
Abstract
Mycobacterium tuberculosis (MTB) is commonly resistant to various drugs. Multidrug-resistant tuberculosis (MDR-MTB) is mostly caused by mutation in drug-binding proteins and protein folding. The aim of the study was to identify the pattern of mutations in embC, inhA and rpoB proteins and investigate its interactions with available drug such as rifampicin, ethambutol and isoniazid, using a computer docking method. The evolution of drugs resistant mechanisms of MTB was analyzed using an in silico approach. The model proteins were considered to be in a protein–protein interaction network among the twenty transmembrane proteins. The changes in structural conformation may describe the significance of the proton pumps system. The docking analysis revealed that unlike isoniazid, both rifampicin and ethambutol, bound to the same residues in mutant and wild forms. Moreover, multiple-sequence alignment (MSA) showed mutational hotspot regions where the substitution of amino acids in these three target proteins was position specific under stress. The molecular basis of drug resistance in M. tuberculosis can be represented by a protein network which is a well-regulated system for efflux pump activation by popularly used drugs. Ethambutol and rifampicin form stable complexes with EmbC and RpoB, respectively. Isoniazid shows no binding affinity to mutant InhA (2015). Analysis of the cellular network associated with drug regulatory proteins suggest that mmpl3, Rv1634 and Rv1258c play a major role by altering the protein pump to remove the active drug compounds from the bacterial cell.
Similar content being viewed by others

Introduction
More than one third of the world’s population is infected by Mycobacterium tuberculosis. Each year, 8–10 million new cases of Mycobacterium tuberculosis (TB) infection are reported and approximately 1.5 million people are killed per year [1,2,3]. The rate of death due to TB has increased by 22% in 2000–2015 and this increase is believed to be primarily because of multidrug resistant (MDR) strains of Mycobacterium tuberculosis [3,4,5]. Generally, MDR strains survive after treatment with anti-mycobacterial drugs either by activating drug efflux pump system or by the production of inactivating/degrading enzymes. The resistance mechanisms of M. tuberculosis are specifically associated with point mutations and deletions/insertions [6,7,8]. Rapid identification of resistant strains is required for proper usage of anti-TB drugs to control MDR strains. Isoniazid (INH), rifampicin (RIF), and their derivatives are key drugs to combat MDR-MTB [9,10,11]. Ethambutol (EMB) is a synthetic compound which inhibits the major cell wall biosynthetic enzymes, arabinosyl transferases like EmbA, EmbB, EmbC [12, 13]. INH mainly targets InhA to stop its function. InhA catalyses the reduction of long chain trans-2-enoyl-ACP which is a major component in the type II fatty acid biosynthesis pathway that is required for the biosynthesis of mycolic acid in M. tuberculosis [14]. Rifampicin inhibits the DNA dependent RNA polymerase of M. tuberculosis [15]. Susceptible strains of M. tuberculosis are transformed into MDR by involving several genes which control several complex genetic systems [16]. Resistance to INH is mostly associated with multiple mutations in InhA, KatG and resistance to RIF involves mutations in RpoB (Leu 511 Pro, Asp 516 Tyr and Leu 533 Pro) [17]. Multidrug resistance in M. tuberculosis can also be due to the activation of efflux pump SMR, MFS, and ABC. These pumps may be directly or indirectly regulated by mutation of some genes like rpoB, rpsl, katG and others [18, 19]. In this paper, several mutational hotspots were identified in the drug binding pockets of regulatory proteins of susceptible and mutant MTB by in silico MSA analysis. Docking of modeled receptors/target proteins with ligands showed a preferential and non-preferential binding affinity in susceptible versus mutant strains, respectively. Structural deviations of twenty regulatory transmembrane proteins were analyzed and finally, Rv1258c, Rv1634 and Mmpl3 represented structural alterations among them.
