
Abstract
Adeno-associated viral vectors (AAV) are unique in their ability to transduce a variety of both dividing and nondividing cells, with significantly lower risk of random genomic integration and with no known pathogenicity in humans, but their role in ex vivo regional gene therapy for bone repair has not been definitively established. The goal of this study was to test the ability of AAV vectors carrying the cDNA for BMP-2 to transduce human mesenchymal stem cells (MSCs), produce BMP-2, and induce osteogenesis in vitro as compared with lentiviral gene therapy with a two-step transcriptional amplification system lentiviral vector (LV-TSTA). To this end, we created two AAV vectors (serotypes 2 and 6) expressing the target transgene; eGFP or BMP-2. Transduction of human MSCs isolated from bone marrow (BMSCs) or adipose tissue (ASCs) with AAV2-eGFP and AAV6-eGFP led to low transduction efficiency (BMSCs: 3.57% and 8.82%, respectively, ASCs: 6.17 and 20.2%, respectively) and mean fluorescence intensity as seen with FACS analysis 7 days following transduction, even at MOIs as high as 106. In contrast, strong eGFP expression was detectable in all of the cell types post transduction with LV-TSTA-eGFP. Transduction with BMP-2 producing vectors led to minimal BMP-2 production in AAV-transduced cells 2 and 7 days following transduction. In addition, transduction of ASCs and BMSCs with AAV2-BMP-2 and AAV6-BMP-2 did not enhance their osteogenic potential as seen with an alizarin red assay. In contrast, the LV-TSTA-BMP-2-transduced cells were characterized by an abundant BMP-2 production and induction of the osteogenic phenotype in vitro (p < 0.001 vs. AAV2 and 6). Our results demonstrate that the AAV2 and AAV6 vectors cannot induce a significant transgene expression in human BMSCs and ASCs, even at MOIs as high as 106. The LV-TSTA vector is significantly superior in transducing human MSCs; thus this vector would be preferable when developing an ex vivo regional gene therapy strategy for clinical use in orthopedic surgery applications.
Similar content being viewed by others

Introduction
The treatment of fracture nonunions and other significant bone defects in various orthopedic surgery settings remains a challenging clinical problem and can result in substantial morbidity, health care costs, and socioeconomic burden [1, 2]. Despite the advances made in multiple aspects of orthopedic clinical care including but not limited to surgical technique, implant material science, and new biologics, there is no consistently satisfactory solution for managing these challenging bone healing problems [2,3,4]. Bone repair strategies incorporating gene therapy regimens are being developed to address the deficits of conventional approaches [2, 5]. Regional gene therapy for bone regeneration focuses on transferring specific genes encoding for osteoinductive proteins or growth factors at a specific anatomic site [5]. As gene therapy has now become a viable clinical regimen for the treatment of various medical conditions such as hematologic disorders [6, 7], ocular conditions [8], spinal muscular atrophy [9], prostate cancer [10], and others [11], it could have the clinical potential to be adapted for the management of challenging bone repair scenarios too.
Gene transfer can be achieved either with in vivo or ex vivo strategies. In in vivo gene therapy the vector is delivered directly to the tissues of the host, either locally by injection or implantation at the region of interest, or systemically via intravenous administration. The potential advantage of this strategy is that it is less invasive, not as technically challenging, and possibly less expensive, as it allows for gene delivery in a single procedure. The major disadvantage in a biologically challenging bone repair scenario is that there may be an insufficient number of host cells available for transduction at the injury site [12, 13]. In addition, the overall transduction efficiency may be low which will further limit protein production [12]. Ex vivo gene therapy involves target cell harvesting, possible culture-expansion, and cell transduction outside of the host before surgical implantation at a specific anatomic site. It is considered more efficient as it allows for the delivery of both osteogenic cells and osteoinductive factors, and safer since it does not involve direct inoculation of viruses [5, 14]. Moreover the transduced cells can be tested prior to delivery to ensure viability and appropriate levels of transgene expression, thus allowing for standardization of the approach, more consistent performance, reproducibility, and quality. The major disadvantages include the additional step of tissue culture expansion and its associated cost. Due to its advantages, our laboratory has been pursuing the development of ex vivo regional gene therapy to address difficult bone repair scenarios.
Ex vivo regional gene therapy strategies using lentiviral-mediated bone morphogenetic protein (BMP) delivery has been well established in the literature for use in bone regeneration. Multiple pertinent studies have demonstrated the ability of lentiviral vectors to transduce different types of rodent [15, 16] and human cells [17, 18], and lead to sustained BMP-2 production that can last up to 12 weeks [16]. This prolonged BMP-2 delivery has been associated with robust bone healing in critical-sized bone defects in animal models [19,20,21,22,23]. However, since there is a theoretical risk for insertional mutagenesis [24] and AAV is already being used clinically we decided that it would be worthwhile to assess its clinical potential to treat challenging bone repair cases.
Adeno-associated viruses (AAVs) are small, non-enveloped, linear single-stranded DNA viruses. The recombinant counterparts of these viruses are unique in their ability to transduce a variety of both dividing and nondividing cells, with significantly lower risk of random genomic integration [13]. Recombinant AAV vectors have not been associated with any cell-mediated immunity and there is no reported pathogenicity in humans to date [25]. Due to their advantages and safety profile, AAV vectors are FDA approved for use in the treatment of biallelic RPE65 mutation-associated retinal dystrophy [26] and spinal muscular atrophy [27]. In addition, AAV vectors have already been evaluated in multiple clinical trials for different applications [28], including but not limited to lipoprotein lipase deficiency [29], hemophilia B [30], and Leber congenital amaurosis [31]. With regards to bone healing, AAVs as part of an in vivo gene therapy approach have been used successfully for bone repair applications [32] in preclinical studies of ectopic bone formation [25], and treatment of calvarial defects [33] and femoral bone defects [34]. However, limited literature exists with regards to the use of AAV vectors for ex vivo gene therapy strategies in bone regeneration. The goal of this study was to test the ability of two types of AAV vectors, type 2 and 6, carrying the cDNA for BMP-2 to successfully transduce human mesenchymal stem cells (MSCs), produce BMP-2, and enhance the osteogenic potential of human MSCs in vitro.
