
Abstract
Organic solar cells (OSCs) utilizing π-conjugated polymers have attracted widespread interest over the past three decades because of their potential advantages, including low weight, thin film flexibility, and low-cost manufacturing. However, their power conversion efficiency (PCE) has been far below that of inorganic analogs. Geminate recombination of charge transfer excitons is a major loss process in OSCs. This paper reviews our recent progress in using transient absorption spectroscopy to understand geminate recombination in bulk heterojunction OSCs, including the impact of polymer crystallinity on charge generation and dissociation mechanisms in nonfullerene acceptor-based OSCs. The first example of a high PCE with a small photon energy loss is also presented. The importance of delocalization of the charge wave function to suppress geminate recombination is highlighted by this focus review.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Introduction
Over the past three decades, organic solar cells (OSCs) utilizing π-conjugated polymers have attracted widespread interest because of their potential advantages, including low weight, thin film flexibility, and low-cost manufacturing [1,2,3,4]. When light is shined on semiconducting polymers, singlet excitons, i.e., Coulombically bound electron–hole (e–h) pairs instead of free charge carriers, are promptly generated because of their small dielectric constants. This is in sharp contrast to inorganic semiconductors such as silicon, in which light absorption directly results in the generation of free electrons and holes in the conduction and valence bands, respectively. OSCs require a donor–acceptor (DA) heterojunction to ionize singlet excitons, where singlet excitons separate into holes on the donor and electrons on the acceptor as a result of the energy offset of the lowest unoccupied molecular orbital (LUMO) or highest occupied molecular orbital (HOMO). If the electron and hole separate further, they become free from Coulombic attraction and hence survive up to nano- or microseconds, long enough to be transported to each electrode. Otherwise, the geminate e–h pairs are likely to recombine to the ground state (Fig. 1).

a Chemical structures of materials highlighted in this focus review. b Photocurrent conversion processes in OSCs: (1) photon absorption to generate singlet excitons, (2) exciton diffusion to the DA interface, (3) charge transfer to generate CT excitons, (4) dissociation of CT excitons into free carriers, and (5) charge collection to each electrode. c Exciton diffusion followed by charge transfer at the interface. d Geminate recombination and charge dissociation of CT excitons
Historically, geminate recombination has been one of the most detrimental processes for OSCs. Singlet excitons in blends of regiorandom poly(3-hexylthiophene) (RRa-P3HT) and the fullerene derivative PCBM undergo efficient charge transfer (CT) at the DA interface (the quantum efficiency of CT at the DA interface is nearly unity); however, CT excitons then experience strong geminate recombination within ~2 ns (~70% of CT excitons geminately decay to the ground state) [5]. Higher-performing OSCs generally consist of crystalline donor polymers such as regioregular P3HT (RR-P3HT) blended with either PCBM or its C70 analogue PC71BM. For example, in blends of RR-P3HT and PCBM, >90% of CT excitons dissociate into free charge carriers [5]. Although recent state-of-the-art OSCs do not display large geminate recombination loss, a few OSCs exhibit ~100% charge dissociation efficiency, falling behind their inorganic analogs. Therefore, it is important to understand how polymer crystallization impacts charge dissociation efficiency.
Nonfullerene acceptors (NFAs) have been considered an alternative to fullerenes. They provide stronger optical absorption in the visible to near-IR region, allowing easier optimization of molecular orbital energies. Until recently, however, NFA-based OSCs routinely lagged in charge generation efficiency and hence power conversion efficiency (PCE) behind their fullerene-based analogs [6,7,8]. One reason for the lower device efficiency of NFA-based OSCs is that most NFA-based OSCs suffer from terrible geminate recombination loss [9,10,11,12,13,14]. For example, blends of PBDTTT-C and a perylene diimide (PDI) monomer lost more than half of the CT excitons through geminate recombination on the sub-nanosecond time scale [14]. As a result, fullerenes were considered “special” electron acceptors for a long time. Thanks to the development of new NFAs, the PCEs for NFA-based OSCs have improved very rapidly [4, 15, 16], and currently, some NFA-based OSCs have a PCE far superior to that of conventional fullerene-based OSCs [17,18,19]. In contrast to the rapid improvement of device efficiency, the fundamental mechanisms for charge generation and recombination in NFA-based OSCs are still being debated. It is important to understand whether the models developed for fullerene acceptor systems are also applicable to efficient NFA systems.
