
Abstract
The 22q11.2 deletion syndrome (22q11DS) is associated with a wide spectrum of cognitive and psychiatric symptoms. Despite the considerable work performed over the past 20 years, the genetic etiology of the neurodevelopmental phenotype remains speculative. Here, we report de novo heterozygous truncating variants in the HIRA (Histone cell cycle regulation defective, S. Cerevisiae, homolog of, A) gene associated with a neurodevelopmental disorder in two unrelated patients. HIRA is located within the commonly deleted region of the 22q11DS and encodes a histone chaperone that regulates neural progenitor proliferation and neurogenesis, and that belongs to the WD40 Repeat (WDR) protein family involved in brain development and neuronal connectivity. To address the specific impact of HIRA haploinsufficiency in the neurodevelopmental phenotype of 22q11DS, we combined Hira knock-down strategies in developing mouse primary hippocampal neurons, and the direct study of brains from heterozygous Hira+/− mice. Our in vitro analyses revealed that Hira gene is mostly expressed during neuritogenesis and early dendritogenesis stages in mouse total brain and in developing primary hippocampal neurons. Moreover, shRNA knock-down experiments showed that a twofold decrease of endogenous Hira expression level resulted in an impaired dendritic growth and branching in primary developing hippocampal neuronal cultures. In parallel, in vivo analyses demonstrated that Hira+/− mice displayed subtle neuroanatomical defects including a reduced size of the hippocampus, the fornix and the corpus callosum. Our results suggest that HIRA haploinsufficiency would likely contribute to the complex pathophysiology of the neurodevelopmental phenotype of 22q11DS by impairing key processes in neurogenesis and by causing neuroanatomical defects during cerebral development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The 22q11.2 deletion syndrome (22q11DS), also known as DiGeorge syndrome or velocardiofacial syndrome, is the most frequent chromosomal microdeletion syndrome. The phenotypic expression of the 22q11DS is highly heterogeneous and includes congenital heart defects, thymus and parathyroid hypoplasia or agenesis leading, respectively, to immune deficiency and hypocalcemia. The phenotypic spectrum also includes a large variety of neurodevelopmental disorders (NDD). Indeed, intellectual disability (ID) is present in about 50% of the patients with a mean total IQ of approximately 70 (Swillen et al. 1997; De Smedt et al. 2007). The frequency of Autism Spectrum Disorders (ASD) is estimated to range between 15 and 50% (Fine et al. 2005; Vorstman et al. 2006; Bruining et al. 2010). Psychiatric disorders are also very frequent as Attention Deficit/Hyperactivity Disorder is present in 37% of affected children and psychotic disorders in 41% of the adult patients (Schneider et al. 2014). This makes the 22q11DS the highest known genetic risk factor for developing schizophrenia and the second most common genetic cause of developmental delay (Rauch et al. 2006). Considerable work has been done over the past 20 years to address the complex pathophysiology of the neurodevelopmental phenotype of the 22q11DS. Nevertheless, until now, the etiology of the cognitive and psychiatric phenotype remains speculative.
Among the 32 protein-coding genes located in the 1.5 Mb proximal 22q11.2 deletion, HIRA (Histone cell cycle regulator [MIM: 600237]) encodes a histone chaperone and plays an important role in the epigenetic regulation of gene expression since its tissue-specific knock-out affects many basic cellular processes including DNA damage, limited de novo methylation, and global aberrant transcription (Nashun et al. 2015; Dilg et al. 2016; Valenzuela et al. 2017). The HIRA protein contains a WD40 repeat (WDR) domain and belongs to the WDR family proteins, recently pointed out for their putative role in brain development and neuronal connectivity (Kannan et al. 2017). Finally, it has been recently shown that HIRA regulates neural progenitor cell proliferation and neurogenesis particularly by controlling the Wnt signaling pathway (Li and Jiao 2017).
Here, we report the identification of de novo truncating variants in the HIRA gene in two unrelated patients referred to the genetic consultation for diagnostic purpose of a neurodevelopmental disorder. To provide evidence for the pathogenicity of these variants, we performed functional analyses combining in vitro mouse neuronal models in which Hira gene expression was decreased, with the neuroanatomical analysis of Hira heterozygous knock-out mice, and the neurodevelopmental expression profile of Hira mRNA. Our findings revealed that HIRA haploinsufficiency leads to abnormal defects in both in vitro and in vivo models, suggesting that this gene would be involved in the regulation of neuronal differentiation and maturation.
Materials and methods
Patients
The study was approved by the local institutional review boards, and written informed consent was obtained from the patients’ parents, including explicit permission to share clinical and identifying information. Genomic DNA and RNA were obtained from the University Hospital of Tours (France) and from the Radboud University Medical Center in Nijmegen (The Netherlands).
Hira mRNA neurodevelopmental expression analyses
Total RNAs were extracted from neuronal cultures or from mouse brains using Trizol (Invitrogen, USA) and purified by the DirectZol kit (Ozyme, France). Complementary DNAs were obtained from 2 µg of total RNAs using the Prime Script RT Reagent commercial kit (Takara Bio, Nojihigashi, Japan). Primers were designed with Primer3 software: forward primer (5′CGTACTGCTGCTGTGCTGTT3′) was located in exon 9 of Hira and the reverse primer (5′CTGGGAGAAGTCGAGGAATG3′) was located in exon 11. qPCR experiments were run in duplicates on the LightCycler 480 (Roche Diagnostics, Meylan, France) using the SYBR Green Master Mix commercial kit (Applied Biosystem, Foster City, CA, USA). Relative expression was assessed using the advanced E-method from LightCycler software (Roche Diagnostics, Meylan, France). Hypoxanthine phosphoribosyltransferase 1 (HPRT1) for human samples, Cyclin C1 (Cyc1) and Beta-2-microglobulin (B2m) for mouse samples were used as reference genes for normalization.
