
Abstract
A deeper understanding of the interaction between tumor cell and the immune microenvironment in bladder cancer may help select predictive and prognostic biomarkers. The current study aims to construct a prognostic signature for bladder cancer by analysis of molecular characteristics, as well as tumor-immune interactions. RNA-sequencing and clinical information from bladder cancer patients were downloaded from the TCGA database. The single sample Gene Sets Enrichment Analysis (ssGSEA) and Cell type Identification by Estimating Relative Subsets of RNA Transcripts (CIBERSORT) were employed to separate the samples into two clusters. Lasso Cox regression was performed to construct an immune gene signature for bladder cancer. The correlation between key target genes of immune checkpoint blockade and the prognostic signature was also analyzed. Dataset from Gene Expression Omnibus (GEO) was retrieved for validation. Two immunophenotypes and immunological characteristics were identified, and a 17-immune gene signature was constructed to provide an independent prognostic signature for bladder cancer. The signature was verified through external validation and correlated with genomic characteristics and clinicopathologic features. Finally, a nomogram was generated from the clinical characteristics and immune signature. Our study reveals a tumor-immune microenvironment signature useful for prognosis in bladder cancer. The results provide information on the potential development of treatment strategies for bladder cancer patients. Prospective studies are warranted to validate the prognostic capability of this model, but these data highlight the role of the microenvironment in the clinical outcome of patients.
Similar content being viewed by others


Introduction
Bladder cancer is the 9th most frequently diagnosed cancer worldwide. Bladder cancer is a heterogeneous disease and classified into muscle-invasive and nonmuscle-invasive disease [1]. Platinum-based chemotherapy has been the standard treatment strategy of metastatic muscle-invasive bladder cancer (MIBC) for many years, based on the results of clinical trials. The inhibition of immune checkpoints reactivates immune cytotoxicity and promotes tumor regression in MIBC [2]. Immune checkpoint inhibitors (ICIs) improve the survival of MIBC patients [3]. However, treatment responses differ among patients, partly due to the interpersonal heterogeneity [4].
Current clinical stratification systems focus on the tumor histopathology and the staging system to inform prognosis. Current predictive biomarkers of immunotherapy in bladder cancer include tumor molecular subtype [5], programmed cell death ligand-1 (PD-L1) expression [6], tumor mutational burden (TMB) [7], and CD8+ tumor-infiltrating lymphocytes (TILs). The molecular and immune characterization of cancer is critical for the prediction of treatment responses [8]. Molecular classification of bladder cancer is facilitated by transcriptome profiling, which will lead to accurate prediction of treatment outcomes. There is great importance of revising these stratification systems to include molecular and immune parameters to guide clinical decisions.
Previous research identified 6 subtypes and molecular features [9] based on The Cancer Genome Atlas (TCGA), UROMOL (nonmuscle-invasive bladder cancer cohort) [10], and IMvigor210 [4]. The subtypes include luminal papillary, luminal unstable, luminal non-specified, basal/squamous, stroma-rich, and neuroendocrine-like subtypes [9, 11]. The immune infiltration, oncogenic mechanisms, clinical characteristics and outcomes differ among the consensus classes.
Recent studies have investigated the immune characteristics of bladder cancer, which has prognostic potential compared with clinical characteristics [12,13,14,15,16]. The studies investigated the characteristics of bladder cancer mainly focusing on immune cell infiltration. However, the tumor-immune interactions and the prognostic value in bladder cancer are largely overlooked. In this study, we aim to explore the molecular characteristics associated with tumor-immune interaction and establish an immune signature for prognosis.
Methods
Data collection
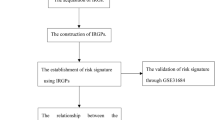
The transcriptomic information and clinical data of bladder cancer patients were retrieved. Normalized gene expression from RNA-sequencing (Fragments Per Kilobase Million value) and somatic mutation of TCGA-BLCA cohort were downloaded from TCGA database on 1 August 2021 (https://portal.gdc.cancer.gov). Then, TMB per Megabase was calculated for each sample. Data of clinical information and normalized gene expression were downloaded from Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo); accession number is GSE13507. Data were analyzed with R (version 4.0.3) (Fig. 1).