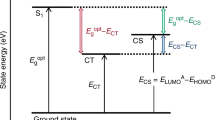
Another remaining challenge in this field is to reduce photon energy loss (Eloss) in OSCs, which is defined as the difference between the optical bandgap (Eg) of the materials and the open-circuit voltage (VOC) (Eloss = Eg − qVOC, where q is an elementary charge). It is well known that the Eloss in OSCs is typically more than 0.7 eV [20], which is much larger than that in silicon-based solar cells (Eloss is only ~0.4 eV). This is partly because OSCs require a DA heterojunction to ionize the excitons, as mentioned above, resulting in a reduction of energy from Eg to the CT state energy ECT. Historically, it has been considered that a LUMO (or HOMO) energetic offset of more than 0.3 eV is necessary for efficient charge generation. For example, Li et al. found a strong correlation between the LUMO energetic offset and the external quantum efficiency (EQE) [21]. The energetic offset was varied by using various donor polymers with fullerene acceptors, and it was found that the EQE dropped sharply when the Eloss was <0.6 eV. This is rationalized by an insufficient driving force for charge generation as a result of the small LUMO energetic offset. OSCs that exhibit efficient charge generation despite a small energetic offset are therefore strongly required.
We have approached the abovementioned challenges by using transient absorption (TA) spectroscopy. TA spectroscopy is one of the most powerful and useful tools for tracking the time evolution of transient species in OSCs, such as excitons and charges, with high temporal resolution [22,23,24,25,26,27,28,29,30,31,32]. In this focus review, our recent findings regarding the dissociation of CT excitons into free charge carriers are summarized. The impacts of polymer crystallization on charge dissociation efficiency [24] and mechanisms of charge dissociation in efficient NFA-based OSCs [31] are discussed. Finally, a first example of efficient charge generation with a small LUMO energetic offset is introduced [28].
Impacts of polymer crystallization on charge dissociation efficiency
Many previous studies have reported that crystallization or aggregation of materials promotes charge dissociation in OSCs [5, 33,34,35,36,37,38]. However, exactly how polymer crystallinity impacts charge dissociation is still unclear. Herein, we discuss the impact of polymer crystallization on charge dissociation in blends of various donor polymers with PCBM [24]. Figure 2a shows the TA spectra of a PSBTBT/PCBM blend film as an example. A positive signal at ~1500 nm observed immediately after photoexcitation is attributable to a photoinduced absorption (PIA) band of PSBTBT singlet excitons. Even just after photoexcitation (t = 0 ps), the TA amplitude of the singlet exciton band is reduced compared with that of the PSBTBT pristine film, indicating that some excitons dissociate to charge pairs within the time resolution of our TA system (~130 fs, full width at half maximum). Instead, another PIA band ascribable to polymer hole polarons was observed ranging from 800 to 1300 nm for the blend film. Note that the PCBM radical anion has a PIA band centered at 1020 nm5, but this PIA band was not observed because of its small absorption cross section. The singlet excitons decayed with a time constant of 0.8 ps, and the polaron band became more pronounced at the same time. At 10 ps after photoexcitation, singlet excitons completely disappeared, and a broad polaron band with peaks at 1000 and 1300 nm was clearly observed. Interestingly, decay kinetics monitored at 1000 nm were faster than those monitored at 1300 nm, indicating that there are at least two distinct charged states in the PSBTBT domains. We found that the band at 1000 nm could be assigned to polarons generated in the PSBTBT disordered phase and that the band at 1300 nm could be assigned to the PSBTBT crystalline phase. Figure 2b shows the decay kinetics of those two polarons after 10 ps. Interestingly, the population of polarons in the crystalline phase still increased with a time constant of ~24 ps, even though singlet excitons had completely disappeared at this point. We found that the increase in polarons in the crystalline phase was accompanied by a decrease in polarons in the disordered phase, indicating that some of the polarons in the disordered phase were transferred to the crystalline phase. Within a nanosecond time scale, approximately half of the polarons in the disordered phase decayed geminately, while geminate recombination was negligible in the crystalline phase.

