
Abstract
With the growing interest and demand in the realm of hyperbranched (hb) polymers, lot of synthesis approaches have already been explored some of which are detailed earlier in the Chap. 2. Both step-growth and chain-growth approaches are widely followed in the synthesis of hb polymers. Step-growth approaches mainly include AB x polycondensation and double monomer (symmetric/asymmetric pairs) methodologies. Whereas chain-growth approaches include radical copolymerization, surface grafting, and other controlled polymerization techniques. Interestingly, both self-condensing vinyl polymerization (SCVP) and self-condensing ring opening polymerization follow step-growth as well as chain-growth routes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others
3.1 Introduction to Simultaneous Step- and Chain-Growth Methodologies
With the growing interest and demand in the realm of hyperbranched (hb) polymers, lot of synthesis approaches have already been explored, some of which are detailed earlier in the Chap. 2. Both step-growth and chain-growth approaches are widely followed in the synthesis of hb polymers. Step-growth approaches mainly include AB x polycondensation and double monomer (symmetric/asymmetric pairs) methodologies. Whereas chain-growth approaches include radical copolymerization, surface grafting, and other controlled polymerization techniques. Interestingly, both self-condensing vinyl polymerization (SCVP) and self-condensing ring opening polymerization follow step-growth as well as chain-growth routes. In step-growth approaches, high DP is achieved only at high monomer conversions (as predicted from Carother’s equation). Hence, often hb polymers with high M.W cannot be obtained as high monomer conversions are avoided in practical cases, which otherwise would encourage gelation. On the contrary, in chain-growth approaches as the polymer chains grow via reactions of the growing chain ends with monomers, polymerization reaction completes in a reasonable time (unlike step growth) and thus ensures high DP/high M.W. However, chain-growth polymerization is highly random in nature which eventually broadens M.W.D of the resultant hb polymers. Hence, it would be highly desirable to perform chain-growth and step-growth polymerization simultaneously in order to ensure hb polymers with controlled properties (especially desirable for biomedical applications). This chapter deals with more recent and controlled synthesis strategies based on both chain-growth and step-growth approaches that are used in the development of hb polymers for biomedical applications.
3.2 Radical Polymerization
In the preparation of hb polymers via free radical polymerization (FRP), chain transfer to monomer, polymer, or other molecules is a guiding parameter and must be significantly high. Transfer to polymer is significant in ethylene and vinyl acetate systems, whereas transfer to monomer is popular with the acrylate systems.
Liu et al. reported synthesis of a hb polystyrene via FRP of styrene and a comonomer 4-vinyl benzyl thiol (which is a chain transfer, CT monomer) [1]. A CT monomer is constituted of a vinyl group and a telogen group (generally a thiol group). During a FRP, low M.W oligomers containing terminal double bonds are generated which subsequently act as macromonomers and allow further polymerization to limited chain lengths (due to telomerization of the telogen groups); Scheme 3.1. This process of the generation of macromonomers and subsequent FRP through telomerization is analogous to AB x polycondensation (which even follows Flory’s branching theory) and thus generates hb polymers [1]. In spite of the high potentiality of CT monomers in developing hb polymers, they are often discouraged. CT monomers are unstable and susceptible to self-condensation by Michael addition. And also the availability of CT monomer is highly limited. In this regard, usage of a balancing amount of a chain transfer agent (CTA); usually a long chained thiol, in the recipe of a vinyl polymerization reaction, facilitates transfer reactions and in turn encourages formation of the hb polymers from a myriad of vinyl monomers in the presence of a multivinyl monomer (MVM); Scheme 3.2. This concept gave birth to a new synthesis strategy, named as “Strathclyde methodology” which was introduced in the year 2000, by the gang of Sherrington from the University of Strathclyde, UK [2]. “Strathclyde methodology” is easy to follow and cost-effective which encourages commercialization of the hb polymers. Hb polymers from different vinyl monomers have already been prepared by following the “Strathclyde methodology” [3,4,5]. In fact, in our lab, we have successfully developed amphiphilic hb copolymers of acrylic acid and propargyl acrylate through the “Strathclyde methodology” and subsequently functionalized them through click reaction with a surfactant [6]. The resultant hb copolymer was capable of self-assembling from polymersomes to aggregates when pH of the medium was decreased from an alkaline phase to an acidic phase which facilitated encapsulation of both hydrophilic and hydrophobic molecules. Typically, in the recipe of “Strathclyde methodology”, MVM is basically a crosslinking agent. Incorporation of MVM into a FRP recipe produces crosslinked networks (as the number of crosslinks per primary chain exceeds unity even at a low concentration of MVM and at low monomer conversions). Depending upon the level of monomer dilution, FRP of a vinyl monomer in the presence of MVM either produces macrogelation (in concentrated solutions) or else causes microgelation (in dilute solutions). Hence, branched polymers are thought to be precursors to crosslinked polymers and the former can be isolated only at an infinite monomer dilution (which is a hypothetical case). One of the ways of preventing gelation and ensuring hb structures is by keeping the monomer conversion incomplete, so that the vinyl groups along the primary polymer chains are not fully consumed; some vinyl groups remain free. However, in a FRP even if controlled, it is impossible to avoid intramolecular cyclization which causes consumption of the free vinyl groups along the primary polymer chains. Hence, pure branched polymers could never be obtained through “Strathclyde methodology” but are popular only due to cost-effectiveness of the process. In this regard, some research groups employed asymmetrical MVMs for the synthesis of hb polymers with pendant vinyl groups [7, 8]. However, again, this approach lacks versatility owing to the limitations in the design and availability of the asymmetrical MVMs. In a MVM system, a critical overlap polymer chain concentration c* determines the nature of the dominant interactions [9]. When the concentration is below c* then intramolecular cyclization is favored whereas above c* intermolecular branching is favored. Raising the primary polymer chain concentration (say by reducing solvent proportion) increases the risk of gelation. In this respect, “Strathclyde methodology” is highly useful as it helps in maintaining a desirable primary polymer chain length to ensure hb structures through controlled chain transfer reactions by thiols. Typically, in a recipe for “Strathclyde methodology”, concentration of MVM should be maintained below 15% and molar ratio of MVM to initiator should not exceed 1 in order to prevent gelation. Unfortunately, due to randomness of the “Strathclyde methodology” (nonliving nature) which adversely affects the properties of the hb polymers, due to usage of toxic thiols/MVMs and also due to lack of variety in desirable functionalities, this approach is hardly followed to synthesize hb polymers for biomedical applications. Due to the nonliving nature of the “Strathclyde methodology”, i.e., occurrence of irreversible chain transfer reactions, near-diffusion controlled radical coupling and disproportionation, hb polymers with broad M.W.D, and uncontrolled/low DB are produced. However, recently, attractive post-polymer functionalization (say via click reaction) is attracting further research in the development of “Strathclyde methodology” in spite of the shortcomings of the process.

Schematic representation of the generation of a hb polymer from a CT monomer. Reprinted (adapted) with permission from Liu et al. [1]. Copyright (2008) Wiley Online Library

Schematic representation of “Strathclyde methodology” for the synthesis of a hb polymer. Reprinted (adapted) with permission from O’Brien et al. [2]. Copyright (2000) Elsevier
In many instances, apart from using thiols, transition metal catalysts (generally low spin cobalt (II) complexes like cobaloxime) have also been used to control chain transfer reactions in FRP for the generation of hb polymers [10]. In fact, catalytic chain transfer polymerization (CCTP) is an extension of “Strathclyde methodology”. CCTP or rather “cascade polymerization” forms macromonomers, i.e., polymers carrying vinyl ω-end groups, which eventually facilitate post-polymerization end group modification. In general, CCTP follows a two-step radical process; Scheme 3.3 [11]. In the first step, Co(II) complex abstracts a ß-hydrogen atom from the propagating radical which forms a Co(III)-H complex and a dead polymer chain containing a vinyl ω-end group. The hydrogen abstraction by Co(II) complexes occurs via Co···H···C transition state. The ß-hydrogen abstraction occurs either at an α-methyl position (in case of methacrylates, α-methyl styrene, methacrylonitile, etc.) or at the backbone (for acrylates, styrenics, acrylonitrile, etc.). It has been found that, hydrogen abstraction at the α-methyl position occurs readily and thus monomers containing α-methyl groups are highly active for the CCT reactions. Finally, in the next step, Co(III)-H complex reacts with the monomer to form back an active Co(II) complex and a monomer radical which is capable of propagation. In this type of polymerization, branching topology is maintained through the competition between propagation and chain transfer reactions. Guan in his novel work used “cascade polymerization” of ethylene glycol dimethacrylate (EGDMA, 1), in the presence of cobalt oxime boron fluoride (COBF) catalyst, to develop hb PEGDMA, 2; Scheme 3.4 [10]. The homopolymerization of EGDMA resulted in repetitive trimerization of the dimethacrylates which eventually formed the hb polymers. In another work, Smeets et al. prepared hb polymers via Co(II) mediated emulsion copolymerization of MMA and EGDMA [12]. They studied how the physiochemical properties of Co(II) complex, the intrinsic chain transfer activity, and the partitioning behavior played an important role in the development of desirable polymer architecture (branched or crosslinked). There are few advantages of CCTP over the “Strathclyde methodology” which include (1) better control in the polymerization conditions, thereby yielding hb polymers with narrow P.D.I/high DB and (2) presence of significant number of pendant vinyl groups which can be functionalized to desirable groups in the further steps. In case of the “Strathclyde methodology”, pendant vinyl groups often undergo organic reactions with thiols and thus their availability decreases [12].

Schematic representation of a catalytic cycle for Co(II) mediated CCT reaction. Reprinted (adapted) with permission from Smeets [11]. Copyright (2013) Elsevier

Scheme showing the synthesis of a hb polymer via CCT reaction of EGDMA. Reprinted (adapted) with permission from Guan [10]. Copyright (2002) American Chemical Society
Another variation of the “Strathclyde methodology” for the synthesis of hb polymers is initiator fragment incorporation radical polymerization (IFIRP) which has been extensively explored by the group of Sato [13,14,15,16]. FRP of divinyl monomer (DVM) generates crosslinked polymers which are believed to exhibit very high or infinite M.Ws. In a conventional FRP, as the concentration of the initiator increases, M.W of the resultant polymer decreases due to faster chain termination. This fact led to the belief that if the initiator concentration in a FRP of DVM system is kept very high such that M.W decreases dramatically which eventually produces soluble hb polymers. This synthesis approach is named as initiator fragment IFIRP because around 30–40 mol% of the initiator fragments get incorporated into the primary polymer chains through initiation and primary radical termination reactions. In one of the early works, Sato et al. developed organic solvent soluble hb copolymers from concentrated divinyl benzene, DVB and ethylstyrene (EtSt), in the presence of high concentrations of an initiator and a retarder [13]. In the following year, the group of Sato further developed soluble hb polymers from a set of EGDMA and N-methylmethacrylamide [14] and another set of EGDMA and α-ethyl β-N-(ά-methylbenzyl) itaconamate [17] via IFIRP, in the presence of high concentration of an initiator. Later Sato and his team further explored IFIRP to synthesize hb polymers from different DVM systems (like DVB, EGDMA and DVA) as compiled in the works of Tai et al. [18]. IFIRP is quite an interesting approach to develop hb polymers for biomedical applications due to the absence of any harmful trace materials. However, IFIRP may be a bit expensive due to the high cost of the initiators.
3.3 Proton Transfer Polymerization
One of the well-recognized methods for the successful generation of hb polymers, especially intended for biomedical applications is proton transfer polymerization (PTP). Chang and Frechet introduced the concept of PTP, which proceeds via proton transfer reaction in each propagation step [19]. They provided a mechanistic pathway for a typical PTP employing an AB x -type monomer; Scheme 3.5. Initially, a catalytic amount of an initiator (an anion) abstracts a proton from a H-AB2-type monomer and generates a reactive nucleophile. The reactive nucleophile further reacts with another AB2 molecule to form a dimer in which one of the B groups act as a nucleophile for the next molecule. The active dimer undergoes a proton transfer reaction with another H-AB2 monomer instead of a propagation reaction because the former is thermodynamically more favorable. Finally, a polymer is formed through subsequent nucleophilic addition and proton transfer reactions. Owing to the multiplicity of the reactive B species in each of the growing molecule, branching is ensured during polymerization. PTP is successful only when the activation of AB2-type monomer or H-A groups of the growing species is significantly faster than the propagation of nucleophile. PTP can be followed using either AB x -type monomers or A2+B3 monomer systems. Generally, PTP is popular with addition and ring opening reactions.

