
Abstract
Developing high-performance multicomponent ceramics, which are promising in solving challenges posed by emerging technologies, shows grand difficulties because of the immense compositional space and complex local distortions. In this work, an accurate machine learning (ML) model built upon an ab initio database is developed to predict the mechanical properties and structural distortions of multicomponent transition metal carbides (MTMCs). The compositional space of MTMCs is thoroughly explored by the well-trained model. Combined with electronic and geometrical analysis, we show that the elemental adaptability to the rock-salt structure elegantly elucidates the mechanical characteristics of MTMCs, and such adaptability can be quantified by local lattice distortions. We further establish new design principles for high-strength MTMCs, and V–Nb–Ta-based MTMCs are recommended, which are validated by the present experiments. The proposed model and design philosophy pave a broad avenue for the rational design of MTMCs with exceptional properties.
Similar content being viewed by others


Introduction
The emerging technologies aiming to solve global challenges, such as energy crises and climate change, impose stringent requirements for the needed structural materials. For instance, superhard and incompressible materials are essential for high-performance cutting tools and wear-protective coatings1. For such purposes, materials with bulk modulus higher than 300 GPa are desirable1. Though metallic transition metal carbides, e.g., ZrC, TiC, and WC, show good performance in some scenarios2, their wide applications face obstacles because of poor sinterability and low fracture toughness3. Due to the heightening demands involved in future applications, new strategies for materials design are required to fulfill the growing needs of advanced techniques.
The concept of high-entropy alloys brings new opportunities for the design of high-strength materials4. Since then, the configurational space for materials development has expanded from the corner region to the center region, where the configurational entropy can be maximized5. The idea of entropy stabilization was successfully introduced to ceramics by Rost et al.6, followed by the discovery of high-entropy borides7, carbides8,9,10, nitrides11, carbonnitrides12,13, and sulfides14. These newly discovered materials with superb properties show great potential in replacing conventional ceramics for demanding conditions. Among these ceramics, multicomponent transition metal carbides (MTMCs), including high- and medium-entropy carbides, stand out as they possess high hardness, good oxidative resistance, and tunable thermal conductivity15,16,17. Experiments also show that the properties of MTMCs may depart from or surpass their rule-of-mixtures (ROM) values12,13,18, making MTMC a flexible platform for designing novel ceramics with desired properties.
Despite the promising opportunities of MTMCs, the vast compositional space makes the exploration and fine-tuning of MTMCs by experiments and ab initio calculations unaffordable. The experimental synthetization generally costs days, even months. On the other hand, ab initio calculations of mechanical properties, such as elastic modulus of MTMCs, are resource-consuming because dozens of computations are required even for one composition based on the energy-strain or stress-strain methods, which is unsuitable for the composition optimization of multicomponent systems. Therefore, an accurate and fast methodology for predicting the mechanical properties of MTMCs is highly desired.
For MTMCs, superior mechanical performance surpassing the ROM values has been found in certain cases9,19, which is usually attributed to the complex compositions and geometrical misfits20, but without fundamental investigations and detailed explanations to the best of our knowledge. Furthermore, challenges were encountered in predicting heat transfer coefficient21, superconducting transition temperature21, and elastic properties22,23 of high-entropy alloys using the mixture rules. Traditionally, valence electron concentration (VEC) is adopted to predict phase formation and mechanical properties of materials successfully24,25,26,27,28. For some MTMCs, VEC also manifests good performance in correlating their mechanical properties27. However, previously studied MTMCs are limited to no more than three principal components27,28. Moreover, VEC cannot precisely estimate the phase29 and ductility30 of high-entropy alloys. Thus, there is still a significant gap in understanding the role of VEC in MTMCs within the high-entropy region.
Machine learning (ML) methods are good at mining the complex correlation between input descriptors (atomic or precursory properties) and target properties in high and non-linear dimensions. Recently, various ML models were successfully utilized for the rational design of high-entropy alloys31,32,33,34,35,36,37, high-entropy ceramics38,39,40,41,42,43, and more44,45,46,47. Artificial neural network (ANN)40, support vector machine40, random forest39, and k-nearest neighbor41 models were used to predict the single-phase synthesizability of high-entropy ceramics. In addition, Zuo et al.38 combined Bayesian optimization and graph neural networks to predict the formation energies and elastic properties of transition metal ceramics. After that, Tang43 et al. incorporated bond parameters (bond order, bond length, and bond ionicity) to train a Gaussian process regression model and predict the mechanical properties of binary and high-entropy ceramics. Jaafreh et al.42 adopted various algorithms to train the ML model for hardness prediction based on a database of experimental results. Notably, all the published models38,43,48 for predicting the mechanical properties of MTMCs were trained based on datasets only comprised of very sparse points in the high-entropy region, making it challenging to generalize the model to the whole compositional space. Specifically, the considered number of cations is usually no more than 438,48, or very few high-entropy samples within extremely small supercells43 are studied, which can hardly incorporate the compositional and geometrical complexity. Although deep-learning models have been applied to fit interatomic potentials for MTMCs, such as (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C49 and (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B250, such deep-learning potentials are not suited for high-throughput predictions within a large compositional space. By now, a database and ML models specialized for predicting the mechanical properties of general MTMCs are still lacking.