a TA spectra of the PSBTBT/PCBM blend film excited at 800 nm with a fluence of 11 μJ cm−2. The broken line represents the PIA band of PSBTBT singlet excitons observed for a PSBTBT pristine film. b Time evolution of polarons in the disordered phase (open triangles) and polarons in the crystalline phase (open squares) in the PSBTBT/PCBM blend film. The closed circles represent the total polaron density. Adapted from Ref. [24] with permission from the PCCP Owner Societies
Charge dissociation efficiencies for other blends are summarized in Table 1 [5, 38,39,40,41,42,43,44,45]. We first focus on the impacts of hole transfer from the disorder phase to the crystalline phase observed in PSBTBT/PCBM blends. A similar hole transfer has been reported for RR-P3HT/PCBM blends [5]. Owing to the hole transfer from the disordered phase to the crystalline phase, the charge dissociation efficiency in the disordered phase (ηCDdis) in blends of crystalline donor polymers is higher than that in amorphous RRa-P3HT blends. The driving force for hole transfer is believed to be energetic cascades of molecular orbitals between the crystalline and interfacial disordered phases. Previous studies have established that most OSCs consist of nanoscale domains of pure polymers and pure fullerenes, with intermixed regions in between [46,47,48,49]. Thus, the three-phase morphology introduced by polymer crystallization would be a key driver for efficient charge dissociation through hole transfer [35] because the ionization potential of conjugated polymers, in general, is reduced with increasing polymer crystallinity, resulting in a HOMO energetic cascade between the intermixed disordered phase and the pure polymer crystalline phase [50].
The charge dissociation efficiency is clearly dependent on the blend morphology. The quantum efficiencies of charge dissociation in crystalline phases (ηCDcry) are nearly unity, which is surprisingly high according to the conventional charge dissociation model based on the Braun–Onsager framework [1]. According to the Braun–Onsager framework, an electron and a hole are tightly bound at the DA interface by strong Coulombic attraction because the dielectric constants of organic materials are low; hence, efficient charge dissociation requires a strong electric field (>107 V m−1) and/or a high temperature. However, this is not true for most OSCs, wherein electric field- and temperature-independent charge dissociation has been observed [33, 51]. We found a positive correlation between dissociation efficiency and crystalline coherent length in the π-stacking direction L010, suggesting that delocalization of hole polarons in the crystalline phase plays a key role in charge dissociation. As shown in Table 1, high charge dissociation efficiencies are observed for crystalline polymers with a correlation length of L010 > 4 nm, which is comparable to the effective Coulombic capture radius at the DA interface, taking into account the entropic contribution to the Gibbs free energy [1]. We therefore conclude that large delocalization of hole wave functions in polymer crystalline phases promotes charge dissociation. It is worth noting that some amorphous polymers display efficient charge generation [36, 52]. In that case, delocalization of the electron wave function as a result of fullerene aggregation is a key driver of ultrafast long-range charge separation for OSCs, as reported previously [36].
Ultrafast charge dissociation in nonfullerene acceptor-based solar cells
The charge dissociation mechanisms in NFA-based OSCs are exemplified in blends of a benchmark donor polymer, PTB7-Th, and an efficient NFA, bay-di-PDI [31]. Figure 3a shows the femtosecond TA spectra for the PTB7-Th/bay-di-PDI blend under the selective excitation of PTB7-Th. The large positive band in the visible region is ascribed to ground-state bleaching (GSB), and a broad PIA tail observed at ~1400 nm is attributable to polymer singlet excitons. Singlet excitons rapidly decayed with a lifetime of 1.3 ps. The intrinsic exciton lifetime of PTB7-Th is ~220 ps, indicating that almost all excitons were converted into charged species. The PIA centered at 1150 nm became dominant within 10 ps, which can be ascribed to hole polarons on the donor polymer. Note that the PIA band of the bay-di-PDI radical anion was not observed because it fully overlaps with the large GSB band [53]. Hole polarons, then, remained almost unchanged until the nanosecond time scale. For comparison, Fig. 3b shows the TA spectra for the PTB7-Th/PC71BM blend film. As in the PDI blend, the polaron band remains unchanged until 1 ns. Note that an additional PIA band was observed at 1300 nm at later times only for the PC71BM blend, which we attribute to polymer triplet absorption formed through bimolecular charge recombination [54,55,56].