Scheme showing the step-by-step mechanism for the synthesis of a hb polymer via PTP. Reprinted (adapted) with permission from Chang et al. [19]. Copyright (1999) American Chemical Society
The first notable application of PTP was followed in the synthesis of hb poly (hydroxyether)s from an AB2-type monomer constituting of two epoxide moieties and one phenolic hydroxyl group [19]. The polymerization proceeded via a step-growth route where deprotonation of the phenol was succeeded by nucleophilic attack onto the epoxide ring. Ring opening of the epoxide groups generated an active dimer constituting of a secondary alkoxide anion. In the subsequent step, the activated dimer underwent a proton transfer reaction with another AB2 molecule to regenerate a phenolate anion and a neutral anion. In this reaction, Chang and Frechet maintained the pKa value of the phenolic group around 10 whereas that of the alkoxide anion was around 17, so that a rapid proton transfer from the phenolic group to the secondary alkoxide group was exclusively possible. The resultant hb poly (hydroxyether)s exhibited a M.W around 2.06 × 105 g mol−1. In another work, Gong and Frechet developed epoxy terminated hb polyesters, 4 from an AB2-type monomer, 3 via PTP; Scheme 3.6 [20].

Scheme showing the synthesis of a hb polyester via PTP of an AB2-type monomer. Reprinted (adapted) with permission from Gong and Frechet [20]. Copyright (2000) American Chemical Society
Chen et al. synthesized a temperature-responsive hb polyether via PTP between 1,2,7,8-diepoxyoctane (DEO) and multiols like ethylene glycol, diethylene glycol, triethylene glycol, 1,2-propane diol and glycerol [21]. The LCSTs of these polymers were tuned in the range of 23.6–67.2 °C just by adjusting the hydrophilic/hydrophobic balance of DEO and multiols, the feed ratio of DEO to multiols and M.Ws. It is worthy to mention at this point that the temperature-responsive polymers are highly useful in drug delivery, separation processes, and sensing applications [22].
In a recent work, Gadwal et al. developed a polythioether-based hb polymer, 6 via base catalyzed PTP of an AB2-type monomer, 5 constituting of a thiol and two epoxides groups; Scheme 3.7 [23]. The hb polymer 6 exhibited DB around 0.65–0.69. The hb polymer 6 contained two reactive sites; hydroxyl units and epoxide units which were distributed throughout the branched structure. The epoxide rings were capable of attaching a lipophilic alkyl, aryl (known for excellent membrane penetration properties), or ethylene oxide group (for excellent stealth properties) through thiol-epoxy reactions while the hydroxyl groups could be attached to the positively charged primary ammonium groups (known for efficient complexation with siRNA molecules). Post-polymer functionalizations of the dual reactive hb polymer 6 yielded dual functionalized hb polymers with high potentiality in gene delivery applications.

Scheme showing the synthesis of a polythioether-based hb polymer via PTP of thiol and epoxide groups. Reprinted (adapted) with permission from Gadwal et al. [23]. Copyright (2014) American Chemical Society
As already mentioned that PTP is explored in A2+B3 synthesis strategies, Emrick et al. developed a hb aliphatic polyether, 8 via PTP of 1,2,7,8- diepoxyoctane (A2) and 1,1,1- tris (hydroxymethyl)ethane (B3) at 120 °C, in the presence of tetra-n-butylammonium chloride as a nucleophilic catalyst; Scheme 3.8 [24]. The hb polymer 8 exhibited a M.W of 15,000 kDa. In another work, Ma et al. synthesized epoxy terminated hb polymers via A2+B3 based PTP, from bisphenol-A (A2) and trimethylolpropane triglycidyl ether (B3) [25]. From the reported studies, it is clear that PTP is quite a welcome approach in the generation of hb polymers as most of them proceed via addition or ring opening reactions for which numerous peripheral functional groups could be included into the structures (for modification to attract biomedical applications).

Scheme showing the synthesis of hb aliphatic polyether via A2+B3 based PTP. Reprinted (adapted) with permission from Emrick et al. [24]. Copyright (1999) American Chemical Society
3.4 Self-Condensing Vinyl Polymerization/Copolymerization, Self-Condensing Ring Opening Polymerization and Controlled/Living Polymerization
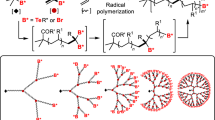
The polymerization of vinyl monomers is still considered as one of the important organic reactions for mass scale productions. However, vinyl monomers cannot be polymerized via AB x polycondensation. Conventional radical polymerization may produce hb polymers (if chain length is maintained below a critical point) but they exhibit uncontrolled topologies and properties. For biological and biomedical applications like drug delivery, tissue engineering, bioimaging, etc., functional polymers with controlled architectures and properties are essential. One such important functional polymer from nature is protein. Surprisingly, protein is an example of highly stereospecific polymer of amino acids, which exhibits controlled polymer compositions, functionalities, chemical properties, M.W, and uniform M.W.D. Inspired from nature, attempts have been made to develop polymers with as much control in the macromolecular engineering as possible. Hence, in order to generate hb polymers (with controlled structures) from vinyl monomers, a new category of reaction named as SCVP was introduced under the guidance of Frechet [27]. SCVP employs AB-type initiator monomers (later called inimers). These monomers possess both initiation and propagation properties. In a typical AB-type monomer, A is a vinyl group and B is a pendant group that can be converted into an initiating center B* by the activation of B group in the presence of an external stimulus (Lewis acid or light). The generated B* is capable of reacting with the vinyl groups of another AB molecule to facilitate the propagation reaction. The first reaction of AB* with a double bond group of another AB molecule generates a dimer which acts an AB2-type intermediate due to the presence of two active initiating sites (A*, B*) and one double bond. Both B* (initiating site) and A* (propagating site) can further react in a repetitive fashion to form three-dimensional polymers, rich in end groups and active centers. Hence, SCVP follows two modes of polymerization––polymerization of double bonds (chain growth) and condensation of the initiating group with double bonds (step growth); Scheme 3.9. Earlier SCVP was believed to follow Flory’s branching theory of equal reactivity as that is followed in AB x polycondensation. However, in recent studies it has been found that in SCVP, the kinetic activities of the chain propagation of the growth sites and the initiating sites differ significantly, which results in a lower DB than those obtained from an AB x polycondensation reaction. Till date, the theoretical maximum DB of a SCVP reaction is 46.5% [28]. In SCVP, often side reactions like chain transfer or recombination reactions cause crosslinking/or gelation and broaden M.W.D of the resultant hb polymers. Thus, as a remedy, living/controlled polymerization techniques like cationic, anionic, group transfer polymerization (GTP), nitroxide mediated radical polymerization (NMRP), atom transfer radical polymerization (ATRP), reversible addition fragmentation chain transfer polymerization (RAFT), and photo-initiated radical polymerization techniques are preferred. The first SCVP approach as reported by Frechet et al. was based on cationic polymerization where they used 3-(1-chloroethyl)-ethenylbenzene as an AB-type monomer [27]. They employed SnCl4 (as a Lewis acid) in CH2Cl2 at −15 or −20 °C, in the presence of tetrabutylammonium bromide to carry out the cationic polymerization. In this study, they found that M.W-time profile resembled that of the polycondensation process; a slow initial increase in M.W was followed by an exponential growth with time. The resultant hb polymer exhibited a M.W of 2.5 × 105 g mol−1 and a P.D.I of 6. Later, SCVP was developed using living cationic copolymerization. Paulo and Puskas synthesized hb polyisobutylenes via living cationic SCVCP of 4-(2-methoxyisopropyl)styrene(p-methoxycumylstyrene) as an AB-type monomer and isobutylene, in the presence of TiCl4 (a co-initiator) in 60:40 mixture of methylcyclohexane and methyl chloride at −80 °C [29]. The obtained hb polymer had a M.W of 8 × 105 g mol−1 and a M.W.D of 1.2.

Schematic representation of the synthesis of a hb polymer via SCVP of an AB-type monomer. Reprinted (adapted) with permission from Zhao [26]. Copyright (2015) Research Repository UCD (http://researchrepository.ucd.ie/handle/10197/6852)
Other variations like SCVP-based anionic polymerization or rather living anionic SCVCP are also followed. Knauss et al. prepared hb polystyrene via living anionic polymerization in the presence of vinylbenzyl chloride (VBC-a coupling agent) and nBuLi; Scheme 3.10 [30]. The synthesis procedure proceeded by slow addition of a chain-growth promoter (constituting of a polymerizable vinyl group and a functionality capable of quantitatively coupling with the living chain end). The addition of a chain-growth promoter to a living chain forms macromonomer which subsequently undergoes addition reactions to generate hb polymers. In another work, Baskaran reported synthesis of a hb polymer via living anionic SCVCP of a vinyl monomer, 1,3-diisopropenylbenzene (in the presence of nBuLi the vinyl monomer formed an AB-type monomer in situ) and a chain-growth promoter, styrene [31]. He observed that addition of styrene in the intermittent stages, during the polymerization reaction, increased M.W of the resultant hb polymer and decreased Mark–Houwink parameter, α significantly. In fact, low α values indicate densely packed three-dimensional structures. Recently, Young et al. developed vinyl functionalized hb polymers via living anionic SCVCP of allyl methacrylate and hydroxyethyl methacrylate. They showed that the presence of free vinyl pendant groups along the polymer backbone could be further functionalized according to needs (say for biomedical applications). Examples of living anionic SCVCP are rare due to instability of the anionic AB-type monomers. The method of slow addition of a stoichiometric quantity of a chain-growth promoter to the living polymer chain facilitates anionic SCVCP in few cases. Another disadvantage of anionic SCVCP is that the polymerization has to be carried out at sub-ambient temperature (below 0 °C). However, anionic SCVP/SCVCP is still considered as a challenging approach. In another variation, GTP technique as introduced by Dupont in the year 1983 is considered as one of the startling procedures to develop architectural polymers (block, graft, hb polymers etc.). GTP uses a trimethylsilyl ketene acetal initiator which is catalyzed by metal free nucleophilic anions. GTP is basically a “quasi-living” oxyanionic polymerization technique (generally follows Mukaiyama–Michael addition reactions), used for the controlled polymerization of α, ß-unsaturated carbonyl compounds. Mukaiyama–Michael addition reaction is a carbon–carbon coupling reaction which ensures high stereo selective control. GTP proceeds by repetitive transfer of a reactive silyl group from a propagating chain end to the monomer and thus produces a living end. Though, in later studies, it was proven that this transfer mechanism is inappropriate. According to new findings, silyl ketene acetals and a Lewis acid/base catalyst should coexist in the reaction medium which actually features group transfer reactions. GTP produces living polymers with controlled topologies/narrow M.W.D/high DB, free of termination or chain transfer reactions just at room temperature and under mild reaction conditions. Recombination terminations are avoided in GTP as trialkylsilyl capped propagating chain ends are electrically neutral. Due to the suppression of undue side reactions, GTP is favored to polymerize functional methacrylates which bear reactive side groups like epoxides, dienes, vinyls, allylics, etc., which otherwise are highly sensitive to the conditions prevailing in conventional radical and ionic polymerization routes. In literature, there are a lot of examples concerning the synthesis of star-branched polymers (i.e., a polymer with branches radiating from a core) via GTP [32]. GTP is quite often explored with the inimer type monomers to generate hb polymers (follow Scheme 3.11). Simon et al. synthesized hb polymethacylates (hb PMMA) via self-condensing group transfer copolymerization (SCGTCP) of an inimer 2-(2-methyl-1-triethylsiloxy-propenyloxy)ethyl methacrylate and MMA, in the presence of a Lewis base catalyst tetrabutylammonium bibenzoate at room temperature [33].

