
Abstract
ARID1A, an SWI/SNF chromatin-remodeling gene, is commonly mutated in cancer and hypothesized to be a tumor suppressor. Recently, loss-of-function of ARID1A gene has been shown to cause intellectual disability. Here we generate Arid1a conditional knockout mice and investigate Arid1a function in the hippocampus. Disruption of Arid1a in mouse forebrain significantly decreases neural stem/progenitor cells (NSPCs) proliferation and differentiation to neurons within the dentate gyrus (DG), increasing perinatal and postnatal apoptosis, leading to reduced hippocampus size. Moreover, we perform single-cell RNA sequencing (scRNA-seq) to investigate cellular heterogeneity and reveal that Arid1a is necessary for the maintenance of the DG progenitor pool and survival of post-mitotic neurons. Transcriptome and ChIP-seq analysis data demonstrate that ARID1A specifically regulates Prox1 by altering the levels of histone modifications. Overexpression of downstream target Prox1 can rescue proliferation and differentiation defects of NSPCs caused by Arid1a deletion. Overall, our results demonstrate a critical role for Arid1a in the development of the hippocampus and may also provide insight into the genetic basis of intellectual disabilities such as Coffin–Siris syndrome, which is caused by germ-line mutations or microduplication of Arid1a.
Similar content being viewed by others


Introduction
The SWItch/Sucrose Non-Fermentable (SWI/SNF)-like chromatin-remodeling complex (BAF complex) is critical for modulation of gene expression, involving many cellular processes such as proliferation and differentiation [1, 2], lineage specification, maintenance of stem cell pluripotency and DNA repair [3,4,5,6]. ARID1A (AT-rich interactive domain-containing protein 1A), also known as BAF250A, is one of the main BAF complex subunits. Loss-of-function of Arid1a disrupts SWI/SNF targeting and nucleosome remodeling and consequently aberrant gene expression [7, 8]. As a tumor suppressor, Arid1a is associated with the development, survival, and progression of cancer cells, and its dysfunction is a key contribution to ovarian, endometrial, gastric, and breast cancers [8,9,10,11,12,13,14].
Recent genetic studies have reported that both germ-line mutations and microduplication of ARID1A cause Coffin–Siris syndrome (CSS), a genetic disorder characteristically exhibiting intellectual disability (ID) and multiple organ abnormalities [2, 15]. Ablation of Arid1a in early mouse embryos results in developmental arrest and absence of the mesodermal layer [16]. Arid1a deficiency compromises embryonic stem (ES) cell pluripotency, severely inhibits self-renewal, and promotes differentiation into primitive endoderm-like cells, indicating a critical role of Arid1a in mammalian embryonic development [16]. Although Arid1a shows constitutive expression in ES cells, neuronal progenitors and post-mitotic neurons, it may have different functions due to the structural divergence of developmental stage-specific BAF assemblies [17]. Therefore, a deeper inquiry into the role and mechanism of Arid1a in neural development and intellectual disability is necessary.
NSPCs generate new neurons throughout life primarily in two regions: the subgranular zone of the hippocampal dentate gyrus (DG) and the subventricular zone (SVZ) of the lateral ventricles [18, 19]. Proliferation and neuronal differentiation of NSPCs are controlled by extrinsic and intrinsic signals, including microenvironment, secreted molecules, transcription factors and chromatin regulators. Among these, chromatin regulators, have been shown to be required for the regulation of NSPCs proliferation and differentiation [20, 21]. PRC2 component EZH2 has been known as a chromatin regulator through targeting miR-203 to promote the proliferation of NSPCs [21]. UTX demethylates H3K27me3 at the Pten promoter and contributes to the NSPCs proliferation and differentiation during embryonic corticogenesis [22]. Our previous study demonstrates that Arid1a regulates the proliferation and differentiation of NSPCs during embryonic cortical development [23]. Overall, these studies clearly suggest that chromatin factors play key roles in NSPCs proliferation and differentiation, whereas the function of many chromatic factors that are highly expressed in postnatal NSPCs is still unclear. Previously published data show that chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice [24]. However, we still have no ideas about the roles of chromatin remodeling underlying ARID1A in NSPCs and neurodevelopment.
Here, we develop a forebrain conditional Arid1a knockout mouse model that exhibits growth retardation phenotypes. We found that the disruption of Arid1a results in postnatal lethality, impaired cell proliferation and differentiation of hippocampus, and smaller DG size. Mechanistically, we demonstrate that Arid1a is required for the maintenance of open chromatin and regulates the development of DG by controlling the expression of Prox1. These results highlight the critical role of Arid1a in a series of essential events that cumulatively orchestrate the developmental formation of the DG, and thus provide molecular evidence supporting that dysregulation of ARID1A contributes to the etiology of CSS.
Results
Arid1a cKO mice exhibit disorganized dentate gyrus
To explore the role of Arid1a in the central nervous system, we first examined its expression patterns in neural stem/progenitor cells, neurons and astrocytes. We performed ARID1A immunostaining of neural lineage cells by using Nestin-GFP and Thy1-GFP transgenic mice in which NSPCs or neurons were labeled with the green fluorescent protein (GFP). The results showed that ARID1A localized mainly in the Nestin-positive NSPCs and Thy1-positive neurons (Supplementary Fig. S1A). We then co-stained ARID1A with astrocyte specific maker GFAP and found that ARID1A was also expressed in the astrocytes (Supplementary Fig. S1A). Next, we examined the expression of Arid1a at different embryonic and postnatal stages and found that Arid1a was highly expressed at the prenatal stage, whereas both mRNA and protein levels of Arid1a were gradually decreased after birth (from P0 to P21) (Supplementary Fig. S1B–D). Therefore, these results suggest that Arid1a involve in NSPCs and neural development.
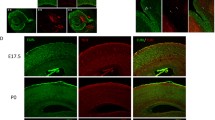
To determine the function of Arid1a in the developing brain, we generated Arid1aflox/flox; Emx1-cre mice (hereafter referred to as Arid1a cKO) by crossing Arid1aflox/flox mice with Emx1-Cre transgenic mice to delete Arid1a specifically within the cortex and hippocampus (Supplementary Fig. 1E). Immunofluorescence staining (Fig. 1A) and western blotting (Fig. 1B, C) analyses in hippocampus at embryonic day 16.5 confirmed that Arid1a was successfully deleted. To study the integrity of SWI/SNF after Arid1a deletion, we tested the expression of BRG1, the central ATPase subunit of SWI/SNF. The western blotting results showed that BRG1 was significantly downregulated after Arid1a specific deletion in the forebrain (Fig. 1B, C). Arid1a cKO mice were born at the expected Mendelian ratios and were indistinguishable from their WT littermates at postnatal day 0 (P0). However, most Arid1a cKO mice (>80%) died at 4–5 weeks after birth (Supplementary Fig. S1F). Histologic analysis of cKO mice revealed striking abnormalities in the hippocampal formation at P21 (Fig. 1D), which lacked the dentate granule cell layer and displayed disorganized dentate hilus and pyramidal layers (Fig. 1D). We then examined the granule cell layer (GCL) in Arid1a cKO mice at prenatal (E16.5 and E18.5) and postnatal stages (P0, P7, P14 and P21). In Nissl-stained sections, the developing pyramidal cell layer in Ammon’s horn of the hippocampus was similar in morphology (such as breadth) between Arid1a cKO mice and their WT littermates (Fig. 1E). By contrast, the DG region in cKO mice was hardly discriminated from E16.5 to E18.5, and displayed a significantly smaller GCL volume at P0 compared with WT mice (Fig. 1E). Moreover, the difference GCL volume between cKO and WT mice further magnified as they grew older (Fig. 1E, F).