TA spectra of the a PTB7-Th/bay-di-PDI blend and b PTB7-Th/PC71BM blend excited at 650 nm with a fluence of 1.6 μJ cm−2. The spectra in the near-IR region (850–1050 nm) were rescaled to that of the IR region (1050–1400 nm) owing to a small change in the pump–probe overlap. Reprinted with permission from Ref. [31]. Copyright 2017 American Chemical Society
Figure 4a, b shows the normalized TA spectra in the visible region. We found that both the peak position and onset of the main GSB band blueshift even after singlet excitons have been fully quenched. This indicates that a small PIA overlap occurs in this region, with dynamics different from both singlet excitons and charges. In line with previous results [36], we attribute this to the electroabsorption (EA) of PTB7-Th. When an exciton dissociates into an e–h pair, a dipole-like local electric field is generated in its surroundings. This results in a Stark shift for surrounding molecules, adding a first-derivative-like spectrum component to the TA data. As the amplitude of the EA signal is a function of the local electric field, i.e., a function of the separation distance of the e–h pair, we can directly quantify the separation distance of the e–h pair. Figure 4c shows the time evolution of the EA amplitude. For the PC71BM blend, the EA amplitude is already large at 200 fs and reaches its maximum by 400–500 fs. As shown in the right axis of Fig. 4c, the EA amplitude was converted into electrostatic energy stored in the field per e–h pair, which was calculated as \(\frac{1}{2}\varepsilon _0\varepsilon _{{r}}{\int} {\left| E \right|^2 {\rm{d}}V}\) and calibrated against quasi-steady-state EA measurements on diode structures. During this short duration, an electrostatic energy of ~200 meV was stored in the field, which is well above the thermal energy at room temperature and on the same order as the CT state binding energy. This indicates that the e–h pair undergoes rapid spatial separation, despite the opposing Coulombic attraction. This picture is again inconsistent with the Braun–Onsager framework but is consistent with ballistic charge separation through the delocalized wave function [36]. Interestingly, the EA amplitude of the PDI blend is as large as that of the PC71BM blend, indicating that charge separation mechanisms for the PDI blend can be rationalized by the ballistic charge separation model. Before our study, ballistic charge separation was only observed in blends that consist of relatively large fullerene aggregates. However, we provided clear experimental evidence that ballistic charge separation can also take place in NFA-based blends, and hence, the model that charges separate coherently through delocalized states can be more widely applied to various types of OSCs.

a, b Normalized TA spectra in the visible region. Broken lines represent the TA spectra of the pristine PTB7-Th film as a reference. c Time evolution of the EA amplitude per unit charge obtained by dividing the EA amplitude by the charge signal. The right axis shows the total electrostatic energy stored in the field per electron–hole pair. The energy is obtained by assuming that half of the field is in the donor phase as \(\frac{1}{2}\varepsilon _0\varepsilon _{{r}}{\int} {\left| E \right|^2{\rm{d}}V}\) and by taking a value of 3 for εr. The spatially integrated square of the electric field was converted from the diode-based EA amplitude. Reprinted with permission from Ref. [31]. Copyright 2017 American Chemical Society
Figure 5a shows the time evolution of polaron signals in the PDI and PC71BM blends on the nanosecond time scale. We found no apparent difference in the charge dynamics between the PDI and PC71BM blends, indicating that there is no intrinsic disadvantage for NFAs in terms of charge generation; i.e., fullerenes are not so “special.” Figure 5b shows the excitation wavelength dependence of charge dynamics in the PDI blend. As clearly seen in the figure, the charge dynamics are independent of the excitation wavelength, indicating that both electron transfer from PTB7-Th to bay-di-PDI (excited at 650 nm, red line) and hole transfer from bay-di-PDI to PTB7-Th (excited at 500 nm, green line) result in efficient charge generation. This is consistent with the EQE spectra for this blend, which follows the absorption spectra. It is interesting to investigate the separate roles of electrons and holes in the charge dissociation process. We find that the charge dissociation process is strongly dependent on the morphology of both the donor and acceptor, indicating that both the donor and acceptor play an important role in the charge dissociation process.

a Normalized time evolution of the polaron band in the PDI (red) and PC71BM (black) blends. b Excitation wavelength dependence of polaron dynamics in the PDI blend. Reprinted with permission from Ref. [31]. Copyright 2017 American Chemical Society
Efficient charge separation with a small driving force
We have demonstrated the first efficient charge generation with small photon energy loss [28]. We used a highly efficient donor polymer, PNTz4T or PNOz4T, blended with PC71BM. The PNTz4T-based device had an Eg of 1.56 eV (based on absorption onset) and showed a PCE of 10.1% with a high short-circuit current density (JSC) of 19.4 mA cm−2 [57]. However, the Eloss was as large as 0.85 eV, resulting in a VOC of only 0.71 V. Such a large Eloss is partly because of the large LUMO energetic offset between PNTz4T and PC71BM. In contrast, the PNOz4T-based device showed an Eg of 1.52 eV [58], which is almost the same as that of the PNTz4T-based device, and a VOC of 0.96 V, resulting in an Eloss of 0.56 eV [28]. Surprisingly, the JSC and PCE were as high as 14.6 mA cm−2 and 8.9%, respectively. This results in our system having the highest known PCE among OSCs with an Eloss <0.6 eV (at least in 2015).