The main steps and bioinformatics tools used for data analysis were displayed.
Estimation of immune and stromal content
ESTIMATE (Estimation of STromal and Immune cells in MAlignant Tumor tissues using Expression data) tool was used to evaluate the infiltration of tumor and normal cells. StromalScore, ImmuneScore and EstimateScore were calculated [17]. CIBERSORT was utilized to determine the abundance of 22 infiltrating immune cells of the cancer sample [18].
ssGSEA and hierarchical clustering analysis
Using R packages (“GSVA”, “GSEABase”, and “limma”), ssGSEA algorithm was performed to investigate the immunological characteristics of each sample based on 29 immune gene sets [19]. The ssGSEA score xi of each sample was transformed into xi’ by deviation standardization. Hierarchical clustering analysis was used to determine the subtypes of the bladder cancer by Euclidean distance and Ward’s linkage. The discrimination and accuracy of the subtypes of the bladder cancer were confirmed by T-distribution stochastic neighbor embedding (tSNE) algorithm [20].
Analysis of prognosis-associated immunity genes
Differentially expressed genes (DEGs) were screened, with filter set as |log2 fold change | > 0.58 and false discovery rate (FDR) < 0.05 between Immunity_H and Immunity_L groups. Differentially expressed immunity genes (DEIGs) were retrieved from Immunology Database and Analysis Portal (ImmPort) [21, 22]. Univariate Cox proportional hazards regression analysis was performed. A gene with p < 0.01 was identified as prognosis-associated immunity genes (PIGs) from the DEIGs, followed by the Benjamini-Hochberg (BH) procedure to control for FDR.
Analysis of functional annotation and regulation network
In order to uncover the signaling pathways of the DEGs, a gene-set enrichment analysis was conducted of TCGA-BLCA cohort by Gene Set Enrichment Analysis (GSEA) [23, 24]. Kyoto Encyclopedia of Genes (KEGG) pathways upregulated in Immunity_H and Immunity_L groups were identified. Screening condition was set as FDR < 0.01. Then, transcription factors associated with tumorigenesis and tumor progression of bladder cancer were obtained from CISTROME project (http://cistrome.org/), and the differentially expressed transcription factors (DETFs) were extracted from the total DEGs. The regulatory network of PIGs and DETFs were constructed using Pearson’s correlation coefficient analysis [25, 26]. The cutoffs for significance were set as |r| > 0.3 and FDR < 0.01. STRING (string-db.org/) was used to analyse protein-protein interaction (PPI).
Construction of immunity gene-associated prognostic model
An optimal immunity gene-associated prognostic model (IGPM) for bladder cancer was generated using Cox regression model with LASSO via R package “glmnet” using PIGs. The risk score was calculated as follows:
where Expri indicates the expression level of gene i, and coefi denotes the regression coefficient of gene i in the signature.
All patients were separated into low- and high-risk groups based on the median value of IGPM-based risk signature. Survival analysis was performed with Kaplan-Meier curve. The log rank test was used to compare the survival status between the groups. The time-dependent receiver operating characteristic curve (ROC) was generated and the area under the curve (AUC) was calculated for 1-, 3-, and 5-year overall survival to determine the predictive value of the risk signature. The relativity between the risk signature and clinical parameters, immune checkpoints, immune cell infiltration, and TMB were analyzed using Pearson’s or Spearman correlation. A p < 0.05 was considered as statistically significant. Finally, IGPM-based risk signature and clinicopathological parameters were analyzed with univariate and multivariate Cox regression. Then, the above factors were used to construct a nomogram. Finally, ROC and calibration curves were used to determine the suitability of the nomogram.
Results
Two subtypes of bladder cancer were identified by immunogenomic profiling
The samples included 414 cancer tissue and 19 adjacent tissue samples. For available clinical information data, only patients with complete information were included. Those with missing data were excluded, including survival time, survival status, age, gender, grade, or TNM classification. Then, 411 cases with data on somatic gene mutations were enrolled (Table 1).