Hira knock-down expression analysis
Plasmids containing a small hairpin RNA (shRNA) targeting HIRA human and murine transcripts, as well as plasmids containing a scramble shRNA were used (Origene, TF500962; Rockville, MD, USA). The target sequence of the effective shRNA (5′GACAGTATGAATGCTACCTCTACTCCTGC3′) overlapped exons 3 and 4 of Hira mRNA. The target sequence of the scramble shRNA is as follows, 5′GCACTACCAGAGCTAACTCAGATAGTACT3′. The expression of the shRNA was driven by a U6 promoter. These constructions contained the chloramphenicol resistance gene and a CMV driven RFP gene, which expresses RFP protein constitutively in mammalian cells. The plasmidic DNAs were extracted using the Endo Free Plasmid Maxi Kit (Qiagen, Venlo, Netherlands). Both effective shRNA and non-effective shRNA were transfected in HEK293T cells to evaluate their efficiency to repress the expression of endogenous HIRA. HEK293T cultures were maintained in Dulbecco’s modified Eagle’s medium (DMEM)-Glutamax (Invitrogen, USA) supplemented with 10% Fetal Bovine Serum (Eurobio, France) and were transfected with the appropriate plasmid using Lipofectamine 2000 (Invitrogen, USA). In brief, 2.5 µg of DNA was combined with 6 µL of Lipofectamine 2000 in 300 µL of DMEM. The DNA/Lipofectamine 2000 mixture was incubated for 20 min at room temperature. Complexes were added dropwise onto cells growing after half of the medium was removed. Next, cells were incubated for 4 h at 37 °C and 5% CO2. The transfection medium was then replaced by DMEM/10% Fetal Bovine Serum, and total RNA lysates were obtained 72 h post-transfection. To measure the expression levels of HIRA in each condition, RT-qPCR analyses were performed on 5 independent transfections.
Primary hippocampal cultures
All mouse experiments were performed according to the protocols approved by the University Francois-Rabelais of Tours and INSERM (Project authorization number 01456.03, Ministry of Research). Hippocampi or cortices were dissected from embryonic day 17.5 C57BL/6 J mouse embryos (Janvier Labs, Le Genest Saint-Isle, France), manually dispersed in cold PBS (Fisher Scientific, Waltham, MA, USA), and triturated with papain (10 U/mL; Worthington Biochemical, Lakewood, NJ, USA) for 22 min at 37 °C. Cells were resuspended in DMEM/F12 (Fisher Scientific) with 10% fetal bovine serum (Eurobio, Courtaboeuf, France) and centrifuged at 250 g for 3 min, and the final pellet was resuspended in primary neuron growth medium (PNGM; Lonza, Basel, Switzerland). Dissociated cells were then plated onto glass coverslips coated in poly-D-lysine (Sigma Aldrich, Saint-Louis, MO, USA) and laminin (Fisher Scientific) at a density of 400 cells per mm2. The cultures were kept in Primary Neuron Growth Medium, and half of the medium was changed twice a week.
Transfections and cellular analyses
Hippocampal neurons were transfected 11 days after plating (Day in vitro 11, DIV11) with the appropriate plasmids (non-effective shRNA and effective shRNA) with the DNAin Neuro transfection Kit (Amsbio, Abingdon, UK). In brief, 500 ng of plasmid DNA was combined with 1 µL of DNAin Neuro in 100 µL of PNBM medium. The DNA and DNAin Neuro mixture was incubated for 15 min at room temperature. Complexes were added dropwise onto cells growing after half of the medium was removed and stored. Next, cells were incubated for 4 h at 37 °C and 5% CO2. After incubation, the medium was changed. Cells were fixed at DIV14 using 4% paraformaldehyde and coverslips were mounted on a glass slide using ProLong Diamond Antifade Mountant with DAPI (Invitrogen). Individual neurons were directly imaged under fluorescence and confocal microscopy (Leica SP8, Leica, Wetzlar, Germany) using RFP (562 nm) labeling as a tracer of morphology. Image analysis was done using Imaris software (Bitplane Scientific Software, Zurich, Switzerland) allowing the evaluation of the number of neurites per neurons, the number of neurites by branching level and the mean neurite length. The neuronal branching was evaluated using Sholl analysis on ImageJ software (Wayne Rasband, NIH, Bethesda, MD, USA). Statistical analysis was performed using the GraphPad Prism 6.0 software (La Jolla, CA, USA). For the neuronal morphological study, the data were first analyzed using the D’Agostino-Pearson normality test. When the result was positive, the data were then analyzed using an Ordinary one-way ANOVA followed by Tukey’s multiple comparisons. P < 0.05 was assumed significant.
Generation of Hira +/− knock-out mice
The construction of the Hira allele was based on the “Knockout-first allele” method (Skarnes et al. 2011). A “critical” exon (exon 4) present on all transcripts was first identified. A promotor-less targeting cassette was inserted in intron 3 in a C57BL/6 N blastocyst, which created a frameshift mutation, thus generating the KO-first allele (also known as the tm1a allele). A heterozygous-by-heterozygous (“het-by-het”) breeding scheme was used to propagate the line. After weaning, animals were housed three–four mice per cage with WT controls housed separately, in specific-pathogen-free environment in individually ventilated cages under 12/12 light/dark cycle with temperature-controlled conditions and free access to food and water with hardwood bedding. All animals were regularly monitored for health and welfare concerns.
Mouse Hira +/− neuroanatomical study
The neuroanatomical study was carried out using a cohort of 10 male mice aged 16 weeks (n = 5 per genotype). Mouse brain samples were fixed in 4% buffered formalin for exactly 48 h. A total of 40 brain parameters, consisting of surface area and length measurements, were taken blind to the genotype across one sagittal section at Lateral + 0.60 mm. These parameters were distributed across 22 developmentally distinct brain regions related to the cerebrum, the cortex, the pons, the cerebellum, the ventricle, the corpus callosum, the thalamus, the caudate putamen, the hippocampus, the fimbria, the anterior commissure, the stria medullaris, the fornix, the optic chiasm, the hypothalamus, the pontine nuclei, the substantia nigra, the transverse fibers of the pons, the cingulate gyrus, the dorsal subiculum, the inferior colliculus and the superior colliculus. These brain regions were further delineated into six main brain categories (brain size, commissures, ventricles, cortex, subcortex and cerebellum). Data were analyzed using a two-tailed Student t-test assuming equal variance to determine whether a brain region is associated with neuroanatomical defects or not.