Materials and methods
The three-dimensional (3D) structural data of target proteins, RpoB, INhA, EmbB and their corresponding sequences of MTB were retrieved from RCSB-PDB (www.rcsb.org) and Uniprot (www.uniprot.org) databases. The structure of ligand molecules was obtained from Pubchem (pubchem.ncbi.nlm.nih.gov). Twenty transmembrane efflux pump proteins sequence and structures were also downloaded from Uniprot (www.uniprot.org) and RCSB-PDB (www.rcsb.org) databases.
MSA Analysis
Multiple sequence alignment (MSA) was performed in Clustal Omega to determine time-dependent mutation (Supplementary 1 Table A, B, C).
Structural model building
The modeling of the 3D structure of target proteins was performed by Swiss Model Server (www.swissmodel.expasy.org). The constructed model structure was validated by Procheck.
Molecular docking
Target proteins and ligands were docked by using AutoDock 1.5.6 [20] (Supplementary 2 Table D, E, F). Ligands were taken from pubchem database and docked with modeled proteins. Ligands were prepared for docking and the grid size (X = 60; Y = 60; Z = 55) and center was defined to run Autodock genetic algorithm [21].
Inter and intra molecular interaction
Discovery Studio (v17.2.0.16349) was used for the visualization and analysis of molecular interactive architecture.
Core free energy of protein
Stability of protein was calculated through free energy calculation by using FoldX (http://foldx.crg.es).
Structural homology analysis
PyMOL was used to calculate the structural deviation of transmembrane proteins based on RMSD algorithm [22].
Protein protein interactive complex
Protein correlation networks of MTB pathway information were accessed by using String (string-db.org) database. The binding affinity including protein activation networks associated with RNA polymerase β subunit (RpoB), Enoyl [acyl-carrier-protein] reductase [NADH] (InhA), Arabinosyl Transferase C (EmbC) were studied.
Visualization of architectural modification
From, the mutational hotspot regions of transmembrane proteins were identified by the MSA analysis. The coordinates of corresponding structural PDB files were downloaded or modeled and then displayed in RasMol.
Result and discussion
Assessment of multiple sequence protein alignment
Changes of amino acid sequence of the proteins (RpoB, InhA, EmbC) from both wild and mutant strains of MTB were identified by Multiple Sequence Alignment. The mutation in the specific residue was identified and represented in Fig. 1. MSA results indicated that the transitions of amino acids occur between susceptible and MDR strain. The result shows how the array of amino acids is changed based on the “R” group and “Polarity”, which actually effect the folding pattern of those particular proteins (Supplementary 1 Table A, B, C). In case of RpoB and InhA mutational blocks indicate that most of the substitution occurred in their drug binding pockets. MSA block of EmbC shows subtle differences in the drug-binding active site.

Block shows multiple sequence alignment of RopB, InhA, EmbC containing susceptible/wild type and resistant or mutant proteins found with maximum substitution
Molecular docking study
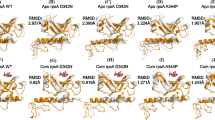
Wild and mutant forms of RpoB, InhA, EmbC proteins isolated from sequential years were modeled and docked with different ligands like RIF, INH and EMB respectively. RpoB1 (RpoB,1994) from a wild type was docked with rifampicin, which had a total six major interactions in its active site at SER428, PHE433, HIS674, and HIS445; ILE491 and LEU452 residues by hydrogen and sigma bonding (Supplementary 2 Table D). The Docking results of the mutant strains from the years 2012, 2014, and 2017 indicated some unusual bonding where the rifampicin did not well fit into the hydrophobic pockets of RpoB through hydrogen bonding or by some pi-sigma bonding (Fig. 2). But in the mutant isolates from the year 2013, 2015, and 2016 rifampicin was exceptionally good fit with hydrophobic pockets of RpoB compared to the previous years. Mutated RpoB from the isolates of 2012 showed the interactions at SER431, ARG16, PHE433, and ASN673. The binding energy is extended from −2.5 to −4.0 kcal/mol, when rifampicin interacts with RpoB protein of the mutant and wild strain, respectively.