Based on the ssGSEA scores and hierarchical clustering algorithm, the samples were separated into two clusters: Immunity_H (High) and Immunity_L (Low) (Fig. 2A, B). The characteristics of tumor microenvironment between the two subtypes were identified based on the results of ESTIMATE. The results indicate that the EstimateScore, ImmuneScore and StromalScore of Immunity_H group are higher than those of Immunity_L group (Wilcox test, p < 0.001) (Fig. 2C).

(A) Bladder cancer patients were separated into Immunity_H and Immunity_L subtypes by hierarchical clustering. (B) Landscape of tumor microenvironment and the immune characteristics in the TCGA-BLCA cohort. (C) EstimateScore, StromalScore, and ImmuneScore between two subtypes. (D) Immune cell infiltrations between two subtypes. (E) Validation of immunophenotype via tSNE. (F) HLA gene expression levels between two subtypes. *p < 0.05, **p < 0.01, ***p < 0.001. tSNE T-distribution stochastic neighbor embedding.
The immune cell infiltration was investigated via CIBERSORT algorithm. There are significant differences of the infiltration of CD8+ T cells, activated memory CD4+ T cells, naive CD4+ T cells, monocytes, naive B cells, M0 macrophages, M1 macrophages, and neutrophils between Immunity_H and Immunity_L groups (Fig. 2D). The immune level clustering of the bladder cancer patients was further confirmed by tSNE algorithm. The classification results were obtained (Fig. 2E). The expression of HLA genes in the two subtypes were investigated. Results show that most HLA genes are highly expressed in Immunity_H than those in Immunity_L group (Wilcox test, p < 0.05) (Fig. 2F).
Tumor-immune interactions
The molecular characteristics of tumor-immune interactions in bladder cancer patients were explored by immunophenotype stratification. After preliminary screening, 2689 genes were identified as DEGs. Of these, 1526 and 1163 genes were upregulated and downregulated in the Immunity_H group, respectively. The expression levels of all DEG were shown in Fig. 3A. Subsequently, 468 genes were identified as DEIGs. Among them, 412 and 56 genes were upregulated and downregulated, respectively (Fig. 3B). Finally, 32 PIGs were identified using univariable Cox proportional hazards regression analysis (Fig. 3C). The correction of FDR by the Benjamini & Hochberg method was shown in Supplementary Table 1. Of these, 12 and 20 gene were upregulated and downregulated in the Immunity_H group, respectively. The log2 fold change values in the DEGs and their FDR values were shown in Fig. 3D.

(A) All DEGs between two subtypes. (B) All DEIGs between two subtypes. (C) PIGs and their hazard ratios based on univariable Cox proportional hazards regression analysis. (D) Volcano plot of DEGs. DEG differentially expressed gene, DEIG differentially expressed immunity gene, PIG prognosis-associated immunity gene.
GSEA was performed to investigate the pathways involved in the DEGs in order to uncover the overall pattern of the immune gene expression. Several biologically sensible themes were obtained in Immunity_H group, indicating that the DEGs were involved in the immune-related biological processes and signaling pathways. For KEGG analysis (Fig. 4A, B and Supplementary Table 2), the immune-associated pathways were highly active in Immunity_H. The pathways include cytokine receptor interaction, hematopoietic cell lineage, natural killer (NK) cell mediated cytotoxicity, chemokine signaling pathway, JAK/STAT signaling pathway, cell adhesion molecules, antigen processing and presentation, and T cell and B cell receptor signaling pathway. Furthermore, various immune disease-associated pathways hyperactivated in Immunity_H were identified, including asthma, autoimmune thyroid disease, type I diabetes mellitus, primary immunodeficiency, systemic lupus erythematosus, and acute myeloid leukemia. Several pathways relating to infectious diseases were also enriched in Immunity_H group, such as viral myocarditis, leishmaniasis, prion diseases, and pathogenic Escherichia coli infection.