A Representative images of fluorescent immunohistochemistry showing deletion of Arid1a in the dentate gyrus of hippocampus in cKO mice. Scale bars, 20 µm. B Western blot analysis of ARID1A and BRG1 in Arid1a cKO mice from hippocampus tissues at embryonic day 16.5. C Quantification of the density of the ARID1A and BRG1 protein bands by normalization to the intensity of β-Actin bands in (B) (n = 6 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, unpaired two-tailed t-test. D Representative images of brain slice stained by Hematoxylin and eosin staining of staining, from WT and Arid1a cKO mice at P21. Upper panel, scale bars, 500 µm; Lower panel, scale bars,100 µm. E Representative images Nissl staining of E16.5, E18, P0, P7 hippocampus sections from WT and Arid1a cKO mice and show the relative levels of defective in the development of the DG after Aird1a loss. White dotted lines indicate the boundaries of the hippocampus DG. Scale bar, 100 μm. F Quantification of the lengths of the upper blades of the dentate gyrus, the lengths of the lower blades of the dentate gyrus and the DG volume in WT and Arid1a cKO mice at different time points in (D-E) (n = 3–4 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpairedi two-tailed t-test.
Development of DG involves various cell types including NSPCs and granule cells, therefore the cell compartments in the DG of Arid1a cKO mice are highly likely altered as their remarkably reduced size. The number of BLBP+ NSPCs in Arid1a cKO mice was significantly decreased at P0 compared with the WT controls (Supplementary Fig. 2A, B). In addition, the numbers of Calretinin+ mossy cells and Calbindin+ granule cells were all significantly reduced in the DG area of cKO mice at P21 (Supplementary Fig. 2C–F). These data suggest that Arid1a is essential for DG development.
Loss of Arid1a damages neuronal cell composition in hippocampus
Distinct lamination appeared in the dentate gyrus at P7 [25]. Dendritic spines begin to increase in density during the first week after birth and with a peak in the third week [26]. We thus performed scRNA-seq (10x Genomics platform) to examine the abnormality in the Arid1a cKO hippocampus at P7 from three mice. After quality control and filtering, 14,894 cells from cKO samples and 15,520 cells from WT samples were used for further analysis. To identify the major cell types in the hippocampus, we used unsupervised clustering and identified 11 major distinct clusters according to the expression of canonical gene markers (Fig. 2A). These cells included progenitor cells (Mki67, Ascl1), astrocytes (Gfap, Aqp4), neurons (Neurod2, Rbfox3), microglia (Cx3cr1, P2ry12), oligodendrocyte precursor cells (OPCs) (Olig2, Pdgfra), Cajal-Retzius cells (Ndnf, Clstn2), ependymal cells (Ccdc153, Dnah11), endothelial cells (Cd93, Arhgap29), blood (Hba-a1, Hbb-bs), mural cells (Col3a1, Arhgap29) (Fig. 2C and Supplementary Fig. 3A). Cells clustered largely by connections between different cell types (Supplementary Fig. 3B).

A UMAP visualization of mouse hippocampal cells at P7. B UMAP visualization of cell types in WT and Arid1a cKO mice. C Violinplot showing the expression of marker genes of astrocytes, blood, Cajal-Retzius cells, endothelial cells, ependymal cells, microglia, mural cells, neurons, OPCs, other and progenitor cells. D The proportion of WT and Arid1a cKO mice in each cell type. E Scatterplot showing differentially expressed genes (DEGs) between WT and Arid1a cKO in progenitor cells. Each red dot denotes an individual gene with p_val_adj<0.05 and abs (average_log2 fold change) > 0 in the WT/Arid1a cKO progenitor cells comparisons. Example genes are labeled with the gene name. F GO analysis showing enriched terms in progenitor cells. Each of the top five pathways was selected for presentation.
To reveal the differences in cell compositions between WT and cKO, we analyzed the relative percentage of the 11 major cell types based on scRNA-seq data. The relative percentage of progenitors decreased 10% in cKO mice. Of note, the percentage of neurons decreased 20% in cKO mice, compared with WT mice. The massive decrease of neurons was in accordance with the smaller volume of hippocampus in cKO mice (Fig. 2B, D). Next, to investigate the transcriptomic changes of different cell types in cKO mice, we performed differentially expressed genes (DEGs) analysis (Supplementary Fig. 3C). We found progenitor cells exhibited differential expression of Ccnd1, Ccnd2, Nrn1, and Npy, which were involved in mitotic cell cycle, cell cycle phase transition and cell differentiation (Fig. 2E, F). These data suggest that the proliferation of NSPCs might be altered in Arid1a cKO hippocampus compared to WT hippocampus.
Arid1a regulates neuronal dynamic changes in DG
To characterize gene expression changes in neurons between WT and cKO mice, we first identified 2 groups according to known cell type and developmental markers (Fig. 3A). Elavl2, Dkk3, Homer3, and Pcp4 which have been identified as markers of cornu ammonis (CA), while Prox1 was expressed in DG (Supplementary Fig. 4A). To further confirm the lineage relationships and neurons in the DG region are most affected, we used Monocle analysis. Monocle 2 identified a bifurcating trajectory with three branches. Progenitor cells (PCs) were the beginning and end of the trajectory at two branches, with neurons in CA or DG were distributed at the other end of the trajectory branch, indicating their neuronal identity (Fig. 3B). Using pseudo-time analysis, we found Elavl2, Pcp4, Prox1 expression were gradually upregulated and Sox2 expression was downregulated along pseudo-temporally ordered paths from progenitor cells to neurons (CA/DG) (Fig. 3C). To further uncover the different regulating modules of gene expression during the development of neurons, we clustered genes using Monocle 2. Three different gene expression modules along with pre-branch(root), cell fate1(state 1–2), cell fate 2(state 3) of neuron development were identified by branched expression analysis modeling (BEAM) for significantly regulated genes. In addition, we defined three distinct development stages based on the three different gene expression modules, including the naïve stage (module 1), intermediate stage (module 2), and mature stage (module 3) (Supplementary Fig. 4B).

A UMAP visualization of neurons with 2 cell subpopulations. B Single-cell trajectory of the development of neurons by Monocle analysis. Two lineages, representing PC lineage and neuronal lineage, were identified. PC, progenitor cells. C Expression of marker genes with pseudo-time. Points in the figure are colored with different cell types. D Expression of marker genes with pseudo-time. Points in the figure are colored with WT and cKO respectively. E Violinplot showing the expression of selected genes between WT and Arid1a cKO in neurons.
To help in determining the biological processes of neurons development, Gene Ontology (GO) term enrichment (top three GO terms) was performed for three different gene expression modules(Supplementary Fig. 4B). Module 1 was mainly comprised of gene sets that were involved in the biological process of the mitotic cell cycle(Mki67, Cdk1, Ccnd2, Hmgb1), DNA packaging (Top2a, Cdk1, Hmgb1), which played a vital part in cell proliferation. The genes enriched in Module 2 were involved in the positive regulation of neuron differentiation (Id4, Hes5, Mmd2), axon ensheathment in central nervous systems (Apoe, Id4, Hes5). In module 3, the enriched genes were upregulated in cell fate 2(state 3), which was involved in synapse maturation (Grbra2, Grin2b, Nrn1, Reln, Neurod2), granule layer formation (Nrxn1, Prox1, Dcx, Nfix,), dentate gyrus development (Prox1, Reln, Neurod6), cognition (Nrxn1, Meis2, Reln, Neurod2, Grin2b). In summary, the common and distinct gene regulation patterns were constructed in neuron development, which contributed to an in-depth understanding of dentate gyrus development processes and underlying regulatory basis.
Genes transiently regulated at the bifurcation point where neuronal lineage separated from PC lineage could have a critical function in promoting the commitment of progenitor cells. Using Monocle 2, we found that the expression of Prox1 was significantly decreased in the neuron of DG in Arid1a cKO mice compared with WT mice, while the markers of CA such as Elavl2, Pcp4 were decreased slightly in Arid1a cKO mice compared with WT mice (Fig. 3D). In addition, we found that neurons of DG displayed differential expression of Prox1, Calb2, Rbfox3, Casp3 and Bax, which were associated with granule cells development, neuron differentiation and cell apoptosis (Fig. 3E and Supplementary Fig. 4C). Together, our data therefore indicate a sequential activation of Elavl2, Pcp4, Prox1 during PC commitment and confirmed a strong change in cell differentiation and cell apoptosis in Arid1a cKO mice DG.
