

Abstract
Different from the traditional two-electron oxidative addition-transmetalation-reductive elimination coupling strategy, visible light has been successfully integrated into transition metal-catalyzed coupling reaction of propargylic alcohol derivatives highly selectively forming allenenitriles: specifically speaking, visible light-mediated Cu-catalyzed cyanation of propargylic oxalates has been realized for the general, efficient, and exclusive syntheses of di-, tri, and tetra-substituted allenenitriles bearing various synthetically versatile functional groups. A set of mechanistic studies, including fluorescence quenching experiments, cyclic voltammetric measurements, radical trapping experiments, control experiments with different photocatalyst, and DFT calculation studies have proven that the current reaction proceeds via visible light-induced redox-neutral reductive quenching radical mechanism, which is a completely different approach as compared to the traditional transition metal-catalyzed two-electron oxidative addition processes.
Similar content being viewed by others
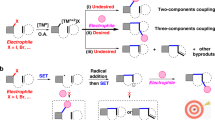

Introduction
Due to the wide existence of allene unit in natural products, bioactive molecules1, and functional materials2, development of methods for efficient allene syntheses is of high current interest3,4,5,6,7,8,9,10,11. A few strategies such as allenylic substitution with 2-halo-1,3-butadienes12,13 or allenyl esters14,15,16,17,18, 1,4-difunctionalization of 1,3-enynes19,20,21,22,23,24,25,26,27,28,29,30,31,32, allenation of the terminal alkynes (ATA) reaction33,34, and coupling reactions involving propargylic substrates35,36,37,38,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53, have been extensively and well established. For the last reaction, in addition to the SN2′-type substitution of propargylic substrates38,39,40,41,42,43,44,45,46, transition metal-catalyzed coupling reaction of propargylic alcohol derivatives with organometallic reagents47,48,49,50,51,52,53 involves a two-electron oxidative addition-transmetalation-reductive elimination process (Fig. 1a). However, scope and selectivity limitation remain due to the issues of the intrinsic two-electron mechanism54,55,56. Allenenitriles have been frequently employed as useful synthetic precursors for various organic motifs57,58,59, while the classic synthetic method relies on stoichiometric amount of CuCN-mediated cyanation of propargylic alcohols with KCN (1.5 equiv) in the presence of HBr (2.5 equiv)60. We envisioned a concept for allenenitrile syntheses via the coupling reaction from propargylic derivatives involving a one-electron process (Fig. 1b). The challenges here (Fig. 1b) are (1) the regioselectivity issue on possible formation of alkyne products61,62, (2) the match of radical reactivity with the transition metal species, and (3) the regeneration of the catalytically transition metal catalyst.

a Traditional transition metal-catalyzed two-electron cross-coupling reactions. b A concept of one-electron process for cross-coupling reactions. c This work: an example of such a concept for allenenitrile synthesis (visible light/transition metal dual catalysis).
In this work, we wish to report such a concept-a radical-based efficient syntheses of allenenitriles from propargylic oxalates and TMSCN under the dual catalysis of photo and copper (Fig. 1c)60.
Results
Optimization of reaction conditions
We began our study on the coupling reaction of propargylic oxalate 1a with trimethylsilyl cyanide (TMSCN) under blue light irradiation in the presence of CuBr and photocatalyst, fac-Ir(ppy)3. The desired allenenitrile 2a was formed in DMF for 24 h in 29% NMR yield with 18% recovery of 1a (Table 1, entry 1). After evaluation of a series of 2,2′-dipyridine ligands, we were glad to find that 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy, L5) was the optimal ligand (Table 1, entries 2–6). Notably, no propargylic isomer 3a was detected in the crude reaction mixture. The reaction performed in CH3CN gave higher yield than other checked solvents such as DMAC, NMP, DMPU, THF, and DCM (Table 1, entry 7, for details on solvent screening, see the Supplementary Information). Increasing the loading of TMSCN (Table 1, entry 8) and running the reaction at a concentration of 0.1 M (Table 1, entry 9) further promoted the formation of 2a, which could be isolated in 89% yield on a 0.5 mmol reaction scale. As expected, CuBr2 was totally ineffective (Table 1, entry 10). No reaction occurred in the absence of the light (Table 1, entries 11 and 12) or photocatalyst fac-Ir(ppy)3 (Table 1, entry 13), suggesting that both the light and photocatalyst were indispensable for the transformation.