(A) Bar plot chart of DEGs through KEGG enrichment analysis. (B) Bubble chart of DEGs through KEGG enrichment analysis. (C) Regulatory network revealed by alluvial diagram of the BCTFs and PIGs. (D) PPI network between BCTFs and PIGs. KEGG Kyoto Encyclopedia of Genes and Genomes, BCTF bladder cancer transcription factor, PIGs prognosis-associated immunity genes.
Differential expression and data from CISTROME database were combined to identify transcription factors related to bladder cancer. A total of 36 upregulated transcription factors were identified in the Immunity_H group. Then, the regulatory relationships of the bladder cancer transcription factor (BCTF)-PIGs were determined from correlation analysis (Supplementary Table 3). The regulatory network of the BCTF-PIGs was shown in Fig. 4C. PPI analysis was performed to investigate the correlation between the BCTF and PIGs, which confirm the interactions between them (Fig. 4D).
Construction and validation of the IGPM-based risk signature
An IGPM-based risk signature was constructed, including 17 genes to predict overall survival in TCGA-BLCA cohort (Fig. 5A, 5B). The 17 genes are HLA-G, AGER, VEGFA, SCG2, IL9R, CTSE, NOX1, S100A10, GBP2, VIM, CD3D, CCL17, PDGFRB, CTSS, PLXNB1, GNLY, and LTBP2.

(A) 10-fold cross-validation for tuning parameter selection in the LASSO model. (B) LASSO coefficient profiles of the 32 PIGs. (C–F) Distribution of the risk score, survival time, and survival status in TCGA-BLCA (C, E) and GSE13507 cohort (D, F). (G, H) Correlation analysis of the risk score and survival time in TCGA-BLCA and GSE13507 cohort. (I, J) Kaplan-Meier survival analysis based on IGPM-based risk signature in TCGA-LCA and GSE13507 cohort. (K, L) The ROC curve and AUC of the predictions for 1-, 3-, and 5-year of IGPM-based risk signature for TCGA-BLCA and GSE13507 cohort. (M, N) The calibration plot of IGPM-based risk signature for TCGA-BLCA and GSE13507 cohort. IGPM immunity gene-associated prognostic model, ROC curve receiver operating characteristic curve, AUC area under the curve, kIG key PIG involved in the construction of IGPM.
The risk score of each patient was calculated, and the patients of TCGA-BLCA cohort and GSE13507 were separated into high- and low-risk groups based on a median risk score (Fig. 5C, D). The clinical characteristics of 165 bladder cancer patients of GEO dataset was shown in Table 2. Survival time in the high-risk group is shorter than that in the low-risk group (Fig. 5E, F). Correlation analysis suggest that the risk score had a significantly negative correlation with survival time (Fig. 5G, H). Results show that high-risk group was significantly associated with poorer overall survival (Log-rank test, p < 0.0001 and p = 0.006, respectively, Fig. 5I, J). The AUCs of the 1-, 3-, and 5-year predictions were 0.742, 0.595, 0.734, 0.623, 0.747, and 0.612, respectively (Fig. 5K, L). The calibration plot was used to determine the discriminative ability of the IGPM-based risk signature. The predicted value of IGPM-based risk signature was in agreement with the actual value (Fig. 5M, N).
Correlation of risk score, clinical features, immune cell infiltration, immune checkpoints and TMB
The pairwise correlations of the risk score were examined with clinical features, immune cell infiltration, immune checkpoints, and TMB, respectively. Differences of risk score among subtypes according to clinical variables were determined. The risk score increased with tumor grade (Fig. 6A) and stage (Fig. 6B), indicating that risk score was positively correlated with tumor progression. Our results show that LTBP2, PDGFRB, SCG2, and VIM have significantly positive correlation with American Joint Committee on Cancer (AJCC) stage and grade (Supplementary Fig. S1), indicating that the PIGs might affect the prognosis of bladder cancer patients.