In this work, we leverage a data-driven approach to design and discover high-performance MTMCs. An ab initio database containing ternary, quaternary, quinary, and senary MTMCs was delicately constructed (see Supplementary Table 1), on which ML models with high accuracy and good generalizability are established. This is distinct from previous works38,42,43 where no or very few MTMCs with more than four principal elements were incorporated. Based on the well-trained ML model, high-throughput prediction was conducted on both equiatomic and non-equiatomic MTMCs. Our findings reveal that the mechanical properties of many MTMCs do not follow the ROM values. Besides, the mechanical strength of MTMCs only weakly correlates with VEC. Through in-depth geometrical and electronic structure analyses, we elucidate that the elemental adaptabilities to the rock-salt structure are the underlying reasons for the mechanical properties of MTMCs. We further delineate a rule for high-performance MTMC design, through which MTMCs containing V, Nb, and Ta in the cation sublattice are recommended as high-modulus carbides. These conclusions are supported by our experiments, indicating the effectiveness of our proposed design principles for high-performance MTMCs.
Result
Machine-learning model
Supervised ML models for predicting material properties are generally comprised of several essential parts: database collection, feature engineering, model training and evaluation, model deployment, and experimental validation. Among these steps, the feature engineering step can be skipped if non-empirical features are adopted51. However, to digest non-empirical inputs, deep layers and a large number of trainable parameters are needed. In this work, we extract physical descriptors to train and predict MTMC properties (Fig. 1). In this case, much fewer variables are required in the learning algorithm thanks to the guidance of physical descriptors.

ANN and PCA refer to artificial neural networks and principal component analysis, respectively. The input features are listed in Supplementary Table 2. The “+”, “×”, “2”, “3”, and “log10” are operators and are explained in Supplementary Table 3. There are five steps for the design of high-performance MTMCs: constructing a database, feature engineering, model training, high-throughput prediction, and experimental validation.
As provided in the workflow illustrated in Fig. 1, we first prepare a database consisting of MTMCs with 2–5 metallic elements. As presented in Supplementary Table 1, 65% of samples consist of no less than four metallic elements, distinct from previous studies38,42,43,48.
The samples are encoded by 13 precursory properties (see Supplementary Table 2 for the details). These descriptors do not require extra experiments or simulations, and thus are suitable for building physics-guided ML models and have the advantage of exploring the whole compositional space51. Note that the mixing enthalpy and volume change induced by mixing are not considered as they need prior DFT calculations, which may limit the high-throughput predictions for non-equiatomic MTMCs.
The mixing entropy is calculated as:
where R and ci are the gas constant and concentration of the ith precursor, respectively. The average values of precursory properties can be used to describe MTMC samples based on the mean-field theory. In contrast, the deviation and the range of properties are used to describe the dissimilarities among constituents. The average (\(\overline{\rm{prop}}\)), deviation (\({\sigma }_{\rm{prop}}\)), and the range of precursory properties are calculated by the following equations.
where propi is the property value of ith precursor.
The range of each property is calculated by:
where \({\rm{prop}}_{\max }\) and \({\rm{prop}}_{\min }\) are the maximum and minimum values of the precursory property, respectively. The collected inputs were then passed to operators (listed in Supplementary Table 3) to further expand the input dimension. After these procedures, the input dimension is enlarged to 224. However, some input features are highly correlated (Supplementary Fig. 1), which slows the training and prediction efficiency. Thus, we further reduce the input dimensionality with the principal component analysis, where most data covariance (0.99) is reserved.
We adopt a global optimization algorithm, i.e., the adaptive moment estimation method52, to train the ML model. During the optimization iterations, the trainable parameters are updated, and the differences between predictions and data labels are minimized. To reduce overfitting, we have explored the effect of regularization. As depicted in Fig. 2a, b, the regularization method reduces the overfitting significantly; the validation error would increase after 500 training epochs without such a regularizer. To make the best use of collected data points, we adopt a cross-validation (CV) method in which the database is divided into independent subsets, and each one of them serves as the validation set for different training folds. We have also optimized the number of hidden layers, number of neurons per layer, number of CV folds, and batch size. The results show that a 5-CV model with four layers and 80 nodes in each layer gives the lowest mean absolute percentage error (MAPE): 3.54%. The interpretation of input features can be seen in Supplementary Note 1.

Learning curves a without and b with L2 regularization. MAPE with varied hyperparameters. c and d are colored with the value of MAPE averaged among all folds.
Deployment of the well-trained model
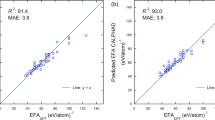
The bulk modulus (B), shear modulus (G), Young’s modulus (E), and Vickers hardness (Hv) are widely employed to evaluate the mechanical performance of structural materials. The elastic constants of C11 and C44 are also used to quantify the material’s ability to withstand uniaxial strain and shearing. Compared to the properties calculated by density functional theory (DFT), the well-trained ML model shows excellent prediction accuracy. As shown in Fig. 3, the estimated mean absolute error (MAE) of B, G, and E is lower than previous reports43,48, which is understandable as only very few high-entropy MTMCs were incorporated into the database of previous works.

a Bulk modulus (B); b Shear modulus (G); c Young’s modulus (E); d Hardness (Hv); e C11; f C44. Units of MAPE and MAE are in % and GPa, respectively.
The well-trained model is then applied to predict the whole compositional space of MTMCs. As shown in Supplementary Note 2, the model accurately predicts the non-equiatomic compositions that are not encountered during the training phase, suggesting good generalizability across the entire compositional space. Additionally, the ternary property diagrams in Fig. 4 show that unexploited non-equiatomic compositions can have superior mechanical properties than equiatomic MTMCs. Especially, B of the Ti–V–Mo system can be enhanced by decreasing the concentration of Mo and increasing the concentration of V. Ti–Nb–Ta and V–Nb–Ta systems (Fig. 4b, e) show similar patterns in shear modulus, indicating that the high-cost and detrimental V element can be replaced by low-cost and environmentally-friendly Ti. In Fig. 4f, a low basin of E can be found in the bottom left region, which should be avoided if one aims to design Hf–V–W-based carbides with high E values. As these diagrams of mechanical properties can be generated efficiently, the ML model effectively guides the design of exceptional MTMCs with economical and eco-friendly constituents.