Ethical considerations in animal use
The care and use of Hira+/− mice in the Wellcome Sanger Institute study was carried out in accordance with UK Home Office regulations, UK Animals (Scientific Procedures) Act of 1986 under two UK Home Office licences (80/2485 and P77453634) that approved this work, which were reviewed regularly by the Wellcome Sanger Institute Animal Welfare and Ethical Review Body.
Results
We identified two cases with neurodevelopmental disorder and de novo genetic variants of HIRA. The patient 1 was a 4-year-old female born to non-consanguineous Algerian parents. She was born at 38 weeks. Birth parameters were unknown except for the weight which was at the 25th percentile. Neonatal period was marked by hypotonia and feeding difficulties with velopharyngeal insufficiency. She had psychomotor retardation, moderate ID and motor stereotypies. She also had growth retardation and mild microcephaly (OFC: − 2.8 SD). At physical examination, facial features were suggestive of 22q11DS as she had narrow and slightly upslanted palpebral fissures, bilateral ptosis, tubular nose with hypoplastic alae nasi. The mouth was small, with a high palate and a short uvula. Ears were posteriorly rotated with small lobes. Brain MRI showed diffuse atrophy of the white matter. Metabolic investigation, blood count and calcemia were normal. Array-CGH (cytoshure ISCA 60 k, Agilent, Santa-Clara, CA, USA) identified a de novo 28 kb intragenic deletion encompassing exons 2–13 of HIRA (Fig. 1). This deletion resulted in a disruption of the open reading frame of HIRA leading to a premature stop codon in exon 14 (Fig. 1).


Molecular genetic investigations in the two patients with HIRA genetic alteration. a Schematic representation of the 22q11.2 region and array CGH profile showing a 28 kb intragenic deletion of the HIRA gene in the DNA from Patient 1. Major candidate genes for the neurodevelopmental phenotype in the literature are written in blue characters. Green boxes represent the low copy repeats (LCR) regions. b Representative graph of qPCR analyses performed on genomic DNA from patient 1 and her respective parents confirming the de novo occurrence of the deletion; c DNA sequencing electrophoregram of the deletion breakpoint; d Schematic representation of the predicted truncated HIRA transcript and protein resulting from patient 1 variant; e Agarose gel electrophoresis of the amplified HIRA transcript from patient 2 compared to control showing the presence of a truncated transcript; f DNA sequencing electrophoregram of the truncated transcript showing the skipping of the exon 4 resulting from the splice site variant carried by patient 2; g Schematic representation of the predicted HIRA transcript and protein resulting from patient 2 variant
The Patient 2 was a 6-year-old female who presented behavioural problems mainly confined to symptoms of ASD. She had average head circumference for age. She also had diabetes mellitus type 1 and recurrent infections since 4 years of age. Array-CGH did not show any pathogenic CNV but whole-exome sequencing revealed a de novo heterozygous splice site variant in the HIRA gene, (hg19, chr22:19394706C > G; NM_003325.4: c.302 + 1G > C). The cDNA analysis confirmed the predicted deletion of exon 4 leading to a frameshift with a premature stop codon in exon 6, p.(Thr101Thrfs*20) (Fig. 1).
We also looked in public databases (DECIPHER, ClinVar, denovoDB) for additional de novo heterozygous HIRA variants potentially associated with pathogenicity. Interestingly, the denovo-db database (release v.1.6.1, May 2020 accessed) included two additional cases. This includes a case from the Deciphering Developmental Disorders (DDD) study (individual DDD4K.03620) (Deciphering Developmental Disorders Study 2017), with NDD associated with a de novo truncating variant in HIRA consisting in a stop-gained mutation, hg19 chr22:19341607G > A, NM_003325.4:c.2596C > T and leading to a truncated HIRA protein, p.(Gln866*). We also found one variant of unknown significance, chr22:19341564A > G; NM_003325.4:c.269 T > C, p.(Met880Thr), carried by a patient for which the phenotype was not clearly described but included in a large autism spectrum disorder cohort (de Rubeis et al. 2014).
Following these observations, we hypothesized that haploinsufficiency of HIRA might cause the phenotype observed in our patients and play a role in the developmental phenotype of the 22q11DS. We, therefore, studied the cerebral pattern of Hira expression and assessed the impact of haploinsufficiency of Hira in the neuronal plasticity and brain neuroanatomy in mice.
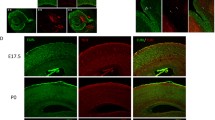
Expression analysis of Hira mRNA by RT-qPCR in mouse total brain extracts revealed a maximal level at birth with an approximately twofold increased expression compared to embryonic and postnatal expression (Fig. 2). Furthermore, RT-qPCR analyses performed in developing primary hippocampal cultures from mouse embryonic brains showed that Hira mRNA expression was maximal at 4 days of culture (Day in Vitro, DIV4) and significantly decreased when neurons become fully mature with functional synapses (DIV10 to DIV21). The pattern of Hira mRNA expression observed in total brain and in primary hippocampal neurons would, thus, correlate with the neuritogenesis and early dendritogenesis stages suggesting that Hira might modulate neuronal differentiation and early maturation processes.

