
Abstract
Corneal and ocular surface diseases (OSDs) carry significant psychosocial and economic burden worldwide. We set out to review the literature on the application of artificial intelligence (AI) and bioinformatics for analysis of biofluid biomarkers in corneal and OSDs and evaluate their utility in clinical decision making. MEDLINE, EMBASE, Cochrane and Web of Science were systematically queried for articles using AI or bioinformatics methodology in corneal and OSDs and examining biofluids from inception to August 2021. In total, 10,264 articles were screened, and 23 articles consisting of 1058 individuals were included. Using various AI/bioinformatics tools, changes in certain tear film cytokines that are proinflammatory such as increased expression of apolipoprotein, haptoglobin, annexin 1, S100A8, S100A9, Glutathione S-transferase, and decreased expression of supportive tear film components such as lipocalin-1, prolactin inducible protein, lysozyme C, lactotransferrin, cystatin S, and mammaglobin-b, proline rich protein, were found to be correlated with pathogenesis and/or treatment outcomes of dry eye, keratoconus, meibomian gland dysfunction, and Sjögren’s. Overall, most AI/bioinformatics tools were used to classify biofluids into diseases subgroups, distinguish between OSD, identify risk factors, or make predictions about treatment response, and/or prognosis. To conclude, AI models such as artificial neural networks, hierarchical clustering, random forest, etc., in conjunction with proteomic or metabolomic profiling using bioinformatics tools such as Gene Ontology or Kyoto Encylopedia of Genes and Genomes pathway analysis, were found to inform biomarker discovery, distinguish between OSDs, help define subgroups with OSDs and make predictions about treatment response in a clinical setting.
摘要
角膜和眼表疾病 (OSDs) 在世界范围内带来了严重的心理和经济负担。我们对人工智能 (AI) 和生物信息学分析角膜和眼表疾病中生物流体标志物的相关文献进行了综述, 评估其在临床决策中的实用性。 本文对MEDLINE、EMBASE、Cochrane和Web of Science数据库进行系统筛选, 找到从建库到2021年8月期间将人工智能或生物信息学方法应用于角膜和眼表疾病并检查了生物体液的文章。共筛选10264篇文献, 纳入23篇, 共1058位受试者。 使用各种人工智能/生物信息学工具发现某些促炎性泪膜细胞因子的表达变化, 例如载脂蛋白、结合珠蛋白、膜联蛋白1、S100A8、S100A9和谷胱甘肽S-转移酶的表达增加, 支持性泪膜成分如脂质运载蛋白-1、催乳素诱导蛋白、溶菌酶C、乳转铁蛋白、胱抑素S的表达减少, 乳球蛋白-B和脯氨酸富集类蛋白等与干眼症、圆锥角膜、睑板腺功能障碍和干燥综合征的发病机制和/或治疗结果相关。总之, 大部分人工智能/生物信息学工具可根据生物液体将疾病亚群进行分类, 从而区分眼表疾病, 识别风险因素或对治疗反应和/或预后进行预测。 总的来说, 人工智能模型如人工神经网络, 分层聚类, 随机森林等, 在进行蛋白质组或代谢组分析中使用的生物信息学工具, 如基因本体论或京都基因和基因组路径分析百科全书, 可以为生物标志物的发现提供信息, 区分眼表疾病, 帮助定义眼表疾病亚群, 并在临床环境中预测治疗反应。
Similar content being viewed by others

Introduction
Ocular surface diseases (OSD) are conditions affecting corneal and conjunctival structures, tear film characteristics and production, and adnexal gland functions [1, 2]. OSDs are not only associated with significant psychological burden and poor self-perceived health status[3], but also poses a significant economic burden to the individual and society, such as decreased work productivity, absenteeism, and costs of physician visits, ocular lubricants, punctual plugs, and more, reported in the United States [4], Canada [5], and China [6].
Tear fluid homeostasis is central to providing lubrication and nutrients to the ocular surface and is composed of various enzymes, growth hormones, lipids, salts, neuropeptides, mucins [7], which are produced by lacrimal glands, meibomian glands, conjunctival goblet cells, corneal epithelial cells, and vascular sources [8]. The complex protein and metabolite tear film content facilitates a dynamic, wide ranging, individually tailored response to infection and other abnormalities affecting the ocular surface. Tears, which can easily and non-invasively be collected in clinic, have been used to discover biomarkers for determining disease aetiology and risk factor, conversion, severity, or prognosis, and treatment strategy and outcomes, using proteomics [7,8,9] and metabolomics [10, 11]. Several studies have made use of differences in tear proteomes of various OSDs and corneal diseases such as aqueous deficient dry eye and Meibomian gland dysfunction (MGD) [12], or keratoconus, pterygium, graft-versus-host-disease, and controls [7], to identify differentially expressed proteins and evaluate them as potential biomarkers for diagnosis and treatment.
Aqueous humour obtained in surgery (e.g. keratoplasty, phakic intraocular lens implantation) has been found to correlate with disease progression in keratoconus [13]. For example, abnormal expression of proteome measured via liquid chromatography with tandem mass spectrometry (LC-MS/MS) and analysed by hierarchical clustering, principal component analysis, functional interaction sub-networks, and Gene ontology (GO) analysis were implicated in corneal proteolysis, regulation of hypoxia, of fibrinolysis, response to calcium ions, platelet activation, etc. (e.g., haemoglobin subunit beta, haptoglobin, Ig kappa chain V-I region EU) [13].
Generally, artificial intelligence (AI) refers to the capability of computing systems for pattern recognition, and for reproducing human cognitive characteristics (e.g., generalize, and learn from experience) in large datasets [14]. Machine learning (ML), a type of AI, can be used to extract generalized principles from data to make predictions or classifications by applying algorithms and mathematical modelling based on explicit rules and instructions about the data [15].