We expected that the origin of the small Eloss in the PNOz4T-based device is a small LUMO energetic offset. To confirm this, we measured the temperature dependence of the VOC, as shown in Fig. 6. The VOC increased linearly with decreasing temperature. It is well known that the effective bandgap (Egeff) obtained from the intersection at 0 K is a good measure for ECT because Egeff is identical to ECT at 0 K [59, 60]. Note that the ECT at room temperature is typically 0.1–0.2 eV above Egeff. The Egeff values for the PNOz4T- and PNTz4T-based devices were 1.38 and 1.07 eV, respectively. Thus, the difference between Eg and ECT in the PNOz4T-based device at room temperature would be <0.14 eV, while it is as large as 0.4–0.5 eV in the PNTz4T-based device. This is consistent with the observation that the electroluminescence spectrum of the PNOz4T/PC71BM device is almost the same as that of the pristine PNOz4T device. This indicates that charge generation in the PNOz4T-based device is efficient even though the LUMO energetic offset is smaller than the empirical threshold of 0.3 eV.

Temperature dependence of the VOC for the PNOz4T and PNTz4T-based devices. Adapted from Ref. [28]. Copyright 2015 Springer Nature
Figure 7a, b shows the TA spectra of the PNTz4T/PC71BM and PNOz4T/PC71BM blend films. Immediately after photoexcitation, PIA bands attributable to singlet excitons were observed at 1100–1400 nm in both blends. In the PNTz4T blend, singlet excitons had almost disappeared within 10 ps, which is a sign of fine morphology. Instead, another PIA band ascribable to PNTz4T polarons centered at 1100 nm was observed, which then slightly decayed on the nanosecond time scale through geminate recombination, probably as a result of the well-mixed disordered morphology. In contrast, singlet excitons decayed slowly in the PNOz4T blend, which implies large phase-separated domains or slow CT at the interface. Interestingly, a contribution from polaron absorption was observed for the TA data even at 0 ps, which indicates immediate CT of some excitons that generated very close to the interface. This suggests that CT itself is still fast and efficient even in the PNOz4T blend, and hence, the slow exciton decay kinetics are attributable to a large domain size. This is consistent with the difference in morphology of the two blends observed in the transmission electron micrography images. What is important in our findings is that no polaron decay was observed in the PNOz4T blend for up to 3 ns, which is clear evidence of efficient charge dissociation. We therefore conclude that, in the PNOz4T-based device, excitons dissociate into charges very efficiently without a large driving force. Although the mechanism of efficient charge generation without a large offset is still unclear, the highly crystalline nature of PNOz4T is probably essential, and this study clearly demonstrates the potential advantages of utilizing small offset systems.

a, b TA spectra of the PNTz4T/PC71BM and PNOz4T/PC71BM blend films. c, d Normalized time evolution of the singlet exciton (blue) and polaron (red) bands in the PNTz4T/PC71BM and PNOz4T/PC71BM blend films. Green squares represent the total density of singlet excitons and polarons. Adapted from Ref. [28]. Copyright 2015 Springer Nature
Conclusion
This focus review highlights our recent studies using TA spectroscopy to investigate charge generation and recombination dynamics in OSCs. The impacts of polymer crystallinity on charge dissociation efficiency, mechanisms of charge dissociation in NFA-based OSCs, and efficient charge generation with a small LUMO energetic offset were described. It was shown that the crystallization or aggregation of materials promotes charge dissociation, which is rationalized by the delocalization of charge wave functions in the crystalline or aggregate domains. Recent state-of-the-art OSCs consist of wide-bandgap donor polymers and low-bandgap NFAs with a small HOMO energetic offset, but understanding charge generation mechanisms with a small energetic offset remains a challenge. Further spectroscopic studies will provide a comprehensive understanding and clear design concepts for materials and devices.
References
Clarke TM, Durrant JR. Charge photogeneration in organic solar cells. Chem Rev. 2010;110:6736–67.
Lu L, Zheng T, Wu Q, Schneider AM, Zhao D, Yu L. Recent advances in bulk heterojunction polymer solar cells. Chem Rev. 2015;115:12666–731.
Inganäs O. Organic photovoltaics over three decades. Adv Mater. 2018;30:1800388.
Hou J, Inganäs O, Friend RH, Gao F. Organic solar cells based on non-fullerene acceptors. Nat Mater. 2018;17:119–28.
Guo JM, Ohkita H, Benten H, Ito S. Charge generation and recombination dynamics in poly(3-hexylthiophene)/fullerene blend films with different regioregularities and morphologies. J Am Chem Soc. 2010;132:6154–64.
