuclease mRNA and AAV6 donor delivery. Nucleic Acids Res. 44, e30 (2016).
MacLeod, D. T. et al. Integration of a CD19 CAR into the TCR alpha chain locus streamlines production of allogeneic gene-edited CAR T cells. Mol. Ther. 25, 949–961 (2017).
Jackson, H. J., Rafiq, S. & Brentjens, R. J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 13, 370–383 (2016).
Fry, T. J. et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat. Med. 24, 20–28 (2018).
Shah, N. N. et al. Minimal residual disease negative complete remissions following anti-CD22 chimeric antigen receptor (CAR) in children and young adults with relapsed/refractory acute lymphoblastic leukemia (ALL). Blood 128, 650 (2016).
Shalabi, H. et al. A prospective evaluation of neurocognitive function and neurologic symptoms in pediatric and young adult patients with relapsed/refractory acute lymphoblastic leukemia (ALL) undergoing anti-CD22 chimeric antigen receptor therapy. Blood 128, 1625 (2016).
Ali, S. A. et al. T cells expressing an anti-B-cell maturation antigen chimeric antigen receptor cause remissions of multiple myeloma. Blood 128, 1688–1700 (2016).
Cohen, A. D. et al. B-cell maturation antigen (BCMA)-specific chimeric antigen receptor T cells (CART-BCMA) for multiple myeloma (MM): initial safety and efficacy from a phase I study. Blood 128, 1147 (2016).
Wang, Y. et al. Effective response and delayed toxicities of refractory advanced diffuse large B-cell lymphoma treated by CD20-directed chimeric antigen receptor-modified T cells. Clin. Immunol. 155, 160–175 (2014).
Till, B. G. et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood 119, 3940–3950 (2012).
Ramos, C. A. et al. Clinical responses with T lymphocytes targeting malignancy-associated κ light chains. J. Clin. Invest. 126, 2588–2596 (2016).
Vera, J. et al. T lymphocytes redirected against the κ light chain of human immunoglobulin efficiently kill mature B lymphocyte-derived malignant cells. Blood 108, 3890–3897 (2006).
Hudecek, M. et al. The B-cell tumor-associated antigen ROR1 can be targeted with T cells modified to express a ROR1-specific chimeric antigen receptor. Blood 116, 4532–4541 (2010).
Berger, C. et al. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunol. Res. 3, 206–216 (2015).
Mackall, C. L. & Miklos, D. B. CNS endothelial cell activation emerges as a driver of CAR T cell-associated neurotoxicity. Cancer Discov. 7, 1371–1373 (2017).
Ghorashian, S. et al. A novel low affinity CD19 CAR results in durable disease remissions and prolonged CAR T cell persistence without severe CRS or neurotoxicity in patients with paediatric ALL. Blood 130, 806 (2017).
Nishio, N. et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res. 74, 5195–5205 (2014).
Bonini, C. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science 276, 1719–1724 (1997).
Maher, J., Brentjens, R. J., Gunset, G., Rivière, I. & Sadelain, M. Human T-lymphocyte cytotoxicity and proliferation directed by a single chimeric TCRζ/CD28 receptor. Nat. Biotechnol. 20, 70–75 (2002).
Pulè, M. A. et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol. Ther. 12, 933–941 (2005).
Hombach, A. A., Heiders, J., Foppe, M., Chmielewski, M. & Abken, H. OX40 costimulation by a chimeric antigen receptor abrogates CD28 and IL-2 induced IL-10 secretion by redirected CD4+ T cells. Oncoimmunology 1, 458–466 (2012).
Finney, H. M., Akbar, A. N. & Lawson, A. D. G. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR chain. J. Immunol. 172, 104–113 (2004).
Guedan, S. et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 124, 1070–1080 (2014).
Song, D. G. et al. CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119, 696–706 (2012).
Shaffer, D. R. et al. T cells redirected against CD70 for the immunotherapy of CD70-positive malignancies. Blood 117, 4304–4314 (2011).
Leen, A. M. et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol. Ther. 22, 1211–1220 (2014).
Tanoue, K. et al. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res. 77, 2040–2051 (2017).
Li, S. et al. Enhanced cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin. Cancer Res. 15, 6982–6992 (2017).
Acknowledgements
This work was supported by a SU2C-St. Baldrick’s Pediatric Cancer Dream Team Translational Research Grant (SU2CAACR-DT1113). C.L.M. is a member of the Parker Institute for Cancer Immunotherapy, which supports the Stanford University Cancer Immunotherapy Program. L.L. is supported by the National Science Foundation Graduate Research Fellowship, Stanford Graduate Fellowship, and Stanford EDGE Fellowship. R.G.M. is supported by a SARC Career Development Award.
Author information
Authors and Affiliations
Contributions
All authors contributed to writing and editing the manuscript.
Corresponding author
Ethics declarations
Competing interests
C.L.M. is an inventor on a patent for a CD22-directed CAR licensed by JUNO Therapeutics, receives research funding from Bluebird Bio and Obsidian Therapeutics, and serves on the advisory boards of Unum Therapeutics, GlaxoSmithKline and Vor Pharmaceuticals. L.L. and R.G.M. declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Labanieh, L., Majzner, R.G. & Mackall, C.L. Programming CAR-T cells to kill cancer. Nat Biomed Eng 2, 377–391 (2018). https://doi.org/10.1038/s41551-018-0235-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41551-018-0235-9
- Springer Nature Limited
This article is cited by
-
Immune evasion in cell-based immunotherapy: unraveling challenges and novel strategies
Journal of Biomedical Science (2024)
-
Modeling the neuroimmune system in Alzheimer’s and Parkinson’s diseases
Journal of Neuroinflammation (2024)
-
Programmable synthetic receptors: the next-generation of cell and gene therapies
Signal Transduction and Targeted Therapy (2024)
-
CAR T-cells targeting FGFR4 and CD276 simultaneously show potent antitumor effect against childhood rhabdomyosarcoma
Nature Communications (2024)
-
CAR-T and CAR-NK as cellular cancer immunotherapy for solid tumors
Cellular & Molecular Immunology (2024)