With its impressive power to identify patterns, classify, cluster, or make predictions from large datasets, AI is well suited to analyse the massive data output produced by continually advancing novel analytical technologies such as proteomics and metabolomics. In proteomics, the data regarding proteins expression in ocular fluids is analysed using AI and compared against databases containing large amounts of labelled protein sequence information [16]. The end result is a proteomic signature or profile of the fluid, which can not only elucidate molecular mechanisms of ocular diseases, but also be used to diagnose disease or monitor the outcome of therapeutics [17,18,19]. Metabolomics involves the large-scale study of endogenous and exogenous metabolites in various tissues as to provide an assessment of the metabolic phenotype of a certain state of disease, and in combination with AI/bioinformatics can be used to obtain putative metabolic pathways and biomarkers associated with disease mechanisms and treatment strategies at the level of the individual patient [10, 11, 20]. The combination of these methods has driven major advancements in precision medicine by allowing examination of individual variability in disease prognosis and individualized treatment strategies [18, 21]. As such, a growing number of ophthalmology studies have adopted these methods to analyse biofluids as biomarkers.
Exploration of biofluids using AI and bioinformatics may offer insight into pathophysiology, prognosis, and fuel the discovery of new therapies of OSDs and corneal diseases. Therefore, the current study aims to systematically review the literature describing application of AI and bioinformatics-based analyses using biofluids as biomarkers in OSDs and corneal diseases. The methodology and findings of eligible studies are summarized and appraised with a focus on assessing the potential of clinical implementation of these approaches.
Methods
Study design and registration
The findings from this systematic review are reported in accordance with the Preferred Reporting Items for a systematic Review and Meta-analysis (PRISMA) guidelines [22]. Study protocol details were prospectively registered on PROSPERO (reg. CRD42020196749). The current systematic review is focused on OSDs and corneal diseases and is a part of a series of systematic reviews on analysis of biofluids using AI for various specific eye conditions in ophthalmology, which are reported elsewhere.
Search strategy
Systematic searches of the literature were conducted in five databases including Embase, MEDLINE, Cochrane Central Register of Controlled Trials, Cochrane Database of Systematic Reviews, and Web of Science from the time of database inception through August 11, 2020, with an update of the search strategy performed on August 1, 2021. A comprehensive set of search terms capturing three categories including ophthalmology, AI/bioinformatics, and proteomics/metabolomics/lipidomics terms were used to construct the search strategy (Appendix A). The search was not restricted by language or study design. Hand-searching of the reference list of included studies was also performed in order to identify relevant articles.
Inclusion and exclusion criteria
Studies were included if they referred to intra-ocular or ocular surface conditions using biofluid marker samples to make AI/bioinformatic-based predictions about disease aetiology or risk factors, treatment outcomes or treatment strategies, and diseases conversion or progression. Samples of biofluids from vitreous, aqueous, or tear fluid, as well as plasma or ophthalmic biopsies were deemed eligible. Studies were excluded if they referred exclusively to paediatric eye diseases, non-human subjects, or included only post-mortem biofluid samples. Additionally, we excluded cross-sectional studies that only used the simplest form of AI (simple regression analysis). Abstracts, reviews, systematic reviews, meta-analyses, single case reports, editorials (without adequate study details and data presentation), and any type of non-peer reviewed article were considered ineligible. Lastly, the subset of studies that met the inclusion criteria, and referred to OSDs or corneal diseases were selected for the current review.
Study selection
The titles and abstracts, and then the full texts were independently screened by two review authors (DRP, AP) for relevant articles. Title and abstract screening included any literature that focused on any OSD and biofluid sampling. At this point, articles were included even if it was not clear if an AI analysis was performed. During full-text screening, any articles that did not meet all our specified inclusion criteria were excluded. If a consensus for conflicts could not be reached between the two reviewers, a third reviewer (SK or TF) resolved the conflict.
Data collection and risk of bias assessment
Data extraction of included studies was undertaken by one reviewer (DRP) using a standardized data abstraction form. To ensure accuracy and consistency of the extraction process, 10% of extractions were randomly double-abstracted by a second independent reviewer (AP or SK). Risk of bias (ROB) and quality assessment of retrieved studies were performed using the Joanna Briggs Institute Critical Appraisal Tools (JBI) [23]. For each article, JBI criteria questions were noted as “yes”, “no”, “unclear”, or “not applicable.” The assessment was performed by one reviewer (DRP) and none of the studies were excluded from the review. Studies that reached up to 49% of questions as “yes” were classified as high ROB; from 50 to 69% as moderate ROB; and more than 70% as low ROB [24].
Data synthesis
There was substantial heterogeneity in biofluid types, AI techniques, and study designs, and consequently a meta-analysis was not undertaken. Means and standard deviations (SD) were used to characterize the study sample(s) age(s). The study characteristics tabulated included study design, location, type of OSD, sample size, sex ratio, study aim, fluid collection methods, and a list of biofluids reported. Articles were further categorized according to OSDs or corneal disease type, statistical model, AI, or bioinformatics analyses performed. Moreover, the AI/Bioinformatics methodology purpose was noted.
Results
Study characteristics
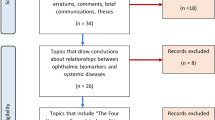
The search strategy resulted in 10,264 articles after removal of duplicates (Fig. 1). Of the 23 articles that were found eligible for inclusion, 7 were prospective (30%), 16 were cross-sectional (70%), and one was a randomized controlled trial (Table 1). There was a global distribution in the country of origin of the included studies with China (4, 17%) and Spain (3, 13%) being the most common. There were 1058 individuals included, with 350 individuals with dry eye, 61 with keratoconus, 43 with pterygium, 179 with meibomian gland dysfunction (MGD), 59 with graft-versus-host-disease (GVHD), 51 with Sjogren, 2 with climatic droplet keratopathy (CDK), 19 with bullous keratopathy, 2 with Fuchs’ endothelial dystrophy, 18 with vernal keratoconjunctivitis (VKC), 12 with various indications for penetrating keratoplasty, and 237 healthy controls, as well as 5 myopic and 20 diabetic individuals as comparator groups.