Material and methods
Cell isolation and culture
Human bone marrow stem cells (BMSC) and adipose-derived stem cells (ASCs) were used in our study, because these two cell types are the most commonly used cellular delivery vehicles in an ex gene therapy strategies. Human bone marrow samples were collected from the femurs of six healthy patients (4 male/2 female), aged 59.8 ± 14 years, undergoing primary total hip arthroplasty for hip osteoarthritis. Lipoaspirates were obtained from six healthy patients (1 male/5 female), 45.8 ± 11 years of age, during liposuction procedures of the abdomen, thigh, and/or buttock for cosmetic purposes. The collected specimens, namely the bone marrow from the femoral medullary canal and the lipoaspirates, are normally discarded after the surgery. All of the patient samples were de-identified; only the patients’ sex and age were documented. Patients with significant co-morbidities, or known history of HIV, HBV, or HCV infection were excluded. The protocol and all pertinent procedures were reviewed and approved by the Institutional Review Board prior to initiating any experiments involving human cells.
Previously published cell isolation protocols were used to obtain the mononuclear cell fraction from bone marrow samples [18, 23] and the stromal vascular fraction (SVF) from lipoaspirates [18, 35]. For the bone marrow, the collected specimen was transferred into sterile tubes and diluted with equal volume of PBS. This suspension was then layered on top of Histopaque 1077 (Sigma–Aldrich, St. Louis, MO), and centrifuged for 30 min at 400 g to isolate the mononuclear cell fraction. Following isolation the cells were washed thoroughly and resuspended in Dulbecco’s modified Eagle medium (DMEM, Corning Mediatech, Manassas, VA, USA) supplemented with 10% fetal bovine serum (FBS, Omega Scientific, Tarzana, CA, USA) as well as antibiotic and antifungal agents (100 IU/mL penicillin, 100 lg/mL streptomycin, and 0.25 lg/mL amphotericin B). Similarly, the collected lipoaspirate samples were washed with an equal volume of Dulbecco’s phosphate-buffered saline (DPBS; Caisson Laboratories, North Logan, UT) and then digested with 0.1% collagenase (Sigma–Aldrich, St. Louis, MO) at 37 °C until the adipose layer was completely liquefied [18, 35]. The adipose layer was subsequently filtered and washed, then incubated with ammonium–chloride–potassium (ACK) lysing buffer (Lonza, Allendale, NJ) to remove contaminating red blood cells. Following a final washing step to eliminate the ACK buffer, the resultant SVF was resuspended in DMEM + 10% FBS.
Both the isolated mononuclear cells and the SVF cells were counted with an automated cell counter (BioRad, Hercules, CA) using trypan blue and then plated for culture expansion at a concentration of 40 × 106 cells and 2–3 × 106 cells per 10 cm plate, respectively. The cells were maintained in a 5% CO2 humidified atmosphere, at 37 °C. The culture medium was replaced every 3–4 days, with all non-adherent cells and contaminating red blood cells being removed. When >90% confluent the adherent cells were further passaged at a density of 0.8–1.0 × 106 cells per 10 cm plate. Both cell types were expanded until they reached passage 3.
Viral vector construction and transduction
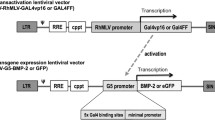
Two different AAV serotypes were used in this study; type 2 and 6 (Fig. 1). AAV6 (pRC6) and AAV2 (pRC2) capsid plasmids, pHelper plasmid, and AAV2 inverted terminal repeats (ITR) plasmid containing the GFP transgene, were purchased through AAV helper-free packaging system (Cell BioLabs, San Diego, CA). Vectors were prepared according to the procedure detailed for production of AAV9 [36]. In brief, 9 × 106 293T AAV cells (CellBioLabs, San Diego, CA) were seeded in a 15-cm diameter dish 1 day before calcium chloride transfection. Transfected cells were then incubated for 16 h prior to PBS wash and media change. Seventy-two hours after cell transfection, the media were discarded and the cells were harvested by gentle scraping. Cell pellets were freeze thawed −80 °C/37 °C, lysed, and treated with bezonase. Vector was then purified by iodixanol density gradient ultra-centrifugation at 59 K rpm for 70 min. The vector was harvested from the 40% iodixanol layer and then buffer exchanged to d-sorbitol PBS (PBS with 5% d-sorbitol and 350 mM NaCl). Vector was further concentrated using Amicon Ultra-50 centrifugal filter tubes (Millipore, Burlington, MA, USA). All vectors were kept at −80 °C until time of use. As previously described [36], AAV titers were determined by a 96-well plate Q-PCR and verified by 293 transduction. To ensure continuity between serotypes/preparations, we performed titers for all AAV preparations on the same 96-well plate in triplicate along with a commercial AAV-GFP preparation used as an internal reference standard. The Q-PCR is designed to amplify only one of ITR regions. We verified these molecular ITR-based titers via 293T transduction with our preparations and used the same commercial preparation included as an internal control to provide a transduction reference standard. For the creation of the AAV-BMP-2 vector, in the AAV plasmid with a CMV promoter carrying the transgene for eGFP, the eGFP cDNA was replaced with BMP-2 in the pAAV backbone plasmid to create two new AAV vectors: AAV2-CMV-BMP-2 and AAV6-CMV-BMP-2. The AAV-BMP-2 vectors were generated in house using the same methodology as in the eGFP vector preparation. Finally, for the GFP transduction with an MOI of 106 core-acquired AAV2-CMV-GFP and AAV6-CMV-BMP-2 vectors were used (Vigene Biosciences, Rockville, MD), due to the inability of our lab to obtain such high MOIs with in-house viral vector production. The AAV-GFP preparations from the vector core were again compared with the in house preparations to ensure similar titers and transduction efficiency.