Substrate scope
With the optimized reaction conditions in hand, we set out to investigate the substrate scope of this method (Fig. 2). Overall, a variety of terminal tertiary propargylic oxalates smoothly underwent cyanation to form trisubstituted allenenitriles as exclusive regioisomer in good to excellent yields. No obvious yield difference among cyclic (2a, 2b, 2c, 2d) and acyclic (2i, 2j, 2m) substrates was observed. Even with the sterically hindered adamantyl-containing oxalate 1l, the yield of 2l was 91% after increasing the catalyst loadings of CuBr and L5 to 15 mol% and 18 mol%, respectively. A wide range of reactive yet synthetic useful functional groups, such as sulfide (2e, easily poisoning Cu catalysis), amide (2f), halogen (2n, 2o, 2p), ester (2k, 2q), ketal (2g, 2s), terminal alkyne (2q), and terminal olefin (2r) were intact under the standard mild reaction conditions. Interestingly, under the standard conditions the propargylic oxalate 1h with a ketone unit was converted to nitrile 2h with the in situ formation of a synthetically useful enol silyl ether entity63,64 in 65% yield. The thiophene unit in substrate 1t was also accommodated. Furthermore, products incorporating Boc-protected L-proline 2u, pentoxyifylline 2v, Boc-protected tropinone 2w and 2w’, and raspberry ketone tetra-O-acetyl-β-D-glucopyranoside 2x, mestranol 2y worked well without affecting the other fragile functionalities. The structure of 2w’ was unambiguously established by its X-ray analysis. The reaction could be easily conducted on gram-scales (2q and 2y), demonstrating the practicality of this protocol. Even the reaction of terminal secondary propargylic oxalates 1z and 1A still afforded 1,3-disubstituted allenenitriles 2z and 2A as the products in decent yields and a very high allene/alkyne selectivity (25:1 and 14:1). 4-Phenylallenenitrile 2J could also be obtained via the current method in 54% yield as the only isomer, and the slightlylower isolated yield may be attributed to its instability.

aCuBr (15 mol%) and L5 (18 mol%) were used. bDue to the difficulty of separating the two regioisomers, the yield value refers to the isolated yield of a mixture of alkyne and allene; the regioselectivity was determined by 1H NMR analysis. cThe reaction was conducted in 10 mL CH3CN.
The reaction could be further extended to non-terminal propargylic oxalates, such as 1B, 1C, and 1K. When trimethylsilyl-substituted alkyne 1C was used, TMS-substituted allenenitrile 2C was produced exclusively in 88% yield, which was not readily accessible by other ways65 and very useful in propargylation reaction66,67. For non-terminal propargylic oxalates with R3 being Ph (1D) and CO2Me (1E), dinitrile products 4a and 4b were obtained, which must be produced from the subsequent conjugate addition of TMSCN with the in situ formed allenenitrile intermediate 2D and 2E, respectively.
Interestingly, when MgCl2 or MgBr2•6H2O replaced TMSCN as the nucleophile, various chloroallene or bromoallene bearing sterically hindered adamantyl (12l or 13l), ketal (12s), ester, or terminal alkyne (13q) could be obtained in decent yields. As a comparison, TMSBr or TMSCl gave inferior results (Fig. 3).

The reaction condition A was used for the synthesize of cholorallenes (present in red color), and the reaction condition B was used for the synthesize of bromoallenes (present in blue color).
Synthetic applications
These allenenitriles are synthetic versatile as shown in Fig. 4: The Cu(I) catalyzed [4 + 2] cycloaddition68 of 2a (R1, R2 = -(CH2)5-) with furan provided 7-oxa-bicyclo-[2.2.1]heptene derivatives endo-5 and exo-5 in 55 and 14% yield, respectively. The configuration of endo-5 was unambiguously identified by X-ray analysis. Conjugate addition of 4-methylbenzenethiol with 2a afforded sulfur-substituted tetrasubstituted alkene 6 in an excellent yield69. Deuteration of α-H of 2m (R1 = Me, R2 = -(CH2)2Ph) with D2O in the presence of K2CO3 and n-Bu4NBr readily yielded d-2m in 96% yield with 96% D-incorporation. Hydrolysis of nitrile group in 2l (R1 = Me, R2 = 1-adamantyl) with a base produced allenyl amide 7 in 64% yield70. In addition, the ethynyl group in 2q underwent the Cu-catalyzed click reaction with anti-HIV drug AZT (Zidovudine)71 while the allenenitrile unit remained unreacted, offering useful handle for further synthetic elaboration.

Reagents and conditions: (a) 2a (0.2 mmol), Cu(CH3CN)4BF4 (20 mol%), freshly distilled furan (2 mL), 50 °C, 2 d; (b) 2a (0.4 mmol), 4-methylbenzenethiol (1.2 equiv), Et3N (2.0 equiv), CHCl3, rt, 24 h; (c) 2 m (0.27 mmol), K2CO3 (5.0 equiv), n-Bu4NBr (1.0 equiv), Toluene/D2O = 9:11, rt, 2.5 d; (d) 2 l (0.2 mmol), NaOH (20 mol%), Na2CO3 (1.0 equiv), H2O2 (3.9 equiv), EtOH/H2O = 5:1, rt, 24 h; (e) 2q (0.4 mmol), AZT (1.0 equiv), CuSO4·5H2O (5 mol%), sodium ascorbate (15 mol%), DCM/H2O = 1:1.