Arid1a is required for the proliferation and differentiation of NSPCs
To further confirm the phenotypes of Arid1a deficiency on DG development, we compared the number of dividing cells (Ki67+ cells) in the DG region in Arid1a cKO and WT littermates. The results showed that Ki67+ cells decreased dramatically in Arid1a cKO mice at all developmental stages (Fig. 4A, B). To examine whether Arid1a deficiency alters the proliferation and division of neural stem and progenitor cells (NSPCs) in DG, the mice received a single intra-peritoneal (i.p.) injection of bromodeoxyuridine (BrdU), and brains were collected 2 h later. The quantification results demonstrated that the number of BrdU+ cells was significantly reduced in SGZ of Arid1a cKO mice from E16.5 to P21 (Fig. 4C, D). These results indicate a pivotal role of ARID1A in the proliferation of NSPCs.

A Representative images of Ki67 (green) staining in the dentate gyrus of P7 from WT and Arid1a cKO mice. DAPI, blue. Scale bar, 50 μm. B Quantification of the number of Ki67 positive cells per area in the dentate gyrus of WT and Arid1a cKO mice at E16.5, P0, P7, P14, P21 (WT, n = 3 mice; cKO, n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired two-tailed t-test. C Immunolabeling of BrdU(red) after 2 h of incorporation in the dentate gyrus of WT and Arid1a cKO mice at P7. Scale bar, 20 μm. D Quantification of BrdU+ cell in the dentate gyrus of WT and Arid1a cKO mice at E16.5, P0, P7, P14, P21(WT, n = 3 mice; cKO, n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired two-tailed t-test. E Representative images the DCX (red) staining in the DG from WT and Arid1a cKO mice at P21. Scale bar, 20 μm. F Quantification of DCX + cell in the DG of WT and Arid1a cKO mice at P21. (WT, n = 3 mice; cKO, n = 3 mice). Data are represented as mean ± SEM. ****p < 0.0001, unpaired two-tailed t-test. G Representative images of TUNEL (red) staining in the dentate gyrus of P21 from WT and Arid1a cKO mice. DAPI, blue. Scale bar, 50 μm. H Quantification of the number of TUNEL positive cells per area in the dentate gyrus of WT and Arid1a cKO mice at E16.5, P0, P7, P21(WT, n = 3 mice; cKO, n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired two-tailed t-test.
To evaluate whether Arid1a cKO also affects neural differentiation in DG, we first analyzed the expression of DCX in the DG of Arid1a WT and cKO mice. In support of this, we also found that Arid1a cKO mice had fewer DCX+ cells (types 2b and 3 NPCs and immature neurons) in the SGZ (Fig. 4E, F). These results clearly suggest that Arid1a is required for neural differentiation and cell fate determination in vivo. Furthermore, we found that the Arid1a cKO mice brains displayed drastically increased apoptosis in the DG compared with their littermate controls (Fig. 4G, H). To further confirm the role of Arid1a in the cell fate decision of DG in vivo, we constructed an inducible Arid1a−/− mouse model by injecting AAV-CRE-GFP virus into the hippocampus of 4–6-week-old Arid1aflox/flox mice to delete Arid1a acutely. In this induced depletion model, we found that Ki67+ GFP+ proliferative cells and DCX+GFP+ differentiated cells in the DG region were largely decreased, meanwhile, massive apoptosis dramatically increased after Arid1a deletion, consistent with our genetic knockout system (Supplementary Fig. 5A–E). Taken together, these data showed that the deletion of Arid1a impaired the proliferation and differentiation of NSPCs in the dentate gyrus and induced cell apoptosis.
To further assess the function of Arid1a in neurogenesis, we isolated hippocampal NSPCs from postnatal Arid1a cKO mice and their WT littermates. To assess the proliferation of NSPCs, we pulse-labeled the cells with BrdU for 6 h (Fig. 5A). Quantification of BrdU+ Nestin+ labeled cells demonstrated that less cKO NSPCs had incorporated BrdU than that of WT NSPCs (Fig. 5A, B). Moreover, in an in vitro assay, we isolated hippocampal NSPCs from Arid1a cKO or WT pups at postnatal, and cultured them in the neural differentiation medium. We observed a decreased number of Tuj1+ cells that was differentiated from Arid1a cKO NSPCs (Fig. 5C, D). These results indicate that Arid1a plays essential role in proliferation and cell differentiation of NSPCs in hippocampus in vitro.

A Representative images of WT and Arid1a cKO neural progenitor/stem cells labeled by BrdU(red) and Nestin(green) from three independent experiments (n = 3 mice). DAPI, blue, Scale bar, 20 μm. B Quantification of BrdU and Nestin-positive cells in WT and Arid1a cKO neural stem cells. Data are represented as mean ± SEM. ***p < 0.001, unpaired two-tailed t-test. C Representative images of differentiated neurons stained by Tuj1 (Red). DAPI, blue. D Quantification of Tuj1 positive cells in WT and Arid1a cKO group. Data are represented as mean ± SEM. **p < 0.01, unpaired two-tailed t-test.
Loss of Arid1a results in profiling changes in histone modifications and abnormal gene transcriptions
To investigate the consequences of altered SWI/SNF targeting induced by Arid1a deletion, we characterized histone modifications associated with cis-regulatory elements (H3K4me3, H3K27me3, and H3K27ac) at SWI/SNF binding sites in the hippocampus of wild-type and Arid1a cKO mice at E16.5. As expected, most of these three histone markers are enriched around the TSS of protein coding genes (Fig. 6A). While, among these three histone markers, H3K4me3 and H3K27ac downregulate the enrichments in TSS, H3K27me3 upregulates the enrichment (Fig. 6A). H3K4me3 and H3K27ac modifications enrichment at the TSS regions is important for gene activation. The Venn diagram (Fig. 6B) and Gene Ontology (GO) enrichment (Supplementary Fig. 6A) analysis of 47 downregulated genes with H3K4me3 and H3K27ac modifications showed enrichment in the pathways involved in dentate gyrus development, neuron differentiation and synapse maturation.

A Average profiles and heatmaps of histone marks and BRG1 in wild-type (WT) and conditional knockout (cKO) mice at peak centers 2500 bp upstream and downstream with decreased H3K27ac binding. B Venn diagram showing the overlap of dysregulated genes among H3K4me3, H3K27ac ChIP-seq peak and scRNA DEGs. C GO enrichment analysis of 46 genes with decreased H3K4me3 and H3K27ac enrichment and scRNA DEGs in neurons and progenitor cells in Arid1a cKO hippocampi. D RT-qPCR analysis confirmed that organ growth, granule cell differentiation and dentate gyrus-associated genes were downregulated in Arid1a cKO P7 hippocampus (n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, unpaired two-tailed t-test. E Genome-browser view at Prox1 gene of different sequencing data sets.
To identify the underlying molecular mechanism, we examined the effect of Arid1a deletion on Brg1 expression. We found that BRG1 protein levels decreased in the cKO group (Fig. 1B, C). Because components of the BAF complex contribute to the specificity of the BAF complex, these results indicate that Brg1 recruitment may be affected by Arid1a deletion. Based on previously published data, we found that Brg1 also accumulates at the TSS region (Fig. 6A). These data suggest that Arid1a may be required for proper recruitment of Brg1 to maintain proper nucleosome configuration for gene expression in the hippocampus.