Neuronal expression study and functional analyses of Hira in mouse brain tissue and primary hippocampal cultures. a Relative Hira mRNA expression levels at different stages of embryonic and postnatal development. E embryonic day, P postnatal day. RT-qPCR analysis of Hira mRNA expression level is shown relative to the Gapdh reference gene. Data are presented as dot plots with mean. For each time point, the experiments were performed using total RNAs extracted from a pool of total brain extracts from 5 different mice (25 mouse brains in total); each dot plot represents a technical replicate (n = 3); b Relative Hira mRNA expression level in developing mouse primary hippocampal neuronal cultures. Expression of Hira mRNA is shown relative to the Gapdh reference gene at the culture days 4, 10, 14 and 21. n = 2 independent cultures, Div Day in vitro, Data are presented as dot plots with mean; c Relative HIRA mRNA expression in HEK293T cultures transfected with the non-effective shRNA and with the effective shRNA (n = 5 independent transfections). Statistical significance was assessed using a non-parametric Mann–Whitney test; d Altered morphology of primary embryonic hippocampal neurons transfected with the effective shRNA or with the non-effective shRNA, observed in confocal microscopy (representative images of 3 transfected neurons that are also Red fluorescent protein (RFP)-labeled); scale bar: 50 µm. n = 2 independent transfections leading to the analysis of 5 neurons with non-effective shRNA and 7 neurons with Hira shRNA). e Graphical representation of the quantification of the number of neurite intersections (i.e., branching numbers) from the transfected hippocampal neurons (each dot indicates data for one transfected neuron); f Graphical representation of the quantification of the total neuritic length (each dot indicates data for one transfected neuron); g Graphical representation of the quantification of the number of neurites per branching level (each dot indicates data for one transfected neuron). Statistical significance was evaluated using Mann–Whitney test for the number of neurite intersections on Scholl analysis, the mean neurite length. The data on the number of neurites per branching level were analyzed using an Ordinary one-way ANOVA followed by Tukey’s multiple comparisons. Data are represented as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001
To address the impact of Hira haploinsufficiency in developing primary hippocampal neuronal cultures, we used a shRNA approach to knock-down endogenous Hira gene expression. We first sought to validate the efficiency of our approach by transfecting of a shRNA expression plasmid targeting HIRA in HEK293T cell lines. Our experiments resulted in a 56% (P = 0.0079) reduction of HIRA mRNA expression level, in accordance with a haploinsufficiency model. As the shRNA targets a sequence of HIRA gene that is identical between human and mouse orthologs, we then assessed the impact of Hira haploinsufficiency in developing mouse neuronal cultures. Primary hippocampal neurons transfected at DIV11 with the effective shRNA displayed morphological impairments compared to controls. Particularly, they displayed a 46% (P = 0.0051) decrease in the number of neuritic intersections per neuron, a 28% (P = 0.0177) decrease in the mean total neurite length per neuron and a 23% decrease in the number of neurites per neuron compared to neurons transfected with the non-effective shRNA (i.e., scramble shRNA). The decrease in the number of neurites per neurons was not statistically significant (P = 0.1490) but the assessment of the number of neurites by branching level showed a 33% (P = 0.0241) decrease in the number of neurites in the third branching level compared to neurons transfected with the non-effective shRNA. These results suggested that a twofold reduction of Hira gene expression dysregulates dendritic arborization in primary neuronal cultures, suggesting that the HIRA protein is essential for dendritic development growth and stability.
Since no previous mouse studies reported the implication of Hira in the context of brain development, and in order to study the role of Hira haploinsufficiency in vivo, we developed a heterozygous mouse model using the knock-out first allele method (Skarnes et al. 2011). The strategy relies on the identification of an exon common to all transcript variants (exon 4), upstream of which a LacZ cassette was inserted. Exon 4 of the Hira allele was flanked by loxP sequences bilaterally. The resulting Hiratm1a(EUCOMM)Wtsi mice were then phenotyped. At weaning age, mouse survival was assessed from 187 successfully genotyped mice originating from several litters and derived from a het-by-het breeding scheme. We obtained no homozygous mice suggesting that Hira is essential for viability. To determine the window of death, we carried out a recessive lethality screen at mouse embryonic day 14.5 (E14.5). We looked at 41 embryos, none of which were homozygous, suggesting that homozygous mice die before E14.5. Using a recently developed and highly robust approach for the assessment of 40 brain parameters distributed across 22 developmentally distinct brain regions (Collins et al. 2018), we analyzed neuroanatomical defects in Hira+/− transgenic mice (data available upon request). These parameters encompass six main brain categories: brain size, commissures (callosal, anterior and stria medullaris), ventricles (lateral and fourth), cortex (motor and cingulate), subcortex (hippocampus, caudate putamen, fimbria, thalamus, hypothalamus, substantia nigra, subiculum and colliculus), and cerebellum (granule layer, cerebellar nuclei, number of folia, pons, nerves, and pontine nuclei). This consisted of a systematic quantification on a single sagittal brain section at Lateral + 0.60 mm. To minimize environmental and genetic variations, mice of each genotype were analyzed at 16 weeks of age and bred on the same genetic background (Bl/6 N). Minor anomalies were identified in Hira+/− mice when compared to wild type, with suggestive evidence of possible convergence with microcephaly in some brain regions. For example, the hippocampus showed reduced height in the molecular layer (− 20%, P = 0.04). The corpus callosum and the fornix showed a marginally reduced area (− 10%, P = 0.04 and − 9%, P = 0.016, respectively) (Fig. 3).

Mouse Hira+/− displays minor reductions in neuroanatomical phenotypes. a Histogram comparing male Hira+/− to WT and showing variation (decreased-minus scale or increased-positive scale) in areas and lengths expressed as percentage of WT together with a color map indicating the significance level. A schematic representation of a section at Lateral + 0.60 mm. White color indicates a P-value higher than 0.05 and gray means that data were not enough to calculate a P-value. Three parameters were failed due to poor quality (folia, caudate putamen and optic chiasm). b Box plot and raw data points plots of the corpus callosum area and length of the lacunosum moleculare layer. c Sagittal sections of the hippocampus and the corpus callosum from WT and Hira+/− adult male mice stained with cresyl violet/luxol blue. Scale bar 0.25 cm. d List of brain parameters measured
Discussion
We report here de novo truncating variants in the HIRA gene, which is located in the commonly deleted region of the 22q11DS, identified in patients presenting with ID or ASD symptoms. These two variants were not found in control databases (GnomAD) and were predicted to be pathogenic and to lead to a loss-of-function, knowing that HIRA is predicted intolerant to loss-of-function (pLI = 1). This observation and the further identification of two additional cases with NDD and a de novo HIRA truncating mutation led us to consider HIRA as a candidate gene for aspects of the neurodevelopmental phenotype of 22q11DS.