1. AAV vector (serotypes 2 or 6) and 2. lentiviral two-step transcriptional amplification (TSTA) system. ITR inverted terminal repeat, polyA polyadenylation signal, LTR long terminal repeat, Ψ packaging signal, RRE rev-responsible element, cPPt central polyprine tract, SIN self-inactivating.
A two-step transcriptional amplification lentiviral vector system (LV-TSTA) encoding BMP-2 or eGFP was used to compare the efficacy of AAV versus lentivirally mediated gene therapy in vitro. The LV-TSTA system [37] involves co-transduction with two different lentiviral vectors; the transactivator vector GAL4-VP16 (LV-RhMLV-GAL4-VP16) and the transgene expression vector encoding BMP-2 or eGFP (Lenti-G5-BMP-2 or LV-G5-eGFP). As previously described [17, 18, 23], viral vectors were generated using transfection of 293T cells (American Type Culture Collection, Manassas, VA), with their titers quantified by p24 ELISA assays (ELISA, Quantikine, R&D Systems, Minneapolis, MN, USA).
Passage 3 human MSCs were transduced overnight with either AAV2 or 6 in serum-free media, or LV-TSTA in media containing FBS and 8 μg/ml polybrene. Non-transduced MSCs were used as negative control. 293T cells were used as a positive control. Transduction with BMP-2-carrying AAV vectors was used to evaluate the cells’ in vitro osteogenic potential. Transduction with AAV2/6-eGFP was performed to assess transduction efficiency. The AAV vectors for BMP-2 and eGFP experiments were identical except for the inserted target gene. All experiments were done in parallel with the LV-TSTA vector for comparison. MOIs of 105 and 106 were used for transduction with the eGFP-expressing AAV vectors. The MOI of 105 was selected based on a prior study, which reported transduction efficiencies up to 99% and duration of gene expression up to 56 days following AAV2-eGFP transduction of ASCs [38]. However, since the MOI of 105 was found to be associated with low transgene expression in our study a tenfold higher MOI was also tested (MOI = 106). MOI of 106 was used for AAV2-BMP-2 and AAV6-BMP-2. Transduction with the LV-TSTA vectors (LV-TSTA-eGFP or LV-TSTA-BMP-2) was done at an MOI of 5 for the LV-RhMLV-GAL4-VP16 and MOI of 25 for the LV-G5-BMP-2 or eGFP vector, based on prior in vitro experiments (MOI of 5/25) [17, 39]. Following transduction the cells were washed thoroughly to eradicate any cell debris and extracellular viral particles.
In vitro eGFP expression
Three samples per tissue type (bone marrow or adipose tissue) were used for this aspect of the study. MSCs were cultured in 10 cm plates until they reached passage 3 and then transduced with either the AAV-eGFP (serotype 2 or 6) or the LV-TSTA-eGFP vectors. 293T cells transduced with the aforementioned vectors were used as a positive control. Two different MOIs were tested for the AAV vectors; MOI of 105 and 106. An MOI of 5/25 was used for the LV-TSTA-eGFP vector. Transduced cells were washed 24 h following transduction to remove any extracellular viruses and then plated again for one more day. Each plate was imaged to detect eGFP positive cells using a Revolve R4 inverted microscope (Echo, San Diego, CA). Half of these cells were then trypsinized and resuspended in PBS to determine transduction efficiency and mean fluorescence intensity (MFI) with fluorescence-activated cell sorting (FACS). Non-transduced target cells were used as control. The rest of the cells were kept in culture for an additional week, then imaged and further analyzed with FACS. FACS analysis was done using a BD LSR II flow cytometer (BD Biosciences). FACS data were analyzed using Flowjo software (FlowJo LLC).
In vitro BMP-2 production
BMSCs and ASCs from four different patients (two per cell type) were plated at a concentration of 105 cells per well and transduced overnight with the BMP-2 expressing vectors (AAV2-BMP-2, AAV6-BMP-2, or LV-TSTA-BMP-2). Each sample was run in triplicate. Only the highest MOI (106) was tested for the AAV vectors. The culture medium was collected at 2 days and 1 week post transduction to evaluate BMP-2 levels in vitro. A commercially available enzyme-linked immunosorbent assay (ELISA) kit (Quantikine, R&D Systems, Minneapolis, MN, USA) was used to quantify BMP-2 production over a 24 h period. All results were standardized by cell number and reported as nanograms of BMP-2 per 1 × 106 cells per 24 h.