Scheme showing the synthesis of hb polystyrene via living anionic SCVCP. Reprinted (adapted) with permission from Knauss and Al-Maullem [30]. Copyright (2000) Wiley online library

Schematic representation of the synthesis of hb polymethacylates via SCGTCP catalyzed by Lewis base. Reprinted (adapted) with permission from Hadjichristidis and Akira [34]. Copyright (2015) Springer publishing house (Japan)
In later studies, Simon and Muller further explored SCGTCP with 2-(2-methyl-1-triethylsiloxy-propenyloxy)ethyl methacrylate and MMA/tBMA in the presence of tetrabutylammonium bibenzoate [35]. In the conventional living ionic polymerization techniques (cationic or anionic), often there is incompatibility between the growing chain ends and the different types of functional groups which subsequently restrict controlled polymerization of many functional vinyl monomers. Further, a living ionic polymerization demands inert atmosphere, exclusion of by products like water during a reaction and usage of extra pure reagents/dry solvents. These complications of living ionic polymerizations restrict their applications in many instances. Hence, it was necessary to develop polymerization techniques which combine the desirable features of both conventional FRP and ionic polymerization. Attempts to develop living radical polymerization systems in the earlier years included usage of iniferters (initiator-transfer agent-terminator) [36]. The basic concept of this approach was reversible termination of the growing polymer chains. However, polymerization carried out in the presence of iniferters yield polymers with broad M.W.D and poor properties (due to very low M.W). In the later years, while searching for various radical trappers (for studying the detailed chemistry of initiation in radical polymerization), nitroxides (like TEMPO and its derivatives) were found to combine with carbon centered radicals to form alkoxyamines and thus were considered as excellent initiator inhibitors. However, nitroxides either do not react or react reversibly with oxygen centers (tert-butoxy, benzoyloxy, etc.). Alkoxyamines (C–ON bonds) are thermally labile (typically at 80 °C or above) and undergo reversible dissociation. This concept was utilized to give birth to a new type of controlled radical polymerization technique based on alkoxyamine chemistry. In fact, with the further advancement in the research of living polymerization techniques, NMP first came into existence as a breakthrough.
The term NMP was introduced by an eminent scientist Ezio Rizzardo of Commonwealth Scientific and Industrial Research Organization, Australia. Moad from Rizzardo’s group introduced the concept of reversible end capping of the propagating chain ends by TEMPO [38]. Typically, during NMP, at first C–ON bonds are formed at the end of the propagating polymer chains which break homolytically under the polymerization condition (in the following steps) to regenerate free nitroxide radicals and polymeric radicals. The polymer radicals extend the growth of the chains through normal propagation reactions and thus DP increases. Again, the polymer radicals (with higher DP) recombine with nitroxide radicals. This cycle of homolysis/monomer addition/recombinations repeats to maintain a living nature of the growing polymer chains. At any instance of NMP, there is a significant presence of inactive polymer chain ends, which substantially decrease the overall concentration of the reactive polymer chain ends and thus reduce the unwanted side reactions like bimolecular radical-radical termination, disproportionation, combination, or cyclic reactions. Moreover, the rate of cross reactions between nitroxide radicals and carbon centered radicals are much faster than the homo coupling reactions between the consecutive carbon-centered radicals [39]. These phenomena of “persistent radical effects” (PRE) enable the polymer chains to grow in a controlled manner. The equilibrium step in NMP is shown in Scheme 3.12. In the realm of hb polymers, NMP has been mostly explored through SCVP techniques. The AB-type monomer that is used in SCVP-based NMP constituted of a polymerizable double bond (A) and an initiating alkoxyamine group (B). In a novel work, Hawker et al. homopolymerized an AB-type styrenic monomer functionalized with an alkoxyamine initiating group, 9 at 130 °C in 72 h, to a hb polymer 10 without any gelation; Scheme 3.13 [40]. The resultant hb polymer exhibited a M.W around 6000 g mol−1 and a P.D.I of 1.4. Interestingly, they further utilized the hb polymer as a macroinitiator for a second step chain extension to develop a hyperbranched-star (hyperstar) polymer with M.W around 3 × 106 g mol−1 and a P.D.I of 4.35.

Schematic representation of activation–deactivation equilibrium in NMP. Reprinted (adapted) with permission from Gigmes [37]. Copyright (2015) Royal society of chemistry

Khan et al. discovered that the presence of pendant polymerizable groups along the polymer backbone may be useful in generating resist materials for UV-imprint lithography; UV-IL applications [42]. The group prepared different hb polymers from a variety of mono- and di-functional monomers using NMP. One of the hb polymer grades was prepared by copolymerization of styrene and a di-functional methacrylate (1,6-hexanediol dimethacrylate) mediated by TIPNO. Using this concept, NMP may be extended to generate hb polymers suitable for biomedical applications just by functionalization. Unfortunately, there is limited reports on the synthesis of hb polymers via NMP-SCVP due to slow polymerization kinetics that require high polymerization temperatures (often 120–145 °C)/lengthy polymerization duration (24–72 h), incompatibility of nitroxides with various functional groups of the vinyl monomers (say in case of MMA) and multistep synthesis of alkoxyamine-based inimers [39]. Additional disadvantages of NMP include homolysis of N–O bonds instead of C–ON bonds, chain transfer to solvent, loss of nitroxide radicals, oxidation of alkyne bearing monomers and high costs of the nitroxide initiators [39]. To overcome these shortcomings, researchers are altering the structures of nitroxides [43]. Unlike initiating radicals, nitroxide radicals are mediating radicals that are thoroughly involved in several activation/deactivation steps. Hence, the structure of the nitroxides definitely puts a significant effect on the polymerization. However, structurally altered nitroxides have still not been used to generate hb polymers and this leaves enough scope for exploration. Rather, other living polymerization techniques like ATRP and RAFT were developed to prepare hb polymers with controlled topologies and properties.
In recent times, ATRP is considered as one of the dominating polymerization techniques to prepare polymers with predictable structures and highly targeted M.W ranges. As the name suggests, atom transfer (from an organic halide to a transition metal complex) is the key step in the reaction responsible for controlled polymer chain growth. From the mechanistic point of view, ATRP resembles radical addition of R-X across an unsaturated C–C bond which is commonly known as atom transfer radical addition (ATRA) reaction. In ATRP, a dynamic equilibrium is maintained between the propagating radicals (R· or P ·n ) and the dormant species (alkyl halides or pseudo halides; R-X or dormant propagating chain ends; R-Pn-X) [45]. Typically, in ATRP, radicals are generated from a halide through a reversible redox reaction, catalyzed by a redox-active, low oxidation state transition metal complex (M zt -Ln where M zt denotes the metal atom or ion in oxidation state z and Ln denotes a ligand in the counter ion form). In this activation step, alkyl halogen bond (R-X) is reversibly and homolytically cleaved by M zt (activator, in the lower oxidation state) to generate an alkyl radical (R·) and M (z+1)t (deactivator, in the higher oxidation state) via an inner sphere electron transfer process. In the subsequent step, R· either attacks vinyl monomers to generate P ·n or is reversibly deactivated by X-M (z+1)t Ln. X-M (z+1)t Ln being a deactivator cleaves heterolytically, then rapidly transfers the X group back to the radicals and transform the radicals into dormant species (Pn-X). Actually, transition metal complexes are highly effective in transferring halogens than R-X. Activation and deactivation steps repeat throughout the polymerization reaction until all the vinyl monomers are consumed. Termination reactions occur by radical coupling and disproportionation. However, the termination step is suppressed to a minimum, but not eradicated, in a well-controlled ATRP. Due to PRE, when the polymerization progresses, the termination step is slowed down significantly as at any instance, the equilibrium is shifted towards the dormant species, i.e., k act ≪ k deact (where k act and k deact are the rate constants for the activation and deactivation steps, respectively). Mechanistically, X-M (z+1)t Ln accumulates due to PRE and maintains a very low concentration of the propagating radicals. This phenomenon reduces the probability of bimolecular termination of the propagating radicals and prolongs the propagation step which eventually produces polymers with high M.W and uniform M.W.D. And also, topology of the polymer is well controlled in ATRP due to the absence of unwanted side reactions. The entire mechanism of ATRP is shown schematically in Scheme 3.14. So far, the most commonly used transition metal complex in protic medium ATRP is those of copper (due to two oxidation states Cu(I) and Cu(II)). The catalyst consists of Cu(I) halide accompanied by a nitrogen donor based complexing ligand. The ligands play a key role in solubilizing the catalysts and determining the redox potential of the catalysts which eventually guide the shifting of the equilibrium in ATRP. The synthesis of hb polymers using ATRP is often extended with SCVP [46]. For ATRP-SCVP, B group must carry a halogen atom that is capable of initiating through a reaction with the copper catalyst.

Schematic representation of the mechanism of ATRP. Reprinted (adapted) with permission from Patten and Matyjaszewski [44]. Copyright (1998) Wiley online library
It was for the first time Gaynor et al. developed a hb polymer 12 from p-(chloromethyl) styrene (11, an AB-type monomer) via ATRP-SCVP; Scheme 3.15. In the following year, Matyjaszewki et al. synthesized hb polyacrylates via ATRP-SCVP of an AB-type monomer, 2-((2-bromopropionyl)oxy)ethyl acrylate (BPEA) [49]. Interestingly, Muthukrishnan et al. synthesized hb glycopolymers via ATRP-SCVCP of BPEA, 13 and a sugar carrying acrylate, 3-O-acryloyl-1,2,5,6-di-O-isopropylidene-α-D-glucofuranoside (AIG1c)- 14, followed by deprotection of isopropylidene protecting groups in the final step; Scheme 3.16 [50]. These polymers have potential prospects in biological, pharmaceutical, and medical applications due to multiple binding sites.


Scheme showing the synthesis of hb glycopolymers via ATRP-SCVCP. Reprinted (adapted) with permission from Muthukrishnan et al. [50]. Copyright (2005) American chemical society
In most cases, three types of AB-type monomer based on polymerizable vinyl groups are explored in ATRP-SCVP which includes acrylate inimers, methacrylate inimers, and styrenic inimers. Amin and Gaffar synthesized hb polyamides, 16 via ATRP-SCVP of an AB-type monomer, 15 designed from p-amino phenol; Scheme 3.17 [51]. Tsarevsky et al. prepared degradable hb polymers with multiple alkyl halide chain ends via ATRP-SCVP of inimers containing esters (2-(2′-bromopropionyloxy)ethyl acrylate) or disulphides (2-(2′-bromoisobutyryloxy)ethyl 2″-methacryloyloxyethyl disulfide) groups [52]. It is worthy to mention that degradable polymers are highly demanded in biomedical applications.