Mechanistic studies
To probe the reaction mechanism, we conducted a set of mechanistic studies. First, several propargylic compounds with different leaving groups 1F (Boc), 1G (Ac), 1H (CO2Me) were prepared. The Cyclic Voltammetry (CV) experiments were performed to measure the reduction potential of these substrates 1d, 1F, 1G, and 1H (Fig. 5a). The half peak potential of redox active oxalate 1d was determined to be Ep/2 [1d/1d•-] = −1.71 V vs SCE (Saturated calomel electrode) in CH3CN. However, under the same measurement conditions for 1F (Boc), 1G (Ac), and 1H (CO2Me), no apparent anodic and cathodic current peaks could be observed in the range of −3.0 to 0 V, suggesting that these were redox-inactive leaving groups. Indeed, when 1F (Boc), 1G (Ac), or 1H (CO2Me) were subjected to the optimal conditions, 100% of the corresponding unreacted starting materials were recovered.

a Experiments and cyclic voltammograms of different propargylic compounds. b Stern-Volmer quenching experiments of fac-Ir(ppy)3. c Reaction with Ph-PTZ photocatalyst. d The radical trapping experiment with TEMPO. e Reaction with Ir(dtbbpy)(ppy)2PF6 or Ir[dF(CF3)ppy]2(dtbbpy)PF6 as photocatalyst.
Two possible reaction pathways for this transformation based on CV data were proposed as shown in Fig. 6a and Supplementary Fig. 5. In oxidative quenching cycle (Supplementary Fig. 5), first, the excited state of fac-Ir(ppy)3* (E1/2red [IrIV/IrIII*] = −1.73 V vs SCE in CH3CN)72 could be quenched with oxalate 1 (Ep/2 [1d/1d•-] = −1.71 V vs SCE in CH3CN) to generate [fac-Ir(ppy)3]+ species and anionic radical intermediate 9, which would form propargylic radical 10 by releasing oxalate anion. Then LCuICN (Ep/2red [CuII/CuI] = +0.15 V vs SCE in CH3CN, see Supplementary Information for details) would be oxidized by [fac-Ir(ppy)3]+ (E1/2red [IrIV/IrIII] = +0.77 V vs SCE in CH3CN)72 to produce LCuIICN, which would further react with TMSCN to yield LCuII(CN)2. Alternatively, in reductive quenching cycle (Fig. 6a), the excited state of fac-Ir(ppy)3* (E1/2red [IrIII*/IrII] = +0.31 V vs SCE in CH3CN)72 could be quenched with LCuICN to generate LCuII(CN) and [fac-Ir(ppy)3]− species (E1/2red [IrIII/IrII] = −2.19 V vs SCE in CH3CN)72. Oxalate 1 could be reduced by [fac-Ir(ppy)3]− to yield anionic radial intermediate 9 via one-electron reduction. Finally, in both pathway the radical intermediate 10 may isomerize to allenyl radical 1173,74, which may bind with LCuII(CN)2, followed by reductive elimination to deliver allenenitrile 2 and regenerate the catalytically active species LCuICN. Another possible pathway, 11 could abstract the CN group from LCuII(CN)2 to afford allenenitrile 232,75,76. The steric effect of R1, R2, and R3 may play an important role in the reaction selectivity for forming 2 or 3.

a Proposed mechanism via reductive quenching cycle. b Free energy profiles calculated for the reaction of L4CuII(CN)2 with Int1. Relative free energies are given in kcal/mol.
To distinguish the two pathways, Stern-Volmer quenching experiments of fac-Ir(ppy)3 were carried out. As shown in Fig. 5b, the excited state of the photocatalyst fac-Ir(ppy)3 was efficiently quenched by the CuBr/L5 catalyst. Furthermore, if the cyanation of 1d would be realized via oxidative quenching cycle, considering the redox-potential window of typical photocatalysis (Ir/Ru/organic-PC etc.)77, the Ph-PTZ was selected as another potential photocatalyst for this transformation. The reaction in the presence of photocatalyst Ph-PTZ instead of fac-Ir(ppy)3 would provide readily radical 10 or 11, the subsequent SET process between the oxidized state of Ph-PTZ+ (E1/2[Ph-PTZ+/Ph-PTZ] = +0.815 V vs SCE in CH3CN)61 and LCuICN (Ep/2red [CuII/CuI] = +0.15 V vs SCE in CH3CN) would form LCuIICN, which could yield 2d. However, such a reaction only afforded 10% of 2d with 90% of 1d being recovered (Fig. 5c). When 2 equiv of TEMPO were used as the radical trapping agent in the reaction of 1d, the formation of 2d was obviously reduced (16% vs 87%), and the TEMPO-trapped product 14 and/or 15 could be detected by LC-HRMS analysis, which supports the involvement of radical intermediates in the current transformation (Fig. 5d). Furthermore, in order to check the possible triplet energy transfer mechanism, other ruthenium- or iridium-based dyes or organic photocatalysts were tested under standard conditions (for details on photocatalyst screening, see the Supplementary Information): Photocatalysts (Ir(dtbbpy)(ppy)2PF6, ET = 49.2 kcal/mol and Ir[dF(CF3)ppy]2(dtbbpy)PF6, ET = 60.8 kcal/mol) with its triplet energy similar to that of fac-Ir(ppy)3 (ET = 57.8 kcal/mol) did not provide 2d at all (Fig. 5e)78,79.