The PRISMA flow diagram for the systematic review presenting the number of studies included and excluded at each screening step, and reasons for exclusion.
Majority of studies focused on biomarker discovery and identification of pathophysiology of OSDs (15, 65%), while eight (35%) assessed treatment outcomes or prognosis, and one assessed risk factors related to OSD.
The risk of bias (ROB) assessment is presented in Appendix B. Two studies were found to have a high ROB [9, 11], nine moderate ROB [7, 13, 25,26,27,28,29,30,31], and twelve low ROB [12, 32,33,34,35,36,37,38,39,40,41]. The main areas of bias identified among cross-sectional studies were: criteria for inclusion in the sample were not clearly defined (n = 5, 31%), the study subjects and setting was not described in detail (n = 9, 56%), confounding factors were not identified (n = 7, 44%), and strategies to deal with confounding factors were not stated (n = 9, 56%). Among cohort studies, none of them had participants that were free of the ocular disease at the start of the study. This represents a source of bias because if the samples were taken after an OSD had occurred, it is not possible to definitively conclude if the biofluids identified are contributory to the OSD and/or a reflection of the downstream consequences of the OSD. Future studies involving long-term collection of samples prior to and following disease onset may provide more definitive evidence for the associations of biomarkers with OSD pathogenesis.
Biomarkers involved in pathogenesis of dry eye disease
Upregulation of apolipoprotein [26, 27], haptoglobin [26, 27], annexin 1 [27, 34], Glutathione S-transferase [26, 27, 32, 34], and downregulation of lipocalin-1 [7, 12, 26, 27, 31, 34], prolactin inducible protein (PIP) [26, 27, 34], lysozyme C [7, 26, 27, 31, 33, 34], lactotransferrin [7, 26, 27, 34], cystatin S [7, 26, 27, 34], and mammaglobin-b [26, 34], proline rich protein [27, 31] were associated with dry eye pathogenesis. AI analyses using bioinformatics databases implicated the upregulated proteins in biological pathways regulating lipid metabolic processes, oxidation reduction, cytokine production, while the downregulated proteins were associated with transportation, and regulation of immune response [7, 12, 26, 27, 34]. A proteomic study of contributing tear film proteins to the pathogenesis of diabetic dry eye using weighted correlation network analysis, GO and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis found three differentially expressed proteins (lysozyme C, zinc-alpha-2-glycoprotein, DNA J homolog subfamily C member 3) in adults with diabetic dry eye compared to controls, and one in children (phosphoglycerate kinase 1) with diabetic dry eye compared to controls [33]. In both adults and children, these proteins were involved in dysregulation of metabolic pathways associated with inflammation and immunity such as glycolysis, pentose phosphate pathway, and proteasomes [33]. In adults, the expression levels of these proteins significantly correlated with tear film break-up time, Schirmer I test, and corneal fluorescein staining.
There were several overlapping biomarkers between MGD [34, 40], Sjögren’s [30, 42], and dry eye [7, 26, 27, 31, 33, 34], associated with pathogenesis (e.g., lipocalin-1, lysozyme C, annexin A1, cystatin S). Additionally, Sjögren’s patients presented increased expression of TNF-a signalling, B cell survival, proteins involved in the Krebs cycle, and in oxidative stress in tear fluid [30], as well as upregulation of elastase, calreticulin, and tripartite motif-containing protein [42], proteins involved in inflammation and complement coagulation cascade [42]. A prospective randomized controlled trial investigating the efficacy of intense pulsed light (IPL) for MGD using proteomic analysis found that tear level of interleukin-1 receptor agonist was significantly lower at 3 months compared to baseline in both sham and IPL groups but there were no differences between the groups [40].
Biomarkers involved in treatment response of dry eye disease
Two articles assessed the role of biofluids in predicting response to treatments for dry eye, specifically punctual occlusion [32], and diquafosol tetrasodium or topical cyclosporine A [8]. By using tear proteomics and clustering analysis of identified proteins (i.e., measured at baseline and after 3 weeks), two distinct patient profiles of treatment response emerged, and each group presented differentially expressed tear proteins (one beneficial, one inflammatory). Patients from the group with a beneficial pattern of protein expression, a reduction in inflammatory proteins (e.g., S100A9) and an increase in lacrimal proteins protective of the ocular surface (e.g., lysozyme), also presented a lower Schirmer score at baseline than the patients from the inflammatory pattern group [32]. Thereby, allowing clinicians to identify patients with low scores who may benefit from punctual occlusion, and potentially change management in those patients less likely to benefit from this treatment. Another proteomic study found that that there were treatment specific differences in tear proteome and associated biological pathways in patients treated with diquafosol tetrasodium or topical cyclosporine A despite similar clinical outcomes, with 49 proteins showing an inverse expression pattern [8].
Biomarkers involved in pathogenesis of keratoconus
Several studies performed proteomic [7, 13, 35] and metabolomic profiling [11] of biofluids to investigate their role in keratoconus pathogenesis. However, there was little overlap in discovered biomarkers. A cross-sectional study of various OSDs, found 8 potential tear biomarkers contributing to keratoconus pathophysiology, and which allowed differentiation between keratoconus, pterygium, and graft-versus-host-disease related dry eye [7].
A proteomic study of aqueous fluid from keratoconus patients obtained during keratoplasty, identified 16 out of 137 proteins related to dysregulation of apoptosis, oxidative stress, response to vitamin D, angiogenesis, as potential markers of pathological changes in keratoconus [13]. A metabolomic analysis [11] of contributing metabolites using gas chromatography and mass spectrometry (GC/MS) and unsupervised hierarchical cluster analysis identified downregulation of 13 out of 377 metabolites related to aberrations in energy production, lipid metabolism, and amino acid metabolism in the corneal buttons of keratoconus patients compared to those of healthy donors [11].