Scheme showing the synthesis of hb polyamide via ATRP-SCVP. Reprinted (adapted) with permission from Amin and Gaffar [51]. Copyright (2008) Taylor and Francis online
The major disadvantage of ATRP in the design of hb polymers for successful biomedical applications is the usage of copper catalysts in high concentrations (~0.1–1 mol% with respect to the monomers). One cannot reduce the amount of Cu species in the initial recipe because the bimolecular termination reactions consume activators and the polymerization does not proceed to completion. Removal of copper contaminants from the system is difficult and expensive (say by extraction, precipitation, immobilization, or by using biphasic systems) [53]. However, a lot of efforts have been put to reduce copper contamination in order to prevent ATRP from dying. Usage of iron catalysts instead of copper catalysts is often favored in biomedical applications due to low toxicity and biocompatibilty. However, iron catalysts are not so efficient like copper catalysts and are quite expensive. An effort was made to diminish the concentration of copper catalysts by the employment of activators regenerated by electron transfer (ARGET) process [53]. Typically in ARGET process, a reducing agent like Sn(II) 2-ethylhexanoate; Sn(EH)2, ascorbic acid, or sugars like glucose (which are generally FDA approved) reduce the accumulating Cu(II) species by transforming them to Cu(I) species. This continuously regenerates activators and thus reduces the catalyst concentration in the initial recipe. In fact, ARGET process can reduce the catalyst concentration in an ATRP recipe to 50 ppm [54]. Interestingly, these redox processes do not generate initiating radicals or initiating species and thus pure copolymers can be synthesized. Another effort is the employment of initiators for continuous activator regeneration (ICAR) process in ATRP-SCVP. Typically, in an ICAR process, conventional initiator radicals (say AIBN) reduce the accumulating Cu(II) species to Cu(I) species. This process definitely diminishes copper loading and avoids the addition of external reducing agents. However, care should be taken that the decomposition of the selected free radical initiator must be sufficiently low in order to discourage bimolecular termination. Both ARGET and ICAR processes leave enough scope for exploration to produce biologically friendly hb polymers by ATRP for commercialization [55]. With the advancement in ATRP, in the most updated cases, in the synthesis of hb polymers, Cu(0) mediated ATRP has also gained impetus. For the first time, Matyjaszewki et al. synthesized hb polyacrylates via Cu(0) mediated ATRP-SCVP [56]. Actually, in SCVP, due to high initiator concentration, often a shift in the equilibrium towards the active radicals is favored. This increases the concentration of Cu(II) deactivator species in the system which eventually prevents polymerization. Cu(0) facilitates reduction of Cu(II) to Cu(I). Cu(I) species generated by Cu(0) are highly active and thus reacts with ATRP initiators so rapidly that the disproportionation to regenerate Cu(0) and Cu(II) species gets suppressed significantly. This approach is indirectly used to activate dormant halide (pseudo halide) species via an inner sphere electron transfer process and is thus named as supplementary activators and reducing agents ATRP (SARA-ATRP); Scheme 3.18. SARA-ATRP also diminishes amounts of the copper catalysts in the initial recipe and improves oxygen tolerance of the catalysts which is often a critical issue in the commercialization of ATRP. To address the issues of toxicity of copper catalysts, Ag(0) and Fe(0) have also been implemented in SARA-ATRP [55]. The mechanism as explained in SARA-ATRP is often a controversial issue. There is a complementary mechanism in Cu(0) mediated ATRP named as single electron transfer living radical polymerization (SET-LRP); Scheme 3.18. In SET-LRP, Cu(0) instead of Cu(I) single-handedly activates dormant halide (pseudo halide) species via an outer sphere electron transfer process. At any instance during a polymerization, Cu(I) spontaneously disproportionates into Cu(0) and Cu(II) as the reaction has a very low activation energy.

Schematic representation of SARA-ATRP and SET-LRP. Bold arrows indicate major reactions, solid arrows indicate supplementary reactions, and dashed arrows indicate minor reactions. Reprinted (adapted) with permission from Konkolewicz et al. [57]. Copyright (2014) Royal society of chemistry
Xue et al. synthesized hb poly (methyl acrylate)-block-poly (acrylic acid)s; HBPMA-b-PAAs via SET-LRP technique [58]. HBPMA-b-PAAs spontaneously formed unimolecular micelles, constituting of hydrophobic cores (PMA) and hydrophilic shells (PAA), in an aqueous environment above pH 3. However, HBPMA-b-PAAs formed regular quadrangular prisms in an aqueous medium having a pH less than 2. Undoubtedly, such pH responsive HBPMA-b-PAAs may prove to be potential candidates for the delivery devices of biomacromolecules.
Finally, another widely exploiting living polymerization technique is RAFT, which is again highly favored in the design of hb polymers with controlled architectures. RAFT is easy to explore due to high tolerance of the reactants to various functional monomers (vinyl acetate, N-vinyl pyrrolidone etc.) and mild reaction conditions (ambient temperature and tolerance to oxygen). Typically, RAFT polymerization proceeds via reversible, degenerative (addition fragmentation) chain transfer processes, unlike in conventional chain transfer processes in FRP where chain transfer occurs only once. CTAs employed in RAFT polymerization are commonly named as RAFT agents like thiocarbonates, thiocarbamates, or dithioesters, all of which have a structure Z(C=S)SR. The RAFT agents maintain equilibrium between the active and the dormant species. In the first step (i.e., initiation step), free radicals are generated which attacks vinyl monomers and generate primary polymer radicals (P ·n ). Just at the start of propagation, P ·n react with the RAFT agents through chain transfer reactions. The chain transfer reaction proceeds via addition of P ·n across C=S and is followed by fragmentation of R group (leaving group). The fragmentation of R group generates R· and oligomeric RAFT agents (macro-RAFT agents). It is reported in literature that all of the RAFT agents are consumed just prior to the propagation step (obviously if appropriate RAFT agents are used) [60]. This is so because due to very high reactivity of C=S bonds, the addition of P ·n across C=S bonds in the RAFT agent is favored over the addition of P ·n across C=C bonds in the vinyl monomers. In the subsequent step, R· reinitiates vinyl monomers and generates the propagating P ·m . P ·m /P ·n further activates the oligomeric RAFT agents and establishes an equilibrium between the active P ·n /P ·m and the dormant oligomeric/polymeric RAFT agents. The established equilibrium limits the termination reactions, enables all the polymer chains to grow at a time, and thus significantly narrows M.W.D of the resultant polymers. Although insignificant, termination reactions still occur via combination or disproportionation mechanisms. The mechanism of RAFT polymerization is schematically shown in Scheme 3.19. When the polymerization is complete, most of the chains in the dead polymers retains S–C=S bonds which can be isolated as stable compounds. Interestingly, even after the formation of a dead polymer by RAFT process, further monomers may be added at the end of the dead polymers which facilitates block copolymerization.

Schematic representation of the mechanism of RAFT polymerization. Reprinted (adapted) with permission from Moad et al. [59]. Copyright (2006) CSIRO (Australia)

Cartoon of a R-transmer and a Z-transmer. Reprinted (adapted) with permission from Wang and Gao [41]. Copyright (2017) MDPI
For the design of hb polymers, RAFT-SCVP is highly popular. In RAFT-SCVP, the AB-type monomer is basically a RAFT agent containing a polymerizable vinyl group and is named as transmer [41, 61]. Unlike the inimers used in NMP-SCVP or ATRP-SCVP, B groups in the inimers for RAFT-SCVP have to be initiated by R· which in turn is cleaved by an external radical initiator. A transmer may be designed in two forms; the polymerizable vinyl group may be present at the R group (R approach) or at the Z group (Z approach); Scheme 3.20. The R approach is more popular than the Z approach because in the latter approach, steric hindrance restricts access to CTA functionalities and the generated branch points are very weak. Actually in the Z approach, the initiating thiocarbonylthio groups are located at the branch points of the hb polymers which increases steric hindrance and eventually results in hydrolytically unstable groups at every branch point. On the contrary, in the R approach, the thiocarbonylthio groups are present at the polymer chain ends and thus results in highly peripheral functional polymers. The first work on RAFT-SCVP was carried out by Yang et al. where they synthesized a hb polymer from an AB-type monomer designed by introducing dithioester groups into the styrene monomers [62]. Unfortunately, this approach being a Z approach, the hb polymers exhibited weak branching points. Interestingly, in a recent work, Kalourkoti et al. synthesized segmented amphiphilic hb block copolymers of styrene and 2-vinylpyridine or 4-vinylpyridine via RAFT-SCVP [63]. In this work, they at first synthesized a hb polymer utilizing one of the monomer. The hb homopolymer contained thiocarbonylthio groups at the branch points which facilitated further polymerization with the second monomer. As the second monomer was inserted at the branch points, it resulted in hb block copolymers. The final amphiphilic hb block copolymer self-assembled into micelles with sizes greater than 10 nm. Heidenreich and Puskas synthesized a hb copolymer via RAFT-SCVP (R approach) of styrene and 4-vinylbenzene dithiobenzoate (an AB-type monomer) in bulk conditions (at 110 °C) [64]. The resultant hb copolymer exhibited a high M.W around 3.7 × 105 g mol−1 and a P.D.I of 2.65. In an interesting work, Carter from Rimmer’s group prepared hb poly (N-isopropylacrylamide); hb PNIPAM (18A/B/C) via RAFT-SCVP like technique from NIPAM––17A, in the presence of a branching monomer that contained imidazole groups––17B which could be effectively transferred to the polymer chain ends through chain transfer reactions; Scheme 3.21 [65]. They found that the hb polymer was temperature responsive and was highly effective in purifying His-tagged BRCA-1 protein fragments by precipitation.

Scheme showing the synthesis of hb PNIPAM rich in imidazole groups via RAFT-SCVP type approach. Reprinted (adapted) with permission from Carter et al. [65]. Copyright (2005) Wiley online library
In an attempt to develop hb polymers with suitable properties for biological and biomedical applications, Ghosh Roy and De synthesized amino acid containing hb polymers, 20 via RAFT-SCVP approach from tert-butyl carbamate (Boc)-L-valine acryloyloxyethyl ester (Boc-Val-HEA), 19A and S-(4-vinyl)benzyl S´-butyltrithiocarbonate (VBBT), 19B; Scheme 3.22 [66]. When Boc was removed, water soluble, pH responsive, biocompatible, cationic hb polymers were obtained.

Scheme showing the synthesis of amino acid containing hb polymer via RAFT-SCVP and deprotection of the Boc groups step to generate water soluble hb polymers. Reprinted (adapted) with permission from Ghosh Roy and De [66]. Copyright (2014) Royal society of chemistry
Han et al. prepared a series of chain segmented hb poly (tertiary amino methacrylate)s; HPTAMs with hydrophilic cores and hydrophobic shells, via RAFT-SCVP of 2-(dimethylamino)ethyl methacrylate (DMAEMA) and 2-((2-(((dodecylthiocarbonothioyl)thio)-2-methylpropanoyl)oxy)-ethyl acrylate (ACDT, an inimer) [67]. HBTAMs possessed regular linear chains between every two adjacent branching points for which they resembled HyperMacs in structures. In an interesting work, Bai et al. developed hb polyacrlamides 22 via redox RAFT-SCVP from a monomer containing reducing groups, in the presence of Cu(III) and Ce(IV) as oxidants; Scheme 3.23 [68]. In A stages, amide group/Cu(III) redox process generated free radicals which rapidly initiated monomers and subsequently continued propagation. In B stages, in the excess of oxidizing agents, the linear polymers got further oxidized to form hb polymers.