a B of V–Mo–Ti system; b G of Nb–Ta–Ti system; c E of Nb–Ta–Ti system; d B of Nb–Ta–V system; e G of Nb–Ta–V system; f E of Hf–W–V system. Units are given in GPa. The elemental combinations are selected based on the results calculated in Supplementary Note 3.
Mechanical properties indicated by VEC and ROM
Based on the DFT and ML predicted results, some phenomena arise: poor correlation of the mechanical properties of MTMCs with VEC (Fig. 5a–c) and the breakdown of the ROM (as shown in Fig. 5d–f). Previously, such deviation was commonly attributed to the complex interactions between constituent species, but without fundamental explanations.

Correlation between VEC and a B, b G, and c E from ML predictions. d–f Parity plots between mechanical properties given by ML and ROM. Group A: all elements in groups 4–6 and periods 4–6; Group B: exclude Cr, Mo, and W; Group C: exclude Cr, Mo, W, V, and Nb.
VEC has been widely used to dictate material properties, such as phase stability26 and mechanical strength27,28. Balasubramanian et al.27 reported that the ductility of binary and ternary ceramics increases with increasing VEC. Jhi et al.28 attributed the highest hardness of transition-metal carbonitrides with VEC = 8.4 to the directional d–p σ bonds. However, for MTMCs, the correlation between modulus and VEC has not been systematically investigated yet because of the huge compositional space and complex distortions in the local environment. Thanks to the well-trained ML model, we can explore the mechanical properties of MTMCs with different compositions efficiently. As shown in Fig. 5a, B of all MTMCs (Group A) increases slightly with an increasing VEC. The E values increase with increasing VEC when VEC is <9.0, and then decrease to lower than 400 GPa until VEC = 10.0, similar to the trend of G. More strikingly, large fluctuations of B, G, and E exist in MTMCs at the same VEC, which differs from ceramics with ordered structures or no more than three components27. For instance, the span of B and E at VEC = 9.0 is around 150 and 250 GPa, respectively, signifying the poor capability of VEC in dictating the modulus of disordered MTMCs.
Since some precursory binary carbides are unstable in the rock-salt structure [B1 or face-centered-cubic (FCC) structures], it is reasonable to postulate that these metallic elements should show lower adaptability to the rock-salt MTMCs. We collected the energy Hull of binary carbides from Materials Project53 and listed them in Supplementary Table 5. B1-type CrC, MoC, and WC show higher energies above the convex Hull (>0.25 eV/atom), declaring their high instability in rock-salt structures. In comparison, VC and TaC exhibit positive hull energies lower than 0.1 eV/atom, which explains their relatively low instability. If these elements with positive energy above the convex hull are excluded (the remaining MTMCs are labeled as Group C), as seen in Fig. 5a–c, strong upward trends for both B, G, and E can be observed. Similar correlations between VEC and mechanical properties are also evident if we plot moduli after excluding highly unstable elements (Group B: exclude Cr, Mo, and W). These trends agree well with the previous reports27,28 for carbides containing one or two low-stability metallic elements, signifying that the elements with low adaptability should be responsible for the deviation from the VEC trends in MTMCs.
The physical intuition tells us that the properties of mixtures may differ or be close to the ROM values depending on the adaptability of individual elements. For example, Zhao20 reported that the general stacking fault energies in selective MTMCs obey the ROM. In contrast, some high-entropy nitride and carbonitride have moduli that surpass ROM estimations9,12,13,18. The comparison of mechanical properties of MTMCs obtained from the ML model and ROM is provided in Fig. 5d–f. Similar to the deviations from the VEC trends, the varied adaptability of different elements accounts for poor ROM predictions compared to ML results. If we exclude Cr, Mo, and W, a clear correlation between the moduli of MTMCs and their ROM approximations can be observed [Group B in Fig. 5d–f]. The correlation is more significant if only considering Ti, Zr, Hf, and Ta elements (highly adaptable), denoting that ROM can only be applied to selected elemental combinations for MTMCs [Group C in Fig. 5]. The above trends are also visible based on our raw ab initio data [Supplementary Fig. 6], and the ML predictions in the whole compositional space make them more salient.
Adaptability and local distortion from electronic and geometrical analysis
The trends of mechanical properties against VEC in MTMCs with more than three metallic elements have not been reported previously. An understanding of the fundamental reasons for this phenomenon can help design MTMCs with excellent properties. From the energy perspective, the energies above the convex Hull in Supplementary Table 5 indicate that the adaptability of constituent elements plays a critical role. To gain deep insight into the mechanism, we further explore the electronic and geometrical structures of MTMCs.