Biomarkers involved in prognosis or treatment response of keratoconus
The release of several key inflammatory (interferon gamma, IFN-y; interleukin-13, IL-13; IL-17A, chemokine C-C motif ligand 5, CCL5; matrix metalloproteinase, MMP-13; and plasminogen activator inhibitor 1, PAI-12) factors at one year follow-up were found to predict keratoconus progression in a group of 42 patients [35]. NGF and IL-13 were found to identify progression with 100% specificity and 88% sensitivity.
Biomarkers involved in other corneal diseases
Other OSDs investigated were ocular graft-versus-host-disease [7, 39], and pterygium [7, 29, 37]. Patients with chronic ocular graft-versus-host-disease experience inflammation and fibrosis of the ocular surface, in addition to severe ocular dryness [43]. A proteomic study of 785 proteins using AI tools such as random forest and penalized logistic regression, and bioinformatics tools such as GO analysis, found that disease severity of ocular graft-versus-host-disease in patients after allogenic hematopoietic cell transplantation (AHCT) could be predicted based on the differential expression of 13 biofluids (i.e., Phosphoglycerate mutase 1, Keratin type I, cytoskeletal 9) [39]. Biochemical pathways highlighted in pathogenesis were related to complement and coagulation cascades (i.e., Clusterin, Complement factor B, Complement C3, plasminogen) [7].
A prospective cohort study of endothelial keratoplasty patients examined the kinetics of their tear profiles over the course of recovery after transplant using a clustering algorithm (i.e., principal component analysis) and found alterations in the level of expression of eleven tear fluid proteins predictive for recovery from corneal haze, with the group of patients with no corneal haze within one month after surgery having significantly lower levels at the pre-transplant baseline timepoint than the group that did develop corneal haze [28]. Several inflammatory cytokines were associated with corneal graft rejection following penetrating keratoplasty in a group of 12 patients followed for 12 – 14 months after surgery [36]. Proteomic tear profiling indicated that IL-6 and IL-8 concentrations were increased in patients with rejection, while IL-10, TNF-α, and IL-12p70 were decreased compared to patients with uncomplicated corneal grafts [36].
Climatic droplet keratopathy, a degenerative disease associated with progressive accumulation of droplets on the cornea, found to be associated with 105 proteins, mainly related to cell junction function, glycolysis, focal adhesion, regulation of cytoskeleton, fibril formation and deposits (e.g., retinal dehydrogenase, aldehyde dehydrogenase, desmoplakin, etc.) when its proteome was analysed with KEGG in a case series [9].
Biomarkers involved in pathogenesis, prognosis and treatment response of VKC
In a small sample of six VKC patients responsive to treatment with cyclosporine or corticosteroids proteomic analysis with isobaric tags for relative and absolute quantification (iTRAQ) technology showed downregulation of Hemopexin, transferrin, mammaglobin B, and secretoglobin 1D [41]. These proteins were suggested to be involved in oxidative stress regulation and inflammatory response regulation [41]. Additionally, expression of tear albumin and transferrin was found to be positively correlated with VKC disease severity, and therefore may be potential biomarkers for disease diagnosis and monitoring [41].
Applications of AI and bioinformatics
As presented in Table 2, there was prominent heterogeneity in the use and reporting of AI methodology. Seventeen articles used AI and/or bioinformatics with classification algorithms, five used predictive models, and four used both classification algorithms and predictive models.
Eight articles used a combination of at least two different AI classes. Most commonly, articles analysed biofluids using conventional AI ML techniques such as (1) clustering analyses, including hierarchical clustering [11, 13, 32, 42], k-nearest neighbour [13, 34], nonlinear iterative partial least squares [12], (2) discriminant analyses [31] including partial least square discriminant analysis [7], feature extraction by stepwise discriminant analysis [12], (3) decision tree algorithms including random forest [34, 39], (4) classification algorithms such as support vector machine, naive bayes [34], and (5) dimensionality reduction algorithms such as principal component analysis [11, 28, 38].
Two articles analysed biofluids using deep learning AI [12, 31]. A prospective case-controlled study of 93 patients aimed at elucidating differences between the tear proteome profile of individuals with dry eye, MGD associated dry eye, and healthy individuals, used a nonlinear iterative partial least squares algorithm to cluster the proteomic data followed up a multilayer perceptron neural network predictive model to distinguish between the three distinct tear proteome profiles. Validation of the model yielded a 89.3% correct assignment [12].
Similarly, a cross-sectional study of 88 individuals with dry eye and 71 healthy individuals, used a combination of univariate regression and multivariate discriminant analysis to identify a seven-biomarker panel of potential tear biofluids that may distinguish between the proteomic profile of individuals with dry eye and healthy individuals. These biomarkers were used to train a multiple-layer feed-forward network with back-propagation training algorithm to classify individuals into one of the two groups. Correct classification was quantified using a receiver operating characteristic curve (ROC) and area under the ROC (AUC), which was reported as 0.93, indicating high accuracy [31].
Bioinformatics methodology description largely consisted of the standard analysis protocol of established software such as GO analysis with database for annotation, visualization, and integrated discovery (DAVID), KEGG pathway analysis, iTRAQ proteomics with MASCOT engine, or STRING database searches. Overall, bioinformatic tools were used to classify biofluids into diseases subgroups [26, 33, 39], distinguish between OSD [7, 34], identify risk factors[29], or make predictions about treatment response, and/or prognosis [28, 32, 35, 36, 39].