Scheme showing the synthesis of a hb polymer via redox RAFT-SCVP. Reprinted (adapted) with permission from Bai et al. [68]. Copyright (2014) Springer
Recently, the combination of RAFT and macromolecular architecture design via interchange of xanthates (MADIX) polymerization technique has gained much attraction to produce polymers with a variety of architectures and controlled properties. RAFT and MADIX follow the same mechanism but differ only by the mediator used. Delduc introduced the concept of degenerative chain transfer of radical species to xanthates; Scheme 3.24 [69]. Zhou et al. synthesized hb poly (vinyl acetate); Hb PVAc, 24 via RAFT/MADIX-SCVP from vinyl acetate, in the presence of vinyl 2-(ethoxycarbonothioylthio) acetate (ECTVA; a xanthate-based RAFT agent), 23; Scheme 3.25 [70]. Hb PVAc 24 on hydrolysis produced hb PVA which is a potential pharmaceutical material.


Scheme showing the synthesis of hb PVAc via RAFT/MADIX-SCVP. Reprinted (adapted) with permission from Zhou et al. [70]. Copyright (2011) Elsevier
One of the disadvantages of RAFT polymerization is that the synthesized polymers are colored (either pink or yellow) due to the presence of S–C=S end groups. Hence, RAFT is often discouraged in commercial processes. There are certain techniques to remove the colorant groups from the polymers like aminolysis to produce thiol terminated polymers and transforming S–C=S groups to terminal hydrogen groups through a reaction with tri-n-butylstannane [72, 73]. Another shortcoming of RAFT-SCVP is that the synthesized hb copolymers exhibited higher M.W than the hb homopolymers. The preparation of hb polymers from cyclic AB-type monomers (cyclic inimers) relies on a new strategy named as ring opening multi-branching polymerization (ROMBP) or self-condensing ring opening polymerization (SCROP). In this approach, a strained, cyclic group generates a branching point only upon ring opening. ROMBP is considered as one of the important synthesis process to develop hb polymers for biomedical applications like hb polyamines, hb polyethers, hb polyesters, and hb poly siloxanes [74]. ROMBP may be further categorized as cationic ROMBP, anionic ROMBP, and catalytic ROMBP [75]. Hauser developed hb poly(ethyleneimine)s via cationic ROMBP of aziridines (three-membered alkylene imines) in the presence of cationic initiators [76]. Herein, intermolecular nucleophilic attack of the secondary amine nitrogens (present along the polymer backbone) on the propagating iminium centers produces tertiary amine groups which eventually cause branching; Scheme 3.26. Hb poly (ethyleneimine)s synthesized from aziridines is a well-known commercial product, sold under the trade name Lupasol®. Other cyclic monomers based on oxiranes and oxetanes have also been polymerized via cationic ROMBP.

Scheme for cationic ROMBP of ethylene imine. Reprinted (adapted) with permission from Wilms et al. [75]. Copyright (2011) Wiley online library
Generally, cyclic amides and esters (lactams and lactones), cyclic ethers or Leuchs’ anhydrides, and other vinyl monomers with electron withdrawing groups (like acrylonitrile, methyl vinyl ketone, etc.) are polymerized via anionic ROMBP. A typical anionic ROMBP mechanistic pathway is shown in Scheme 3.27. Among them, synthesis of hb polyglycerols (hbPGs) is considered as one of the important anionic ROMBP approaches. Vandenberg reported anionic ROMBP of glycidol using KOH as an initiator, for the first time [77]. Unfortunately, he obtained branched PG oligomers only. This happened because alkoxide anions tend to form aggregated species (through both inter- and intramolecular associations) in polar solvent which eventually caused a slow propagation. Sunder et al. improved the propagation in ROMBP of glycidol by slow monomer addition technique and obtained hbPGs with M.W around 6000 g mol−1 and P.D.I < 1.3 [78]. Goodwin and Baskaran prepared hbPGs via epoxy inimer mediated ROMBP where they used glycol as an initiator in the presence of potassium counter ion [79]. They employed batch monomer addition in order to improve the propagation step. This reaction proceeded via propagation at two centers––one that originated from glycidol proton transfer leading to epoxy anion inimer and the other that undergoes hyperbranching without any transfer to monomers. In this approach, equilibrium was maintained between oligomers and high M.W hbPGs.

Schematic representation of the mechanism of anionic ROMBP of glycidol. Reprinted (adapted) with permission from Wilms et al. [75]. Copyright (2011) Wiley online library
Rockicki et al. designed hb aliphatic polyethers with hydroxyl end groups from glycerol carbonate (4-hydroxymethyl-1,3-dioxolan-2-one) via anionic ROMBP and was accompanied by CO2 liberation [80]. In this approach, 1,1,1-tris(hydroxymethyl)propane was used as a trifunctional initiator and a core of the polyether. At this point, it is worthy to mention that hbPGs are often clinically tried as human serum albumin substitutes [81], as drug carriers [82], as colloids for cold preservation of cells or organs [83] and for use in biomineralization [84]. In fact, Du et al. observed that hbPGs are better substrates than glucose for peritoneal dialysis and cause minimum damage to the peritoneal membranes in rats [85]. Anionic ROMBP being living in nature often encourages synthesis of hb polymers with controlled architectures and properties. Another approach catalytic ROMBP is quite popular in the design of hb polymers with well-defined tacticity and narrow M.W.D. However, this approach is unsuitable for the generation of products for biomedical applications owing to the presence of toxic metal traces and thus discouraged.
Both SCVP (normal or controlled) and ROMBP are highly acknowledged in the design of hb polymers but the availability of any type of inimer or their stringent preparation processes restricts industrialization. Often the slow monomer addition techniques are followed in SCVP/ROMBP to obtain hb polymers with higher DB. In future, further research has to be done in developing easier methods to synthesize inimers from the conventional vinyl monomers so that commercialization of SCVP and ROMBP approaches are possible.
3.5 Hypergrafting
Hypergrafting, i.e., covalent attachment of hb polymers to different substrates generates hybrid structures with complex architectures which is often favored in many biomedical applications [87]. There are two categories of hypergrafting––homogeneous hypergrafting (if the substrates are soluble like hydrophilic polymers) and heterogeneous hypergrafting (if the substrates are insoluble like metal nanoparticles or silicon wafers); Scheme 3.28.

Cartoon showing various substrates available for hypergrafting; a homogeneous hypergrafting and b heterogeneous hypergrafting. Reprinted (adapted) with permission from Schull and Frey [86]. Copyright (2013) Elsevier
3.5.1 Homogeneous Grafting––Hyperbranched-Graft-Hyperbranched Copolymers
When hb polymers are grafted from the surface of hb macroinitiators (like low M.W hbPGs) then hyperbranched-graft-hyperbranched copolymers (hb-g-hb) are generated. Hb-g-hb copolymers have core–shell structures which are highly suitable for biomedical transport applications [67, 88]. Xu et al. synthesized amphiphilic hb-g-hb copolymers via cationic ROMBP of 3-ethyl-3-hydromethyl oxetane (EHMO) and glycidol [89]. The resultant hb-g-hb copolymers were composed of hydrophobic Hb-PEHMO cores and hydrophilic HbPG shells. Popeney et al. developed a dendritic core–shell nanostructures via a two-step process––generation of nonpolar dendritic PE cores by late transition metal catalyzed chain walking polymerization which was followed by anionic ROMBP of glycidol to graft hbPG shells [88]. The core–shell nanoparticles exhibited high internal polarity gradient which facilitated the transport of poorly water soluble molecules. Unfortunately, there is very limited works on hb-g-hb copolymers and therefore it leaves enough scope for research.
3.5.2 Homogeneous Grafting––Linear-Graft-Hyperbranched Copolymers
When hb polymers are grafted from the surface of linear macroinitiators, then linear-graft-hyperbranched copolymers (lin-g-hb) are generated. Lin-g-hb copolymers have attracted much attention in the realm of macromolecules design because they have a high number of functional groups at the side chains and also exhibit cylindrical nano conformations in bulk or solution (just like DenPols). Kuo et al. developed hb poly (ethylene imine); hb PEI from poly (allyl amine) macroinitiators using 2-chloroethylamine hydrochloride [90]. The resultant polymer constituted of linear allylic polymer chains with pendant hb PEI and thus was considered as excellent multident chelates. Schull and Frey synthesized lin-g-hb copolymers of linear (4-hydroxy styrene)-graft-hyperbranched PGs (PHOS-g-hbPG), via three steps anionic and oxy-anionic polymerizations [91].
In an effort to increase M.Ws of the lin-g-hb copolymers, a grafting to strategy was developed; Scheme 3.29. Schull et al. prepared hbPGs dendron analogues, 25 containing exactly one focal amino group by ROMBP of glycidol in the presence of a trifunctional initiator (N,N-dibenzyl tris (hydroxylmethyl) amino methane) [92]. Then they attached hbPGs dendron analogues to poly (pentafluorophenol methacrylate), 26 to develop poly (hbPGs methacrylamide-g-pentafluorophenol methacrylate); hbPGMA-g-PFPMA, 27. Lin-g-hb cpolymers 27 could be further functionalized with drugs or biomedical imaging compounds due to the presence of multifunctionality of hbPG dendrons in the structures. Here again, we have obtained only few works on lin-g-hb polymers.

Scheme showing the synthesis of lin-g-hb copolymer via grafting to strategy. Reprinted (adapted) with permission from Schull et al. [92]. Copyright (2012) American chemical society
3.5.3 Homogeneous Grafting––Linear-Block-Hyperbranched Copolymers
Linear-block-hyperbranched copolymer (lin-b-hb) is a block copolymer which consists of a hyperbranched/dendrimer block linked to the chain ends of a linear block. The hb block adds many functional groups to the linear blocks and thus develops polymers with many new interesting properties. There are three ways in which lin-b-hb copolymers may be synthesized: (1) “chain first”––hypergrafting from the linear segments which bear initiating groups for the generation of the hb blocks through polymerization, (2) “coupling strategy”––hypergrafting through the reactions between monofunctional hb blocks and end functional linear blocks and (3) “core first”––hypergrafting from the hb blocks (cores) which bears a single initiating group for divergent polymerization [93]. There are numerous examples of lin-b-hb copolymers due to the ease of synthesis.
Barriau et al. synthesized an amphiphilic block copolymer constituting of a linear apolar block and a hydrophilic hb block; Scheme 3.30 [94]. Initially, they prepared a diblock copolymer of polystyrene and linear hydroxylated polybutadiene (PS-b-PBOH), 28 via anionic polymerization and subsequent hydroboration reaction. PS-b-PBOH acted as a macroinitiator for glycidol (an AB2-type monomer), in the post-polymerization grafting step which generated PS-b-PB-b-hbPG, 29. Lin-b-hb copolymer 29 formed self-assembled micelles in various apolar solvents. These micelles appeared to be suitable for the generation of nano-reactors on the surfaces which may facilitate biomineralization and other biomedical applications. Marcos et al. used PS-b-PBOH macroinitiator for the grafting of hb poly (carbosilane); hb PCS blocks from methyldiundec-10-enylsilane (an AB2-type monomer) via catalytic hydrosilylation reaction [95]. In a later work, Wurm et al. developed a double hydrophilic block copolymer of linear polyethylene oxide (PEO) and hbPG via anionic polymerization of ethylene oxide and subsequently with ethoxyethyl glycidyl ether [96]. In another work, Wurm et al. synthesized amphiphilic block copolymer of linear PEO and hb PCS in a three-step process by combining anionic polymerization of allyl glycidyl ether onto PEO-OH and subsequent catalytic hydrosilylation polyaddition of an AB2-type carbosilane monomer [97]. One of the disadvantages of “chain first” technique is that a certain fraction of hb homopolymer may be generated which cannot be attached to the linear blocks. However, extensive purification by precipitation may help in this regard. The “coupling strategy” is rarely employed in the synthesis of lin-b-hb copolymer because of tedious purification steps and uncontrolled coupling reactions. Limited availability of the hb dendrons with a single focal functionality reduces the probability of coupling and thus makes the synthesis procedure challenging. Yet Tao from Yan’s group prepared an amphiphilic lin-b-hb copolymer through non-covalent and host–guest coupling interactions of adamantine functionalized, long alkyl chained hbPGs from ß cyclodextrin [98]. These lin-b-hb copolymers could self-assemble into unimolecular vesicles and disassembled upon the addition of a competitive host for ß-cyclodextrin. Similarly, the “core first” technique is also an unexplored strategy. However, Nuhn et al. explored the “core first” strategy to develop lin-b-hb copolymers via ROMBP of glycidol and subsequent RAFT polymerization of the hb macro-RAFT agents of functional thermoresponsive methacrylate or biocompatible methacrylamide monomers [99].