In the B1-type carbides, bands of different orbitals play different roles against shear distortion28. In specific, the eg–p σ bands are highly directional and responsible for the shearing-resistant bonding interactions, but the π bonding between p and t2g orbitals is insensitive to shear distortion. The high-energy σ bonding between intermetallic d orbitals is stronger after shearing, resulting in lowered shear and Young’s modulus if it is filled by electrons28. The calculated band structure of TiC is shown in Supplementary Fig. 7a, which is in excellent agreement with literature28,54. For other binary carbides belonging to IV–VI groups, their electronic structures are similar to TiC. Such similarities among B1-type binary carbides indicate that the valence states are occupied successively with increased VEC, and the above-mentioned interactions (eg–p, t2g–p, and t2g–t2g) become effective successively. Therefore, clear trends of the mechanical moduli of binary carbides are found as VEC increases monotonically. To compare the difference between binary carbides and MTMCs, the electronic band structures of selective MTMCs are calculated. Those selected MTMCs all have VEC = 9, but exhibit large, medium, and low modulus, respectively (Supplementary Table 6). As evidenced in Fig. 6a–c, the electronic structures of these MTMCs show varied patterns, and the interactions between different bands are not involved in a similar order as in binary carbides (Supplementary Fig. 7). Thus, even with the same VEC, the fundamental electronic interactions are different. Therefore, the selected systems show distinct mechanical properties. Moreover, as discussed above, Nb and Ta are more adaptable to the rock-salt structure than Cr, Mo, and W. Consequently, the band structure of (NbTa)C2 exhibits more similarities to binary carbides than (CrZr)C2 and (MoTiWZr)C4, which leads to the smaller deviation of (NbTa)C2 from trends of group B elements shown in Fig. 5. We further calculate the band structure of MTMCs consisting only of group-C elements, and the electronic band structures in Supplementary Fig. 8 show features akin to the binary carbides (Supplementary Fig. 7). Hence, the large fluctuations in mechanical properties at the same VEC are attributed to the elements which refuse to redistribute their electronic band structure to dissolve into rock-salt MTMCs. In short, VEC is a good indicator of the mechanical properties of MTMCs with high electronic adaptability to the B1 structure and binary carbides, but not for MTMCs consisting of Cr, Mo, and W elements.

a–c Band structures of three selected MTMCs with VEC = 9.0 but with distinct E values: a 311 GPa, b 425 GPa, and c 545 GPa. d Local distortion of different elements in all the studied MTMCs. e–g Correlations between the magnitude of local distortion of MTMC and their mechanical properties.
To quantify the adaptability of different elements, we collect the atomic displacement (ΔR) from the octahedral center in the FCC lattice: \(\Delta {{R}}={{\Bigg|\Bigg|}\frac{\mathop{\sum }\nolimits_{{{i}}}^{{{n}}}{{{R}}}_{1\text{NN}}}{{{n}}}-{{R}}{\Bigg|\Bigg|}}_{2}\), where R and R1NN are the atom position and the positions of its first-nearest neighboring (1NN) atoms, respectively. The number of 1NN atoms is n. Additionally, as shown in Supplementary Fig. 9, the magnitude of ΔR exhibits high consistency across independent special quasi-random structures (SQSs). As shown in Fig. 6d, the investigated elements relocate their positions in the rock-salt lattice in the order of Cr~Mo~W > V~Nb > Ti~Zr~Hf~Ta. Such an order agrees well with the analysis of the energy above convex Hull (Supplementary Table 5) and electronic band structures [Fig. 6a–c and Supplementary Fig. 8]. Interestingly, the magnitude of local distortion shows a good ability to predict deviation magnitudes of elastic moduli from ROM values, as reflected in Fig. 6e–g. For MTMCs with low geometrical adaptabilities (high local distortions), the mechanical properties calculated by DFT are much lower than the ROM values, and consequently, the ROM values fall short in predicting the mechanical properties. Trends are also observed between the local distortion and elastic moduli (Supplementary Fig. 10), signifying that the local distortion is a better descriptor than VEC and ROM for MTMCs. Furthermore, large distortions in local environments are concurrently observed with large volume expansions (Supplementary Fig. 11), designating that the elements with low adaptability expand their atomic volume in the rock-salt structure. We additionally fit the linear relationship between B and local distortion with the formula: B = ROM − ∑ke∆Re + b, and found that Cr, Mo, and W decrease B significantly (Supplementary Fig. 12). As the geometrical distortion can effectively quantify the adaptabilities of constituent elements and dictate the moduli, it plays a pivotal role in bridging the underlying band structures and mechanical performance of MTMCs, including high- and medium-entropy ceramics.
Rational design of high-performance MTMCs
According to the adaptability to the rock-salt structure, the investigated elements are split into three groups. Ti, Zr, Hf, and Ta readily dissolve into the B1 lattice without dramatic distortion of local environments. In contrast, Cr, Mo, and W display large atomic dislocations in the FCC structure. For the other elements (V and Nb), moderate adaptability is found. The magnitude of local distortion in MTMCs is obtained with DFT calculations in the above discussions, which may limit the high-throughput screening to identify excellent MTMCs. To address this dilemma, an ANN model is trained and applied to predict the local distortion of MTMCs. As shown in Supplementary Fig. 13, the ANN model accurately predicts the local distortions in MTMCs, effectively alleviating resource-demanding DFT calculations.