To further elucidate gene regulatory mechanisms underlying the regulation of Arid1a on the hippocampus development, we integrate H3K4me3 ChIP-seq, H3K27ac ChIP-seq, and scRNA-seq DEGs in progenitor cells and neurons to enrich the target genes under the epigenetic modulations (Fig. 6B). Go analysis (Biological of Process) show that these 46 overlapping DEGs are involved in the regulation of organ growth, dentate gyrus development, granule cells differentiation (Fig. 6C). Especially Prox1 [27], Tmem108 [28] have been reported to be involved in the regulation of DG development. During all the examined targets (Fig. 6D), we mainly focus on the homeobox gene Prox1, which is highly expressed in several brain regions (i.e., cortex, DG, thalamus, hypothalamus, cerebellum) during prenatal and postnatal stages of development [27, 29]. Published data have shown that Prox1 is expressed through all the stages of DG formation and is required for the maturation of granule cells during DG development [27]. Next, we assayed the binding of three histone markers on Prox1. The results showed decreases in H3K4me3 and H3K27ac and an increase in the H3K27me3 marker at the promoter region of Prox1(Fig. 6E). We also found BRG1 has binding on the Prox1 promoter (Fig. 6E). Consistent with this, the expression level of Prox1 was downregulated in cKO mice. Therefore, these results indicate Arid1a mediates BAF functions to establish the poised chromatin configuration, which is essential for the proper DG development.
Overexpression of Prox1 rescues the proliferation and differentiation defects of neural stem/progenitor cells in Arid1a cKO hippocampus
To examine a functional relationship between Arid1a and Prox1 in mediating neural development, we then examined the expression of Prox1. Immunostaining results showed that the expression of Prox1 is indeed reduced in the DG of Arid1a cKO mice at prenatal and postnatal stages (Fig. 7A, B). To determine whether Arid1a regulates Prox1 expression directly, we chose 3000 base pairs (bp) promoter according to reference and cloned every 1000 bp of the Prox1 promoter. The dual-luciferase reporter assay suggested that ARID1A was bound to Prox1 promoter-1(Supplementary Fig. 7A). To further validate this interaction, we performed ChIP-qPCR assays in E16.5 NSPCs isolated from WT and Arid1a cKO mouse hippocampus. We verified that Arid1a directly bound to Prox1 promoter (Locus −128) (Fig. 7C). These data suggest that Arid1a directly regulated Prox1 expression via binding to Prox1 promoter.

A Representative images of Prox1(green) staining on P21 WT and Arid1a cKO hippocampus coronal sections. Scale bar, 50 μm. B Quantification of Prox1+ cell in the DG of WT and Arid1a cKO mice at E16.5, P0, P14, P21 (WT, n = 3 mice; cKO, n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, unpaired two-tailed t-test. C ChIP analysis for Arid1a binding to Prox1 gene in chromatin prepared from E16.5 NSPCs. Data are represented as mean ± SEM. **p < 0.01, unpaired two-tailed t-test. D Infection of NSPCs from WT and Arid1a cKO postnatal pups with control (CTRL), Prox1-OE virus co-expressing GFP under the CMV promoter (green). The proliferation ability of postnatal NSPCs was assessed by BrdU+ cells (red). DAPI, blue. Scale bar, 20 μm. E Quantification of BrdU+GFP+ cells as a fraction of all GFP+cells in Arid1a cKO NSPCs that were infected with Prox1-OE virus compared with those in Arid1a cKO NSPCs that were infected with CTRL virus (n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, unpaired two-tailed t-test. F Infection of NSPCs from WT and Arid1a cKO postnatal pups with CTRL, Prox1-OE virus co-expressing GFP under the CMV promoter (green). The differentiation ability of postnatal NSPCs was assessed by Tuj1+ cells (red). DAPI, blue. Scale bar, 20 μm. G Quantification of Tuj1+GFP+ cells as a fraction of all GFP+cells in Arid1a cKO NSPCs that were infected with Prox1-OE virus compared with those in Arid1a cKO NSPCs that were infected with CTRL virus (n = 3 mice). Data are represented as mean ± SEM. *p < 0.05, unpaired two-tailed t-test.
Given the substantial decrease of Prox1 in the hippocampal upon the loss of Arid1a, we thus reasoned whether Prox1 gain-of-function could ameliorate the defects of Arid1a cKO hippocampal development. To test this, we first constructed Prox1 overexpression virus and infected cultured Arid1a cKO hippocampal NSPCs and assessed the impact of Prox1 overexpression on the abnormal proliferation and differentiation phenotypes associated with the loss of Arid1a. Western blotting showed increased PROX1 protein levels in the Prox1 overexpression group (Supplementary Fig. 7B). Prox1 overexpression could reverse the reduced NSPCs proliferation and differentiation induced by Arid1a deletion measured by the number of BrdU+GFP+ cells (Fig. 7D, E) and the number of Tuj1+GFP+ cells (Fig. 7F, G). These results suggest that Prox1 is a functional downstream target of Arid1a in modulating the proliferation and differentiation of NSPCs.
Discussion
The dentate gyrus (DG) plays a major role in the formation of hippocampal memory. DG lesions impair most of hippocampus-dependent mnemonic functions [30]. In this study, we provide direct evidences showing that ARID1A is required for proper development of the DG at prenatal and postnatal stages. Loss of Arid1a results in the decreased numbers of neural progenitors and granule neurons, due to the failure of NSPCs proliferation and neural differentiation, and massive apoptosis in the DG. Meanwhile, we using high-throughput scRNA-seq and ChIP-seq identify Arid1a deletion disrupts histone marks and deregulates of a set of genes including Prox1 involved in the development of DG. Thus, we propose that Arid1a promotes the establishment and proliferation of NSPCs in the DG as well as their derivative neural progenitor’s differentiation into immature neurons by mediating histone modifications at the promoter of Prox1.
One of our most striking findings is that Arid1a cKO mice showed a severe morphological disorganization in the DG from E16 to P21. Microcephaly has been reported as a sign of CSS [15, 31]. It is understood that the brain’s size at birth is dependent on the ability of neural progenitor cells to proliferate and self-renew [32, 33]. Therefore, a slight perturbation in the number of cell divisions of progenitor and stem cells can have dramatic effects on brain size and may lead to microcephaly. Our data suggested Arid1a deletion leading to abnormal proliferation and neuronal differentiation of NSPCs and the resulting increased cell death may at least in part contribute to microcephaly, which is frequently observed in individuals with CSS [34]. One of the clinical criteria for the diagnosis of CSS is bilateral hippocampal atrophy [35]. In the present study, we found a smaller size of hippocampal dentate gyrus in Arid1a cKO mice, which partially mimics bilateral hippocampal atrophy in human CSS patient [36]. Mice heterozygous for either Brg1 or BAF155/Srg3, two subunits of the BAF complex, are predisposed to anencephaly, possibly by generation of too few neurons [5, 37]. Brg1 and Prox1 have been proved to be essential for neuronal differentiation [38, 39]. In this study, both Brg1 and Prox1 were downregulated in Arid1a cKO mice, and loss of Arid1a results in a marked reduction in number of dividing progenitors in all matrices of the DG with a major increase in cell death, suggesting the Arid1a function is required for the generation and expansion of DG progenitors possibly through targeting Brg1 and Prox1.
Our previous work indicated that the expression of BRG1 is significantly downregulated, while the expression of BAF155 and BAF170 (another two subunits of cBAF/pBAF/ncBAF) remained unchanged in neural stem/progenitor cells after Arid1a deletion [23]. Consistently, Shang et al. reported that BRG1 is the most obviously downregulated subunit in liver cancer cells upon ARID1A depletion [40]. Interestingly, Wang et al. reported that dual ARID1A/ARID1B deletion does not lead to degradation of other SWI/SNF subunits in ARID1-less cells, except for a dramatic loss of DPF2 protein in H2.35 liver cell line [41]. Therefore, we speculate whether BRG1 is a target of ARID1A-cBAF transcriptional activity may depend on cell type and/or stress condition. We hope that more detailed analysis of the regulatory relationship of individual subunit loss on complex composition in neural cells will be included in future studies.