(A) Correlation of risk score with tumor grade and tumor stage. (B) Infiltrating immune cell subpopulations and levels between low-/high-risk groups. (C) Correlation of the IGPM-based risk signature with kIGs and immune cell infiltration. (D) Association analyses between risk score and immune checkpoints. Association between risk score and PDCD1LG2 (E), PDCD1 (F), and CTLA4 (G). (H) Comparison of expression levels of ICI-related genes between low-/high-risk groups. (I) Comparison of TMB between IGPM-high and IGPM-low groups. (K) Kaplan-Meier survival analysis based on the TMB in the TCGA-BLCA cohort. (L) Kaplan-Meier survival analysis for groups stratified by combining the TMB and the IGPM-based risk signature in the. *p < 0.05, **p < 0.01.
Results of CIBERSORT algorithm indicate that proportion of CD8 T cells, follicular helper T cells, memory activated CD4 T cells, regulatory T cells (Tregs), and activated dendritic cells (DCs) were negatively associated with risk score (Fig. 6C). SCG2 had a negative correlation with the follicular helper T cells, CD8 T cells, and activated dendritic cells, whereas GNLY had a positive correlation with CD8 T cells, memory activated CD4 T cells, resting NK cells, and M1 macrophages. Besides, CD3D, CTSS, GBP2, and HLA-G had positive correlation with CD8 T cells and memory activated CD4 T cells, but there is a negative correlation with M0 macrophages. LTBP2, VIM, and PDGFRB are positively correlated with M0 macrophages and M2 macrophages, but are negatively correlated with follicular helper T cells and activated dendritic cells (Fig. 6D). These results suggest that the prognostic signature is a novel approach to reveal immunity regulatory network in bladder cancer. The correlation between the immune subtypes and clinical characteristics in the TCGA-BLCA cohort was shown in Table 3.
Furthermore, 6 key immune checkpoint blockade genes (PDCD1, CD274, PDCD1LG2, CTLA4, HAVCR2 and IDO1) were correlated. The correlation between immune checkpoint blockade key targets and the prognostic signature was analyzed (Fig. 6E). The results indicate that the prognostic signature is positively correlated to PDCD1LG2 (r = 0.19; p = 0.00017) (Fig. 6F); negatively correlated to PDCD1 (r = −0.15; p = 0.0032) and CTLA4 (r = −0.13; p = 0.011) (Fig. 6G, H). Further correlation analysis revealed that the expression levels of 16 out of 47 (i.e., PDCD1, CTLA4, etc.,) immune check blockade-associated gene were significantly upregulated in patients of low risk (Fig. 6I), suggesting the prognostic signature could predict clinical outcome for patients receiving immunotherapy.
We further found that there are no significant differences in TMB between high and low IGPM group (Fig. 6J). However, higher TMB was associated with better overall survival (Log-rank test, p < 0.001, Fig. 6K). IGPM and TMB were integrated to stratify all the samples into TMBhigh/IGPMlow, TMBlow/IGPMlow, TMBhigh/ IGPMhigh, and TMBlow/IGPMhigh groups. Significant differences are found among all groups (Log-rank test, p < 0.0001, Fig. 6L), and patients in the TMBhigh/ IGPMlow group have the best overall survival. These results demonstrate that the risk score was positively correlated with tumor malignancy.
Construction and validation of a nomogram
Univariate and multivariate Cox regression analysis was performed to determine whether IGPM-based risk signature is an independent prognostic factor. The IGPM-based risk signature, together with gender, age and AJCC stage were used as covariates. The results suggest that p values of the risk score in two analyses were <0.001, and hazard ratios which were 3.651 [95% confidence interval (CI): 2.713–4.915] and 2.993 (95% CI: 2.187–4.094), confirming that the IGPM-based risk signature could inform the prognosis of bladder cancer patients (Fig. 7A–B).

(A) Univariate Cox regression results. (B) Multivariate Cox regression results. (C) Nomogram constructed in conjunction with the IGPM-based risk signature and clinical characterization. (D) The ROC curve and AUC of the predictions for 1-, 3-, and 5 years of the nomogram. (E) The calibration plot of the nomogram.
Finally, a nomogram was constructed to extend the clinical application of IGPM-based risk signature (Fig. 7C). The ROC curve suggests that the nomogram has good predictive accuracy for survival (Fig. 7D). Furthermore, the calibration plot shows that the nomogram has similar performance to that of an ideal model (Fig. 7E).