3D and 2D interaction images representing the docked complexes of wild type RpoB including mutant proteins with Rifampicin (RpoB1:wild and RpoB2-7:mutants)
Docking calculations help to recognize the hypothetical binding mode of EmbC with ethambutol. The complex structure of arabinosyl transferase C and ethambutol showed the conserved interaction with the residues (ASN 21, LEU 329) in the binding site (Fig. 3). The binding energy of ethambutol with EmbC protein of mutant and wild strains ranges between −2.0 to −3.5 kacl/mol, respectively. The results of this theoretical molecular docking represent the higher binding affinity with each of the MDR strains of MTB. The docking of drug and receptor produced a more stable complexes by maintaining the conserved positions (GLY 14, SER 20 94, ILE 21 95 122 194, PHE 41 149, ALA 191, LYS 165, ASP 64, VAL 65, LEU 63) (Supplementary 2 Table E). The binding conformation of ethambutol with EmbC in both wild and resistance strains showed a stable complex. The drug binding pockets of RpoB and EmbC from wild and resistant strains remained unchanged for two decades. A resistance strain, “InhA3” does not show (Fig. 4) any significant binding affinity with isoniazid (Supplementary 2 Table F). InhA protein of the mutant and wild strains interact with isoniazid having the binding energy in between −3.3 to −3.5 kcal/mol. In this case the inhibition constant (ki) of InhA3 is lower than the susceptible strain.

3D and 2D interaction images representing the docked complexes of wild-type EmbC including mutant proteins with Ethambutol (EmbC2:wild and EmbC1 & 3:mutants)

3D and 2D interaction images representing the docked complexes of wild type InhA including mutant proteins with Isoniazid (InhA1:wild and InhA2 & 3:mutants)
Interactome
Access of all the nodes in a string database is required to build a functional network. Correlation of the network topology information for each drug regulatory protein was performed to analyze the degree of connectivity between all of them. The results lead to the interesting conclusion that the drug regulatory proteins are directly associated with the function of membrane embedded proteins. The robust information from these networks suggests that more than twenty correlative proteins contribute some positive role to evading drug susceptibility. The network topology of all these three regulatory proteins (RpoB, InhA, EmbC) displays (Fig. 5) that it mainly induces or suppresses the Fas and KasA regulatory proteins. The overexpression of these three proteins directly or indirectly stimulates the Fas to active other membrane bound transporter proteins. EmbC is the positive regulator of atfB which in-turn negatively regulates the Fas protein. Mutation in EmbC characterized from resistant strains apart from the drug binding pocket may results negative regulation of aftB (Fig. 5a) which could result in the over-expression of the Fas protein. The activated Fas protein may modulate structural changes in v1258c, v1634 and mmpl3; as well as mycolic acid production. In addition, the sequence substitution in InhA (Fig. 5b) and RpoB (Fig. 5c) (mutated) could also indirectly leads to the structural alteration of efflux pumps. The protein network of RpoB and InhA directly or indirectly activates the efflux pump system to escape from the effect of the drugs on the bacterial cell. It was also noted that EmbC promotes some force to change the peptidal architectural pattern of Rv1258c, Rv1634, and mmpl3 for activating the efflux pump system.

3D protein network of Embc (a), InhA (b), and Rpob (c) along with the other drug regulatory proteins representation with nodes. Positive interaction is represented in Green, Negative interaction in Red and moderate or no interaction is represented by Blue Color (see color figures online)
Visualization of architectural modification
The effect of residual substitution mutations on the function of efflux pump system was visualized. The protein family of MFS [18], SMR [18], and ABC [19] efflux pump contains mostly positive charged residues which are important to transport negative ions and anti-port positive charged ions. RMSD analysis presented that only Rv1258c, Rv1634, and Mmpl3 among these twenty regulatory transmembrane proteins showed the structural alteration (Fig. 6). The network study reveals that Rv1258c and Rv1634 change their structural conformation to deactivate influx and activate efflux mechanism by substituting charge residues. Although the structural change of mmpl3 commonly indicates the amino acid transition from charged to neutral residues (Supplementary 3 Table H). Mutant mmpl3 may indirectly monitor the expression of Rv1258c and Rv1634.The striking feature of these proteins is that it transits towards neutral to positive (Supplementary 3 Table G, I). The structural changes of three transporter proteins may accelerate the detoxification process of mycobacteria through drug evading efflux mechanism.