Osteogenic differentiation potential
Osteogenic differentiation profiles of transduced BMSCs and ASCs were assessed 7 days after exposure to BMP-2, using alizarin red staining and quantification with spectrophotometry as previously described [18, 40]. Non-transduced cells were used as control. Cells were seeded in duplicate for each experiment. In brief, BMSCs and ASCs were cultured in ostegenic media containing DMEM + 10% FBS, antibiotics/antimycotics, 0.1 μM of dexamethasone, 50 μg/mL of l-ascorbic acid, and 10 mM of β-glycerophosphate to induce osteogenic differentiation. Seven days after transduction the medium was aspirated and cells were carefully washed using DPBS. A solution of 10% formaldehyde was added to the plate to fix the cells for 10 min, followed by a wash with distilled water. The cellular monolayer was then stained with 2% Alizarin red S solution for 30 min to detect extracellular calcium deposits. Stained MSCs were imaged using an EVOS XL inverted microscope (AMG, Mill Creek, WA) and then stored at −20 °C for further processing with dye extraction and quantification of alizarin red. A colorimetric assay was performed to accurately quantify the amount of alizarin red in each well. The stained cell monolayer in each well was incubated at the room temperature with 10% acetic acid (800 μL) for 30 min with gentle shaking, then transferred to 1.5 mL eppendorf tubes, heated at 85 °C for 10 min, followed by a 5-min cooling on ice. The resultant cell slurry was centrifuged for 15 min at 20,000 g. The supernatant was mixed with ammonium hydroxide (200 μL) into a new 1.5 mL eppendorf tube. Aliquots (150 μL) were transferred to a 96-well plate (triplicates) and absorbance was read at 405 nm by a plate reader (Model 680, BioRad, Hercules, CA, USA).
Statistical analysis
Statistical analysis was performed using the IBM SPSS Statistics 22.0 software, with the significance level set at 0.05 for all comparisons. Data for all performed experiments are reported as mean and standard deviation. Equality of variances was also confirmed before performing any further analysis. After assessing the data for normality using Shapiro–Wilk test, one-way ANOVA, and post hoc analysis with Tukey’s range test were selected for comparisons in GFP expression and BMP-2 production between vectors and cell types. One-way ANOVA was also used to compare extracellular calcium deposits between groups.
Results
In vitro eGFP expression
We evaluated the ability of two different AAV vector serotypes to transduce 293T cells, BMSCs, and ASCs, using GFP reporter genomes. Both AAV2-eGFP and AAV6-eGFP were able to transduce all cell types, although at different success rates. In general, in BMSCs and ASCs, GFP signal was noted as early as 48 h post transduction, with levels of expression peaking at 5–7 days post transduction, and then steadily decreasing to minimal, if any, GFP expression past day 11 (Supplementary Fig. 1). Therefore, comparisons with regards to transduction efficiency are presented for the 7-day time point.
Transduced 293T cells showed strong eGFP expression 1 week post transduction as seen with fluorescent microscopy and FACS analysis (97.6% and 99%, respectively). In contrast, transduction with AAV2 and 6 even at a high MOI of 106 in human MSCs was associated with minimal eGFP expression at the same time point (BMSCs: 3.57% and 8.82%, respectively, ASCs: 6.17% and 20.2%, respectively). Strong eGFP expression was detectable in all of the cell types following transduction with the lentiviral system LV-TSTA-eGFP (Fig. 2). Moreover, the MFI associated with AAV transduction in BMSCs and ASCs was significantly lower compared with LV-TSTA-BMP transduction (p < 0.001) (Table 1, Fig. 3).

Non-transduced cells were used as control. AAV-eGFP showed strong eGFP expression in 293T cells, whereas few GFP+ cells were noted in BMSC and ASC cultures. LV-TSTA-GFP consistently transduced the majority of cells for all of the studied cell types.

Non-transduced cells are used as control.
BMP-2 production in vitro
Transduction with BMP-2 producing AAV vectors at 106 viral genomes/cell led to minimal BMP-2 production in both BMSCs and ASCs 2 and 7 days following transduction. In contrast, LV-TSTA-BMP-2 induced a robust in vitro BMP-2 production by both human MSC cell types, with protein levels being 100–600 times higher compared with AAV transductions (Table 2). No differences with regards to BMP-2 production were noted between AAV2 and 6 (p > 0.05) or between AAV-transduced cells and negative control (non-transduced cells).
In vitro osteogenic differentiation potential
Transduction of ASCs and BMSCs with AAV2-BMP-2 or AAV6-BMP-2 did not enhance the cells in vitro osteogenic differentiation potential as seen with the alizarin red staining (Fig. 4). AAV-BMP-2-transduced cells demonstrated levels of calcium deposits similar to that of non-transduced cells (Fig. 5). In contrast, the LV-TSTA-BMP-2-transduced MSCs were characterized by induction of the osteogenic phenotype in vitro as noted in both qualitative alizarin red stained wells and quantitative colorimetric assay data (p < 0.001 vs. AAV2 and 6) (Figs. 4,5). As expected, no calcified extracellular matrix was seen in non-transduced cell cultures.

Minimal alizarin red staining in AAV2 and 6 transduced MSCs, with abundant extracellular deposition of calcium in LV-TSTA transduced cells. Non-transduced cells were used as a control; no alizarin red staining was noted in non-transduced cells cultures in osteogenic media.

The LV-TSTA-BMP-2-transduced cells had a higher mineralization potential compared with AAV-BMP-2-transduced cells (p < 0.001 vs. AAV2 and 6). Non-transduced BMSCs and ASCs show no extracellular calcium deposition. Non non-transduced cells.
Discussion
The potential of ex vivo gene therapy to revolutionize the treatment of fracture nonunions and bone loss in trauma, revision joint replacement surgery, and spinal fusion using different viral vectors and target genes has been demonstrated [5, 14, 41]. However, before regional gene therapy can be adapted for clinical use it is crucial to assess the osteoinductive capacity of the most promising viral vector candidates and choose the vector that demonstrates the optimal clinical and safety profiles.