Several genes have already been proposed as candidate genes for the neuropsychiatric phenotype of the 22q11DS, such as COMT, PRODH, RANBP1 or even ZDHHC8 (Goodman et al. 2000; Jacquet et al. 2002; Bassett et al. 2007; Raux et al. 2007; Mukai et al. 2008, 2015; Beemer et al. 2009; Vorstman et al. 2009; Philip and Bassett 2011; Gothelf et al. 2013; Meechan et al. 2015; Paronett et al. 2015; Moutin et al. 2017). Nevertheless, the pathophysiology of the neurodevelopmental phenotype remains unexplained. The role of HIRA in development has already been suggested as the homozygous KO is lethal in mice, which we also show in this study, and the heterozygous KO results in gastrulation defects (Roberts et al. 2002). Moreover, as a chromatin organization regulator HIRA is supposed to be necessary during brain development. Indeed, the chromatin remodeling process is frequently impaired in neurodevelopmental disorders and genes involved in this process are known to be responsible for multiple conditions associated with ID or ASD. A recent study described that genes located in the 22q11.2 region are significantly associated with networks involved in brain development and associated with pathogenesis of schizophrenia and ASD (Forsyth et al. 2020). In particular, HIRA was highlighted as a candidate driver of neurodevelopmental disorders such as schizophrenia and ASD.
Here, we bring new evidence of the implication of HIRA in brain development and disorder. We observed that mouse Hira was significantly expressed in both total brain and primary hippocampal cultures during times corresponding to neuritogenesis and early dendritogenesis stages. Furthermore, we demonstrated that a twofold reduction in Hira gene expression induced alteration of neuronal morphology in primary hippocampal cultures by impairing both dendritic growth and branching. Interestingly, primary hippocampal neurons from mouse models of 22q11.2 deletion also have impaired dendritic growth (Mukai et al. 2008; Fénelon et al. 2013; Moutin et al. 2017). Although it has been demonstrated that ZDHHC8 is involved in this neuronal phenotype, our results suggest that HIRA is also involved in the impaired dendritic growth observed in the 22q11DS mouse model. Additionally, it has been demonstrated that HIRA regulates neurogenesis by controlling the Wnt signaling pathway (Li and Jiao 2017). Indeed, HIRA regulates β-catenin levels by recruiting the H3K4 trimethyltransferase SETD1A to the β-catenin promoter. In humans, loss-of-function variants in SETD1A have been associated with schizophrenia and developmental disorders (Singh et al. 2016; Kummeling et al. 2020). Moreover, HIRA defects inhibit β-catenin expression and result in altered neurogenesis. Years of studies have revealed that the Wnt signaling pathway is necessary for the guidance and branching of the axon and dendrites, as well as synapses formation and their structural remodeling. Therefore, we could hypothesize that the impaired dendritic growth and branching observed in our neuronal model results from the alteration of the Wnt pathway caused by the haploinsufficiency of HIRA.
As shown in our study, it has been previously demonstrated that homozygous KO of Hira is lethal in mice (Roberts et al. 2002). Although conditional KO models in heart or muscle tissues have been described, there is no mouse model studying the impact of Hira haploinsufficiency on neurological phenotype (Dilg et al. 2016; Valenzuela et al. 2017). Therefore, we developed and analyzed a heterozygous Hira+/− mouse model to assess the impact of Hira haploinsufficiency on brain anatomy. We showed that heterozygous Hira knock-out mice resulted in subtle neuroanatomical defects with some evidence of possible microcephaly in some brain regions. In particular, the corpus callosum and the molecular layer of the hippocampus displayed a statistically significant reduction in surface area and height, respectively. It is likely that neuroanatomical anomalies exhibited in mice at the tissue level might be partially caused by abnormal neuritogenesis seen in primary neurons. It is noteworthy that nearly 50% of 22q11DS patients have microcephaly. Furthermore, MRI studies in 22q11DS patients have noted a decrease in total brain volume by 9–11% (Kates et al. 2004; Simon et al. 2005). Hypoplastic corpus callosum, abnormalities of the septum pellucidi and hippocampal abnormalities have also been reported (Ryan et al. 1997; Andrade et al. 2013; Bohm et al. 2017). Although mild, the neuroanatomical findings of the Hira+/− mouse model should be put in the context of the neuroradiographic abnormalities reported in 22q11DS patients, potentially sharing common features such as corpus callosum hypoplasia and hippocampal malformations.
In conclusion, neurobiological and genetic analyses have provided important insights into the genetic basis of psychiatric and cognitive symptoms observed in patients carrying 22q11.2 microdeletions. Nevertheless, the neurodevelopmental pathogenesis of this syndrome seems to be extremely complex and may probably require the effects of reduced dosage for multiple genes within the 22q11.2 deletion, possibly interacting with permissive variants in modifier genes elsewhere in the genome. Our results demonstrate that the haploinsufficiency of HIRA impairs dendritic growth, neuronal branching and neuroanatomical development suggesting that HIRA should be considered as a major gene in the landscape of the complex pathophysiology of the neurodevelopmental phenotype of 22q11DS.
Data availability
The data and materials used and analyzed during the current study are available from the corresponding author on reasonable request.
Code availability
Not applicable.