Scheme showing the synthesis of a lin-b-hb copolymer via “chain first” technique. Reprinted (adapted) with permission from Barriau et al. [94]. Copyright (2005) Wiley online library
3.5.4 Heterogeneous Grafting
Often functional polymers are attached to the inorganic substrates (2D planar surfaces or spherical particles) to develop smart materials. There are four different heterogeneous hypergrafting techniques––(1) step-by-step approach, (2) graft-on-graft approach, (3) grafting from approach, and (4) grafting to approach [100]. In the step-by-step approach, which is analogous to the divergent synthesis of dendrimers, oligomeric ABm-type branching building blocks are attached to the functionalized substrates in multiple steps. Tsubokawa et al. attached dendritic poly (amido amine); PAMAM to amino functionalized silica surfaces via an iterative alternating Michael addition of MMA to amines and amidation of the resulting esters with ethylenediamine [101]. In a similar way, Tsubokawa et al. attached PAMAM to chitosan [102]. Following their work, functional methacrylates were attached to the silica templates [103] or PAMAM to carbon blacks [104]. Unfortunately, in a step-by-step approach, only limited generations of polymers can be grown. On the contrary, in the graft-on-graft approach, as macromolecular building blocks are attached to the substrates functionalized with macromolecular groups in multiple steps, thick layers of polymers may be grown. Zhou et al. designed hb poly (acrylic acid)s; hb PAA on self-assembled organomercaptan monolayers (which is basically functionalized by reaction with α,ω-diamino-terminated poly (tert-butyl acrylate) [105]. Owing to the growing demands for one-pot synthesis strategies, grafting to and grafting from approaches have gained much attention. In the grafting to/grafting from approaches, a preformed hb polymer is attached to/from the substrate in one step, respectively, either through a single focal point or multiple end groups on the hb polymers. Mikhaylova et al. attached hydroxyl and carboxyl terminated hb aromatic polyesters to epoxy terminated silicon wafers via grafting to strategy [106]. In a similar fashion, Sidorenko et al. attached epoxy terminated hb polyesters to a silicon oxide substrate [107]. Paez et al. extended the grafting to strategy to gold surfaces [108]. In this novel work, in the initial step, they synthesized hbPGs with disulfide groups and different loadings of amino groups. Then, they attached these functionalized hbPGs to gold surfaces. Gold surface grafted hbPGs are potential protein resistant materials [109] and the presence of amino groups facilitated exclusive cell targeting [108].
In our lab, we are trying to develop another variation in the grafting technique by growing hb polymer chains from the surface of different substrates like polysaccharides, proteins, hydrophilic polymers, and inorganic matrices through the “Strathclyde methodology” and condensation techniques. These works are in the budding stage and thus still under consideration. We hope in future, these techniques will open up new areas of research for the design of a variety of architectural polymers.
3.5.5 Hypergrafting onto Living Cells
Of the various substrates, these days, living cells are considered as potential substrates for the attachment of hb polymers. Earlier adhesion of the hydrophilic macromolecules to cell surfaces posed serious difficulties due to repulsion between the hydrophilic components [110]. To overcome this difficulty, Rossi et al. utilized cell compatible, nonreactive additive polymers like dextran, hbPG, primary amine reactive succinimidyl succinate functionalized PEG, etc., to modify surfaces of the cells with high grafting efficiency [111]. These polymers exhibit improved penetration into the glycocalyx of the cell membranes. They carried the grafting of hbPGs on four different types of cells- RBCs, WBCs, platelets, and Jurkat cells. Interestingly, hbPG-g-live cells exhibited minimal accumulation in the organs (except liver and spleen) and reduced degree of cell membrane deformation with significant higher levels of CD47 self-protein markers which enhance their in vivo survival [112, 113]. In an attempt to improve targeting efficiency of the therapeutic stem cells to the target tissues (via intravascular injection), Jeong et al. synthesized bioactive hbPG associated stem cells [114]. They modified hbPGs with octyl chains and vasculature binding peptides (VBPs). Then they allowed modified hbPGs to bind with the cell membranes of stem cells by hydrophobic interactions and used VBPs to selectively target inflammatory endothelium or other desired tissues. Owing to the attractive features of the hb polymers and improvements in the grafting technologies, various hb polymers along with drugs/genes may be conjugated to different cells and thus may be used to improve therapies. In fact, this concept may be utilized in personalized therapies.
3.6 Conclusion
This chapter has covered some of the notable works on advanced polymerization techniques to develop hb polymers with controlled architectures and properties. The journey from FRP to SCVP, living polymerization, hypergrafting, and combination of various polymerization techniques has been discussed in lengths. Each technique has its own merits and demerits, however, as discussed; these were being selected for synthesis of explicit architectures for specific biomedical applications.
Abbreviations
- ACDT:
-
2-((2-(((dodecylthiocarbonothioyl)thio)-2-methylpropanoyl)oxy)-ethyl acrylate
- AIBN:
-
Azobisisobutyronitrile
- AIG1c:
-
3-O-acryloyl-1,2,5,6-di-O-isopropylidene-α-d-glucofuranoside
- ARGET:
-
Activators regenerated by electron transfer
- ATRA:
-
Atom transfer radical addition
- ATRP:
-
Atom transfer radical polymerization
- BPEA:
-
2-((2-bromopropionyl)oxy)ethyl acrylate
- BuLi:
-
Butyl lithium
- CT:
-
Chain transfer
- CTA:
-
Chain transfer agent
- DB:
-
Degree of branching
- DMAEMA:
-
2-(dimethylamino)ethyl methacrylate
- DVA:
-
Divinyl adipate
- DVB:
-
Divinyl benzene
- DVM:
-
Divinyl monomer
- ECTVA:
-
Vinyl 2-(ethoxycarbonothioylthio) acetate
- EGDMA:
-
Ethylene glycol dimethacrylate
- EHMO:
-
3-ethyl-3-hydromethyl oxetane
- FRP:
-
Free radical polymerization
- GTP:
-
Group transfer polymerization
- ICAR:
-
Initiators for continuous activator regeneration
- IFIRP:
-
Initiator fragment incorporation radical polymerization
- MMA:
-
Methyl methacrylate
- MVM:
-
Multivinyl polymerization
- M.W:
-
Molecular weight
- M.W.D:
-
Molecular weight distribution
- NMP/NMRP:
-
Nitroxide mediated radical polymerization
- PB:
-
Poly (buta-1,2-diene)
- PAMAM:
-
Poly (amido amine)
- PCS:
-
Poly (carbosilane)
- P.D.I:
-
Poly dispersity index
- PE:
-
Polyethylene
- PEG:
-
Polyethylene glycol
- PEGDMA:
-
Poly ethylene glycol dimethacrylate
- PEI:
-
Poly (ethylene imine)
- PEO:
-
Polyethylene oxide
- PG:
-
Polyglycerol
- PMA:
-
Poly (mecthacrylate)
- PS:
-
Polystyrene
- PVA:
-
Poly (vinyl alcohol)
- PVAc:
-
Poly (vinyl acetate)
- PRE:
-
Persistant radical effect
- PTP:
-
Proton transfer polymerization
- RAFT:
-
Reversible addition fragmentation chain transfer
- RBC:
-
Red blood cell
- ROMBP:
-
Ring opening multi-branching polymerization
- SARA-ATRP:
-
Supplementary activators and reducing agents ATRP
- SCGTCP:
-
Self-condensing group transfer copolymerization
- SCROP:
-
Self-condensing ring opening polymerization
- SCVP:
-
Self-condensing vinyl polymerization
- SCVCP:
-
Self-condensing vinyl copolymerization
- tBMA:
-
Tert-butyl methacrylate
- TEMPO:
-
2,2,6,6-tetramethylpiperidin-1-oxyl
- TIPNO:
-
2,2,5-trimethyl-4-phenyl-3-azahexane-3-nitroxide
- VBC:
-
Vinylbenzyl chloride
- VBP:
-
Vasculature binding peptide
- WBC:
-
White blood cell or leukocyte
References
Liu J, Wang Y, Fu Q, Zhu X, Shi W (2008) Branched polymer via free radical polymerization of chain transfer monomer: a theoretical and experimental investigation. J Polym Sci A Polym Chem 46(4):1449–1459
O’Brien N, McKee A, Sherrington DC, Slark AT, Titterton A (2000) Facile, versatile and cost effective route to branched vinyl polymers. Polymer 41(15):6027–6031
Graham S, Cormack PAG, Sherrington DC (2005) One-pot synthesis of branched poly(methacrylic acid)s and suppression of the rheological “Polyelectrolyte Effect”. Macromolecules 38(1):86–90
Liu Y, Haley JC, Deng K, Lau W, Winnik MA (2008) Synthesis of branched poly(butyl methacrylate) via semicontinuous emulsion polymerization. Macromolecules 41(12):4220–4225
Baudry R, Sherrington DC (2006) Synthesis of highly branched poly(methyl methacrylate)s using the “Strathclyde Methodology” in aqueous emulsion. Macromolecules 39(4):1455–1460
Das T, Sengupta S, Ghorai UK, Dey A, Bandyopadhyay A (2015) Sequential amphiphilic and pH responsive hyperbranched copolymer: influence of hyper branching/pendant groups on reversible self assembling from polymersomes to aggregates and usefulness in waste water treatment. RSC Adv 5(124):102932–102941
Dong ZM, Liu XH, Lin Y, Li YS (2008) Branched polystyrene with abundant pendant vinyl functional groups from asymmetric divinyl monomer. J Polym Sci, Part A: Polym Chem 46(18):6023–6034
Dong Zm, Liu Xh, Tang Xl, Li Ys (2009) Synthesis of hyperbranched polymers with pendent norbornene functionalities via RAFT polymerization of a Novel asymmetrical divinyl monomer. Macromolecules 42, (13):4596–4603
Zhao T, Zhang H, Zhou D, Gao Y, Dong Y, Greiser U, Tai H, Wang W (2015) Water soluble hyperbranched polymers from controlled radical homopolymerization of PEG diacrylate. RSC Adv 5(43):33823–33830
Guan Z (2002) Control of polymer topology through transition-metal catalysis: synthesis of hyperbranched polymers by cobalt-mediated free radical polymerization. J Am Chem Soc 124(20):5616–5617
Smeets NMB (2013) Amphiphilic hyperbranched polymers from the copolymerization of a vinyl and divinyl monomer: the potential of catalytic chain transfer polymerization. Eur Polym J 49(9):2528–2544