The mechanical performance of MTMCs can be explained from two aspects: the elemental adaptability to the FCC structure and the intrinsic properties of different species. The adaptability, as discussed above, can be quantified by local distortions, and the inherent properties are given by the moduli of precursory carbides. For MTMCs with minor local distortions, the ROM accurately estimates their mechanical moduli (Fig. 6e–g). Thus, exceptional MTMCs can be discovered with high-moduli precursors. As an example, the model is deployed to predict the local distortion of Ti–Zr–Hf, V–Nb–Ta, and Cr–Mo–W systems. As shown in Fig. 7a, b and Supplementary Fig. 14, (Ti–Zr–Hf)C displays the strongest adaptation to the FCC structure. Nonetheless, as their precursory carbides show low E, the predicted E of (Ti–Zr–Hf)C (Supplementary Fig. 14c) is relatively low. In contrast, the (V–Nb–Ta)C system presents high modulus as it has low local distortion Fig. 7a and is comprised of binary carbides with high moduli. Specifically, the E values of VC, NbC, and Ta are 518, 497, and 530 GPa, respectively, significantly higher than other binary carbides. Thus, many MTMCs in the (V–Nb–Ta)C compositional space exhibit high E values (Fig. 7c). Finally, large local distortion is observed in the (Cr–Mo–W)C system, which gives rise to lower mechanical performance compared to ROM approximations (Fig. 6e–g). Since the E values of B1-type CrC, MoC, and WC are lower than 420 GPa, MTMCs in the (Cr–Mo–W)C system generally have inadequate mechanical performance (as predicted in Supplementary Fig. 14d). As summarized in Fig. 7e, V–Nb–Ta-based carbides stand out as they are consisted of superior precursors and have strong adaptability to the FCC lattice. Since the local distortions of Ti and Hf are small in MTMC, and the E values of TiC and HfC are medium, we additionally explored the (VNbTa)–Ti–Hf-based carbides as Ti and Hf have strong adaptability and moderate E values. Figure 7b shows small local distortions of this system, as expected. The obtained E values increase with the increased concentration of (VNbTa)C3. Thus, if a compromise is made between elemental adaptability and their intrinsic mechanical performance, the V–Nb–Ta-based MTMCs are corroborated to have excellent mechanical performance. As indicated by the white dots in Fig. 7c, d, (Nb0.33Ta0.67)C, (V0.40Nb0.15Ta0.45)C, (HfNbTaTiV)C5, and (Hf0.10Nb0.18Ta0.18Ti0.36V0.18)C are recommended, and their mechanical properties are presented in Fig. 7f and Table 1.

Local distortions a, b and c, d E of V–Nb–Ta and (VNbTa)–Hf–Ti systems predicted by the ANN model, respectively. e Principles of designing high-performance MTMCs, and the color is for E values. White dots in c and d denote the recommendations, and their compositions are I: (Nb0.33Ta0.67)C, II: (V0.40Nb0.15Ta0.45)C, III: (HfNbTaTiV)C5, and IV: (Hf0.10Nb0.18Ta0.18Ti0.36V0.18)C, respectively. f Mechanical properties of recommended compositions from ML model and experiments (8 mN). The additionally synthesized compositions are V: (TiZrHfTaMo)C5, VI: (TiZrHfVTa)C5, and VII: (MoNbTaVW)C5, respectively. Detailed experimental results are shown in Table 1.
Experimental validations
Nanoindentation experiments are broadly adopted to measure the elastic modulus and hardness of ceramics. However, large fluctuations were reported in prior literature. For example, (HfNbTaTiZr)C59,12,55,56,57,58,59 and (HfNbTaZr)C48,55,60,61,62,63 were prepared and tested by independent experiments, but their Young’s modulus distributes from 333 ± 14 GPa to 576 ± 39 GPa and 441 ± 88 GPa to 615 ± 11 GPa, respectively. Therefore, consistent experiments with the same protocol are essential to validate the predictions of DFT calculations and the ML model. For this purpose, we have performed batch experiments on synthetization and nanoindentation testing of these theoretically predicted MTMC compositions. The experimental details are described in the Methods section. As shown in Table 1 and Fig. 7f, reasonable agreements between DFT/ML and experiments are obtained, signifying the good accuracy of DFT calculations and the established ML model. Generally, the elastic modulus and hardness given by nanoindentation are higher than that given by DFT and ML, but the trend of ML predictions is successfully reproduced, proving the effectiveness of our proposed design principles. The comparison with literature results is shown in Supplementary Note 4.
Discussion
The huge compositional space has been the critical factor impeding the high-throughput screening and rational design of outstanding MTMCs since their first discovery. Experiments and ab initio simulations are exceedingly expensive, and they are normally adopted to probe limited compounds only. In this respect, the ML model developed in this work predicts mechanical properties and geometrical distortion of local environments of equiatomic and non-equiatomic MTMCs rapidly and accurately, which considerably accelerates the design process.
VEC and ROM are widely used to pinpoint the properties of alloys and ceramics. However, with synergistic efforts of DFT calculations and ML predictions, this work shows that VEC is insufficient for describing the moduli of MTMCs, and the moduli of many MTMCs deviate from the ROM estimations. Previous works commonly attribute these phenomena to the complexity of disordered multicomponent materials. In contrast, as depicted in Fig. 8, this work dives into the fundamental reasons by deciphering elemental adaptability from perspectives of energy above convex Hull, electronic structures, and geometrical structures. Elements with strong adaptability to the FCC lattice easily redistribute their electronic densities, leading to a lower Hull energy in the precursory carbides, similar band structures, and small local distortions in MTMCs. Such adaptability also elegantly explains the poor dictation ability of ROM and VEC for different elemental combinations.

Elemental adaptability is the root cause of mechanical properties displayed by MTMCs.
Though elemental adaptability is effective in explaining the mechanical performance of MTMCs, it is not a quantifiable descriptor for designing MTMCs with exceptional strength. Our analysis suggests that the local distortion plays a key role in bridging the underlying electronic structures and mechanical performance of MTMCs, and thus reasonably representing the elemental adaptability. By considering the intrinsic properties and local distortions of different species in MTMCs together, V–Nb–Ta-based MTMCs are predicted to be excellent candidates as high-performance carbides, which is validated by our experiments. Therefore, based on the fundamental understandings and established design principles, the developed model provides versatile tools for designing and discovering superior MTMCs.