In this project we aimed to examine the clinical potential of AAV vectors in ex vivo gene therapy for bone repair using human BMSCs and ASCs. AAV vectors were selected because they are already being used to treat patients. An ex vivo gene therapy approach was chosen due to its ability to deliver both cells with osteogenic capacity and an osteoinductive growth factor to the anatomic site of interest. The transgene expression associated with AAV vectors was compared with our established LV-TSTA vector, which has been previously shown to induce bone formation in rodent critical-sized femoral defects [17, 20, 23]. Our hypothesis was that AAV vectors encoding BMP-2 would be able to successfully transduce human MSCs leading to BMP-2 production and an increase in the transduced cells’ osteogenic differentiation capacity in vitro, thus offering an alternative strategy that would facilitate the transition from the lab to the clinic.
To this end, we created two AAV vectors (serotypes 2 and 6) expressing the target transgene. Transduction of human MSCs isolated from adipose tissue or bone marrow with AAV-eGFP led to only low GFP expression following transduction, even at MOIs as high as 106. Moreover, AAV2 and AAV6 produced significantly less BMP-2 compared with the lentiviral vector. Finally AAV-BMP-2 did not enhance the osteogenic potential of the MSCs in vitro, consistent with the levels of BMP-2 production being barely detectable. In contrast, transduction with LV-TSTA-BMP-2 led to high levels of BMP-2 expression and a robust osteogenic response from both BMSCs and ASCs.
Recombinant AAV vectors lack the Rep and Cap sequences that are responsible for viral replication, thus losing the ability to integrate into the host cell genome. Twelve primate serotypes have been described, with AAV2 being the most commonly used. Overall, AAV vectors have considerable promise to overcome limitations of other gene delivery vehicles due to their reported ability to transduce a wide variety of dividing and nondividing cells, and their lack of immunogenicity and pathogenicity [42,43,44]. Moreover, approval of the first-gene therapy product Glybera, an AAV vector for the treatment of lipoprotein lipase deficiency, by the European Medicines Agency was a ground-breaking step in gene therapy development, though it has since be withdrawn from market due to the increased cost [45]. Recently two AAV-mediated gene therapy products have been FDA approved; Luxtera for use in the treatment of biallelic RPE65 mutation-associated retinal dystrophy [30] and Zolgensma for spinal muscular atrophy [31].
The use of AAV vectors has been described in animal studies for bone regeneration as part of an in vivo gene therapy approach [32]. AAV2-BMP-4 and AAV6-BMP-4 successfully induced ectopic bone formation as demonstrated by plain radiographs and histologic analysis either implanted directly into a rat hindlimb model [46] or delivered on a gelfoam carrier into a mouse muscle pouch [33], respectively. In vivo gene delivery using AAV vectors carrying the transgene for BMP-2 or BMP-6 has also been associated with osteogenesis in ectopic models of bone formation in immunocompetent or nude rats [47, 48]. In a different approach, namely allograft revitalization, implantation of murine bone allografts loaded with AAV vectors encoding for vascular endothelial growth factor and/or receptor activator of nuclear factor-kappa B ligand (RANKL) in mouse critical-sized femoral defects was associated with angiogenesis, new bone formation and allograft resorption [34]. Similar results were seen with AAV vectors carrying the cDNA for BMP-2 [49] or activin receptor-like kinase-2 [50]. However, none of these models are as rigorous as a critical-sized femoral defect in a rat or larger animal models.
Ex vivo gene therapy studies evaluating the efficacy of AAV vectors for bone healing have demonstrated mixed outcomes [51]. Both Ito et al. and Ju et al. transduced cells with rAAV-TGFbeta1-IRES-eGFP [52] or AAV-Luciferase [53], respectively and noted poor transduction efficiency. In an in vitro study using human BMSCs and 293T cells, Stender et al. demonstrated that AAV2-eGFP vectors were able to transduce MSCs with transduction efficiencies ranging from 1 to 65% 4 days following transduction depending on the MOI. However, when comparing MFIs between AAV2-eGFP-transduced 293T cells and hBMSCs, 293T cells were associated with a significantly higher eGFP expression (~200-fold for 293T cells versus fourfold for hBMSCs upon increasing the viral dose 10,000-fold) [54]. In other studies, comparison of various viral vectors in rat, rabbit and human MSCs showed that different serotypes of AAV vectors were the least effective in accomplishing gene delivery [55, 56]. Finally, AAV2-mediated gene delivery in fibroblasts has been associated with ~30-fold lower levels of AAV2 DNA replication when compared with 293T or HeLa cells [57, 58].
Similarly in our study, human BMSCs and ASCs were successfully transduced with AAV2-eGFP or AAV6-eGFP, but transduction was associated with low GFP expression, even at MOIs as high as 106. In contrast, transduction of 293T cells with the same vectors was associated with a 16–27 times higher transduction efficiency for AAV2 and 5–12 times higher transduction efficiency for AAV6. There was also a tenfold increase in MFI for both vectors and cell types. Transduction of human MSCs with BMP-2 producing AAV vectors at 106 viral genomes/cell led to minimal BMP-2 production in both BMSCs and ASCs. This indicates that although AAV2 and AAV6 can transduce human MSCs, levels of transgene expression remain severely limited. A lack of the primary cell surface receptor for AAV, heparin sulfate proteoglycan, on the cellular surface of MSCs might be a limiting step for AAV transduction [59]. Uncoating of virions following cellular uptake has been described as an important barrier to efficient AAV transduction [60].