Smeets NMB, Freeman MW, McKenna TFL (2011) Polymer architecture control in emulsion polymerization via catalytic chain transfer polymerization. Macromolecules 44(17):6701–6710
Sato T, Sato N, Seno M, Hirano T (2003) Initiator-fragment incorporation radical polymerization of divinylbenzene in the presence of glyoxylic oxime ether: formation of soluble hyperbranched polymer. J Polym Sci A Polym Chem 41(19):3038–3047
Sato T, Ihara H, Hirano T, Seno M (2004) Formation of soluble hyperbranched polymer through the initiator-fragment incorporation radical copolymerization of ethylene glycol dimethacrylate with N-methylmethacrylamide. Polymer 45(22):7491–7498
Sato T, Arima Y, Seno M, Hirano T (2005) Initiator-fragment incorporation radical polymerization of divinyl adipate with dimethyl 2,2′-Azobis(isobutyrate): kinetics and formation of soluble hyperbranched polymer. Macromolecules 38(5):1627–1632
Sato T, Nomura K, Hirano T, Seno M (2006) Initiator-fragment incorporation radical polymerization of diallyl phthalate: kinetics, formation of hyperbranched polymer, and iridescent porous film thereof. J Appl Polym Sci 102(1):408–415
Sato T, Hashimoto M, Seno M, Hirano T (2004) Soluble hyperbranched polymer through initiator-fragment incorporation radical copolymerization of ethylene glycol dimethacrylate and α-ethyl β-N-(ά-methylbenzyl) itaconamate in benzene. Eur Polym J 40(2):273–282
Tai H, Zheng Y, Wang W (2011) Hyperbranched copolymers synthesized by cocondensation and radical copolymerization. In: Hyperbranched polymers. Wiley, New York, pp 203–226
Chang HT, Frechet JMJ (1999) Proton-transfer polymerization: a new approach to hyperbranched polymers. J Am Chem Soc 121(10):2313–2314
Gong C, Frechet JMJ (2000) Proton transfer polymerization in the preparation of hyperbranched polyesters with epoxide chain-ends and internal hydroxyl functionalities. Macromolecules 33(14):4997–4999
Chen H, Jia Z, Yan D, Zhu X (2007) Thermo-responsive highly branched polyethers by proton-transfer polymerization of 1,2,7,8-diepoxyoctane and multiols. Macromol Chem Phys 208(15):1637–1645
Gil ES, Hudson SM (2004) Stimuli-responsive polymers and their bioconjugates. Prog Polym Sci Jpn 29(12):1173–1222
Gadwal I, Binder S, Stuparu MC, Khan A (2014) Dual-reactive hyperbranched polymer synthesis through proton transfer polymerization of thiol and epoxide groups. Macromolecules 47(15):5070–5080
Emrick T, Chang HT, Frechet JMJ (1999) An A2+B3 approach to hyperbranched aliphatic polyethers containing chain end epoxy substituents. Macromolecules 32(19):6380–6382
Ma Lj, Wang Hq, He Lf, Li Xy (2016) Hyperbranched epoxy resins prepared by proton transfer polymerization from an A2+B3 system. Chin J Polym Sci 29(3):300–307
Zhao T (2015) Controlled/living radical polymerization of multi-vinyl monomer towards hyperbranched polymers for biomedical applications (Thesis)
Frechet JMJ, Henmi M, Gitsov I, Aoshima S (1995) Self-condensing vinyl polymerization: an approach to dendritic materials. Science 269(5227):1080
Yan D, Muller AHE, Matyjaszewski K (1997) Molecular parameters of hyperbranched polymers made by self-condensing vinyl polymerization. 2. Degree of branching. Macromolecules 30(23):7024–7033
Paulo C, Puskas JE (2001) Synthesis of hyperbranched polyisobutylenes by inimer-type living polymerization. 1. Investigation of the effect of reaction conditions. Macromolecules 34(4):734–739
Knauss DM, Al-Muallem HA (2000) Polystyrene with dendritic branching by convergent living anionic polymerization. II. Approach using vinylbenzyl chloride. J Polym Sci A Polym Chem 38(23):4289–4298
Baskaran D (2001) Synthesis of hyperbranched polymers by anionic self-condensing vinyl polymerization. Macromol Chem Phys 202(9):1569–1575
Mishra M, Kobayashi S (1999) Star and hyperbranched polymers, vol. 53. CRC Press
Simon PFW, Radke W, Müller AHE (1997) Hyperbranched methacrylates by self-condensing group transfer polymerization. Macromol Rapid Commun 18(9):865–873
Chen Y, Fuchise K, Satoh T, Kakuchi T (2015) Group transfer polymerization of acrylic monomers. In: Hadjichristidis N, Hirao A (eds) Anionic polymerization: principles, practice, strength, consequences and applications. Tokyo, Springer Japan, pp 451–494
Simon PFW, Muller AHE (2001) Synthesis of hyperbranched and highly branched methacrylates by self-condensing group transfer copolymerization. Macromolecules 34(18):6206–6213
Otsu T (2000) Iniferter concept and living radical polymerization. J Polym Sci A Polym Chem 38(12):2121–2136
Gigmes D (2015) Nitroxide mediated polymerization: from fundamentals to applications in materials science. R Soc Chem
Moad G, Rizzardo E, Solomon DH (1982) Selectivity of the reaction of free radicals with styrene. Macromolecules 15(3):909–914
Grubbs RB (2011) Nitroxide-mediated radical polymerization: limitations and versatility. Polym Rev 51(2):104–137
Hawker CJ, Frechet JMJ, Grubbs RB, Dao J (1995) Preparation of hyperbranched and star polymers by a “Living”, self-condensing free radical polymerization. J Am Chem Soc 117(43):10763–10764
Wang X, Gao H (2017) Recent progress on hyperbranched polymers synthesized via radical-based self-condensing vinyl polymerization. Polymers 9(6):188
Khan A, Malkoch M, Montague MF, Hawker CJ (2008) Synthesis and characterization of hyperbranched polymers with increased chemical versatility for imprint lithographic resists. J Polym Sci A Polym Chem 46(18):6238–6254
Matyjaszewski K, Gaynor SG, Greszta D, Mardare D, Shigemoto T, Wang J-S (1995) Unimolecular and bimolecular exchange reactions in controlled radical polymerization. Macromol Symp 95(1):217–231
Patten TE, Matyjaszewski K (1998) Atom transfer radical polymerization and the synthesis of polymeric materials. Adv Mater 10(12):901–915
Matyjaszewski K, Tsarevsky NV (2014) Macromolecular engineering by atom transfer radical polymerization. J Am Chem Soc 136(18):6513–6533
Gao H, Matyjaszewski K (2009) Synthesis of functional polymers with controlled architecture by CRP of monomers in the presence of cross-linkers: from stars to gels. Prog Polym Sci 34(4):317–350
Gaynor SG, Edelman S, Matyjaszewski K (1996) Synthesis of branched and hyperbranched polystyrenes. Macromolecules 29(3):1079–1081
Graff RW, Wang X, Gao H (2015) Exploring self-condensing vinyl polymerization of inimers in microemulsion to regulate the structures of hyperbranched polymers. Macromolecules 48(7):2118–2126
Matyjaszewski K, Gaynor SG, Kulfan A, Podwika M (1997) Preparation of hyperbranched polyacrylates by atom transfer radical polymerization. 1. Acrylic AB* monomers in “Living” radical polymerizations. Macromolecules 30(17):5192–5194
Muthukrishnan S, Jutz G, Andre X, Mori H, Muller AHE (2005) Synthesis of hyperbranched glycopolymers via self-condensing atom transfer radical copolymerization of a sugar-carrying acrylate. Macromolecules 38(1):9–18
Amin A, El-Gaffar MA (2008) Synthesis of novel polyamide-hyperbranched polymers via self condensing atom transfer radical polymerization. Polym Plast Technol 47(10):984–990
Tsarevsky NV, Huang J, Matyjaszewski K (2009) Synthesis of hyperbranched degradable polymers by atom transfer radical (Co)polymerization of inimers with ester or disulfide groups. J Polym Sci A Polym Chem 47(24):6839–6851
Jakubowski W, Matyjaszewski K (2006) Activators regenerated by electron transfer for atom-transfer radical polymerization of (Meth)acrylates and related block copolymers. Angew Chem Int Ed 45(27):4482–4486
Elsen AM, Burdynska J, Park S, Matyjaszewski K (2013) Activators regenerated by electron transfer atom transfer radical polymerization in miniemulsion with 50 ppm of copper catalyst. ACS Macro Lett 2(9):822–825
Boyer C, Corrigan NA, Jung K, Nguyen D, Nguyen TK, Adnan NNM, Oliver S, Shanmugam S, Yeow J (2016) Copper-mediated living radical polymerization (atom transfer radical polymerization and copper(0) mediated polymerization): from fundamentals to bioapplications. Chem Rev 116(4):1803–1949
Matyjaszewski K, Pyun J, Gaynor SG (1998) Preparation of hyperbranched polyacrylates by atom transfer radical polymerization, 4. The use of zero-valent copper. Macromol Rapid Commun 19(12):665–670
Konkolewicz D, Wang Y, Krys P, Zhong M, Isse AA, Gennaro A, Matyjaszewski K (2014) SARA ATRP or SET-LRP. End of controversy? Polym Chem 5(15):4396–4417
Xue X, Li F, Huang W, Yang H, Jiang B, Zheng Y, Zhang D, Fang J, Kong L, Zhai G, Chen J (2014) Quadrangular prism: a unique self-assembly from amphiphilic hyperbranched PMA-b-PAA. Macromol Rapid Commun 35(3):330–336
Moad G, Rizzardo E, Thang SH (2006) Living radical polymerization by the RAFT—a first update. Aust J Chem 59(10):669–692
McLeary JB, Calitz FM, McKenzie JM, Tonge MP, Sanderson RD, Klumperman B (2005) A 1H NMR investigation of reversible addition-fragmentation chain transfer polymerization kinetics and mechanisms. Initialization with different initiating and leaving groups. Macromolecules 38(8):3151–3161
Alfurhood JA, Bachler PR, Sumerlin BS (2016) Hyperbranched polymers via RAFT self-condensing vinyl polymerization. Polym Chem 7(20):3361–3369
Wang Z, He J, Tao Y, Yang L, Jiang H, Yang Y (2003) Controlled chain branching by RAFT-based radical polymerization. Macromolecules 36(20):7446–7452
Rikkou-Kalourkoti M, Elladiou M, Patrickios CS (2015) Synthesis and characterization of hyperbranched amphiphilic block copolymers prepared via self-condensing RAFT polymerization. J Polym Sci Part A Polym Chem 53(11):1310–1319
Heidenreich AJ, Puskas JE (2008) Synthesis of arborescent (Dendritic) polystyrenes via controlled inimer-type reversible addition-fragmentation chain transfer polymerization. J Polym Sci A Polym Chem 46(23):7621–7627
Carter S, Rimmer S, Sturdy A, Webb M (2005) Highly branched stimuli responsive poly[(N-isopropyl acrylamide)-co-(1,2-propandiol-3-methacrylate)]s with protein binding functionality. Macromol Biosci 5(5):373–378