In summary, an ANN model is developed to predict the mechanical properties and local distortion of MTMCs with high accuracy and efficiency. As the input features are extracted from properties of precursory carbides, the well-trained model can be deployed to rapidly explore the whole compositional space, including equiatomic and non-equiatomic MTMCs. Based on DFT calculations and ML predictions, we found that VEC and ROM cannot be used to correlate with the mechanical performance of MTMCs, especially for those with large local distortions. Further analysis of convex Hull, geometrical distortions, and electronic structures underlines that the adaptability to the FCC lattice of different elements (quantified by local distortion) is the root cause of deviations from estimations made by VEC and ROM. Specifically, the mechanical properties of MTMCs composed of strongly adaptable elements can follow the mixing rule and VEC trend. By considering the elemental adaptabilities and intrinsic mechanical properties of precursory carbides, V-Nb-Ta-based MTMCs are predicted to have excellent performance. (Nb0.33Ta0.67)C, (V0.40Nb0.15Ta0.45)C, (HfNbTaTiV)C5, and (Hf0.10Nb0.18Ta0.18Ti0.36V0.18)C are recommended after a judicious design, and experiments are carried out and successfully validate ML predictions and recommendations. With the help of the ML model and design philosophy established in this work, a broad path for developing high-performance multicomponent ceramics is paved.
Methods
DFT calculations
The density functional calculations were carried out by the Vienna ab initio simulation package (VASP)64. The energy cutoff of 600 eV was specified for the plane-wave-basis to reach high accuracies. The termination conditions for self-consistent electronic loop and ionic relaxations were set to be 10−4 eV and lower than 0.05 eV/Å, respectively. The exchange-correlation functional proposed by Perdew, Burke, and Ernzerhof (PBE)65 was used with generalized gradient approximations (GGA). Projector augmented wave (PAW) potentials66,67 recommended by the VASP tutorial were employed. The tetrahedron method with Blöchl corrections was adopted to treat the partial occupancies of electrons. For the geometrical optimization of bulk structures, all the cell and atomic degrees of freedom were fully relaxed. For the elastic constant calculations, only atomic positions were optimized, while the cell shape was fixed. The simulated supercell consists of 32 cations and 32 anions within the rock-salt structure. The special quasi-random structures (SQSs) were generated by optimizing the Warren–Cowley short-range order (SRO) parameters68,69 toward zero (examples are provided in Supplementary Table 8). A gamma-centered 5 × 5 × 5 k-point mesh was used to sample the Brillouin zone with Bloch vectors. Elastic constants were calculated by the energy-strain method, where seven points were considered to fit the energy curve for each shearing mode.
Experimental procedure
Raw constituent powders NbC (~1.20 μm), TaC (~1.10 μm), TiC (~1.50 μm), HfC (~2.0 μm), VC (~1.0 μm) were individually weighed according to the designed ratio of metal elements. The powder mixture was then ball milled (250 rpm, 10 h) in a cemented carbide jar using cemented carbide balls and a ball-to-powder mixture ratio of 10:1. The jar was filled with Ar gas before grinding to prevent possible oxidation. Densification and consolidation of the powder mixture were fulfilled using spark plasma sintering (SPS, LABOX-325R, Japan). During sintering, the powder mixture was heated to 2000 °C with a rate of 100 °C/min and a soaking time of 10 min. The pressure was maintained at 40 MPa during the sintering process. The nanohardness of MTMCs was determined using a nanoindentation hardness tester (Agilent Nano Indenter G200). Loads of 8 mN and 50 mN were used to perform a 3*3 matrix on each sample, the load was maintained for 10 s at the peak load, and the loading rate was 10−3 mN/s. The interval between each indent was 60 μm. The indentation tip type is Berkovich, with a radius of 20 nm.
Data availability
The authors declare that the experimental data supporting the results of this study can be found in the paper and its Supplementary Information file. The detailed data for the study is available from the corresponding authors upon request.
Code availability
The Python codes that support the findings of this study are available on GitHub.
References
Yeung, M. T., Mohammadi, R. & Kaner, R. B. Ultraincompressible Superhard Materials. Annu. Rev. Mater. Res. 46, 465–485 (2016).
Kral, C., Lengauer, W., Rafaja, D. & Ettmayer, P. Critical review on the elastic properties of transition metal carbides, nitrides and carbonitrides. J. Alloy. Compd. 265, 215–233 (1998).
Feng, L., Fahrenholtz, W. G., Hilmas, G. E. & Zhou, Y. Synthesis of single-phase high-entropy carbide powders. Scr. Mater. 162, 90–93 (2019).
Yeh, J. W. et al. Nanostructured high-entropy alloys with multiple principal elements: novel alloy design concepts and outcomes. Adv. Eng. Mater. 6, 299–303 (2004).
George, E. P., Raabe, D. & Ritchie, R. O. High-entropy alloys. Nat. Rev. Mater. 4, 515–534 (2019).
Rost, C. M. et al. Entropy-stabilized oxides. Nat. Commun. 6, 8485 (2015).
Gild, J. et al. High-entropy metal diborides: a new class of high-entropy materials and a new type of ultrahigh temperature ceramics. Sci. Rep. 6, 37946 (2016).
Castle, E., Csanádi, T., Grasso, S., Dusza, J. & Reece, M. Processing and properties of high-entropy ultra-high temperature carbides. Sci. Rep. 8, 8609 (2018).
Sarker, P. et al. High-entropy high-hardness metal carbides discovered by entropy descriptors. Nat. Commun. 9, 4980 (2018).