Discussion
In recent years, the classification of bladder cancer has moved from classical clinical characteristics to multi-omics profiling. Novel biomarkers and drug targets for bladder cancer are being investigated [9, 27]. With the emergence of ICI in the treatment of multiple cancer types, there is a growing interest in the tumor immunology. The stratification of bladder cancer patients due to their immunological characteristics have been demonstrated to improve patient survival [28]. However, previous studies have not incorporated the tumor-immune interactions.
In our study, ssGSEA was used to separate bladder cancer patients to immunity_H and immunity_L subtypes. In immunity_H group, high infiltration of immune cells in tumors were observed, as well as positive antitumor activities, which is consistent with previous findings [29, 30]. Immune control of tumor growth is mediated by the cytotoxic T lymphocytes or NK cells. CTLs have shown prognostic value in multiple tumor types, including bladder cancer [31]. NK cells exert antitumor effects under low major histocompatibility complex (MHC) conditions. Intratumoral CD56bright NK cells are correlated with better survival outcome in bladder cancer [32, 33]. Among various immune cells, B cells have a role in tumor progression and impact on the bladder cancer metastasis [34]. Moreover, both proinflammatory and tumoricidal macrophages which inhibit inflammation are involved in the process of immune response [35]. M2 macrophage is “protumoral macrophage”, which is associated with poor prognosis [36]. Additionally, DCs can initiate an immune response to adaptive immune system [37]. Consistently, patients in the immunity_H group have better survival outcome than those in the immunity_L group.
The results of ssGSEA combined with bioinformatics analysis demonstrate that the immunity_H group has an activated status of immunity. The immunity_H subtype is abundant with immune-related signatures, but also associated with various cancer-related pathways, such as JAK/STAT, apoptosis, pancreatic cancer, and MARK signaling. The results are in accordance with previous reports demonstrating that immune signatures are associated with the JAK-STAT signaling pathways [38]. Of note, our results suggest that signaling pathway and immune response are correlated in bladder cancer.
IGPM-based risk signature was constructed based on the 17-immune genes. It is a robust prognostic tool, which can predict outcome of the bladder cancer patient, and result was confirmed in two independent datasets. Furthermore, the IGPM-based risk signature has similar value of clinical variables and infiltration of immune cells.
Most of the 17 genes have been associated with tumor initiation. Previous trials have reported that VIM (vimentin) is an epigenetic biomarker for bladder cancer. Hypermethylated VIM is associated with worse relapse-free survival (RFS) in bladder cancer [39,40,41,42]. However, study of Hu et al. reveals that high VIM expression level is associated with worse overall survival [43]. HLA-G (Human leukocyte antigen-G), an immune checkpoint molecule known for its tolerogenic role, is expressed by various cancer types, including bladder cancer [44, 45]. Expression of HLA-G has been associated with higher grade and worse prognosis in multiple cancers [46, 47]. VEGFA (Vascular Endothelial Growth Factor A) can promote angiogenesis and contribute to tumor metastasis [48, 49]. VEGFA overexpression in stage T3-T4 bladder cancer is a predictive factor for antiangiogenic therapy [50]. Compared with MIBC, CTSE (cathepsin E) is overexpressed in nonmuscle-invasive bladder cancer (NMIBC) [51]. During a long-term follow-up study of 693 patients of NMIBC, low CTSE expression was confirmed to be significantly related to the progression of NMIBC to MIBC [52]. SCG2 (secretogranin II) is a secretory protein, which could regulate the functions of leucocyte, endothelial, and mesenchymal cell [53]. Previous study found that SCG2 is secreted by malignant pheochromocytomas, which can serve as a potential diagnostic marker [54, 55]. The expansion of CD8+ T cells is promoted by Th9 cell in colorectal cancer in an IL-9R-dependent manner [56, 57]. Saiki et al. summarized its biological functions and prognostic value of S100A10 in cancer [58]. GNLY encodes granulysin, which causes tumor lysis [59]. High CD3D/CD4 ratio predicts longer survival of MIBC, and the CD3D/CD4 ratio is considered as a prognostic marker [3]. CCL17 induced CCR4 production, and CCR4 expression was a prognostic risk factor in bladder cancer [60]. The level of urinary PDGFRB was correlated with the risk of recurrence of NMIBC [61]. All these genes were closely related to the prognosis of bladder cancer.