References
Andrade DM, Krings T, Chow EWC, Kiehl T-R, Bassett AS (2013) Hippocampal malrotation is associated with chromosome 22q11.2 microdeletion. Can J Neurol Sci 40:652–656. https://doi.org/10.1017/s0317167100014876
Bassett AS, Caluseriu O, Weksberg R, Young DA, Chow EWC (2007) Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol Psychiatry 61:1135–1140. https://doi.org/10.1016/j.biopsych.2006.07.038
Beemer FA, Emanuel BS, Kahn RS, van Engeland H, Kemner C (2009) Proline affects brain function in 22q11DS children with the low activity COMT 158 allele. Neuropsychopharmacology 34:739–746. https://doi.org/10.1038/npp.2008.132
Bohm LA, Zhou TC, Mingo TJ, Dugan SL, Patterson RJ, Sidman JD, Roby BB (2017) Neuroradiographic findings in 22q11.2 deletion syndrome. Am J Med Genet A 173:2158–2165. https://doi.org/10.1002/ajmg.a.38304
Bruining H, de Sonneville L, Swaab H, de Jonge M, Kas M, van Engeland H, Vorstman J (2010) Dissecting the clinical heterogeneity of autism spectrum disorders through defined genotypes. PLoS ONE 5:e10887. https://doi.org/10.1371/journal.pone.0010887
Collins SC, Wagner C, Gagliardi L, Kretz PF, Fischer MC, Kessler P, Kannan M, Yalcin B (2018) A method for parasagittal sectioning for neuroanatomical quantification of brain structures in the adult mouse. Curr Protoc Mouse Biol 8:e48. https://doi.org/10.1002/cpmo.48
De Rubeis S, He X, Goldberg AP, Poultney CS, Samocha K, Cicek AE, Kou Y, Liu L, Fromer M, Walker S, Singh T, Klei L, Kosmicki J, Shih-Chen F, Aleksic B, Biscaldi M, Bolton PF, Brownfeld JM, Cai J, Campbell NG, Carracedo A, Chahrour MH, Chiocchetti AG, Coon H, Crawford EL, Curran SR, Dawson G, Duketis E, Fernandez BA, Gallagher L, Geller E, Guter SJ, Hill RS, Ionita-Laza J, Jimenz Gonzalez P, Kilpinen H, Klauck SM, Kolevzon A, Lee I, Lei I, Lei J, Lehtimäki T, Lin CF, Ma’ayan A, Marshall CR, McInnes AL, Neale B, Owen MJ, Ozaki N, Parellada M, Parr JR, Purcell S, Puura K, Rajagopalan D, Rehnström K, Reichenberg A, Sabo A, Sachse M, Sanders SJ, Schafer C, Schulte-Rüther M, Skuse D, Stevens C, Szatmari P, Tammimies K, Valladares O, Voran A, Li-San W, Weiss LA, Willsey AJ, Yu TW, Yuen RK, Cook EH, Freitag CM, Gill M, Hultman CM, Lehner T, Palotie A, Schellenberg GD, Sklar P, State MW, Sutcliffe JS, Walsh CA, Scherer SW, Zwick ME, Barett JC, Cutler DJ, Roeder K, Devlin B, Daly MJ, Buxbaum JD, DDD Study; Homozygosity Mapping Collaborative for Autism; UK10K Consortium (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515:209–215. https://doi.org/10.1038/nature13772
De Smedt B, Devriendt K, Fryns J-P, Vogels A, Gewillig M, Swillen A (2007) Intellectual abilities in a large sample of children with velo-cardio-facial syndrome: an update. J Intellect Disabil Res 51:666–670. https://doi.org/10.1111/j.1365-2788.2007.00955.x
Deciphering Developmental Disorders Study (2017) Prevalence and architecture of de novo mutations in developmental disorders. Nature 542:433–438. https://doi.org/10.1038/nature21062
Dilg D, Saleh RN, Phelps SE, Rose Y, Dupays L, Murphy C, Mohun T, Anderson RH, Scambler PJ, Chapgier AL (2016) HIRA is required for heart development and directly regulates Tnni2 and Tnnt3. PLoS ONE 11:e0161096. https://doi.org/10.1371/journal.pone.0161096
Fénelon K, Xu B, Lai CS, Mukai J, Markx S, Stark KL, Hsu PK, Gan WB, Fischbach GD, MacDermott AB, Karayiorgou M, Gogos JA (2013) The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J Neurosci 33:14825–14839. https://doi.org/10.1523/JNEUROSCI.1611-13.2013
Fine SE, Weissman A, Gerdes M, Pinto-Martin J, Zackai EH, McDonald-McGinn DM, Emanuel BS (2005) Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11.2 deletion syndrome. J Autism Dev Disord 35:461–470. https://doi.org/10.1007/s10803-005-5036-9
Forsyth JK, Nachun D, Gandal MJ, Geschwind DH, Anderson AE, Coppola G, Bearden CE (2020) Synaptic and gene regulatory mechanisms in schizophrenia, autism, and 22q11.2 CNV mediated risk for neuropsychiatric disorders. Biol Psychiatry 87:150–163. https://doi.org/10.1016/j.biopsych.2019.06.029
Goodman BK, Rutberg J, Lin WW, Pulver AE, Thomas GH (2000) Hyperprolinaemia in patients with deletion (22)(q11.2) syndrome. J Inherit Metab Dis 23:847–848. https://doi.org/10.1023/a:1026773005303
Gothelf D, Schneider M, Green T, Debbané M, Frisch A, Glaser B, Zilkha H, Schaer M, Weizman A, Eliez S (2013) Risk factors and the evolution of psychosis in 22q11.2 deletion syndrome: a longitudinal 2-site study. J Am Acad Child Adolesc Psychiatry 52:1192-1203.e3. https://doi.org/10.1016/j.jaac.2013.08.008
Jacquet H, Raux G, Thibaut F, Hecketsweiler B, Houy E, Demilly C, Haouzir S, Allio G, Fouldrin G, Drouin V, Bou J, Petit M, Campion D, Frébourg T (2002) PRODH mutations and hyperprolinemia in a subset of schizophrenic patients. Hum Mol Genet 11:2243–2249. https://doi.org/10.1093/hmg/11.19.2243
Kannan M, Bayam E, Wagner C, Rinaldi B, Kretz PF, Tilly P, Roos M, McGillewie L, Bär S, Minocha S, Chevalier C, Po C, Chelly J, Mandel JL, Borgatti R, Piton A, Kinnear C, Loos B, Adams DJ, Hérault Y, Collins SC, Friant S, Godin JD, Yalcin B, Sanger Mouse Genetics Project (2017) WD40-repeat 47, a microtubule-associated protein, is essential for brain development and autophagy. Proc Natl Acad Sci USA 114:E9308–E9317. https://doi.org/10.1073/pnas.1713625114