To further elucidate the reaction mechanism, density functional theory (DFT) calculations were preformed to survey the reaction of 1B using ligand L4 (For details on DFT calculations, see the Supplementary Information and Supplementary Data 1). As proposed by Fig. 6a, radical intermediate Int1 could be formed from oxalate 1B. Mulliken atomic spin density analysis of Int1 suggests that the single electron distributes on C1 and C2 with a similar spin density (0.64 and 0.47, Fig. 6b), indicating Int1 is a combination of resonance forms of allenyl radical and propargylic radical. As an allenyl radical, Int1 reacts with L4CuII(CN)2 via a singlet diradical transition structure TS1_a with a free energy barrier of 10.1 kcal/mol, providing a closed-shell propargyl-Cu(III) complex Int2_a reversibly. Subsequent reductive elimination produces the final allenenitrile product 2B with a very low barrier of 1.0 kcal/mol (TS2_a). Furthermore, the concerted radical cyanation process is also investigated. A triplet transition structure TS_a was obtained with a much higher free energy barrier of 30.3 kcal/mol, which indicates that the stepwise pathway via a Cu(III) intermediate is more favorable. On the other hand, the possibility of Int1 acting as a propargyl radical has also been considered. A similar oxidation/reductive elimination process is obtained, but more energy demanding, due to the steric effect caused by the cyclohexyl group with the ligand. Thus, allenenitriles 2B were generated as the only products.
These above results definitely confirmed that the reductive quenching cycle in Fig. 6a was the dominant pathway in the current transformation, which is different from the well-established oxidative quenching mechanism61,75,76.
In conclusion, we have developed a general and efficient method for the highly selective synthesis of di-, tri-, and tetra-substituted allenenitriles from readily available propargylic oxalates and TMSCN under photoredox conditions. This reaction featured mild conditions and a broad functional group compatibility. Excellent regioselectivities were achieved in both terminal and internal propargylic oxalates. Even for secondary substrates, allenenitriles were still the predominant products. The current method was further extended to the synthesis of cholorallenes or bromoallenes by using MgCl2 or MgBr2•6H2O as the nucleophile. Stern-Volmer quenching experiments, cyclic voltammetric measurements, radical trapping experiments, control experiments with different photocatalysts, and DFT calculation studies indicated that propargylic radical and allenyl radical generated via light-induced one-electron process were involved via the reductive quenching cycle. This protocol for allenenitrile syntheses involving one-electron mechanistic pathway is very different from the traditional transition metal-catalyzed two-electron coupling reactions and will surely overcome the scope limitation of the known protocols and enjoy scopes for the efficient syntheses of differently functionalized allenes due to the powerful catalytic activity of copper80,81. Further studies on highly selective allene synthesis via such one-electron process and other photocatalysts are being actively pursued in this laboratory.
Methods
General procedure for the copper-catalyzed cyanation of propargylic oxalates
To a flame-dried 10 mL Schlenk tube were added fac-Ir(ppy)3 (3.3 mg, 5 μmol), CuBr (7.3 mg, 0.05 mmol), 4,4′-di-tert-butyl-2,2′-bipyridine L5 (16.4 mg, 0.06 mmol), 1a (105.4 mg, 0.5 mmol)/CH3CN(2.5 mL), and TMSCN (157.2 mg, 1.5 mmol)/CH3CN(2.5 mL) sequentially under Ar atmosphere. The resulting mixture was irradiated with a 50 W blue LED lamp (2-3 cm away, with cooling fan to keep the reaction temperature at 35–40 °C) for 24 h with stirring and monitored by TLC. The resulting mixture was filtrated through a short pad of silica gel eluted with ethyl ether (30 mL). After evaporation, the residue was purified by chromatography on silica gel to afford the pure product 2a.