As presented in Table 2, GO analysis was used by eleven articles, and KEGG pathway analysis was utilized by five articles. One article applied a weighted correlation network analysis (WGCNA), a data mining method, in conjunction with GO analysis and KEGG pathway analysis to identify key hub genes and proteins associated with diabetes and dry eye in adults and children. The GO and KEGG analyses pointed to differentially expressed proteins involved in various metabolic pathways in the tear proteome of adults and children with diabetes and dry eye [33]. MASCOT was used to identify proteins by four articles [12, 13, 26, 41], and STRING was used to build functional protein association networks by three articles [8, 27, 30].
Discussion
This is the first systematic review, to our knowledge, to describe the applications of AI and bioinformatics-based analyses including proteomics and metabolomics using biofluids as markers in various types of corneal and ocular surface diseases. The potential of these technologies to identify candidate biomarkers for diagnosis or potential drug targets to halt disease progression was explored. Risk factors were investigated by one cross-sectional study on pterygium using proteomics in combination with GO analysis with DAVID and KEGG pathway analysis [29]. However, most studies focused on biomarker discovery and identification of biofluids to elucidate aetiology and identify candidate markers for diagnosis, discriminate between OSDs [7, 12, 34], and even identify different subgroups within an OSD [26, 33, 39, 44].
It is increasingly recognized that inflammation of the ocular surface or cornea, specifically change in tear film cytokines, is involved in multiple OSDs including dry eye (e.g. increased expression of apolipoprotein [26, 27], haptoglobin[26, 27], annexin 1 [27, 34], S100A8, S100A9 [32], Glutathione S-transferase [26, 27, 32, 34], and decreased expression of lipocalin-1 [7, 12, 26, 27, 31, 34], prolactin inducible protein [26, 27, 34], lysozyme C [7, 26, 27, 31, 33, 34], lactotransferrin [7, 26, 27, 34], cystatin S [7, 26, 27, 34], and mammaglobin-b [26, 34], proline rich protein [27, 31], IFN-y [28, 36, 45], TNF-a [28, 36, 45]). Several of these are differentially expressed in MGD [34, 40], and Sjögren’s [30, 42] (e.g. lipocalin-1, lysozyme C, annexin A1, cystatin S), as well as keratoconus (e.g. proline rich protein). The biological function of these proteins explains the overlapping pathophysiology, as lactotransferrin and lysozyme have antibacterial properties and support the epithelium [1], while S100A8 and S100A9 are proinflammatory [32], and proline rich protein may be involved in androgen mediated lipolysis [45].
There is a clear need for advancements to the stage of direct clinical applications such as treatment response prediction or monitoring. Several studies have emphasized that tear protein profiling has the potential to provide a diagnostic signature for various OSDs and corneal diseases, specifically the tear film can differentiate between OSDs [7, 12, 46], and also provide measures of disease severity, as well as treatment effectiveness, and thereby be useful for longitudinal monitoring [28, 35, 39].
For example, in a longitudinal study of dry eye patients, hierarchical clustering revealed distinct patient profiles based on clusters of tear protein expression after 3 weeks of punctual occlusion [32]. In cluster 1 (i.e., beneficial response) patients (n = 10) showed increased expression of proteins protective of the ocular surface (e.g., prolactin-inducible protein, lactoferrin) and decreased expression of pro-inflammatory proteins (e.g., alpha enolase 1), while in cluster 2 (nonresponse), patients (n = 13), the opposite trends were observed [32]. These patient profiles correlated with baseline Schirmer scores and may be used in clinic by ophthalmologists to identify patients most at benefit from punctal plugs (i.e., low Schirmer score).
Proteomic analyses in combination with AI, may provide objective tests for evaluating treatment effectiveness for OSDs. For example, a pilot study on dry eye patients used proteomics with GO with DAVID, KEGG and functional annotation clustering, and protein-network analysis to identify 54 and 106 differential expressed biomarkers indicative of disease severity and treatment effectiveness of CsA or DQS, respectively, at 4 weeks [8]. While both treatments were found to be equally effective, tear protein expression profiles indicated distinct regulatory patterns with CsA treated tears showing upregulation of wound healing, endopeptidase activity, and protein metabolism pathways, and DQs showing upregulation of proteins involved in regulation of stress response, tissue homeostasis, and defence response [8]. Following validation in larger samples, expression levels of proteins such as phospholipase A2 group IIA, which was upregulated 2.1-fold pre-treatment in dry eye compared to control and downregulated to 0.58- and 0.78-fold after treatment with CsA and DQS, may be used as metrics indicative of treatment effectiveness with topical agents [8].
This systematic review highlighted several limitations and challenges associated with the included studies. Importantly, the quality and robustness of the AI and bioinformatics-based biofluid analyses is highly dependent on the selection of ML algorithm and the preprocessing of the data. Clustering algorithms, such as hierarchical clustering, an unsupervised ML technique, and k-nearest neighbour, a supervised technique, are useful for identifying subgroups on the basis of similarities between proteomic profiles [11, 13, 32, 34, 42]. Major disadvantages are related to data preprocessing, as clustering algorithms are sensitive to missing values, outliers, data transformation (i.e., to logarithmic scale), and selection of cluster size[47]. Although these parameters directly affect clustering results, we found that they were not consistently described, and consequently may introduce error, reduce reproducibility, and limit validation. These clustering algorithms are less accurate at datasets with more than 400 features (i.e., input variables)[47]. However, this disadvantage can be mitigated by projecting a large number of proteins or metabolites onto a smaller number of features, a procedure known as dimensionality reduction [47]. The main advantage of classifiers such as principal component analysis, random forest algorithm, partial least squares, and support vector machine in biofluid analyses with large datasets of biomarkers is that dimensionality reduction can remove irrelevant features, reduce noise or extraneous variables, and can account for highly correlated variables [47]. The major disadvantage of dimensionality reduction is that it generally requires the selection of a subset of guiding features, a step with a variable level of subjectivity. Relevant data can erroneously be labelled as noise, and this can lead to the loss of important data [47]. The deep learning algorithm, multi-layer perceptron neural network, was implemented as a “black-box model” by two articles in this review, meaning that its different layers and complex architecture was not described in sufficient detail to allow the reader to map the process from variable input to prediction [12, 31]. Therefore, despite advantages such as better handling of highly dimensional data, complex (non-linear) associations, noise, and incomplete or missing values, the interpretation and generalizability of the results are limited [48].