Kates WR, Burnette CP, Bessette BA, Folley BS, Strunge L, Jabs EW, Pearlson GD (2004) Frontal and caudate alterations in velocardiofacial syndrome (deletion at chromosome 22q11.2). J Child Neurol 19:337–342. https://doi.org/10.1177/088307380401900506
Kummeling J, Stremmelaar DE, Raun N, Reijnders MRF, Willemsen MH, Ruiterkamp-Versteeg M, Schepens M, Man CCO, Gilissen C, Cho MT, McWalter K, Sinnema M, Wheless JW, Simon MEH, Genetti CA, Casey AM, Terhal PA, van der Smagt JJ, van Gassen KLI, Joset P, Bahr A, Steindl K, Rauch A, Keller E, Raas-Rothschild A, Koolen DA, Agrawal PB, Hoffman TL, Powell-Hamilton NN, Thiffault I, Engleman K, Zhou D, Bodamer O, Hoefele J, Riedhammer KM, Schwaibold EMC, Tasic V, Schubert D, Top D, Pfundt R, Higgs MR, Kramer JM, Kleefstra T (2020) Characterization of SETD1A haploinsufficiency in humans and Drosophila defines a novel neurodevelopmental syndrome. Mol Psychiatry. https://doi.org/10.1038/s41380-020-0725-5
Li Y, Jiao J (2017) Histone chaperone HIRA regulates neural progenitor cell proliferation and neurogenesis via β-catenin. J Cell Biol 216:1975–1992. https://doi.org/10.1083/jcb.201610014
Meechan DW, Maynard TM, Tucker ES, Fernandez A, Karpinski BA, Rothblat LA, LaMantia A-S (2015) Modeling a model: Mouse genetics, 22q11.2 Deletion Syndrome, and disorders of cortical circuit development. Prog Neurobiol 130:1–28. https://doi.org/10.1016/j.pneurobio.2015.03.004
Moutin E, Nikonenko I, Stefanelli T, Wirth A, Ponimaskin E, De Roo M, Muller D (2017) Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 deletion syndrome. Cereb Cortex 27:3618–3629. https://doi.org/10.1093/cercor/bhw183
Mukai J, Dhilla A, Drew LJ, Stark KL, Cao L, MacDermott AB, Karayiorgou M, Gogos JA (2008) Palmitoylation-dependent neurodevelopmental deficits in a mouse model of 22q11 microdeletion. Nat Neurosci 11:1302–1310. https://doi.org/10.1038/nn.2204
Mukai J, Tamura M, Fénelon K, Rosen AM, Spellman TJ, Kang R, MacDermott AB, Karayiorgou M, Gordon JA, Gogos JA (2015) Molecular substrates of altered axonal growth and brain connectivity in a mouse model of schizophrenia. Neuron 86:680–695. https://doi.org/10.1016/j.neuron.2015.04.003
Nashun B, Hill PW, Smallwood SA, Dharmalingam G, Amouroux R, Clark SJ, Sharma V, Ndjetehe E, Pelczar P, Festenstein RJ, Kelsey G, Hajkova P (2015) Continuous histone replacement by hira is essential for normal transcriptional regulation and de novo dna methylation during mouse oogenesis. Mol Cell 60:611–625. https://doi.org/10.1016/j.molcel.2015.10.010
Paronett EM, Meechan DW, Karpinski BA, LaMantia A-S, Maynard TM (2015) Ranbp1, deleted in DiGeorge/22q11.2 deletion syndrome, is a microcephaly gene that selectively disrupts layer 2/3 cortical projection neuron generation. Cereb Cortex 25:3977–3993. https://doi.org/10.1093/cercor/bhu285
Philip N, Bassett A (2011) Cognitive, behavioural and psychiatric phenotype in 22q11.2 deletion syndrome. Behav Genet 41:403–412. https://doi.org/10.1007/s10519-011-9468-z
Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, Zenker M, Hüffmeier U, Thiel C, Rüschendorf F, Nürnberg P, Reis A, Trautmann U (2006) Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 140:2063–2074. https://doi.org/10.1002/ajmg.a.31416
Raux G, Bumsel E, Hecketsweiler B, van Amelsvoort T, Zinkstok J, Manouvrier-Hanu S, Fantini C, Brévière GM, Di Rosa G, Pustorino G, Vogels A, Swillen A, Legallic S, Bou J, Opolczynski G, Drouin-Garraud V, Lemarchand M, Philip N, Gérard-Desplanches A, Carlier M, Philippe A, Nolen MC, Heron D, Sarda P, Lacombe D, Coizet C, Alembik Y, Layet V, Afenjar A, Hannequin D, Demily C, Petit M, Thibaut F, Frebourg T, Campion D (2007) Involvement of hyperprolinemia in cognitive and psychiatric features of the 22q11 deletion syndrome. Hum Mol Genet 16:83–91. https://doi.org/10.1093/hmg/ddl443
Roberts C, Sutherland HF, Farmer H, Kimber W, Halford S, Carey A, Brickman JM, Wynshaw-Boris A, Scambler PJ (2002) Targeted mutagenesis of the hira gene results in gastrulation defects and patterning abnormalities of mesoendodermal derivatives prior to early embryonic lethality. Mol Cell Biol 22:2318–2328. https://doi.org/10.1128/mcb.22.7.2318-2328.2002
Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ (1997) Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet 34:798–804. https://doi.org/10.1136/jmg.34.10.798
Schneider M, Debbané M, Bassett AS, Chow EW, Fung WL, van den Bree M, Owen M, Murphy KC, Niarchou M, Kates WR, Antshel KM, Fremont W, McDonald-McGinn DM, Gur RE, Zackai EH, Vorstman J, Duijff SN, Klaassen PW, Swillen A, Gothelf D, Green T, Weizman A, Van Amelsvoort T, Evers L, Boot E, Shashi V, Hooper SR, Bearden CE, Jalbrzikowski M, Armando M, Vicari S, Murphy DG, Ousley O, Campbell LE, Simon TJ, Eliez S, International Consortium on Brain and Behavior in 22q11.2 Deletion Syndrome (2014) Psychiatric disorders from childhood to adulthood in 22q11.2 deletion syndrome: results from the international consortium on brain and behavior in 22q11.2 deletion syndrome. Am J Psychiatry 171:627–639. https://doi.org/10.1176/appi.ajp.2013.13070864
Simon TJ, Ding L, Bish JP, McDonald-McGinn DM, Zackai EH, Gee J (2005) Volumetric, connective, and morphologic changes in the brains of children with chromosome 22q11.2 deletion syndrome: an integrative study. NeuroImage 25:169–180. https://doi.org/10.1016/j.neuroimage.2004.11.018