Halls JJM, Walsh CA, Greenham NC, Marseglia EA, Friend RH, Moratti SC, et al. Efficient photodiodes from interpenetrating polymer networks. Nature. 1995;376:498–500.
Zhan C, Zhang X, Yao J. New advances in non-fullerene acceptor based organic solar cells. RSC Adv. 2015;5:93002–26.
McAfee SM, Topple JM, Hill IG, Welch GC. Key components to the recent performance increases of solution processed non-fullerene small molecule acceptors. J Mater Chem A. 2015;3:16393–408.
McNeill CR, Westenhoff S, Groves C, Friend RH, Greenham NC. Influence of nanoscale phase separation on the charge generation dynamics and photovoltaic performance of conjugated polymer blends: balancing charge generation and separation. J Phys Chem C. 2007;111:19153–60.
Westenhoff S, Howard IA, Hodgkiss JM, Kirov KR, Bronstein HA, Williams CK, et al. Charge recombination in organic photovoltaic devices with high open-circuit voltages. J Am Chem Soc. 2008;130:13653–8.
Hodgkiss JM, Campbell AR, Marsh RA, Rao A, Albert-Seifried S, Friend RH. Subnanosecond geminate charge recombination in polymer-polymer photovoltaic devices. Phys Rev Lett. 2010;104:177701.
Moore JR, Albert-Seifried S, Rao A, Massip S, Watts B, Morgan DJ, et al. Polymer blend solar cells based on a high-mobility naphthalenediimide-based polymer acceptor: device physics, photophysics and morphology. Adv Energy Mater. 2011;1:230–40.
Schubert M, Collins BA, Mangold H, Howard IA, Schindler W, Vandewal K, et al. Correlated donor/acceptor crystal orientation controls photocurrent generation in all-polymer solar cells. Adv Funct Mater. 2014;24:4068–81.
Gehrig DW, Roland S, Howard IA, Kamm V, Mangold H, Neher D, et al. Efficiency-limiting processes in low-bandgap polymer:perylene diimide photovoltaic blends. J Phys Chem C. 2014;118:20077–85.
Zhang GY, Zhao JB, Chow PCY, Jiang K, Zhang JQ, Zhu ZL, et al. Nonfullerene acceptor molecules for bulk heterojunction organic solar cells. Chem Rev. 2018;118:3447–507.
Wadsworth A, Moser M, Marks A, Little MS, Gasparini N, Brabec CJ, et al. Critical review of the molecular design progress in non-fullerene electron acceptors towards commercially viable organic solar cells. Chem Soc Rev. 2019;48:1596–625.
Yuan J, Zhang YQ, Zhou LY, Zhang GC, Yip HL, Lau TK, et al. Single-junction organic solar cell with over 15% efficiency using fused-ring acceptor with electron-deficient core. Joule. 2019;3:1140–51.
Lin YB, Adilbekova B, Firdaus Y, Yengel E, Faber H, Sajjad M, et al. 17% efficient organic solar cells based on liquid exfoliated WS2 as a replacement for PEDOT:PSS. Adv Mater. 2019;31:9.
Cui Y, Yao HF, Zhang JQ, Zhang T, Wang YM, Hong L, et al. Over 16% efficiency organic photovoltaic cells enabled by a chlorinated acceptor with increased open-circuit voltages. Nat Commun. 2019;10:8.
Menke SM, Ran NA, Bazan GC, Friend RH. Understanding energy loss in organic solar cells: toward a new efficiency regime. Joule. 2018;2:25–35.
Li WW, Hendriks KH, Furlan A, Wienk MM, Janssen RAJ. High quantum efficiencies in polymer solar cells at energy losses below 0.6 eV. J Am Chem Soc. 2015;137:2231–4.
Tamai Y, Ohkita H, Shimada J, Benten H, Ito S, Yamanaka S, et al. Dynamical excimer formation in rigid carbazolophane via charge transfer state. J Phys Chem A. 2013;117:7776–85.
Tamai Y, Ohkita H, Benten H, Ito S. Singlet fission in poly(9,9′-di-n-octylfluorene) films. J Phys Chem C. 2013;117:10277–84.
Tamai Y, Tsuda K, Ohkita H, Benten H, Ito S. Charge-carrier generation in organic solar cells using crystalline donor polymers. Phys Chem Chem Phys. 2014;16:20338–46.
Tamai Y, Ohkita H, Benten H, Ito S. Triplet exciton dynamics in fluorene-amine copolymer films. Chem Mater. 2014;26:2733–42.
Tamai Y, Matsuura Y, Ohkita H, Benten H, Ito S. One-dimensional singlet exciton diffusion in poly(3-hexylthiophene) crystalline domains. J Phys Chem Lett. 2014;5:399–403.