Data availability
The X-ray crystallographic coordinates for structures of 2w’ and endo-5 reported in this study have been deposited in the Cambridge Crystallographic Data Centre (CCDC) under deposition numbers CCDC 2047907 (2w’), and CCDC-2047908 (endo-5). These data can be obtained free of charge from http://www.ccdc.cam.ac.uk/data_request/cif. The experimental procedures and characterization of the new compounds in this study are provided in the Supplementary Information. All other data are available from the authors upon request.
References
Hoffmann-Röder, A. & Krause, N. Synthesis and properties of allenic natural products and pharmaceuticals. Angew. Chem. Int. Ed. 43, 1196–1216 (2004).
Rivera-Fuentes, P. & Diederich, F. Allenes in molecular materials. Angew. Chem. Int. Ed. 51, 2818–2828 (2012).
Brummond, K. M. & DeForrest, J. E. Synthesizing allenes today (1982–2006). Synthesis 2007, 795–818 (2007).
Yu, S. & Ma, S. How easy are the syntheses of allenes? Chem. Commun. 47, 5384–5418 (2011).
Neff, R. K. & Frantz, D. E. Recent advances in the catalytic syntheses of allenes: a critical assessment. ACS Catal. 4, 519–528 (2014).
Xiao, Q., Xia, Y., Li, H., Zhang, Y. & Wang, J. Coupling of N-tosylhydrazones with terminal alkynes catalyzed by copper(I): synthesis of trisubstituted allenes. Angew. Chem. Int. Ed. 50, 1114–1117 (2011).
Crouch, I. T., Neff, R. K. & Frantz, D. E. Pd-catalyzed asymmetric β-hydride elimination en route to chiral allenes. J. Am. Chem. Soc. 135, 4970–4973 (2013).
Friese, F. W. & Studer, A. Deoxygenative borylation of secondary and tertiary alcohols. Angew. Chem. Int. Ed. 58, 9561–9564 (2019).
Wei, X.-F. et al. Catalytic regio- and enantioselective proton migration from skipped enynes to allenes. Chem 5, 585–599 (2019).
Lu, R. et al. Enantioselective copper-catalyzed radical cyanation of propargylic C-H bonds: easy access to chiral allenyl nitriles. J. Am. Chem. Soc. 143, 14451–14457 (2021).
Zhou, Z. Z. et al. Photoredox/nickel dual-catalyzed regioselective alkylation of propargylic carbonates for trisubstituted allenes. Chem. Commun. 57, 9390–9393 (2021).
Ogasawara, M., Ikeda, H. & Hayashi, T. π-Allylpalladium-mediated catalytic synthesis of functionalized allenes. Angew. Chem. Int. Ed. 39, 1042–1044 (2000).
Ogasawara, M., Ikeda, H., Nagano, T. & Hayashi, T. Palladium-catalyzed asymmetric synthesis of axially chiral allenes: a synergistic effect of dibenzalacetone on high enantioselectivity. J. Am. Chem. Soc. 123, 2089–2090 (2001).
Imada, Y., Ueno, K., Kutsuwa, K. & Murahashi, S.-I. Palladium-catalyzed asymmetric alkylation of 2,3-alkadienyl phosphates. Synthesis of optically active 2-(2,3-alkadienyl)malonates. Chem. Lett. 31, 140–141 (2002).
Trost, B. M., Fandrick, D. R. & Dinh, D. C. Dynamic kinetic asymmetric allylic alkylations of allenes. J. Am. Chem. Soc. 127, 14186–14187 (2005).
Petrone, D. A., Isomura, M., Franzoni, I., Rӧssler, S. L. & Carreira, E. M. Allenylic carbonates in enantioselective iridium-catalyzed alkylations. J. Am. Chem. Soc. 140, 4697–4704 (2018).
Song, S., Zhou, J., Fu, C. & Ma, S. Catalytic enantioselective construction of axial chirality in 1,3-disubstituted allenes. Nat. Commun. 10, 507 (2019).
Zhang, J. et al. Enantio- and diastereodivergent construction of 1,3-nonadjacent stereocenters bearing axial and central chirality through synergistic Pd/Cu catalysis. J. Am. Chem. Soc. 143, 12622–12632 (2021).
Poutsma, M. L. & Ibarbia, P. A. Radical addition of tert-butyl hypochlorite to conjugated enynes. J. Org. Chem. 35, 4038–4047 (1970).
Matsumoto, Y., Naito, M., Uozumi, Y. & Hayashi, T. Axially chiral allenylboranes: catalytic asymmetric synthesis by palladium-catalysed hydroboration of but-1-en-3-ynes and their reaction with an aldehyde. J. Chem. Soc., Chem. Commun. 19, 1468–1469 (1993).