We noted that AAV-mediated gene expression was transient. We observed a peak of GFP-positive cells and MFI at 5–7 days following transduction, succeeded by a rapid decline to near control levels after day 11. These results are consistent with what has been reported in the literature. In a prior study using human BMSCs transduced with AAV2-eGFP, transgene expression peaked at 4 days followed by significant decline in gene expression 16 days following transduction [54]. Moreover, human ASCs transduced with rAAV1 and rAAV2 or rAAV5 lost gene expression by day 4 or day 8, respectively [61]. The decline of GFP-positive MSCs over time could be explained by the replication deficiency of the AAV vector and the fact that AAV does not integrate in the host cells’ chromosomes which prohibits vertical vector transmission in dividing cell cultures. The duration of transgene expression is a highly clinically relevant parameter for bone repair applications. Although in the bone repair scenarios lifelong gene expression is not necessary, there is a concern that 2–3 weeks of protein production may not induce adequate bone repair in biologically stringent environments. The management of these bone defects may require at least several months of trangene expression. Previous studies in our laboratory have shown the superiority of lentiviral gene therapy as compared with adenoviral gene therapy in bone formation due to more prolonged duration (up to 12 weeks) of target gene expression [15, 16, 62]. Therefore, it is doubtful that the low, transient (~2 weeks) BMP-2 production associated with AAV gene delivery will be adequate to induce an appropriate biologic effect in these patients.
Our study had a few limitations. First, only two AAV serotypes were tested; AAV2 and AAV6. These two serotypes were selected since they are the most commonly studied and clinically relevant. However, it is possible that other serotypes wound have resulted in different outcomes. Second, different promoters were used in the lentivirus and AAV vectors. The expression construct as designed for the LV-TSTA viral vector is too large for efficient packaging into rAAV. Therefore, we used a highly promiscuous CMV promoter that has had several publications showing efficacy in cell lines derived from the same tissue types we intended to target. Third, a single-stranded AAV construct was selected for use in our study instead of self-complementary AAV genome, since BMP-2 is too large to deliver in a self-complementary AAV vector construct. To ensure the functionality of our AAV-GFP delivery, transduction efficiency was verified using 293 cell lines, which neared 100% transduction rates using the single-stranded GFP vector, thus demonstrating that our delivery construct was functional and highly efficacious in control lines.
In conclusion, our study compares the AAV and LV-TSTA viral vectors in an ex vivo gene therapy strategy, with regards to gene expression and osteoinductive ability when transducing human BMSCs and ASCs. Our results demonstrate that the AAV2 and AAV6 vector cannot induce a significant transgene expression in human BMSCs and ASCs, even at MOIs as high as 106. The LV-TSTA vector is significantly superior in transducing human MSCs and in inducing the osteogenic phenotype in vitro. Thus, the LV-TSTA vector would be preferable when developing an ex vivo gene therapy strategy for clinical use in bone repair applications.
Change history
30 July 2024
There was wrong details in Figure 4.
01 August 2024
A Correction to this paper has been published: https://doi.org/10.1038/s41434-024-00472-y
References
Brinker MR. Non-unions: evaluation and treatment. In: Browner BD, Levine AM, Jupiter JB, Trafton PG, editors, Skeletal Trauma: basic science, management, and reconstruction, 4th Ed. Philadelphia: W.B. Saunders; 2009. p. 615–707, Chapter 22.
Schlundt C, Bucher CH, Tsitsilonis S, Schell H, Duda GN, Schmidt-Bleek K. Clinical and research approaches to treat non-union fracture. Curr Osteoporos Rep. 2018;16:155–68.
Sen MK, Miclau T. Autologous iliac crest bone graft: should it still be the gold standard for treating nonunions? Injury. 2007;38:S75–80.
Garrison KR, Donell S, Ryder J, Shemilt I, Mugford M, Harvey I, et al. Clinical effectiveness and cost-effectiveness of bone morphogenetic proteins in the non-healing of fractures and spinal fusion: a systematic review. Health Technol Assess. 2007;11:1–150.
Bougioukli S, Evans CH, Alluri RK, Ghivizzani SC, Lieberman JR. Gene therapy to enhance bone and cartilage repair in orthopaedic surgery. Curr Gene Ther. 2018;18:154–70.
Singh N, Frey NV, Grupp SA, Maude SL. CAR T cell therapy in acute lymphoblastic leukemia and potential for chronic lymphocytic leukemia. Curr Treat Options Oncol. 2016;17:28.
Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N Engl J Med. 2017;377:2531–44.
Voelker R. Gene therapy for vision loss. JAMA. 2018;319:434.
Hoy SM. Onasemnogene abeparvovec: first global approval. Drugs. 2019;79:1255–62.
Higano CS, Armstrong AJ, Sartor AO, Vogelzang NJ, Kantoff PW, McLeod DG, et al. Real-world outcomes of sipuleucel-T treatment in PROCEED, a prospective registry of men with metastatic castration-resistant prostate cancer. Cancer. 2019;125:4172–80.
Approved Cellular and Gene Therapy Products https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/approved-cellular-and-gene-therapy-products. Accessed 7 Dec 2019.
Kofron MD, Laurencin CT. Bone tissue engineering by gene delivery. Adv Drug Deliv Rev. 2006;58:555–76.
Phillips JE, Gersbach CA, García AJ. Virus-based gene therapy strategies for bone regeneration. Biomaterials. 2007;28:211–29.
Evans CH. Gene delivery to bone. Adv Drug Deliv Rev. 2012;64:1331–40.
Sugiyama O, An DS, Kung SP, Feeley BT, Gamradt S, Liu NQ, et al. Lentivirus-mediated gene transfer induces long-term transgene expression of BMP-2 in vitro and new bone formation in vivo. Mol Ther. 2005;11:390–8.
Feeley BT, Conduah AH, Sugiyama O, Krenek L, Chen IS, Lieberman JR. In vivo molecular imaging of adenoviral versus lentiviral gene therapy in two bone formation models. J Orthop Res. 2006;24:1709–21.