The hippocampus develops and shows extensive maturation postnatally. The volume of the hippocampus increases slowly before P7, develops rapidly from P7, reaches its peak at P14 and finally the rate of development becomes stabilize [42]. The DG is a region of hippocampus formation in that most of its granule neurons are born on postnal day 6–7, and neurogenesis persists throughout life [43]. Our study revealed that loss of Arid1a at P7 using single-cell transcriptome globally reduced cell compositions in hippocampi. Progenitor cells and neurons were key cell types affected following Arid1a ablation. Upon formation of the radial glial scaffolding, the earliest born granule neurons begin their radial migration to form the primordial granule cell layer during early development of the hippocampus [44]. In contrast, Arid1a deletion decreased NSPCs pool, impaired the proliferation and differentiation and increased cell apoptosis of NSPC, which leads to no enough cells for migration to the dentate gyrus, so that they form a loosely packed group of cells instead of a compact upper blade. The Arid1a mutant mice are remarkably similar to those lacking CXCR4 [45] and SDF1 [46] in terms of dentate gyrus hypoplasia.
Furthermore, we demonstrate that Prox1 is dramatically downregulated after Arid1a deletion in DG neurons at single-cell level using cell fate trajectory analysis. Prox1 is expressed in neuroepithelial cells adjacent to the cortical hem and in DG granule cells throughout embryonic development and into adulthood. The results have shown that Prox1 is necessary for the maturation of granule cells in the dentate gyrus during development and for the maintenance of intermediate progenitors during adult neurogenesis and are required for adult neural stem cell self-maintenance in the subgranular zone [27]. All these imply that Arid1a regulate the process of DG formation through modulating a bunch of important genes and pathways. In addition, our combined scRNA-seq and ChIP assays clearly demonstrated that, in addition to Prox1, Arid1a may also directly regulate dozens of other genes that are worthy to further explore their regulatory mechanisms in NSPCs.
Epigenetic factors have emerged as important regulators for the development of neuronal morphology through modulation of chromatin structure and gene expression [47]. we observed an increased H3K27me3 and decreased H3K4me3, H3K27ac enrichment at genomic sites. Arid1a has been proposed to maintain chromatin accessibility and active histone H3K4me/H3K27ac marks at enhancer regions [48]. However, recent studies have found that ARID1A only play a marginal role in accessibility and proposed that ARID1A controls the pausing of RNA polymerase near transcriptional start sites to enable robust transcription during cell homeostasis [49]. Based on these molecular findings, we propose that the Arid1a-containing SWI/SNF complex may exert a previously undiscovered function in gene repression via differentially regulating H3K27ac, H3K4me3 and H3K27me3 modifications in forebrain.
Moreover, it is speculated that alterations of H3K4me3 and H3K27me3 levels may indicate a specific repressive role of Arid1a in the developing forebrain. In support of this notion, altered SWI/SNF targeting in Arid1a−/− cells correlated well with the transcriptional activity of the nearest genes, as demonstrated by scRNA-seq. Taken together, our molecular and functional analyses suggest that Arid1a epigenetically controls a subset of genes that are essential for cell differentiation and central nervous system development. These mechanistic findings provide strong biological basis for the phenotypes observed in Arid1a conditional knockout mice. Studies are underway to investigate the possibility of rescuing the abnormal phenotypes observed in Arid1a cKO mice with novel molecular candidates.
Learning and memory are frequently associated with morphological alterations of dendrites in dentate gyrus [50, 51]. Epigenetic factors have emerged as important regulators of dendritic morphogenesis [51, 52] and abnormal these epigenetic regulations often lead to ID phenotypes. Explorations of intervene methods catch more attentions. In Arid1a cKO mice, Prox1 overexpression can rescue the proliferation and differentiation defects of NSPCs from hippocampus caused by Arid1a deletion in vitro. Based on the references and our results, we speculate that Prox1 ectopic expression in DG may promote neuronal differentiation, produce more mature neurons and then restore the neurogenesis defects in Arid1a ablation mice. Of course, more investigations are needed to understand the potential mechanisms and find more potential ways for ameliorating CSS patients, like ID phenotype. Our previous published work reveals that Arid1a haploinsufficiency in excitatory neurons leads to deficits in learning and memory [53]. Moreover, cortical deletion of Arid1a shows axon misrouting defects, a dysregulation of subplate neuron-enriched gene transcription, disruption of axon projections and leads to altered brain development [54]. Given that disturbed DG cell compartments and malformation of the hippocampus in Arid1a mutant mice recapitulate a few symptoms of CSS patients, we therefore believe that Arid1a mutant mice represent a valuable model to study the underlying mechanisms of the etiology of CSS and to develop novel clinical implications for intellectual disability disorders. However, the Arid1a mutant mice cannot accurately recapitulate all, or even most, of the features in the CSS patients. Given that Arid1a is ubiquitously expressed in all tissues, it will be important to generate various animal models with tissue-specific deletion of Arid1a and to explore its role as well as evaluating possible crosstalk between different tissues in the pathogenesis of CSS.
In conclusion, comprehensively combining molecular, circuit, and transcriptome analyses, this study firstly uncovers the essential roles of ARID1A in governing neural progenitor cells to make the epigenetic transition to the neuronal lineage by mediating histone marks at specific loci. dentate gyrus. ARID1A is a chromatin-remodeling factor, evidence shows ARID1A globally enhances chromatin remodeling on many transcription factors or other important genes in a cell type specific patterns depending on cell context [55,56,57,58], therefore, we will focus on investigating the exact roles in more specific neural lineage cells, such as NSPCs and find more drug targets in treating CSS in the future.
Methods
Mice
The Arid1aflox/flox was kindly provided by Dr. Zhong Wang at Harvard Medical School. Arid1aflox/flox mice and Emx1-Cre mice (JAX 005628) were crossed to generate conditional forebrain-specific Arid1a knockout mice. All mice were from a C57BL6 genetic background. All mice experiments were approved by the Animal Committee of the Institute of Zoology, Chinese Academy of Sciences. All mice were tail genotyped using tail lysis. PCR primers used were: Arid1a loxP sequence forward, 5′-TGGGCAGGAAAGAGTAATGG-3′, Arid1a loxP sequence reverse, 5′-AACACCACTTTCCCATAGGC-3′; Emx1-Cre oIMR1084, 5′-GCGGTCTGGCAGTAAAAACTATC-3′; Emx1-Cre oIMR1085, 5′-GTGAAACAGCATTGCTGTCACTT-3′, Emx1-Cre oIMR4170, 5′-AAGGTGTGGTTCCAGAATCG-3′, Emx1-Cre oIMR4171, 5′-CTCTCCACCAGA AGGCTGAG-3′.
Quantification analysis of hippocampus
To measure the hippocampus size, brain sections were DAPI-stained and imaged with a fluorescent microscope. The hippocampal length and area, including the dentate gyrus and CA region, were measured along their internal sides.
Primary cell culture
Primary hippocampal neural stem/progenitor cells (NSPCs) were dissected from postnatal ≤P7 Arid1a cKO and WT mice and cultured as previously described [20, 51]. Briefly, hippocampus tissues were digested by trypsin-EDTA for 10 min at 37 °C. Samples were washed and pipetted (20 rounds per time) three times with Neurobasal medium. Isolated cell suspension was separated into plates coated with poly-D-lysine (100 μg/mL) for neuron culture or with EGF and FGF (both in 0.02%) for NSCs culture. NSCs were cultured in IPM medium, neurobasal medium (Invitrogen) supplemented with 2% B27(Invitrogen), 2 mM GlutaMAX (Invitrogen), and penicillin/streptomycin solution and then passaged in N2 medium, DMEM/F12 medium with listed nutrition factors. For NSCs proliferation, primary cells were incubated with 20 μM BrdU for 6–8 h on day 3 after culture. Cells were performed immunostaining as followed protocol. The number of BrdU-positive cells was counted as proliferative NSCs. For the cell differentiation, the differentiation medium was used to replace the proliferation medium 24 h after cells were plated and were changed every 2 d for a total of 10 d [59]. All the coverslips should be coated with 10 μg/ml poly-Lysine (Sigma) and 5 μg/ml laminin (Sigma).