Huang, Y., Del Pozo, J., Torker, S. & Hoveyda, A. H. Enantioselective synthesis of trisubstituted allenyl-B(pin) compounds by phosphine-Cu-catalyzed 1,3-enyne hydroboration. Insights regarding stereochemical integrity of Cu-allenyl intermediates. J. Am. Chem. Soc. 140, 2643–2655 (2018).
Wang, F. et al. Divergent synthesis of CF3-substituted allenyl nitriles by ligand-controlled radical 1,2- and 1,4-addition to 1,3-enynes. Angew. Chem. Int. Ed. 57, 7140–7145 (2018).
Yu, S., Sang, H. L., Zhang, S.-Q., Hong, X. & Ge, S. Catalytic asymmetric synthesis of chiral trisubstituted heteroaromatic allenes from 1,3-enynes. Commun. Chem. 1, 64 (2018).
Adamson, N. J., Jeddi, H. & Malcolmson, S. J. Preparation of chiral allenes through Pd-catalyzed intermolecular hydroamination of conjugated enynes: enantioselective synthesis enabled by catalyst design. J. Am. Chem. Soc. 141, 8574–8583 (2019).
Bayeh-Romero, L. & Buchwald, S. L. Copper hydride catalyzed enantioselective synthesis of axially chiral 1,3-disubstituted allenes. J. Am. Chem. Soc. 141, 13788–13794 (2019).
Zhang, K. F. et al. Nickel-catalyzed carbofluoroalkylation of 1,3-enynes to access structurally diverse fluoroalkylated allenes. Angew. Chem. Int. Ed. 58, 5069–5074 (2019).
Zhu, X. et al. Copper-catalyzed radical 1,4-difunctionalization of 1,3-enynes with alkyl diacyl peroxides and N-fluorobenzenesulfonimide. J. Am. Chem. Soc. 141, 548–559 (2019).
Zeng, Y. et al. Copper-catalyzed enantioselective radical 1,4-difunctionalization of 1,3-enynes. J. Am. Chem. Soc. 142, 18014–18021 (2020).
Chen, Y., Wang, J. & Lu, Y. Decarboxylative 1,4-carbocyanation of 1,3-enynes to access tetra-substituted allenes via copper/photoredox dual catalysis. Chem. Sci. 12, 11316–11321 (2021).
Dong, X. Y. et al. Copper-catalyzed asymmetric coupling of allenyl radicals with terminal alkynes to access tetrasubstituted allenes. Angew. Chem. Int. Ed. 60, 2160–2164 (2021).
Xu, T. et al. Dual photoredox/Nickel-catalyzed 1,4-sulfonylarylation of 1,3-enynes with sulfinate salts and aryl halides: entry into tetrasubstituted allenes. Org. Lett. 23, 8455–8459 (2021).
Fu, L., Greßies, S., Chen, P. & Liu, G. Recent advances and perspectives in transition metal-catalyzed 1,4-functionalizations of unactivated 1,3-enynes for the synthesis of allenes. Chin. J. Chem. 38, 91–100 (2020).
Crabbé, P., Fillion, H., André, D. & Luche, J.-L. Efficient homologation of acetylenes to allenes. J. Chem. Soc., Chem. Commun. 19, 859–860 (1979).
Huang, X. & Ma, S. Allenation of terminal alkynes with aldehydes and ketones. Acc. Chem. Res. 52, 1301–1312 (2019).
Tsuji, J. & Mandai, T. Palladium-catalyzed reactions of propargylic compounds in organic synthesis. Angew. Chem. Int. Ed. 34, 2589–2612 (1995).
Ma, S. Pd-catalyzed coupling reactions involving propargylic/allenylic species. Eur. J. Org. Chem. 2004, 1175–1183 (2004).
O’Broin, C. Q. & Guiry, P. J. Advances in decarboxylative palladium-catalyzed reactions of propargyl electrophiles. J. Org. Chem. 85, 10321–10333 (2020).
Rona, P. & Crabbé, P. A novel allene synthesis. J. Am. Chem. Soc. 90, 4733–4734 (1968).
Ito, H., Sasaki, Y. & Sawamura, M. Copper(I)-catalyzed substitution of propargylic carbonates with diboron: selective synthesis of multisubstituted allenylboronates. J. Am. Chem. Soc. 130, 15774–15775 (2008).
Li, H., Muller, D., Guenee, L. & Alexakis, A. Copper-catalyzed enantioselective synthesis of axially chiral allenes. Org. Lett. 14, 5880–5883 (2012).
Wu, S. et al. A C-H bond activation-based catalytic approach to tetrasubstituted chiral allenes. Nat. Commun. 6, 7946 (2015).
Kessler, S. N. & Bäckvall, J. E. Iron-catalyzed cross-coupling of propargyl carboxylates and Grignard reagents: synthesis of substituted allenes. Angew. Chem. Int. Ed. 55, 3734–3738 (2016).