In the current study, we found that bladder cancer patients of higher TMB had a better survival outcome, which was similar with other studies [62, 63]. However, there was no statistically significant correlation between TMB and the IGPM-based risk signature. The stratified survival analysis suggested that the prognostic value of IGPM-based risk signature was not interfered by TMB.
There are several limitations of our study. This study aimed to develop a nomogram to predict the overall survival of patients of bladder cancer. However, for NMIBC patients, time to recurrence or recurrence-free survival is the suitable primary endpoint, while overall survival is used as a potential surrogate endpoint [64]. Furthermore, the analysis was based on TCGA and GEO databases; validation using larger datasets is required to emulate the real-world environment. Future studies should investigate the utility of prognostic tools.
In conclusion, these findings demonstrated that the IGPM-based risk signature could be utilized to predict the prognosis of bladder cancer patients.
Conclusion
In conclusion, the comprehensive landscape of tumor-immune interactions in bladder cancer was elucidated, and an IGPM-based risk signature was constructed to inform the prognosis of bladder cancer patients through computational algorithms. The 17-gene signature should be validated in larger prospective cohorts of clinical samples. Further investigations on the molecular mechanisms are needed.
Data availability
Publicly available datasets were analyzed in this study.
References
Pang KH, Esperto F, Noon AP. party EAUYAU-UCW. Opportunities of next-generation sequencing in non-muscle invasive bladder cancer outcome prediction. Transl Androl Urol. 2017;6:1043–8.
Bellmunt J, Powles T, Vogelzang NJ. A review on the evolution of PD-1/PD-L1 immunotherapy for bladder cancer: The future is now. Cancer Treat Rev. 2017;54:58–67.
Jiang W, Zhu D, Wang C, Zhu Y. An immune relevant signature for predicting prognoses and immunotherapeutic responses in patients with muscle-invasive bladder cancer (MIBC). Cancer Med. 2020;9:2774–90.
Balar AV, Galsky MD, Rosenberg JE, Powles T, Petrylak DP, Bellmunt J, et al. Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelial carcinoma: a single-arm, multicentre, phase 2 trial. Lancet. 2017;389:67–76.
Song BN, Kim SK, Mun JY, Choi YD, Leem SH, Chu IS. Identification of an immunotherapy-responsive molecular subtype of bladder cancer. EBioMedicine. 2019;50:238–45.
Zhou TC, Sankin AI, Porcelli SA, Perlin DS, Schoenberg MP, Zang X. A review of the PD-1/PD-L1 checkpoint in bladder cancer: From mediator of immune escape to target for treatment. Urol Oncol. 2017;35:14–20.
Lv J, Zhu Y, Ji A, Zhang Q, Liao G. Mining TCGA database for tumor mutation burden and their clinical significance in bladder cancer. Biosci Rep. 2020;40:BSR20194337.
McConkey DJ, Choi W, Shen Y, Lee IL, Porten S, Matin SF, et al. A Prognostic Gene Expression Signature in the Molecular Classification of Chemotherapy-naive Urothelial Cancer is Predictive of Clinical Outcomes from Neoadjuvant Chemotherapy: A Phase 2 Trial of Dose-dense Methotrexate, Vinblastine, Doxorubicin, and Cisplatin with Bevacizumab in Urothelial Cancer. Eur Urol. 2016;69:855–62.
Tan TZ, Rouanne M, Tan KT, Huang RY, Thiery JP. Molecular Subtypes of Urothelial Bladder Cancer: Results from a Meta-cohort Analysis of 2411 Tumors. Eur Urol. 2019;75:423–32.