TUNEL and proliferation assays
TUNEL assay was performed on brain sections according to the manufacturer’s instructions (Beyotime). For proliferation assays at embryonic stages, time mated female mice were injected with BrdU (100 mg/g body weight, intraperitoneally), and embryos were harvested 2 h later. Embryos were fixed in 4% PFA and cryoprotected in 30% sucrose. For proliferation assays at early postnatal stages, pups were injected with BrdU (100 mg/g body weight, intraperitoneally) 2 h before harvest. Brains were perfused with 4% PFA and cryoprotected in 30% sucrose. Rat anti-BrdU (1:1000; Abcam) antibody was used. Sections were counterstained with DAPI.
Immunohistochemistry
Nissl staining was carried out with cresyl violet according to standard procedures (Beyotime). For immunohistochemistry of brains, the sections were fixed with 4% PFA for 15 min, washed in PBS for 10 min three times, permeated with 0.5% Triton X-100 for 15 min, and blocked at room temperature for 2 h. The primary antibodies were incubated at 4 °C overnight. Immunohistochemical and immunocytochemical staining was carried out with the antibodies listed below: anti-Arid1a (Rabbit, 1:1000, HPA005456, Sigma), anti-Nestin (Chicken, 1:500, NEB, Aves labs), anti-Ki67 (1:1000, RM-9106-S, Thermo), anti-BrdU (Rat, 1:1000, ab6326, Abcam), anti-Prox1 (Rabbit, 1:25,000, AB5475, Millipore), anti-Dcx (Rabbit, 1:500, 4604s, Cell Signaling), anti-NeuN (Mouse, 1:1000, MAB377, Millipore), anti-BLBP (Rabbit, 1:1000, ab32423, Abcam), anti-Calbindin (Rabbit, 1:5000, D28K-300, Swant), anti-Calretinin (Rabbit, 1:500, MAB1568, Millipore). After washing with PBS for 15 min, the brain slices were incubated with the secondary antibodies conjugated to Alexa Fluor 488 or 568(1:500, Life Technology) at room temperature. Photomicrographs were taken using a laser scanning confocal microscope (LSM710, Zeiss).
Luciferase assays
Every 1000 bp of the Prox1 promoter was cloned into the luciferase reporter plasmid. Plasmids of promoter-1(P-1), promoter2(P-2), promoter3(P-3) and Arid1a-overexpression plasmid (OE) were cotransfected in 293T cells. After 24 or 48 h, cells were harvested using lysis buffer (Promega). The luciferase activity was carried out for each sample (20 μl) using the Dual-Luciferase Assay System (Promega, 117 # E1910 a) with a luminometer (Bioscan) according to the manufacturer’s instructions.
Western blot analysis
Western blotting was performed using tissues (cortex, hippocampus) from both control mice and conditional knockout mice (Arid1aflox/flox; Emx1-Cre). Tissues were lysed in RIPA buffer with 2 mM PMSF. Proteins were separated on 6–10% Bis-Tris SDS polyacrylamide gel (Bio-Rad) and transferred to PVDF membranes (Millipore). Blots were sequentially immunostained with rabbit anti-ARID1A antibody (1:1000, HPA005456, Sigma), rabbit anti-Prox1 (1:1000, AB5475, Millipore), rabbit anti-BRG1 antibody (1:1000, ab4081, Abcam) followed by horseradish peroxidase-conjugated secondary antibody (1:3000, Pplygen Co. Ltd) and detect with enhance chemiluminescence reagent (ECL, Pierce). Anti-Actin (Mouse, 1:10,000, Sigma) western blots were used as controls. Original western blots for all relevant figures are shown in “Supplemental file-original western blots”.
Quantitative real-time PCR
To clarify the expression of Arid1a in different stages, total RNA was extracted from the cortex, hippocampus, or neural stem cells according to procedures with Trizol reagent (Invitrogen), and was reverse-transcribed into cDNA using the reverse transcriptions reagents (Roche). Quantitative real-time PCR using SYBR Green was performed according to the manufacturer’s guideline (Roche). The primers used were:
Arid1a forward, 5′-GCCACAAACTCCTCAGTCAACC-3′, Arid1a reverse, 5′-GCATCCTGGATTCCGACTGAGT-3′; Nfix forward, 5′-GGCTTACTTTGTCCACACTCCG-3′, Nfix reverse, 5′-CGTCACAAAGCAGTCCTGGAAAC-3′; Prox1 forward, 5′-CAGCGGACTCTCTAGCACAG-3′, Prox1 reverse, 5′-GCCTGCCAAAAGGGGAAAGA-3′; Zfpm2 forward, 5′-ATGGCAAGGAGTGGAAGACAGC-3′, Zfpm2 reverse, 5′-AAGTCCACCACAAAGGCGACGA-3′; Ncam1 forward, 5′-GGTTCCGAGATGGTCAGTTGCT-3′, Ncam1 reverse, 5′-CAAGGACTCCTGTCCAATACGG-3′; Tmem108 forward, 5′-CCTGAGCTACTGGAACAATGCC-3′, Tmem108 reverse, 5′-CAGTGTCTCGATAGTCGCCATTG-3′; Gapdh forward, 5′-CATCACTGCCACCCAGAAGACTG-3′, Gapdh reverse, 5′-ATGCCAGTGAGCTTCCCGTTCAG-3′.
Single-cell RNA-seq analysis
Cellranger(6.0.2) was used for alignment, filtering, barcode counting, and UMI counting of the single-cell FASTQs. The reads were mapped to the mm10 genome reference. Then, Seurat (4.1.1) was used for cluster analysis. The scRNA-seq data were merged and the cells were filtered as below: gene numbers < 500, gene numbers > 6000; mitochondrial gene percentage of greater than 15. After quality control, 14,894 cells from cKO samples and 15,520 cells from WT samples remained. The NormalizeData function (normalization. method = “LogNormalize”, scale. factor = 10,000) was used to normalize the data to eliminate the influence of sequencing library size. Cluster found using the following functions in order: FindVariableFeatures with 2000 genes, ScaleData, RunPCA, FindNeighbors with the first 18 PCs and FindClusters(resolution = 0.5). The DEGs (Differentially expressed genes) of each cluster were identified using the FindAllMarkers function (‘wilcox’), and genes with p > 0.05 were removed. The 30 clusters of cells were identified by gene expression into 11 cell types, including astrocytes, blood, Cajal-Retzius cells, endothelial cells, ependymal cells, microglia, mural cells, neurons, OPCs, other and progenitor cells. The DEGs in progenitor/neuron between WT and cKO were identified using the FindMarkers function. ClusterProfiler package (4.2.2) was used for Gene Ontology (GO) term enrichment. The Monocle package was used to analyse single-cell trajectories to discover developmental transitions. We used differentially expressed genes identified by monocle to sort cells in pseudo-time order. “DDRTree” was applied for dimensionality reduction analysis. Then we used the plot_cell_trajectory function and the plot_genes_in_pseudotime function for visualization. The function BEAM was used to calculate the Branch-dependent different expression genes.
Chromatin immunoprecipitation (ChIP)
A total of 1 × 107 NSPC cells isolated from Arid1a WT and cKO mice hippocampus at E16.5 were cross-linked with 1% formaldehyde for 10 min at room temperature. ChIP was performed as previously described [53]. Antibodies used include normal rabbit IgG (ChIP grade, 2729, Cell Signaling), rabbit anti-ARID1A (ChIP grade, 04-080, Millipore). rabbit anti-BRG1 (ChIP grade, #49360, Cell Signaling). The primers were used to amplify the genomic loci from mouse Prox1 as follows. Forward: 5′-GTTCTCTTGCCTCGCTATCC-3′; Reverse: 5′-CTCCGCTCCACAACAAGATT-3′.