Ruchti, J. & Carreira, E. M. Rh-catalyzed stereospecific synthesis of allenes from propargylic benzoates and arylboronic acids. Org. Lett. 18, 2174–2176 (2016).
Lu, Q., Gressies, S., Klauck, F. J. R. & Glorius, F. Manganese(I)-catalyzed regioselective C-H allenylation: direct access to 2-allenylindoles. Angew. Chem. Int. Ed. 56, 6660–6664 (2017).
Ng, J. S. & Hayashi, T. Asymmetric synthesis of fluorinated allenes by rhodium-catalyzed enantioselective alkylation/defluorination of propargyl difluorides with alkylzincs. Angew. Chem. Int. Ed. 60, 20771–20775 (2021).
O’Connor, T. J., Mai, B. K., Nafie, J., Liu, P. & Toste, F. D. Generation of axially chiral fluoroallenes through a copper-catalyzed enantioselective β-fluoride elimination. J. Am. Chem. Soc. 143, 13759–13768 (2021).
Ruitenberg, K., Kleijn, H., Elsevier, C. J., Meijer, J. & Vermeer, P. Palladium(0)-promoted synthesis of functionally substituted allenes by means of organozinc compounds. Tetrahedron Lett. 22, 1451–1452 (1981).
Tsuji, Y., Taniguchi, M., Yasuda, T., Kawamura, T. & Obora, Y. Palladium-catalyzed cyanation of propargylic carbonates with trimethylsilyl cyanide. Org. Lett. 2, 2635–2637 (2000).
Wang, Y., Zhang, W. & Ma, S. A room-temperature catalytic asymmetric synthesis of allenes with ECNU-phos. J. Am. Chem. Soc. 135, 11517–11520 (2013).
Scheipers, I., Mück-Lichtenfeld, C. & Studer, A. Palladium-catalyzed decarboxylative γ-arylation for the synthesis of tetrasubstituted chiral allenes. Angew. Chem. Int. Ed. 58, 6545–6548 (2019).
Zheng, W.-F. et al. Tetrasubstituted allenes via the palladium-catalysed kinetic resolution of propargylic alcohols using a supporting ligand. Nat. Cat. 2, 997–1005 (2019).
Teng, S., Jiao, Z., Chi, Y. R. & Zhou, J. S. Asymmetric wacker-type oxyallenylation and azaallenylation of cyclic alkenes. Angew. Chem. Int. Ed. 59, 2246–2250 (2020).
Wang, H. et al. Pd-catalyzed enantioselective syntheses of trisubstituted allenes via coupling of propargylic benzoates with organoboronic acids. J. Am. Chem. Soc. 142, 9763–9771 (2020).
Marshall, J. A. & Wolf, M. A. Amination, aminocarbonylation, and alkoxycarbonylation of allenic/propargylic Pd intermediates derived from nonracemic propargylic mesylates: synthesis of nonracemic propargyl amines, allenic amides, and butenolides. J. Org. Chem. 61, 3238–3239 (1996).
Kjellgren, J., Sundén, H. & Szabó, K. J. Palladium pincer complex-catalyzed trimethyltin substitution of functionalized propargylic substrates. An efficient route to propargyl- and allenyl-stannanes. J. Am. Chem. Soc. 126, 474–475 (2004).
Tsuji, H. & Kawatsura, M. Transition-metal-catalyzed propargylic substitution of propargylic alcohol derivatives bearing an internal alkyne group. Asian J. Org. Chem. 9, 1924–1941 (2020).
Fomum, Z. T., Landor, P. D. & Landor, S. R. Novel synthesis of imidazolines and imidazoles by Michael addition to allenic or acetylenic nitriles. J. Chem. Soc., Chem. Commun. 17, 706 (1974).
Ao, Y. F., Wang, D. X., Zhao, L. & Wang, M. X. Biotransformations of racemic 2,3-allenenitriles in biphasic systems: synthesis and transformations of enantioenriched axially chiral 2,3-allenoic acids and their derivatives. J. Org. Chem. 79, 3103–3110 (2014).
Kondo, M., Omori, M., Hatanaka, T., Funahashi, Y. & Nakamura, S. Catalytic enantioselective reaction of allenylnitriles with imines using chiral bis(imidazoline)s palladium(II) pincer complexes. Angew. Chem. Int. Ed. 56, 8677–8680 (2017).
Greaves, P. M., Landor, S. R. & Laws, D. R. J. The preparation of 1-cyanoallenes. Chem. Commun. 321–322, https://doi.org/10.1039/C19650000321 (1965).
Lu, F. D. et al. Asymmetric propargylic radical cyanation enabled by dual organophotoredox and copper catalysis. J. Am. Chem. Soc. 141, 6167–6172 (2019).