Comparison of wild with mutated structure based on their RMSD deviation. The color variation shows the structural conformation based on their charge residues (see color figures online)
Conclusion
A computational approach was employed to study the alternative metabolic protein profile which is associated with resistance of MTB to various drugs. MSA analysis of the major drug binding proteins suggests that the amino acid residues of RpoB change from polar to non-polar when acquiring resistance. In contrast, resistant InhA and EmbC (Fig. 1) showed non-polar to polar transitions. Amino acid substitutions that occur in these proteins are well regulated and position specific in course of evolution. In silico docking results showed that all the mutated strains were effective in binding perfectly to rifampicin (Fig. 2), ethambutol (Fig. 3) and isoniazid (Fig. 4), except that the mutated strains in the year 2015 showed no such binding affinity with isoniazid (Fig. 4). Therefore, the binding site of InhA3 needed a critical investigation because it had no effective bonding to INH in the resistant strain (POLMtbc.2951, 2015).
The protein networks of RpoB, InhA and EmbC show that they effectively alter the major efflux pumps of MDR-MTB. The overexpression of Fas is associated with the higher amount of mycolic acid production in resistant strains [23, 24]. Therefore, mycolic acid production may be correlated with structural alterations of Rv1258c, Rv1634 pump regulatory proteins which could play a key role in regulating drug transport. The influx mechanism of Mmpl3 is deactivated by changes in charged residues to neutral residues. Structural comparisons (Fig. 6) reveal that the transmembrane proteins, by substituting amino acids, increase efflux of the drugs, isoniazid, rifampicin & ethambutol. The findings in this study, could pave the way to identify new bioactive compounds [25] against these two efflux proteins for the treatment of tuberculosis.
References
WHO. Global tuberculosis control. https://www.who.int/entity/tb/publications/global_report/2011/gtbr11_full.pdf. (2011).
Wirth T, Hildebrand F, Allix-Beguec C, Wolbeling F, Kubica T, Kremer K, et al. Origin, spread and demography of the Mycobacterium tuberculosis complex. PLoS Pathog. 2008;4:e1000160.
Raviglione MC, Snider DE, Kochi A. Globalepide-tion elongation. Science 289, 619–625. miology of tuberculosis. Morb Mortal a Worldw epidemic JAMA. 1995;273:220–6.
Stop TB Partnership. End TB infographic: General facts and information. 2016. https://www.stoptb.org/events/world_tb_day/2016/assets/docs/EndTB_Infographic_GENERAL.jpg. Accessed 2016.
World Health Organisation. Global tuberculosis report 2016. 2016. https://www.who.int/tb/publications/global_report/en/. Accessed 20 Dec 2016.
Vester B, Douthwaite S. Macrolide resistance conferred by base substitutions in 23S rRNA. Antimicrob Agents Chemother. 2001;45:1–12.
Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, et al. A small-molecule nitroimi-dazopyran drug candidate for the treatment of tuberculosis. Nature. 2000;405:962–6.
Zhang Y, Telenti A. Genetics of drug resistance in Mycobacterium tuberculosis. In: Hatful GF, Jacobs WR Jr, editors. Molecular genetics of mycobacteria. Washington DC: ASM Press; p. 235–54. (2000).
Cavusoglu C, Hilmioglu S, Guneri S, Bilgic A. Characterization of rpoB mutations in rifampin-resistant clinical isolates of Mycobacterium tuberculosis from Turkey by DNA sequencing and line probe assay. J Clin Microbiol. 2002;40:4435–8.