Small sample sizes and large datasets of proteins increase the likelihood of finding spurious associations due to the inter-day and inter-individual variability of normal tears [49]. Moreover, the reporting of large proteomic data, up to 2,733 proteins, is challenging considering reporting limits set by various scientific journals, four articles in the current review only reported them in figure format, one only reported significant proteins, and two did not report the full list of measured proteins at all. Online repositories (e.g. The Global Proteome Machine Database, PeptideAtlas, etc.) could be used for data mining in future studies [50]. Other limitations of bioinformatics-based analyses of biofluid data relate to the annotation databases (e.g., GO and KEGG) used to perform the ontological analysis necessary to map the function of input proteins and construct protein-protein interaction networks using clustering, classification, and significance analyses. These databases are manually curated by researchers and may be incomplete, imprecise, variability in the identifiers used by various research groups, and be impacted by annotation bias (i.e., well-studied biological processes are more represented) [51].
The multifactorial pathogenesis of OSDs and corneal diseases, the overlap of symptoms, and lack of concordance between clinical parameters and symptoms reported by patients [34], present a challenge to the identification of unique biomarkers for discriminating between pathologies, or monitoring treatment response. The challenge is not only compounded by small sample sizes, but also lack of healthy controls, and technical variability associated with proteomic and metabolomic studies (e.g., tear collection methods, sample preparation, pre-processing steps for mass spectrometry, lack of reporting of all investigated proteins). We found that most studies used Schirmer strips (n = 12, 52%) or micropipette/glass capillaries (n = 5, 22%) as collection methods. Generally, both collection methods, Schirmer test and capillary, are reported to produced similar results [31, 52]. However, Schirmer test was found to allow for better discrimination between dry eye and healthy samples [31, 34]. Only a handful of studies implemented a statistical validation process of the discovered proteins, with the goal of using the area under the curve (AUC) from multivariate receiver operating characteristic (ROC) analyses to calculate specificity and sensitivity, and estimate the clinical applicability of candidate biofluids [12, 42, 53]. Validation on large samples is crucial particularly considering the lack of a priori hypotheses or pre-selection of a panel of biomarkers characteristic of many proteomic and metabolomic studies, as well as the physiologic heterogeneity of OSD. Without this step, both the generalizability and the predictive specificity of candidate biomarkers remains limited. For example, dysregulation of several candidate biomarkers for pathogenesis in dry eye were also found in MGD (e.g., lipoprotein-1, lysozyme C, lactotransferrin) [7, 26, 31, 34], VKC (e.g. lactotransferrin) [41] or in pterygium and climatic droplet keratopathy (e.g., alcohol dehydrogenase) [9, 26, 37].
The combination of biofluids and imaging metrics obtained from optical coherence tomography (OCT), and analysed using AI, may compound the clinical predictive value of these techniques [54]. For example, wide corneal epithelial mapping using OCT in dry eye analysed with random forest AI, showed that superior intermediate epithelial thickness in dry eye compared to controls, was a promising marker for diagnosing dry eye (sensitivity 86.4%, specificity 91.7%) [54]. Introducing biofluids as covariates in these types of analyses would increase the robustness and validity of these analyses and bring them to clinical standards.
This systematic review appraised the use of AI or bioinformatics tools to analyse biofluid markers in OSDs and corneal disease. These tools implicated various tear film proteins in biological pathways regulating lipid metabolomic processes, oxidative stress regulation, cytokine production, vesicular transportation, and regulation of the immune response. Several studies have suggested that tear protein profiling has the potential of providing a diagnostic signature for various OSDs, may be used to identify patients most at benefit from treatments, or provide indications for treatment effectiveness and be useful for longitudinal monitoring.
References
von Thun und Hohenstein-Blaul N, Funke S, Grus FH. Tears as a source of biomarkers for ocular and systemic diseases. Exp Eye Res. 2013;117:126–37.
Zhou L, Beuerman RW. Tear analysis in ocular surface diseases. Progr Retinal Eye Res. 2012;31:527–50.
Wang MTM, Muntz A, Wolffsohn JS, Craig JP. Association between dry eye disease, self-perceived health status, and self-reported psychological stress burden. Clin Exp Optom. 2021;104:835–40.
Yu J, Asche CV, Fairchild CJ. The economic burden of dry eye disease in the United States: A decision tree analysis. Cornea. 2011;30:379–87.
Chan C, Ziai S, Myageri V, Burns JG, Prokopich CL. Economic burden and loss of quality of life from dry eye disease in Canada. BMJ Open Ophthalmol. 2021;6:e000709.
Yang W, Luo Y, Wu S, Niu X, Yan Y, Qiao C, et al. Estimated annual economic burden of dry eye disease based on a multi-center analysis in china: a retrospective study. Front Med (Lausanne). 2021;8:771352.
de Almeida Borges D, Alborghetti MR, Franco Paes Leme A, Ramos Domingues R, Duarte B, Veiga M, et al. Tear proteomic profile in three distinct ocular surface diseases: keratoconus, pterygium, and dry eye related to graft-versus-host disease. Clin Proteomics [Internet]. 2020;17:1–16.
Ji YW, Kim HM, Ryu SY, Oh JW, Yeo A, Choi CY, et al. Changes in human tear proteome following topical treatment of dry eye disease: Cyclosporine a versus diquafosol tetrasodium. Invest Ophthalmol Vis Sci. 2019;60:5035–44.