Bougioukli S, Sugiyama O, Alluri RK, Yoho R, Oakes DA, Lieberman JR. In vitro evaluation of a lentiviral two-step transcriptional amplification system using GAL4FF transactivator for gene therapy applications in bone repair. Gene Ther. 2018;25:260–8.
Bougioukli S, Sugiyama O, Pannell W, Ortega B, Tan MH, Tang AH, et al. Gene therapy for bone repair using human cells: superior osteogenic potential of bone morphogenetic protein 2-transduced mesenchymal stem cells derived from adipose tissue compared to bone marrow. Hum Gene Ther. 2018;29:507–19.
Hsu WK, Sugiyama O, Park SH, Conduah A, Feeley BT, Liu NQ, et al. Lentiviral mediated BMP-2 gene transfer enhances healing of segmental femoral defects in rats. Bone. 2007;40:931–8.
Virk MS, Sugiyama O, Park SH, Gambhir SS, Adams DJ, Drissi H, et al. “Same day” ex-vivo regional gene therapy: a novel strategy to enhance bone repair. Mol Ther. 2011;19:960–8.
Pensak M, Hong S, Dukas A, Tinsley B, Drissi H, Tang A, et al. The role of transduced bone marrow cells overexpressing BMP-2 in healing critical-sized defects in a mouse femur. Gene Ther. 2015;22:467–75.
Bougioukli S, Jain A, Sugiyama O, Tinsley BA, Tang AH, Tan MH, et al. Combination therapy with BMP-2 and a systemic RANKL inhibitor enhances bone healing in a mouse critical-sized femoral defect. Bone. 2016;84:93–103.
Bougioukli S, Alluri R, Pannell W, Sugiyama O, Vega A, Tang A, et al. Ex vivo gene therapy using human bone marrow cells overexpressing BMP-2: “Next-day” gene therapy versus standard “two-step” approach. Bone. 2019;128:115032.
Bokhoven MC. Measurement of insertional mutagenesis by retroviral and lentiviral vectors. Doctoral thesis. University of London; 2008. https://discovery.ucl.ac.uk/id/eprint/1444113/1/U591415.pdf.
Chen Y, Luk KD, Cheung KM, Xu R, Lin MC, Lu WW, et al. Gene therapy for new bone formation using adeno-associated viral bone morphogenetic protein-2 vectors. Gene Ther. 2003;10:1345–53.
FDA approves novel gene therapy to treat patients with a rare form of inherited vision loss. https://www.fda.gov/news-events/press-announcements/fda-approves-novel-gene-therapy-treat-patients-rare-form-inherited-vision-loss. Accessed 31 Dec 2019.
FDA Approves Zolgensma, Landmark AAV-Delivered Gene Therapy to Treat Spinal Muscular Atrophy. 2019 Annual ASGCT meeting. https://www.asgct.org/research/news/may-2019/fda-approves-zolgensma-gene-therapy. Accessed 31 Dec 2019.
The US National Institute of Health. Adeno-associated virus. https://clinicaltrials.gov/ct2/results?term=adeno+associated. Accessed 22 Dec 2019.
Ferreira V, Petry H, Salmon F. Immune responses to AAV-vectors, the glybera example from bench to bedside. Front Immunol. 2014;5:82.
Nathwani AC, Reiss UM, Tuddenham EG, Rosales C, Chowdary P, McIntosh J, et al. Long-term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371:1994–2004.
Weleber RG, Pennesi ME, Wilson DJ, Kaushal S, Erker LR, Jensen L, et al. Results at 2 years after gene therapy for RPE65-deficient leber congenital amaurosis and severe early-childhood-onset retinal dystrophy. Ophthalmology. 2016;123:1606–20.
Ke J, Zheng LW, Cheung LK. Orthopaedic gene therapy using recombinant adeno-associated virus vectors. Arch Oral Biol. 2011;56:619–28.
Tian K, Qi M, Wang L, Li Z, Xu J, Li Y, et al. Two-stage therapeutic utility of ectopically formed bone tissue in skeletal muscle induced by adeno-associated virus containing bone morphogenetic protein-4 gene. J Orthop Surg Res. 2015;10:86.
Ito H, Koefoed M, Tiyapatanaputi P, Gromov K, Goater JJ, Carmouche J, et al. Remodeling of cortical bone allografts mediated by adherent rAAV-RANKL and VEGF gene therapy. Nat Med. 2005;11:291–7.
Zhu M, Heydarkhan-Hagvall S, Hedrick M, Benhaim P, Zuk P. Manual isolation of adipose-derived stem cells from human lipoaspirates. J Vis Exp. 2013;79:e50585.
Rogers GL, Chen H, Morales H, Cannon PM. Homologous recombination-based genome editing by clade F AAVs is inefficient in the absence of a targeted DNA break. Mol. Ther. 2019;27:1–11.
Iyer M, Wu L, Carey M, Wang Y, Smallwood A, Gambhir SS. Two-step transcriptional amplification as a method for imaging reporter gene expression using weak promoters. Proc Natl Acad Sci USA. 2001;98:14595–600.
Kang Y, Liao WM, Yuan ZH, Sheng PY, Zhang LJ, Yuan XW, et al. In vitro and in vivo induction of bone formation based on adeno-associated virus-mediated BMP-7 gene therapy using human adipose-derived mesenchymal stem cells. Acta Pharmacol Sin. 2007;28:839–49.
Vakhshori V, Bougioukli S, Sugiyama O, Tang A, Yoho R, Lieberman JR. Cryopreservation of human adipose-derived stem cells for use in ex vivo regional gene therapy for bone repair. Hum Gene Ther Methods. 2018;29:269–77.
Gregory CA, Gunn WG, Peister A, Prockop DJ. An Alizarin red-based assay of mineralization by adherent cells in culture: comparison with cetylpyridinium chloride extraction. Anal Biochem. 2004;329:77–84.