Zhou, J. et al. High-entropy carbide: a novel class of multicomponent ceramics. Ceram. Int. 44, 22014–22018 (2018).
Jin, T. et al. Mechanochemical-assisted synthesis of high-entropy metal nitride via a soft urea strategy. Adv. Mater. 30, 1707512 (2018).
Wen, T., Ye, B., Nguyen, M. C., Ma, M. & Chu, Y. Thermophysical and mechanical properties of novel high-entropy metal nitride-carbides. J. Am. Ceram. Soc. 103, 6475–6489 (2020).
Dippo, O. F., Mesgarzadeh, N., Harrington, T. J., Schrader, G. D. & Vecchio, K. S. Bulk high-entropy nitrides and carbonitrides. Sci. Rep. 10, 21288 (2020).
Zhang, R.-Z., Gucci, F., Zhu, H., Chen, K. & Reece, M. J. Data-driven design of ecofriendly thermoelectric high-entropy sulfides. lnorg. Chem. 57, 13027–13033 (2018).
Wei, X. F. et al. High-entropy carbide ceramics with refined microstructure and enhanced thermal conductivity by the addition of graphite. J. Eur. Ceram. Soc. 41, 4747–4754 (2021).
Wang, Z., Li, Z. T., Zhao, S. & Wu, Z. G. High-entropy carbide ceramics: a perspective review. Tungsten 3, 131–142 (2021).
Oses, C., Toher, C. & Curtarolo, S. High-entropy ceramics. Nat. Rev. Mater. 5, 295–309 (2020).
Moskovskikh, D. et al. Extremely hard and tough high entropy nitride ceramics. Sci. Rep. 10, 19874 (2020).
Harrington, T. J. et al. Phase stability and mechanical properties of novel high entropy transition metal carbides. Acta Mater. 166, 271–280 (2019).
Zhao, S. Lattice distortion in high-entropy carbide ceramics from first-principles calculations. J. Am. Ceram. Soc. 104, 1874–1886 (2021).
Miracle, D. B. & Senkov, O. N. A critical review of high entropy alloys and related concepts. Acta Mater. 122, 448–511 (2017).
Yao, H. W. et al. Mechanical properties of refractory high-entropy alloys: experiments and modeling. J. Alloy. Compd. 696, 1139–1150 (2017).
Tian, L.-Y. et al. Alloying effect on the elastic properties of refractory high-entropy alloys. Mater. Des. 114, 243–252 (2017).
Guo, S. & Liu, C. T. Phase stability in high entropy alloys: formation of solid-solution phase or amorphous phase. Prog. Nat. Sci. 21, 433–446 (2011).
Yang, T. et al. Ductilizing brittle high-entropy alloys via tailoring valence electron concentrations of precipitates by controlled elemental partitioning. Mater. Res. Lett. 6, 600–606 (2018).
Guo, S., Ng, C., Lu, J. & Liu, C. T. Effect of valence electron concentration on stability of fcc or bcc phase in high entropy alloys. J. Appl. Phys. 109, 103505 (2011).
Balasubramanian, K., Khare, S. V. & Gall, D. Valence electron concentration as an indicator for mechanical properties in rocksalt structure nitrides, carbides and carbonitrides. Acta Mater. 152, 175–185 (2018).
Jhi, S.-H., Ihm, J., Louie, S. G. & Cohen, M. L. Electronic mechanism of hardness enhancement in transition-metal carbonitrides. Nature 399, 132–134 (1999).
Senkov, O. N., Miller, J. D., Miracle, D. B. & Woodward, C. Accelerated exploration of multi-principal element alloys for structural applications. Calphad 50, 32–48 (2015).
Sheikh, S. et al. Alloy design for intrinsically ductile refractory high-entropy alloys. J. Appl. Phys. 120, 164902 (2016).
Kaufmann, K. & Vecchio, K. S. Searching for high entropy alloys: a machine learning approach. Acta Mater. 198, 178–222 (2020).
Hart, G. L. W., Mueller, T., Toher, C. & Curtarolo, S. Machine learning for alloys. Nat. Rev. Mater. 6, 730–755 (2021).
Zhang, L. et al. Machine learning reveals the importance of the formation enthalpy and atom-size difference in forming phases of high entropy alloys. Mater. Des. 193, 108835 (2020).
Li, Y. & Guo, W. Machine-learning model for predicting phase formations of high-entropy alloys. Phys. Rev. Mater. 3, 095005 (2019).
Huang, W., Martin, P. & Zhuang, H. L. Machine-learning phase prediction of high-entropy alloys. Acta Mater. 169, 225–236 (2019).
Vazquez, G. et al. Efficient machine-learning model for fast assessment of elastic properties of high-entropy alloys. Acta Mater. 232, 117924 (2022).
Zhang, J. et al. Design high-entropy electrocatalyst via interpretable deep graph attention learning. Joule 7, 1–20 (2023).
Zuo, Y. et al. Accelerating materials discovery with Bayesian optimization and graph deep learning. Mater. Today 51, 126–135 (2021).
Kaufmann, K. et al. Discovery of high-entropy ceramics via machine learning. npj Comput. Mater. 6, 42 (2020).
Zhang, J. et al. Design high-entropy carbide ceramics from machine learning. npj Comput. Mater. 8, 5 (2022).
Mitra, R., Bajpai, A. & Biswas, K. Machine learning based approach for phase prediction in high entropy borides. Ceram. Int. 48, 16695–16706 (2022).
Jaafreh, R., Kang, Y. S., Kim, J.-G. & Hamad, K. Machine learning guided discovery of super-hard high entropy ceramics. Mater. Lett. 306, 130899 (2022).