Menegay M, Lee DM, Tabbara KF, Cafaro TA, Urrets-Zavalía JA, Serra HM, et al. Proteomic analysis of climatic keratopathy droplets. Invest Ophthalmol Vis Sci. 2008;49:2829–37.
Jiang Y, Yang C, Zheng Y, Liu Y, Chen Y. A set of global metabolomic biomarker candidates to predict the risk of dry eye disease. Front Cell Dev Biol. 2020;8:344.
Wojakowska A, Pietrowska M, Widlak P, Dobrowolski D, Wylegała E, Tarnawska D. Metabolomic signature discriminates normal human cornea from Keratoconus—A pilot GC/MS study. Molecules. 2020;25:2933.
González N, Iloro I, Soria J, Duran JA, Santamaría A, Elortza F, et al. Human tear peptide/protein profiling study of ocular surface diseases by SPE-MALDI-TOF mass spectrometry analyses. EuPA Open Proteom. 2014;3:206–15.
Soria J, Villarrubia A, Merayo-Lloves J, Elortza F, Azkargorta M, de Toledo JA, et al. Label-free LC–MS/MS quantitative analysis of aqueous humor from keratoconic and normal eyes. Mol Vis. 2015;21:451–60.
Keskinbora K, Güven F. Artificial intelligence and ophthalmology. Turk J Ophthalmol. 2020;50:37–43.
Schmidt-Erfurth U, Sadeghipour A, Gerendas BS, Waldstein SM, Bogunović H. Artificial intelligence in retina. Prog Retin Eye Res. 2018;67:1–29.
Yu LR, Stewart NA, Veenstra TD. Chapter 8 - Proteomics: The Deciphering of the Functional Genome. In: Ginsburg GS, Willard HFBTE of G and PM, editors. San Diego: Academic Press; 2010:89–96
Larrañaga P, Calvo B, Santana R, Bielza C, Galdiano J, Inza I, et al. Machine learning in bioinformatics. Brief Bioinform. 2006;7:86–112.
Cryan LM, O’Brien C. Proteomics as a research tool in clinical and experimental ophthalmology. Proteomics Clin Appl. 2008;2:762–75.
Schmidt A, Forne I, Imhof A. Bioinformatic analysis of proteomics data. BMC Syst Biol. 2014;8(Suppl 2):S3–S3. 2014/03/13
Tan SZ, Begley P, Mullard G, Hollywood KA, Bishop PN. Introduction to metabolomics and its applications in ophthalmology. Eye (Basingstoke). 2016;30:773–83.
Hopkins JJ, Keane PA, Balaskas K. Delivering personalized medicine in retinal care: From artificial intelligence algorithms to clinical application. Curr Opin Ophthalmol. 2020;31:329–36.
Page MJ, McKenzie JE, Bossuyt PM, Boutron I, Hoffmann TC, Mulrow CD, et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021;372:n71.
Munn Z, Moola S, Riitano D, Lisy K. The development of a critical appraisal tool for use in systematic reviews addressing questions of prevalence. Int J Health Policy Manag. 2014;3:123–8.
Valesan LF, Da-Cas CD, Réus JC, Denardin ACS, Garanhani RR, Bonotto D, et al. Prevalence of temporomandibular joint disorders: a systematic review and meta-analysis. Clin Oral Investig. 2021;25:441–53.
Liang JT, Huang J, Chen TC, Hung JS. The Toldt fascia: A historic review and surgical implications in complete mesocolic excision for colon cancer. Asian J Surg. 2019;42:1–5.
Srinivasan S, Thangavelu M, Zhang L, Green KB, Nichols KK. iTRAQ quantitative proteomics in the analysis of tears in dry eye patients. Invest Ophthalmol Vis Sci. 2012;53:5052–9.
Huang Z, Du CX, Pan XD. The use of in-strip digestion for fast proteomic analysis on tear fluid from dry eye patients. PLoS One. 2018;13:e0200702.
Yawata N, Awate S, Liu YC, Yuan S, Woon K, Siak J, et al. Kinetics of tear fluid proteins after endothelial keratoplasty and predictive factors for recovery from corneal haze. J Clin Med. 2020;9:1–14.
Linghu D, Guo L, Zhao Y, Liu Z, Zhao M, Huang L, et al. iTRAQ-based quantitative proteomic analysis and bioinformatics study of proteins in pterygia. 2018;1600094:7–8.
Aqrawi LA, Galtung HK, Vestad B, Øvstebø R, Thiede B, Rusthen S, et al. Identification of potential saliva and tear biomarkers in primary Sjögren’s syndrome, utilising the extraction of extracellular vesicles and proteomics analysis. Arthritis Res Ther. 2017;19:1–15.
Grus FH, Podust VN, Bruns K, Lackner K, Fu S, Dalmasso EA, et al. SELDI-TOF-MS ProteinChip array profiling of tears from patients with dry eye. Invest Ophthalmol Vis Sci. 2005;46:863–76.
Tong L, Zhou L, Beuerman R, Simonyi S, Hollander DA, Stern ME. Effects of punctal occlusion on global tear proteins in patients with dry eye. Ocular Surface. 2017;15:736–41.
Zou X, Wang S, Zhang P, Lu L, Zou H. Quantitative proteomics and weighted correlation network analysis of tear samples in adults and children with diabetes and dry eye. Transl Vis Sci Technol. 2020;9:1–15.
Soria J, Durán JA, Etxebarria J, Merayo J, González N, Reigada R, et al. Tear proteome and protein network analyses reveal a novel pentamarker panel for tear film characterization in dry eye and meibomian gland dysfunction. J Proteomics. 2013;78:94–112.
Fodor M, Vitályos G, Losonczy G, Hassan Z, Pásztor D, Gogolák P, et al. Tear mediators NGF along with IL-13 predict keratoconus progression. Ocul Immunol Inflamm. 2021;29:1090–101.