Pensak MJ, Lieberman JR. Gene therapy for bone regeneration. Curr Pharm Des. 2013;19:3466–73.
Zhang X, Godbey WT. Viral vectors for gene delivery in tissue engineering. Adv Drug Deliv Rev. 2006;58:515–34.
Ellis BL, Hirsch ML, Barker JC, Connelly JP, Steininger RJ 3rd, Porteus MH. A survey of ex vivo/in vitro transduction efficiency of mammalian primary cells and cell lines with Nine natural adeno-associated virus (AAV1-9) and one engineered adeno-associated virus serotype. Virol J. 2013;10:74.
Kumar SR, Markusic DM, Biswas M, High KA, Herzog RW. Clinical development of gene therapy: results and lessons from recent successes. Mol Ther Methods Clin Dev. 2016;3:16034.
http://www.genetherapynet.com/glybera.html. Accessed 10 Nov 2019.
Luk KD, Chen Y, Cheung KM, Kung HF, Lu WW, Leong JC. Adeno-associated virus-mediated bone morphogenetic protein-4 gene therapy for in vivo bone formation. Biochem Biophys Res Commun. 2003;308:636–45.
Chen Y, Luk KD, Cheung KM, Lu WW, An XM, Ng SS, et al. Combination of adeno-associated virus and adenovirus vectors expressing bone morphogenetic protein-2 produces enhanced osteogenic activity in immunocompetent rats. Biochem Biophys Res Commun. 2004;317:675–81.
Li JZ, Li H, Hankins GR, Lieu AS, Noh E, Jacobson L, et al. Different osteogenic potentials of recombinant human BMP-6 adeno-associated virus and adenovirus in two rat strains. Tissue Eng. 2006;12:209–19.
Ben Arav A, Pelled G, Zilberman Y, Kimelman-Bleich N, Gazit Z, Schwarz EM, et al. Adeno-associated virus-coated allografts: a novel approach for cranioplasty. J Tissue Eng Regen Med. 2012;6:43–50.
Koefoed M, Ito H, Gromov K, Reynolds DG, Awad HA, Rubery PT, et al. Biological effects of rAAV-caAlk2 coating on structural allograft healing. Mol Ther. 2005;12:212–8.
Brown N, Song L, Kollu NR, Hirsch ML. Adeno-associated virus vectors and stem cells: friends or foes? Hum Gene Ther. 2017;28:450–63.
Ito H, Goater JJ, Tiyapatanaputi P, Rubery PT, O’Keefe RJ, Schwarz EM. Light-activated gene transduction of recombinant adeno-associated virus in human mesenchymal stem cells. Gene Ther. 2004;11:34–41.
Ju XD, Lou SQ, Wang WG, Peng JQ, Tian H. Effect of hydroxyurea and etoposide on transduction of human bone marrow mesenchymal stem and progenitor cell by adeno-associated virus vectors. Acta Pharmacol Sin. 2004;25:196–202.
Stender S, Murphy M, O’Brien T, Stengaard C, Ulrich-Vinther M, Søballe K, et al. Adeno-associated viral vector transduction of human mesenchymal stem cells. Eur Cell Mater. 2007;13:93–9.
McMahon JM, Conroy S, Lyons M, Greiser U, O’shea C, Strappe P, et al. Gene transfer into rat mesenchymal stem cells: a comparative study of viral and nonviral vectors. Stem Cells Dev. 2006;15:87–96.
Alaee F, Sugiyama O, Virk MS, Tang Y, Wang B, Lieberman JR. In vitro evaluation of a double-stranded self-complementary adeno-associated virus type2 vector in bone marrow stromal cells for bone healing. Genet Vaccines Ther. 2011;9:4.
Hansen J, Qing K, Kwon HJ, Mah C, Srivastava A. Impaired intracellular trafficking of adeno-associated virus type 2 vectors limits efficient transduction of murine fibroblasts. J. Virol. 2000;74:992–6.
Li M, Jayandharan GR, Li B, Ling C, Ma W, Srivastava A, et al. High-efficiency transduction of fibroblasts and mesenchymal stem cells by tyrosine-mutant AAV2 vectors for their potential use in cellular therapy. Hum Gene Ther. 2010;21:1527–43.
Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72:1438–45.
Hauck B, Zhao W, High K, Xiao W. Intracellular viral processing, not single-stranded DNA accumulation, is crucial for recombinant adeno-associated virus transduction. J Virol. 2004;78:13678–86.
Sharma P, Wimalawansa SM, Gould GC, Johnson RM, Excoffon KJ. Adeno-associated virus 5 transduces adipose-derived stem cells with greater efficacy than other adeno-associated viral serotypes. Hum Gene Ther Methods. 2016;27:219–27.
Virk MS, Conduah A, Park SH, Liu N, Sugiyama O, Cuomo A, et al. Influence of short-term adenoviral vector and prolonged lentiviral vector mediated bone morphogenetic protein-2 expression on the quality of bone repair in a rat femoral defect model. Bone. 2008;42:921–31.
Acknowledgements
This project was supported by an NIH RO1 research grant to JRL [R01AR057076].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
SB, MC, VV, HM, OS, DAO, DL, and PC have no conflicts to report. JRL has received royalties from Depuy Inc, and is a shareholder in Hip Innovation Technologies, Inc.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bougioukli, S., Chateau, M., Morales, H. et al. Limited potential of AAV-mediated gene therapy in transducing human mesenchymal stem cells for bone repair applications. Gene Ther 28, 729–739 (2021). https://doi.org/10.1038/s41434-020-0182-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-020-0182-4
- Springer Nature Limited