Tamai Y, Ohkita H, Benten H, Ito S. Exciton diffusion in conjugated polymers: from fundamental understanding to improvement in photovoltaic conversion efficiency. J Phys Chem Lett. 2015;6:3417–28.
Kawashima K, Tamai Y, Ohkita H, Osaka I, Takimiya K. High-efficiency polymer solar cells with small photon energy loss. Nat Commun. 2015;6:10085.
Kasai Y, Tamai Y, Ohkita H, Benten H, Ito S. Ultrafast singlet fission in a push-pull low-bandgap polymer film. J Am Chem Soc. 2015;137:15980–3.
Tamai Y, Ohkita H, Namatame M, Marumoto K, Shimomura S, Yamanari T, et al. Light-induced degradation mechanism in poly(3-hexylthiophene)/fullerene blend solar cells. Adv Energy Mater. 2016;6:1600171.
Tamai Y, Fan Y, Kim VO, Ziabrev K, Rao A, Barlow S, et al. Ultrafast long-range charge separation in nonfullerene organic solar cells. ACS Nano. 2017;11:12473–81.
Umeyama T, Igarashi K, Sasada D, Tamai Y, Ishida K, Koganezawa T, et al. Efficient light-harvesting, energy migration, and charge transfer by nanographene-based nonfullerene small-molecule acceptors exhibiting unusually long excited-state lifetime in the film state. Chem Sci. 2020;11:3250–7.
Mauer R, Howard IA, Laquai F. Effect of nongeminate recombination on fill factor in polythiophene/methanofullerene organic solar cells. J Phys Chem Lett. 2010;1:3500–5.
Howard IA, Mauer R, Meister M, Laquai F. Effect of morphology on ultrafast free carrier generation in polythiophene:fullerene organic solar cells. J Am Chem Soc. 2010;132:14866–76.
Shoaee S, Subramaniyan S, Xin H, Keiderling C, Tuladhar PS, Jamieson F, et al. Charge photogeneration for a series of thiazolo-thiazole donor polymers blended with the fullerene electron acceptors PCBM and ICBA. Adv Funct Mater. 2013;23:3286–98.
Gélinas S, Rao A, Kumar A, Smith SL, Chin AW, Clark J, et al. Ultrafast long-range charge separation in organic semiconductor photovoltaic diodes. Science. 2014;343:512–6.
Dimitrov SD, Durrant JR. Materials design considerations for charge generation in organic solar cells. Chem Mater. 2014;26:616–30.
Etzold F, Howard IA, Forler N, Cho DM, Meister M, Mangold H, et al. The effect of solvent additives on morphology and excited-state dynamics in PCPDTBT:PCBM photovoltaic blends. J Am Chem Soc. 2012;134:10569–83.
Motaung DE, Malgas GF, Arendse CJ. Correlation between the morphology and photo-physical properties of P3HT:Fullerene blends. J Mater Sci. 2010;45:3276–83.
Salleo A, Kline RJ, DeLongchamp DM, Chabinyc ML. Microstructural characterization and charge transport in thin films of conjugated polymers. Adv Mater. 2010;22:3812–38.
Chen H-Y, Hou J, Hayden AE, Yang H, Houk KN, Yang Y. Silicon atom substitution enhances interchain packing in a thiophene-based polymer system. Adv Mater. 2010;22:371–5.
Chen D, Nakahara A, Wei D, Nordlund D, Russell TP. P3HT/PCBM bulk heterojunction organic photovoltaics: correlating efficiency and morphology. Nano Lett. 2011;11:561–7.
Agostinelli T, Ferenczi TAM, Pires E, Foster S, Maurano A, Müller C, et al. The role of alkane dithiols in controlling polymer crystallization in small band gap polymer:fullerene solar cells. J Polym Sci B Polym Phys. 2011;49:717–24.
Yamamoto S, Ohkita H, Benten H, Ito S. Role of interfacial charge transfer state in charge generation and recombination in low-bandgap polymer solar cell. J Phys Chem C. 2012;116:14804–10.
Guilbert AAY, Frost JM, Agostinelli T, Pires E, Lilliu S, Macdonald JE, et al. Influence of bridging atom and side chains on the structure and crystallinity of cyclopentadithiophene–benzothiadiazole polymers. Chem Mater. 2014;26:1226–33.
Mayer AC, Toney MF, Scully SR, Rivnay J, Brabec CJ, Scharber M, et al. Bimolecular crystals of fullerenes in conjugated polymers and the implications of molecular mixing for solar cells. Adv Funct Mater. 2009;19:1173–9.