Fodor M, Gogolák P, Rajnavölgyi É, Berta A, Kardos L, Módis L, et al. Long-term kinetics of cytokine responses in human tears after penetrating keratoplasty. J Interferon Cytokine Res. 2009;29:375–9.
Kim SW, Lee J, Lee B, Rhim T. Proteomic analysis in pterygium; upregulated protein expression of ALDH3A1, PDIA3, and PRDX2. Mol Vis. 2014;20:1192–202.
Sembler-Møller ML, Belstrøm D, Locht H, Pedersen AML. Proteomics of saliva, plasma, and salivary gland tissue in Sjögren’s syndrome and non-Sjögren patients identify novel biomarker candidates. J Proteomics. 2020;225:103877.
O’leary OE, Schoetzau A, Amruthalingam L, Geber-Hollbach N, Plattner K, Jenoe P, et al. Tear proteomic predictive biomarker model for ocular graft versus host disease classification. Transl Vis Sci Technol. 2020;9:1–15.
Piyacomn Y, Kasetsuwan N, Reinprayoon U, Satitpitakul V, Tesapirat L. Efficacy and safety of intense pulsed light in patients with meibomian gland dysfunction-a randomized, double-masked, sham-controlled clinical trial. Cornea. 2020;39:325–32.
Leonardi A, Palmigiano A, Mazzola EA, Messina A, Milazzo EMS, Bortolotti M, et al. Identification of human tear fluid biomarkers in vernal keratoconjunctivitis using iTRAQ quantitative proteomics. Allergy: Eur J Allergy Clin Immunol. 2014;69:254–60.
Sembler-Møller ML, Belstrøm D, Locht H, Pedersen AML. Proteomics of saliva, plasma, and salivary gland tissue in Sjögren’s syndrome and non-Sjögren patients identify novel biomarker candidates. J Proteomics. 2020;225:103877.
Inamoto Y, Valdés-Sanz N, Ogawa Y, Alves M, Berchicci L, Galvin J, et al. Ocular graft-versus-host disease after hematopoietic cell transplantation: Expert review from the Late Effects and Quality of Life Working Committee of the CIBMTR and Transplant Complications Working Party of the EBMT. Bone Marrow Transpl. 2019;54:662–73.
Yam GHF, Fuest M, Zhou L, Liu YC, Deng L, Chan ASY, et al. Differential epithelial and stromal protein profiles in cone and non-cone regions of keratoconus corneas. Sci Rep. 2019;9:1–17.
Sharif R, Bak-Nielsen S, Sejersen H, Ding K, Hjortdal J, Karamichos D. Prolactin-induced protein is a novel biomarker for keratoconus. Exp Eye Res. 2019;179:55–63.
Soria J, Acera A, Merayo-Lloves J, Durán JA, González N, Rodriguez S, et al. Tear proteome analysis in ocular surface diseases using label-free LC-MS/MS and multiplexed-microarray biomarker validation. Sci Rep. 2017;7:17478.
Karimpour-Fard A, Elaine Epperson L, Hunter LE. A survey of computational tools for downstream analysis of proteomic and other omic datasets. Hum Genomics. 2015;9:28.
Lancashire LJ, Lemetre C, Ball GR. An introduction to artificial neural networks in bioinformatics—Application to complex microarray and mass spectrometry datasets in cancer studies. Briefings in Bioinformatics. 2009;10:315–29.
González N, Iloro I, Durán JA, Elortza F, Suárez T. Evaluation of inter-day and inter-individual variability of tear peptide/protein profiles by MALDI-TOF MS analyses. Mol Vis. 2012;18:1572–82.
Vizcaíno JA, Foster JM, Martens L. Proteomics data repositories: Providing a safe haven for your data and acting as a springboard for further research. J Proteomics. 2010;73:2136.
Khatri P, Drǎghici S. Ontological analysis of gene expression data: Current tools, limitations, and open problems. Bioinformatics. 2005;21:3587–95.
Green-Church KB, Nichols KK, Kleinholz NM, Zhang L, Nichols JJ. Investigation of the human tear film proteome using multiple proteomic approaches. Mol Vis. 2008;14:456–70.
Grus FH, Sabuncuo P, Augustin AJ. Analysis of tear protein patterns of dry-eye patients using fluorescent staining dyes and two-dimensional quantification algorithms. Electrophoresis. 2001;22:1845–50.
Edorh NA, el Maftouhi A, Djerada Z, Arndt C, Denoyer A. New model to better diagnose dry eye disease integrating OCT corneal epithelial mapping. Br J Ophthalmol. 2021;106:1488–95.
Acknowledgements
This study was in-part funded by a Fighting Blindness Canada research grant awarded to Dr. Tina Felfeli. The authors would like to acknowledge Arshpreet Bassi, Shaily Brahmbhatt, Priyanka Singh, Ishita Aggarwal, Amy Basilious, Jasmine Bhatti, and Karthik Manickavachagam, who participated in article screening.
Author information
Authors and Affiliations
Contributions
TF, RNM: conceptualization, supervision, methodology, writing, review and editing of manuscript. DRP: conceptualization, data collection, analysis and interpretation of results, draft manuscript preparation. SHK, methodology, data collection. AP, data collection, analysis and interpretation of the results. All authors reviewed the results, and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Appendix A
Search strategy used to survey databases.
Appendix B
Assessment of risk of bias and quality of included studies based on criteria from Joanna Briggs Institute Critical Appraisal Tools.
Supplementary information
41433_2022_2307_MOESM2_ESM.docx
Assessment of risk of bias and quality of included studies based on criteria from Joanna Briggs Institute Critical Appraisal Tools
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Pur, D.R., Krance, S.H., Pucchio, A. et al. Current uses of artificial intelligence in the analysis of biofluid markers involved in corneal and ocular surface diseases: a systematic review. Eye 37, 2007–2019 (2023). https://doi.org/10.1038/s41433-022-02307-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-022-02307-9
- Springer Nature Limited