ChIP-seq data analysis
ChIP-seq libraries were sequenced generating 50-bp single reads. Raw reads data were filtered by using Trimmomatic (v.0.36) and quality-controlled using FastQC (v. 0.11.7) [60]. High-quality reads were aligned using Bowtie 2 (v2.3.5.1) to the mouse reference genome using default parameters [61]. Samtools (v.1.9) was then used to convert files to bam format and filter reads mapped with parameters “-F 1804 -q 30” for single-end sequencing data [62]. After removing PCR duplicates using the Mark Duplicates function in Picard (v.2.21.2) (http://broadinstitute.github.io/picard/) and mitochondrial reads. MACS (v.2.2.5) was used to call peaks (-q 0.01) relative to the input sample [63].
MAnorm (v.1.2.0) was then used for quantitative comparison of ChIP-Seq data [64]. Different binding of H3K27ac peaks was defined by P-values < 0.05 and absolute M-values more than 1.5. Peak annotation was performed using ChIPseeker (v.1.22.1) at the gene level and promoter regions were defined as ±1000 bp of TSS [65]. DeepTools (v. 3.3.1) “computeMatrix,” “plotHeatmap,” and “plotProfile” functions were used to generate heatmaps and profile plots [66]. For genome-browser representation, data in bigwig files generated by deepTools were visualized using IGV (v. 2.4.10) [67]. The mouse reference genome sequence (vM23) and gene annotation (vM23) were downloaded from GENCODE (https://www.gencodegenes.org/). The public BRG1 ChIP-seq data of E11.5 forebrain was downloaded from GSE37151 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37151).
In situ cell counts
Immunopositive signals for BrdU, Ki67 were measured within 1 mm3 or 1 mm2 from WT or cKO mice in at least three pairs of brain sections. Similar quantification methods were applied to TUNEL in the DG. For the number of Prox1, Calretinin and Dcx-positive cells per 100 μm in the upper and lower blades of the dentate gyrus of WT or cKO hippocampus was then counted. In these cases, the ratio of positive signals was calculated using ImageJ and analyzed using GraphPad Prism V8 software.
Experimental design and statistical analysis
Experiments were conducted in three or more biological replicates for each group. Before all statistical analyses, data were examined for normality of variance using the Kolmogorov–Smirnov test. All data were presented as mean ± SEM, and statistically significant was defined as ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001. For statistical analyses, unpaired two-tailed Student’s t-tests (two groups) or ANOVA (three or more groups) were performed using GraphPad Prism 8.0 software. All data were presented as mean ± SE. Differences of p < 0.05 were considered statistically significant.
Data availability
The scRNA-seq data used in this study have been deposited in the Genome Sequence Archive in the National Genomics Data Center, Beijing Institute of Genomics, Chinese Academy of Sciences with accession number GSA: CRA010586. The ChIP-seq data used in this study have been deposited in the Gene Expression Omnibus (GEO) under accession number GSE169562.
References
Bartsocas CS, Tsiantos AK. Mental retardation with absent fifth fingernail and terminal phalanx. Am J Dis Child. 1970;120:493–4.
Tsurusaki Y, Okamoto N, Ohashi H, Kosho T, Imai Y, Hibi-Ko Y, et al. Mutations affecting components of the SWI/SNF complex cause Coffin-Siris syndrome. Nat Genet. 2012;44:376–8.
Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–92.
Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304.
Bultman S, Gebuhr T, Yee D, La Mantia C, Nicholson J, Gilliam A, et al. A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol Cell. 2000;6:1287–95.
Diana CH, Gerald RC. ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 2011;21:396–420.
Chandler RL, Brennan J, Schisler JC, Serber D, Patterson C, Magnuson T. ARID1a-DNA interactions are required for promoter occupancy by SWI/SNF. Mol Cell Biol. 2013;33:265–80.
Kadoch C, Hargreaves DC, Hodges C, Elias L, Ho L, Ranish J, et al. Proteomic and bioinformatic analysis of mammalian SWI/SNF complexes identifies extensive roles in human malignancy. Nat Genet. 2013;45:592–601.
Takeda T, Banno K, Okawa R, Yanokura M, Iijima M, Irie-Kunitomi H, et al. ARID1A gene mutation in ovarian and endometrial cancers (Review). Oncol Rep. 2016;35:607–13.
Zang ZJ, Cutcutache I, Poon SL, Zhang SL, McPherson JR, Tao J, et al. Exome sequencing of gastric adenocarcinoma identifies recurrent somatic mutations in cell adhesion and chromatin remodeling genes. Nat Genet. 2012;44:570–4.
Wu JN, Roberts CWM. ARID1A mutations in cancer: another epigenetic tumor suppressor? Cancer Discov. 2013;3:35–43.
Ogiwara H, Takahashi K, Sasaki M, Kuroda T, Yoshida H, Watanabe R, et al. Targeting the vulnerability of glutathione metabolism in ARID1A-deficient cancers. Cancer Cell. 2019;35:177–190.e178.
Mathur R, Alver BH, San Roman AK, Wilson BG, Wang X, Agoston AT, et al. ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat Genet. 2017;49:296–302.
Nagarajan S, Rao SV, Sutton J, Cheeseman D, Dunn S, Papachristou EK, et al. ARID1A influences HDAC1/BRD4 activity, intrinsic proliferative capacity and breast cancer treatment response. Nat Genet. 2020;52:187–97.
Bidart M, El Atifi M, Miladi S, Rendu J, Satre V, Ray PF, et al. Microduplication of the ARID1A gene causes intellectual disability with recognizable syndromic features. Genet Med. 2017;19:701–10.
Gao XL, Tate P, Hu P, Tjian R, Skarnes WC, Wang ZES. cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci USA. 2008;105:6656–61.
Son EY, Crabtree GR. The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am J Med Genet C. 2014;166:333–49.
Goncalves JT, Schafer ST, Gage FH. Adult neurogenesis in the hippocampus: from stem. Cells Behav Cell. 2016;167:897–914.
Miller SM, Sahay A. Functions of adult-born neurons in hippocampal memory interference and indexing. Nat Neurosci. 2019;22:1565–75.
Liu C, Teng ZQ, Santistevan NJ, Szulwach KE, Guo W, Jin P, et al. Epigenetic regulation of miR-184 by MBD1 governs neural stem cell proliferation and differentiation. Cell Stem Cell. 2010;6:433–44.
Liu PP, Tang GB, Xu YJ, Zeng YQ, Zhang SF, Du HZ, et al. MiR-203 interplays with polycomb repressive complexes to regulate the proliferation of neural stem/progenitor cells. Stem Cell Rep. 2017;9:190–202.
Lei X, Jiao J. UTX affects neural stem cell proliferation and differentiation through PTEN signaling. Stem Cell Rep. 2018;10:1193–207.
Liu X, Dai SK, Liu PP, Liu CM. Arid1a regulates neural stem/progenitor cell proliferation and differentiation during cortical development. Cell Prolif. 2021;54:e13124.
Han L, Madan V, Mayakonda A, Dakle P, Woon TW, Shyamsunder P, et al. Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia. 2019;33:2291–305.
Abdelrahim EA, Eltony SA. Postnatal development of the hippocampal formation in male albino rats. Egypt J Histol. 2011;34:346–64.
Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–57.
Lavado A, Lagutin OV, Chow LML, Baker SJ, Oliver G. Prox1 is required for granule cell maturation and intermediate progenitor maintenance during brain neurogenesis. PLoS Biol. 2010;8:16.
Yu Z, Lin D, Zhong Y, Luo B, Liu S, Fei E, et al. Transmembrane protein 108 involves in adult neurogenesis in the hippocampal dentate gyrus. Cell Biosci. 2019;9:9.
Zhong SJ, Ding WY, Sun L, Lu YF, Dong H, Fan XY, et al. Decoding the development of the human hippocampus. Nature. 2020;577:531.
Hainmueller T, Bartos M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat Rev Neurosci. 2020;21:153–68.