Ghosh Roy S, De P (2014) Facile RAFT synthesis of side-chain amino acids containing pH-responsive hyperbranched and star architectures. Polym Chem 5(21):6365–6378
Han J, Li S, Tang A, Gao C (2012) Water-soluble and clickable segmented hyperbranched polymers for multifunctionalization and novel architecture construction. Macromolecules 45(12):4966–4977
Bai Lb, Zhao K, Wu Yg, Li Wl, Wang Sj, Wang Hj, Ba Xw, Zhao Hc (2014) A new method for synthesizing hyperbranched polymers with reductive groups using redox/RAFT/SCVP. Chin J Polym Sci 32(4):385–394
Delduc P, Tailhan C, Zard SZ (1988) A convenient source of alkyl and acyl radicals. J Chem Soc Chem Commun 4:308–310
Zhou X, Zhu J, Xing M, Zhang Z, Cheng Z, Zhou N, Zhu X (2011) Synthesis and characters of hyperbranched poly (vinyl acetate) by RAFT polymeraztion. Eur Polym J 47(10):1912–1922
Perrier S, Takolpuckdee P (2005) Macromolecular design via reversible addition–fragmentation chain transfer (RAFT)/xanthates (MADIX) polymerization. J Polym Sci A Polym Chem 43(22):5347–5393
Moad G (2006) The emergence of RAFT polymerization. Aust J Chem 59(10):661–662
Postma A, Davis TP, Moad G, O’Shea MS (2005) Thermolysis of RAFT-synthesized polymers. A convenient method for trithiocarbonate group elimination. Macromolecules 38(13):5371–5374
Gao C, Yan D (2004) Hyperbranched polymers: from synthesis to applications. Prog Polym Sci 29(3):183–275
Wilms D, Nieberle J, Frey H (2011) Ring-opening multibranching polymerization. In: Hyperbranched Polymers. Wiley, New York, pp 175–202
Hauser M (1969) Alkylene imines. In: Frisch KC, Reegen SL, Dekker M (eds) Ring-opening polymerization. New York
Vandenberg EJ (1985) Polymerization of glycidol and its derivatives: a new rearrangement polymerization. J Polym Sci Polym Chem Ed 23(4):915–949
Sunder A, Hanselmann R, Frey H, Mulhaupt R (1999) Controlled synthesis of hyperbranched polyglycerols by ring-opening multibranching polymerization. Macromolecules 32(13):4240–4246
Goodwin A, Baskaran D (2012) Inimer mediated synthesis of hyperbranched polyglycerol via self-condensing ring-opening polymerization. Macromolecules 45(24):9657–9665
Rokicki G, Rakoczy P, Parzuchowski P, Sobiecki M (2005) Hyperbranched aliphatic polyethers obtained from environmentally benign monomer: glycerol carbonate. Green Chem 7(7):529–539
Kainthan RK, Janzen J, Kizhakkedathu JN, Devine DV, Brooks DE (2008) Hydrophobically derivatized hyperbranched polyglycerol as a human serum albumin substitute. Biomaterials 29(11):1693–1704
Gao X, Zhang X, Zhang X, Wang Y, Sun L, Li C (2011) Amphiphilic polylactic acid-hyperbranched polyglycerol nanoparticles as a controlled release system for poorly water-soluble drugs: physicochemical characterization. J Pharm Pharmacol 63(6):757–764
Gao S, Guan Q, Chafeeva I, Brooks DE, Nguan CYC, Kizhakkedathu JN, Du C (2015) Hyperbranched polyglycerol as a colloid in cold organ preservation solutions. PloS One 10(2):e0116595
Wilms D, Stiriba SE, Frey H (2010) Hyperbranched polyglycerols: from the controlled synthesis of biocompatible polyether polyols to multipurpose applications. Acc Chem Res 43(1):129–141
Liu J, Wu X, Liu Y, Xu Y, Huang Y, Xing C, Wang X (2016) High-glucose-based peritoneal dialysis solution induces the upregulation of VEGF expression in human peritoneal mesothelial cells: the role of pleiotrophin. Int J Mol Med 32
Schull C, Frey H (2013) Grafting of hyperbranched polymers: from unusual complex polymer topologies to multivalent surface functionalization. Polymer 54(21):5443–5455
Bergbreiter DE, Kippenberger AM (2006) Hyperbranched surface graft polymerizations. In: Jordan R (ed) Surface-Initiated Polymerization II. Springer, Berlin Heidelberg: Berlin, Heidelberg, pp 1–49
Popeney CS, Lukowiak MC, Bottcher C, Schade B, Welker P, Mangoldt D, Gunkel G, Guan Z, Haag R (2012) Tandem coordination, ring-opening, hyperbranched polymerization for the synthesis of water-soluble core-shell unimolecular transporters. ACS Macro Lett 1(5):564–567
Xu Y, Gao C, Kong H, Yan D, Luo P, Li W, Mai Y (2004) One-pot synthesis of amphiphilic core-shell suprabranched macromolecules. Macromolecules 37(17):6264–6267
Kuo PL, Ghosh SK, Liang WJ, Hsieh YT (2001) Hyperbranched polyethyleneimine architecture onto poly(allylamine) by simple synthetic approach and the chelating characters. J Polym Sci A Polym Chem 39(17):3018–3023
Schull C, Frey H (2012) Controlled synthesis of linear polymers with highly branched side chains by “Hypergrafting”: poly(4-hydroxy styrene)-graft-hyperbranched polyglycerol. ACS Macro Lett 1(4):461–464
Schull C, Nuhn L, Mangold C, Christ E, Zentel R, Frey H (2012) Linear-hyperbranched graft-copolymers via grafting-to strategy based on hyperbranched dendron analogues and reactive ester polymers. Macromolecules 45(15):5901–5910
Wurm F, Frey H (2011) Linear-dendritic block copolymers: the state of the art and exciting perspectives. Prog Polym Sci 36(1):1–52
Barriau E, García Marcos A, Kautz H, Frey H (2005) Linear-hyperbranched amphiphilic AB Diblock copolymers based on polystyrene and hyperbranched polyglycerol. Macromol Rapid Commun 26(11):862–867
Marcos AGa, Pusel TM, Thomann R, Pakula T, Okrasa L, Geppert S, Gronski W, Frey H (2006) Linear-hyperbranched block copolymers consisting of polystyrene and dendritic poly(carbosilane) block. Macromolecules 39(3):971–977
Wurm F, Nieberle Jr, Frey H (2008) Double-hydrophilic linear-hyperbranched block copolymers based on poly(ethylene oxide) and poly(glycerol). Macromolecules 41(4):1184–1188
Wurm F, Schule H, Frey H (2008) Amphiphilic linear-hyperbranched block copolymers with linear poly(ethylene oxide) and hyperbranched poly(carbosilane) block. Macromolecules 41(24):9602–9611
Tao W, Liu Y, Jiang B, Yu S, Huang W, Zhou Y, Yan D (2012) A linear-hyperbranched supramolecular amphiphile and its self-assembly into vesicles with great ductility. J Am Chem Soc 134(2):762–764
Nuhn L, Schull C, Frey H, Zentel R (2013) Combining ring-opening multibranching and RAFT polymerization: multifunctional linear-hyperbranched block copolymers via hyperbranched macro-chain-transfer agents. Macromolecules 46(8):2892–2904
Peleshanko S, Tsukruk VV (2008) The architectures and surface behavior of highly branched molecules. Prog Polym Sci 33(5):523–580
Tsubokawa N, Ichioka H, Satoh T, Hayashi S, Fujiki K (1998) Grafting of ‘dendrimer-like’ highly branched polymer onto ultrafine silica surface. React Funct Polym 37(1):75–82
Tsubokawa N, Takayama T (2000) Surface modification of Chitosan powder by grafting of ‘dendrimer-like’ hyperbranched polymer onto the surface. React Funct Polym 43(3):341–350
Wang G, Fang Y, Kim P, Hayek A, Weatherspoon MR, Perry JW, Sandhage KH, Marder SR, Jones SC (2009) Layer-by-layer dendritic growth of hyperbranched thin films for surface sol-gel syntheses of conformal, functional, nanocrystalline oxide coatings on complex 3D (Bio)silica templates. Adv Funct Mater 19(17):2768–2776
Tsubokawa N, Satoh T, Murota M, Sato S, Shimizu H (2001) Grafting of hyperbranched poly(amidoamine) onto carbon black surfaces using dendrimer synthesis methodology. Polym Adv Tech 12(10):596–602
Zhou Y, Bruening ML, Bergbreiter DE, Crooks RM, Wells M (1996) Preparation of hyperbranched polymer films grafted on self-assembled monolayers. J Am Chem Soc 118(15):3773–3774
Mikhaylova Y, Pigorsch E, Grundke K, Eichhorn KJ, Voit B (2004) Surface properties and swelling behaviour of hyperbranched polyester films in aqueous media. Macromol Symp 210(1):271–280
Sidorenko A, Zhai XW, Greco A, Tsukruk VV (2002) Hyperbranched polymer layers as multifunctional interfaces. Langmuir 18(9):3408–3412
Paez JI, Brunetti V, Strumia MC, Becherer T, Solomun T, Miguel J, Hermanns CF, Calderon M, Haag R (2012) Dendritic polyglycerolamine as a functional antifouling coating of gold surfaces. J Mater Chem 22(37):19488–19497
Siegers C, Biesalski M, Haag R (2004) Self-assembled monolayers of dendritic polyglycerol derivatives on gold that resist the adsorption of proteins. Chem Eur J 10(11):2831–2838
Dhalluin C, Ross A, Leuthold LA, Foser S, Gsell B, Muller F, Senn H (2005) Structural and biophysical characterization of the 40 kDa PEG−Interferon-Î ± 2a and its individual positional isomers. Bioconjug Chem 16(3):504–517
Rossi NAA, Constantinescu I, Brooks DE, Scott MD, Kizhakkedathu JN (2010) Enhanced cell surface polymer grafting in concentrated and nonreactive aqueous polymer solutions. J Am Chem Soc 132(10):3423–3430
Chapanian R, Constantinescu I, Brooks DE, Scott MD, Kizhakkedathu JN (2012) In vivo circulation, clearance, and biodistribution of polyglycerol grafted functional red blood cells. Biomaterials 33(10):3047–3057
Chapanian R, Constantinescu I, Rossi NAA, Medvedev N, Brooks DE, Scott MD, Kizhakkedathu JN (2012) Influence of polymer architecture on antigens camouflage, CD47 protection and complement mediated lysis of surface grafted red blood cells. Biomaterials 33(31):7871–7883
Jeong JH, Schmidt JJ, Kohman RE, Zill AT, DeVolder RJ, Smith CE, Lai M-H, Shkumatov A, Jensen TW, Schook LG, Zimmerman SC, Kong H (2013) Leukocyte-mimicking stem cell delivery via in situ coating of cells with a bioactive hyperbranched polyglycerol. J Am Chem Soc 135(24):8770–8773
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Copyright information
© 2018 Springer Nature Singapore Pte Ltd.
About this chapter
Cite this chapter
Das, T., Sengupta, S., Bandyopadhyay, A. (2018). Part II––Synthesis of Hyperbranched Polymers: Mixed Chain-Growth and Step-Growth Methods. In: Hyperbranched Polymers for Biomedical Applications . Springer Series on Polymer and Composite Materials. Springer, Singapore. https://doi.org/10.1007/978-981-10-6514-9_3
Download citation
DOI: https://doi.org/10.1007/978-981-10-6514-9_3
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-10-6513-2
Online ISBN: 978-981-10-6514-9
eBook Packages: Chemistry and Materials ScienceChemistry and Material Science (R0)