Zhou, Z. Z., Jiao, R. Q., Yang, K., Chen, X. M. & Liang, Y. M. Photoredox/palladium co-catalyzed propargylic benzylation with internal propargylic carbonates. Chem. Commun. 56, 12957–12960 (2020).
Brownbridge, P. Silyl enol ethers in synthesis—part I. Synthesis 1983, 1–28 (1983).
Brownbridge, P. Silyl enol ethers in synthesis—part II. Synthesis 1983, 85–104 (1983).
Marshall, J. A., Gung, B. W. & Grachan, M. L. in Modern Allene Chemistry Vol. 1, 493–592 (Wiley-VCH, 2004).
Danheiser, R. L. & Carini, D. J. (Trimethylsilyl)allenes as propargylic anion equivalents: synthesis of homopropargylic alcohols and ethers. J. Org. Chem. 45, 3925–3927 (1980).
Jin, J., Smith, D. T. & Weinreb, S. M. Novel intramolecular ene reactions of allenyl silanes. J. Org. Chem. 60, 5366–5367 (1995).
Reymond, S. & Cossy, J. Copper-catalyzed Diels-Alder reactions. Chem. Rev. 108, 5359–5406 (2008).
Sugita, T. et al. Regioselectivity of addition of thiols and amines to conjugated allenic ketones and esters. J. Org. Chem. 52, 3789–3793 (1987).
Greaves, P. M., Landor, P. D., Landor, S. R. & Odyek, O. Synthesis and reactions of allenic amides. Tetrahedron 30, 1427–1430 (1974).
Rostovtsev, V. V., Green, L. G., Fokin, V. V. & Sharpless, K. B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 41, 2596–2599 (2002).
Flamigni, L., Barbieri, A., Sabatini, C., Ventura, B. & Barigelletti, F. Photochemistry and photophysics of coordination compounds: Iridium. Top. Curr. Chem. 281, 143–203 (2007).
Hansmann, M. M., Melaimi, M. & Bertrand, G. Crystalline monomeric allenyl/propargyl radical. J. Am. Chem. Soc. 139, 15620–15623 (2017).
Muresan, M., Subramanian, H., Sibi, M. P. & Green, J. R. Propargyl radicals in organic synthesis. Eur. J. Org. Chem. 2021, 3359–3375 (2021).
Chandra, P., Choudhary, N., Lahiri, G. K., Maiti, D. & Mobin, S. M. Copper mediated chemo- and stereoselective cyanation reactions. Asian J. Org. Chem. 10, 1897–1937 (2021).
Patel, R. I., Sharma, S. & Sharma, A. Cyanation: a photochemical approach and applications in organic synthesis. Org. Chem. Front. 8, 3166–3200 (2021).
Kelly, C. B. et al. Preparation of visible-light-activated metal complexes and their use in photoredox/nickel dual catalysis. Nat. Protoc. 12, 472–492 (2017).
Strieth-Kalthoff, F., James, M. J., Teders, M., Pitzer, L. & Glorius, F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 47, 7190–7202 (2018).
Zhou, Q. Q., Zou, Y. Q., Lu, L. Q. & Xiao, W. J. Visible-light-induced organic photochemical reactions through energy-transfer pathways. Angew. Chem. Int. Ed. 58, 1586–1604 (2019).
Krause, N. (ed). Modern Organocopper Chemistry (Wiley-VCH, 2002).
Evano, G. & Blanchard, N. (ed). Copper-Mediated Cross-Coupling Reactions (Wiley-VCH, 2013).
Acknowledgements
Financial support from National Key R&D Program of China (2021YFA1500100) and National Science Foundation of China Program (22101252) are greatly appreciated. We thank Prof. Tiansheng Mei for cyclic voltammetric measurements. We thank Prof. Xiaogang Peng’s group at Zhejiang University for sharing of Fluorescence Spectrophotometer. We also thank Mr. Yaqi Shi and Mr. Yifan Cui in this group for reproducing the syntheses of 2e, 2z, and 2B in Fig. 2 presented in the text.
Author information
Authors and Affiliations
Contributions
S.M. directed the research and developed the concept of the reaction with Q.L., who also performed the experiments and prepared the Supplementary Information. J.Z. performed the Stern-Volmer quenching experiments. X.Z. performed the computational studies. Q.L. and S.M. wrote the manuscript with contributions from the other author.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Ming Hu, Zhao-Zhao Zhou, and the other, anonymous, reviewer for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Q., Zheng, J., Zhang, X. et al. Photo and copper dual catalysis for allene syntheses from propargylic derivatives via one-electron process. Nat Commun 13, 3302 (2022). https://doi.org/10.1038/s41467-022-30655-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-022-30655-3
- Springer Nature Limited