Tang, Y., Zhang, D., Liu, R. & Li, D. Designing high-entropy ceramics via incorporation of the bond-mechanical behavior correlation with the machine-learning methodology. Cell Rep. Phys. Sci. 2, 100640 (2021).
Chen, C. et al. A critical review of machine learning of energy materials. Adv. Energy Mater. 10, 1903242 (2020).
Jablonka, K. M., Ongari, D., Moosavi, S. M. & Smit, B. Big-data science in porous materials: materials genomics and machine learning. Chem. Rev. 120, 8066–8129 (2020).
Liu, H., Fu, Z., Yang, K., Xu, X. & Bauchy, M. Machine learning for glass science and engineering: a review. J. Non-Cryst. Solids 557, 119419 (2021).
Tao, Q., Xu, P., Li, M. & Lu, W. Machine learning for perovskite materials design and discovery. npj Comput. Mater. 7, 23 (2021).
Nam, C. Prediction of mechanical properties of high-entropy ceramics by deep learning with compositional descriptors. Mater. Today Commun. 35, 105949 (2023).
Dai, F. Z., Wen, B., Sun, Y., Xiang, H. & Zhou, Y. Theoretical prediction on thermal and mechanical properties of high entropy (Zr0.2Hf0.2Ti0.2Nb0.2Ta0.2)C by deep learning potential. J. Mater. Sci. Technol. 43, 168–174 (2020).
Dai, F. Z., Sun, Y., Wen, B., Xiang, H. & Zhou, Y. Temperature dependent thermal and elastic properties of high entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)B2: molecular dynamics simulation by deep learning potential. J. Mater. Sci. Technol. 72, 8–15 (2020).
Zhang, J. et al. Rational design of high-entropy ceramics based on machine learning—ā critical review. Curr. Opin. Solid State Mater. Sci. 27, 101057 (2023).
Kingma D. P. & Ba J. Adam: A method for stochastic optimization. CoRR. 1412.6980, (2014).
Jain, A. et al. Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013).
Jhi, S. H. & Ihm, J. Electronic structure and structural stability of TiCxN1-x alloys. Phys. Rev. B 56, 13826–13829 (1997).
Vorotilo, S. et al. Phase stability and mechanical properties of carbide solid solutions with 2–5 principal metals. Comput. Mater. Sci. 201, 110869 (2022).
Lu, K. et al. Microstructures and mechanical properties of high-entropy (Ti0.2Zr0.2Hf0.2Nb0.2Ta0.2)C ceramics with the addition of SiC secondary phase. J. Eur. Ceram. Soc. 40, 1839–1847 (2020).
Guan, S. et al. The effect of pressure tuning on the structure and mechanical properties of high-entropy carbides. Scr. Mater. 216, 114755 (2022).
Feng, L., Chen, W. T., Fahrenholtz, W. G. & Hilmas, G. E. Strength of single-phase high-entropy carbide ceramics up to 2300°C. J. Am. Ceram. Soc. 104, 419–427 (2021).
Tan, Y. et al. Compositional effect on mechanical properties of transition-metal carbide solid solutions. Ceram. Int. 47, 16882–16890 (2021).
Wang, Y., Csanádi, T., Zhang, H., Dusza, J. & Reece, M. J. Synthesis microstructure, and mechanical properties of novel high entropy carbonitrides. Acta Mater. 231, 117887 (2022).
Li, Z. et al. Phase, microstructure and related mechanical properties of a series of (NbTaZr)C-based high entropy ceramics. Ceram. Int. 47, 14341–14347 (2021).
Song, J. et al. Regulating the formation ability and mechanical properties of high-entropy transition metal carbides by carbon stoichiometry. J. Mater. Sci. Technol. 121, 181–189 (2022).
Wang, Y. et al. The role of Cr addition on the processing and mechanical properties of high entropy carbides. J. Eur. Ceram. Soc. 42, 5273–5279 (2022).
Kresse, G. & Furthmüller, J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mater. Sci. 6, 15–50 (1996).
Perdew, J. P., Burke, K. & Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996).
Kresse, G. & Joubert, D. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys. Rev. B 59, 1758–1775 (1999).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Zhao, S., Stocks, G. M. & Zhang, Y. Stacking fault energies of face-centered cubic concentrated solid solution alloys. Acta Mater. 134, 334–345 (2017).
Zhang, J., Ma, S., Xiong, Y., Xu, B. & Zhao, S. Elemental partitions and deformation mechanisms of L12-type multicomponent intermetallics. Acta Mater. 219, 117238 (2021).
Acknowledgements
This work is supported by the National Key R&D Program of China (No. 2022YFE0200900), the Research Grants Council of Hong Kong (No. 11200421), the Hong Kong Innovation and Technology Commission (No. MHP/ 098/21). The computational time provided by the National Supercomputer Center in Tianjin and the CityU Burgundy Supercomputer is highly acknowledged.
Author information
Authors and Affiliations
Contributions
J.Z. and S.Z. conceived and conducted density functional theory calculations and developed machine-learning models. L.H. and Z.W. performed the experiment. J.Z. and L.H. co-wrote the paper. Y.X., S.H., B.X., S.M., X.X., and H.F. contributed to the analysis of the results. J.Z., L.H., Z.W., J.K., and S.Z. reviewed and revised the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, J., He, L., Xiong, Y. et al. Local-distortion-informed exceptional multicomponent transition-metal carbides uncovered by machine learning. npj Comput Mater 10, 162 (2024). https://doi.org/10.1038/s41524-024-01351-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41524-024-01351-1
- Springer Nature Limited