Cates NC, Gysel R, Beiley Z, Miller CE, Toney MF, Heeney M, et al. Tuning the properties of polymer bulk heterojunction solar cells by adjusting fullerene size to control intercalation. Nano Lett. 2009;9:4153–7.
Hammond MR, Kline RJ, Herzing AA, Richter LJ, Germack DS, Ro H-W, et al. Molecular order in high-efficiency polymer/fullerene bulk heterojunction solar cells. ACS Nano. 2011;5:8248–57.
Liu F, Gu Y, Jung JW, Jo WH, Russell TP. On the morphology of polymer-based photovoltaics. J Polym Sci B Polym Phys. 2012;50:1018–44.
Sweetnam S, Graham KR, Ndjawa GON, Heumueller T, Bartelt JA, Burke TM, et al. Characterization of the polymer energy landscape in polymer:fullerene bulk heterojunctions with pure and mixed phases. J Am Chem Soc. 2014;136:14078–88.
Jamieson FC, Agostinelli T, Azimi H, Nelson J, Durrant JR. Field-independent charge photogeneration in PCPDTBT/PC70BM solar cells. J Phys Chem Lett. 2010;1:3306–10.
Ie Y, Morikawa K, Zajaczkowski W, Pisula W, Kotadiya NB, Wetzelaer G-JAH, et al. Enhanced photovoltaic performance of amorphous donor–acceptor copolymers based on fluorine-substituted benzodioxocyclohexene-annelated thiophene. Adv Energy Mater. 2018;8:1702506.
Shivanna R, Shoaee S, Dimitrov S, Kandappa SK, Rajaram S, Durrant JR, et al. Charge generation and transport in efficient organic bulk heterojunction solar cells with a perylene acceptor. Energy Environ Sci. 2014;7:435–41.
Rao A, Chow PC, Gélinas S, Schlenker CW, Li CZ, Yip HL, et al. The role of spin in the kinetic control of recombination in organic photovoltaics. Nature. 2013;500:435–9.
Chow PC, Gélinas S, Rao A, Friend RH. Quantitative bimolecular recombination in organic photovoltaics through triplet exciton formation. J Am Chem Soc. 2014;136:3424–9.
Gehrig DW, Howard IA, Laquai F. Charge carrier generation followed by triplet state formation, annihilation, and carrier recreation in PBDTTT-C/PC60BM photovoltaic blends. J Phys Chem C. 2015;119:13509–15.
Vohra V, Kawashima K, Kakara T, Koganezawa T, Osaka I, Takimiya K, et al. Efficient inverted polymer solar cells employing favourable molecular orientation. Nat Photonics. 2015;9:403–8.
Kawashima K, Osaka I, Takimiya K. Effect of chalcogen atom on the properties of naphthobischalcogenadiazole-based π-conjugated polymers. Chem Mater. 2015;27:6558–70.
Vandewal K, Tvingstedt K, Manca JV, Inganäs O. Charge-transfer states and upper limit of the open-circuit voltage in polymer:fullerene organic solar cells. IEEE J Sel Top Quantum Electron. 2010;16:1676–84.
Vandewal K, Tvingstedt K, Gadisa A, Inganäs O, Manca JV. Relating the open-circuit voltage to interface molecular properties of donor: acceptor bulk heterojunction solar cells. Phys Rev B. 2010;81:125204.
Acknowledgements
The author thanks Prof Sir Richard Friend, Dr S. Matthew Menke, Prof Itaru Osaka, Prof Shinzaburo Ito, and Prof Hideo Ohkita for fruitful discussions. The author also thanks all other collaborators. This work was partly supported by JSPS Postdoctoral Fellowships for Research Abroad, JSPS KAKENHI Grant-in-Aid for Young Scientists (B) No. 17K14527, and JST PRESTO program Grant Number JPMJPR1874. The author also acknowledges financial support from Trycom Advance Co., Ltd.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Tamai, Y. Delocalization boosts charge separation in organic solar cells. Polym J 52, 691–700 (2020). https://doi.org/10.1038/s41428-020-0339-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41428-020-0339-4
- Springer Nature Limited
This article is cited by
-
Delocalization suppresses nonradiative charge recombination in polymer solar cells
Polymer Journal (2022)
-
Triplet sensitization via charge recombination at organic heterojunction for efficient near-infrared to visible solid-state photon upconversion
Communications Materials (2022)
-
Ultrafast charge generation in a homogenous polymer domain
Scientific Reports (2022)