Edwards KJ, Metherell LA, Yates M, Saunders NA. Detection of rpoB mutations in Mycobacterium tuberculosis by biprobe analysis. J Clin Microbiol. 2001;39:3350–2.
ElHajj HH, Marras SAE, Tyagi S, Kramer FR, Alland D. Detection of rifampin resistance in Mycobacterium tuberculosis in a single tube with molecular beacons. J Clin Microbiol. 2001;39:4131–7.
Guillermo M, Rodrigo T, Daniel M, Samuel N, Pran D, Marta B, et al. Docking studies of binding of ethambutol to the C-terminal domain of the Arabinosyltransferase from Mycobacterium tuberculosis. J Chem Hindawi. 2013;2013:1–5, https://doi.org/10.1155/2013/601270.
Forbes M, Kuck NA, Peets EA. Mode of action of ethambutol. J Bacteriol. 1962;84:1099–103.
Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101.
Hartmann G, Honikel KO, Knusel F, Nuesch J. The specific inhibition of the DNA-directed RNA synthesis by rifamycin. Biochim Biophys Acta. 1967;145:843–4.
Slayden RA, Barry CE III. The genetics and biochemistry of isoniazid resistance in Mycobacterium tuberculosis. Microbes Infect. 2000;2:659–69.
Somoskovi A, et al. Use of molecular methods to identify the Mycobacterium tuberculosis complex (MTBC) and other mycobacterial species and to detect rifampin resistance in MTBC isolates following growth detection with the BACTEC MGIT 960 system. J Clin Microbiol. 2003;41:2822–6.
Wu X, Zhang J, Zhuang Y, Zhang X, Li G, He X. Molecular mechanisms of drug resistance in Mycobacterium tuberculosis clinical isolates. Chin Med J. 1999;112:524–8.
Braibant M, Gilot P, Content J. The ATP binding cassette (ABC) transport systems of Mycobacterium tuberculosis. FEMS Microbiol Rev. 2000;24:449–67.
Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J Comput Chem. 2009;30:2785–91.
Iscla I, et al. A new antibiotic with potent activity targets MscL. J Antibiot. 2015;68:453–62.
DeLano WL. Pymol: an open-source molecular graphics tool. CCP4 Newsl Protein Crystallogr. 2002;40:82–92.
Cantrell SA, Michael DL, Olivera M, Anthony T, Julie AL, Lee WR. Regulated alteration of mycolic acid structure in the cell wall of Mycobacterium tuberculosis. Mycobacterial Dis. 2012;2:3. https://doi.org/10.4172/2161-1068.1000108.
Ying Y, Zhu Y, Deborah DC, Clifton EB. The effect of oxygenated mycolic acid composition on cell wall function and macrophage growth in Mycobacterium tuberculosis. Mol Microbiol. 1998;29:1449–58.
Mohamad S, Ismail N, Parumasivam T, Ibrahim P, Osman H, Wahab HA. Antituberculosis activity, phytochemical identification of Costus speciosus (J. Koenig) Sm., Cymbopogon citrates (DC. Ex Nees) Stapf., and Tabernaemontana coronaria (L.) Wild. And their effects on the growth kinetics and cellular integrity of Mycobacterium tuberculosis H37Rv. BMC Complement Altern Med. 2018;18:5.
Author information
Authors and Affiliations
Contributions
BB and both RB helped in designing experiments and analyzing the data; method development, material preparation and experiments performed by ASP, BB and AS interpretation of results; ASP wrote the manuscript with contributions from BB and both RB. All authors approved the manuscript before submission.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

{kind=link}
Cite this article
Panja, A.S., Sarkar, A., Biswas, R. et al. Modification of drug-binding proteins associated with the efflux pump in MDR-MTB in course of evolution: an unraveled clue based on in silico approach. J Antibiot 72, 282–290 (2019). https://doi.org/10.1038/s41429-019-0146-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41429-019-0146-3
- Springer Japan KK
This article is cited by
-
Alterations in molecular response of Mycobacterium tuberculosis against anti-tuberculosis drugs
Molecular Biology Reports (2022)