Singh T, Kurki MI, Curtis D, Purcell SM, Crooks L, McRae J, Suvisaari J, Chheda H, Blackwood D, Breen G, Pietiläinen O, Gerety SS, Ayub M, Blyth M, Cole T, Collier D, Coomber EL, Craddock N, Daly MJ, Danesh J, DiForti M, Foster A, Freimer NB, Geschwind D, Johnstone M, Joss S, Kirov G, Körkkö J, Kuismin O, Holmans P, Hultman CM, Iyegbe C, Lönnqvist J, Männikkö M, McCarroll SA, McGuffin P, McIntosh AM, McQuillin A, Moilanen JS, Moore C, Murray RM, Newbury-Ecob R, Ouwehand W, Paunio T, Prigmore E, Rees E, Roberts D, Sambrook J, Sklar P, St Clair D, Veijola J, Walters JT, Williams H, Sullivan PF, Hurles ME, O’Donovan MC, Palotie A, Owen MJ, Barrett JC, Swedish Schizophrenia Study; INTERVAL Study; DDD Study; UK10 K Consortium (2016) Rare loss-of-function variants in SETD1A are associated with schizophrenia and developmental disorders. Nat Neurosci 19:571–577. https://doi.org/10.1038/nn.4267
Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A (2011) A conditional knockout resource for the genome-wide study of mouse gene function. Nature 474:337–342. https://doi.org/10.1038/nature10163
Swillen A, Devriendt K, Legius E, Eyskens B, Dumoulin M, Gewillig M, Fryns JP (1997) Intelligence and psychosocial adjustment in velocardiofacial syndrome: a study of 37 children and adolescents with VCFS. J Med Genet 34:453–458. https://doi.org/10.1136/jmg.34.6.453
Valenzuela N, Soibam B, Li L, Wang J, Byers LA, Liu Y, Schwartz RJ, Stewart MD (2017) HIRA deficiency in muscle fibers causes hypertrophy and susceptibility to oxidative stress. J Cell Sci 130:2551–2563. https://doi.org/10.1242/jcs.200642
Vorstman JAS, Morcus MEJ, Duijff SN, Klaassen PWJ, Heineman-de Boer JA, Beemer FA, Swaab H, Kahn RS, van Engeland H (2006) The 22q11.2 deletion in children: high rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry 45:1104–1113. https://doi.org/10.1097/01.chi.0000228131.56956.c1
Vorstman JA, Turetsky BI, Sijmens-Morcus ME, de Sain MG, Dorland B, Sprong M, Rappaport EF, Beemer FA, Emanuel BS, Kahn RS, van Engeland H, Kemner C (2009) Proline affects brain function in 22q11DS children with the low activity COMT 158 allele. Neuropsychopharmacology 34:739–746. https://doi.org/10.1038/npp.2008.132
Acknowledgments
We would like to thank the patients and their families. We also thank the « IBiSA Electron Microscopy Facility » of the University of Tours for management and access to the confocal microscopy platform. This work was funded by the Association pour le Développement de la Neurogénétique (ADN) and The Fondation de France (to F.L.), and the Institut National de la Santé et de la Recherche Médicale (Inserm) for providing material support. The authors thank staff at the Research Support Facility (Sanger Institute) for their excellent care of the mice and the members of the genome engineering, genotyping, phenotyping and database teams for their contribution to this work.
Funding
This work was funded by the Association pour le Développement de la Neurogénétique (ADN) and The Fondation de France (to F.L.), and the Institut National de la Santé et de la Recherche Médicale (Inserm) for providing material support.
Author information
Authors and Affiliations
Contributions
MJ, FL, BY, and AT contributed to the study conception and design. Clinical and genetic data collection and analysis were performed by MJ, M-LV, SW, DH, NC, RP, JK, M-PM, TK and AT. Material preparation, in vitro and in vivo functional experiments and analyses were performed by MJ, DCU, VEV, CW, SC, SM, BY and FL. The first draft of the manuscript was written by MJ, M-LV, BY, FL and AT, and all authors commented on previous versions of the manuscript. All authors have reviewed and approved the finalized manuscript.
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Ethical approval
The study was approved by the local institutional review boards, and written informed consent was obtained from the patients’ parents, including explicit permission to share clinical and identifying information. All mouse experiments performed at the University of Tours/INSERM were approved by the French Ministry of Research (Project authorization number 01456.03). The care and use of Hira± mice in the Wellcome Sanger Institute study was carried out in accordance with UK Home Office regulations, UK Animals (Scientific Procedures) Act of 1986 under two UK Home Office licences (80/2485 and P77453634) that approved this work, which were reviewed regularly by the Wellcome Sanger Institute Animal Welfare and Ethical Review Body.
Consent to participate
The study was approved by the local institutional review boards, and written informed consent was obtained from the patients’ parents, including explicit permission to share clinical and identifying information.
Consent for publication
The study was approved by the local institutional review boards, and written informed consent was obtained from the patients’ parents, including explicit permission to share clinical and identifying information.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Jeanne, M., Vuillaume, ML., Ung, D.C. et al. Haploinsufficiency of the HIRA gene located in the 22q11 deletion syndrome region is associated with abnormal neurodevelopment and impaired dendritic outgrowth. Hum Genet 140, 885–896 (2021). https://doi.org/10.1007/s00439-020-02252-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-020-02252-1