Schrier SA, Bodurtha JN, Burton B, Chudley AE, Chiong MAD, D'Avanzo MG, et al. The Coffin-Siris syndrome: A proposed diagnostic approach and assessment of 15 overlapping cases. Am J Med Genet Part A. 2012;158A:1865–76.
Rakic P. A small step for the cell, a giant leap for mankind: a hypothesis of neocortical expansion during evolution. Trends Neurosci. 1995;18:383–8.
Barbelanne M, Tsang WY. Molecular and cellular basis of autosomal recessive primary microcephaly. BioMed Res Int. 2014;2014:547986.
Kosho T, Miyake N, Carey JC. Coffin-Siris syndrome and related disorders involving components of the BAF (mSWI/SNF) complex: historical review and recent advances using next generation sequencing. Am J Med Genet C Semin Med Genet. 2014;166C:241–51.
Bilha SC, Teodoriu L, Velicescu C, Caba L. Pituitary hypoplasia and growth hormone deficiency in a patient with Coffin-Siris syndrome and severe short stature: case report and literature review. Arch Clin Cases. 2022;9:121–5.
Barish S, Barakat TS, Michel BC, Mashtalir N, Phillips JB, Valencia AM, et al. BICRA, a SWI/SNF complex member, is associated with BAF-disorder related phenotypes in humans and model organisms. Am J Hum Genet. 2020;107:1096–112.
Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, et al. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol. 2001;21:7787–95.
Matsumoto S, Banine F, Struve J, Xing RB, Adams C, Liu Y, et al. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev Biol. 2006;289:372–83.
Stergiopoulos A, Elkouris M, Politis PK. Prospero-related homeobox 1 (Prox1) at the crossroads of diverse pathways during adult neural fate specification. Front Cell Neurosci 2014;8:454.
Shang XY, Shi Y, He DD, Wang L, Luo Q, Deng CH, et al. ARID1A deficiency weakens BRG1-RAD21 interaction that jeopardizes chromatin compactness and drives liver cancer cell metastasis. Cell Death Dis. 2021;12:990.
Wang Z, Chen K, Jia Y, Chuang JC, Sun X, Lin YH, et al. Dual ARID1A/ARID1B loss leads to rapid carcinogenesis and disruptive redistribution of BAF complexes. Nat Cancer. 2020;1:909–22.
Xu P, Xu J, Li Z, Yang Z. Expression of TRPC6 in renal cortex and hippocampus of mouse during postnatal development. PLoS ONE. 2012;7:e38503.
Ciric T, Cahill SP, Snyder JS. Dentate gyrus neurons that are born at the peak of development, but not before or after, die in adulthood. Brain Behav. 2019;9:e01435.
Li GN, Pleasure SJ. Morphogenesis of the dentate gyrus: What we are learning from mouse mutants. Dev Neurosci. 2005;27:93–99.
Lu M, Grove EA, Miller RJ. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc Natl Acad Sci USA. 2002;99:7090–5.
Bagri A, Gurney T, He XP, Zou YR, Littman DR, Tessier-Lavigne M, et al. The chemokine SDF1 regulates migration of dentate granule cells. Development. 2002;129:4249–60.
Armand EJ, Li J, Xie F, Luo C, Mukamel EA. Single-cell sequencing of brain cell transcriptomes and epigenomes. Neuron. 2021;109:11–26.
Kelso TWR, Porter DK, Amaral ML, Shokhirev MN, Benner C, Hargreaves DC. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. Elife. 2017;6:e30506
Trizzino M, Barbieri E, Petracovici A, Wu S, Welsh SA, Owens TA, et al. The tumor suppressor ARID1A controls global transcription via pausing of RNA polymerase II. Cell Rep. 2018;23:3933–45.
Beique J-C, Lin D-T, Kang M-G, Aizawa H, Takamiya K, Huganir RL. Synapse-specific regulation of AMPA receptor function by PSD-95. Proc Natl Acad Sci USA. 2006;103:19535–40.
Liu PP, Xu YJ, Dai SK, Du HZ, Wang YY, Li XG, et al. Polycomb protein EED regulates neuronal differentiation through targeting SOX11 in hippocampal dentate gyrus. Stem Cell Rep. 2019;13:115–31.
Gao J, Wang WY, Mao YW, Graff J, Guan JS, Pan L, et al. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature. 2010;466:1105–9.
Liu PP, Dai SK, Mi TW, Tang GB, Wang Z, Wang H, et al. Acetate supplementation restores cognitive deficits caused by ARID1A haploinsufficiency in excitatory neurons. EMBO Mol Med. 2022;14:e15795.
Doyle DZ, Lam MM, Qalieh A, Qalieh Y, Sorel A, Funk OH, et al. Chromatin remodeler Arid1a regulates subplate neuron identity and wiring of cortical connectivity. Proc Natl Acad Sci USA. 2021;118.
Liu J, Liu S, Gao H, Han L, Chu X, Sheng Y, et al. Genome-wide studies reveal the essential and opposite roles of ARID1A in controlling human cardiogenesis and neurogenesis from pluripotent stem cells. Genome Biol. 2020;21:169.
Bitler BG, Wu S, Park PH, Hai Y, Aird KM, Wang Y, et al. ARID1A-mutated ovarian cancers depend on HDAC6 activity. Nat Cell Biol. 2017;19:962–73.
Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med. 2015;21:231–8.
Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med. 2018;24:556–62.
Zhang M, Chen D, Xia J, Han W, Cui X, Neuenkirchen N, et al. Post-transcriptional regulation of mouse neurogenesis by Pumilio proteins. Genes Dev 2017;31:1354–69.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–9.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, et al. The sequence alignment/map format and SAMtools. Bioinformatics. 2009;25:2078–9.
Feng JX, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–40.
Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 2012;13:R16.
Yu G, Wang LG, He QY. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics. 2015;31:2382–3.
Ramírez F, Ryan D, Grüning B, Bhardwaj V, Kilpert F, Richter A, et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 2016;44:W160–165.
Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–92.
Acknowledgements
The authors gratefully acknowledge and thank Dr. Zhong Wang at the University of Michigan for providing Arid1aflox/flox mice.
Funding
This work was supported by grants from the National Key Research and Development Program of China Project (2021YFA1101402), the Informatization Plan of Chinese Academy of Sciences (CAS-WX2021SF-0301), and the National Science Foundation of China (82271428), and the Open Project Program of State Key Laboratory of Stem Cell and Reproductive Biology.
Author information
Authors and Affiliations
Contributions
P-PL: Conceptualization; formal analysis; validation; methodology; writing-original draft; approval of draft; S-PL: Data curation; methodology; validation; approval of draft. XLi: Formal analysis; methodology; validation; G-BT: Conceptualization; formal analysis, writing-original draft; XLiu: Data curation; Formal analysis; validation. S-KD: Formal analysis; validation; L-FJ: Formal analysis; validation; X-WL: Formal analysis; validation; X-GL: approval of draft; B-YH: re-analyze seq-data; approval of draft; J-WJ: re-analyze seq-data; approval of draft; Z-QT: Data curation; formal analysis and interpretation; funding acquisition; approval of draft. C-SH: Conceptualization; Project administration. C-ML: Conception and design; data curation; formal analysis and interpretation; supervision; funding acquisition; approval of draft.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Our studies did not include human participants, human data or human tissue.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, PP., Lu, SP., Li, X. et al. Abnormal chromatin remodeling caused by ARID1A deletion leads to malformation of the dentate gyrus. Cell Death Differ 30, 2187–2199 (2023). https://doi.org/10.1038/s41418-023-01199-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41418-023-01199-w
- Springer Nature Limited
This article is cited by
-
Neuropathic pain development following nerve injury is mediated by SOX11-ARID1A-SOCS3 transcriptional regulation in the spinal cord
Molecular Biology Reports (2024)
-
SWI/SNF Complex Connects Signaling and Epigenetic State in Cells of Nervous System
Molecular Neurobiology (2024)