Hedegaard J, Lamy P, Nordentoft I, Algaba F, Hoyer S, Ulhoi BP, et al. Comprehensive Transcriptional Analysis of Early-Stage Urothelial Carcinoma. Cancer Cell. 2016;30:27–42.
Kamoun A, de Reynies A, Allory Y, Sjodahl G, Robertson AG, Seiler R, et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur Urol. 2020;77:420–33.
Liu J, Ma H, Meng L, Liu X, Lv Z, Zhang Y, et al. Construction and External Validation of a Ferroptosis-Related Gene Signature of Predictive Value for the Overall Survival in Bladder Cancer. Front Mol Biosci. 2021;8:675651.
Wang X, Pan L, Lu Q, Huang H, Feng C, Tao Y, et al. A combination of ssGSEA and mass cytometry identifies immune microenvironment in muscle-invasive bladder cancer. J Clin Lab Anal. 2021;35:e23754.
Zhao K, Zhang Q, Zeng T, Zhang J, Song N, Wang Z. Identification and validation of a prognostic immune-related lncRNA signature in bladder cancer. Transl Androl Urol. 2021;10:1229–40.
Wu Y, Zhang L, He S, Guan B, He A, Yang K, et al. Identification of immune-related LncRNA for predicting prognosis and immunotherapeutic response in bladder cancer. Aging (Albany NY). 2020;12:23306–25.
Quan J, Zhang W, Yu C, Bai Y, Cui J, Lv J, et al. Bioinformatic identification of prognostic indicators in bladder cancer. Biomark Med. 2020;14: 1243–54.
Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun. 2013;4:2612.
Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7.
Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7. Jan 16
Kobak D, Berens P. The art of using t-SNE for single-cell transcriptomics. Nat Commun. 2019;10:5416.
Bhattacharya S, Dunn P, Thomas CG, Smith B, Schaefer H, Chen J, et al. ImmPort, toward repurposing of open access immunological assay data for translational and clinical research. Sci Data. 2018;5:180015.
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47.
Mootha VK, Lindgren CM, Eriksson K-F, Subramanian A, Sihag S, Lehar J, et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–73.
Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA. 2005;102:15545–50.
Zheng R, Wan C, Mei S, Qin Q, Wu Q, Sun H, et al. Cistrome Data Browser: expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2019;47:D729–D735.
Mei S, Qin Q, Wu Q, Sun H, Zheng R, Zang C, et al. Cistrome Data Browser: a data portal for ChIP-Seq and chromatin accessibility data in human and mouse. Nucleic Acids Res. 2017;45:D658–D662.
Robertson AG, Groeneveld CS, Jordan B, Lin X, McLaughlin KA, Das A, et al. Identification of Differential Tumor Subtypes of T1 Bladder Cancer. Eur Urol. 2020;78:533–7.
Hu B, Wang Z, Zeng H, Qi Y, Chen Y, Wang T, et al. Blockade of DC-SIGN(+) Tumor-Associated Macrophages Reactivates Antitumor Immunity and Improves Immunotherapy in Muscle-Invasive Bladder Cancer. Cancer Res. 2020;80:1707–19.
He Y, Jiang Z, Chen C, Wang X. Classification of triple-negative breast cancers based on Immunogenomic profiling. J Exp Clin Cancer Res. 2018;37:327.
Liu Z, Jiang Z, Wu N, Zhou G, Wang X. Classification of gastric cancers based on immunogenomic profiling. Transl Oncol. 2021;14:100888.
van Wilpe S, Gerretsen ECF, van der Heijden AG, de Vries IJM, Gerritsen WR, Mehra N. Prognostic and Predictive Value of Tumor-Infiltrating Immune Cells in Urothelial Cancer of the Bladder. Cancers (Basel). 2020;12:2692.
Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, et al. Human CD56bright NK Cells: An Update. J Immunol. 2016;196:2923–31.
Mukherjee N, Ji N, Hurez V, Curiel TJ, Montgomery MO, Braun AJ, et al. Intratumoral CD56(bright) natural killer cells are associated with improved survival in bladder cancer. Oncotarget. 2018;9:36492–502.
Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. 2016;17:174.
Takeuchi H, Tanaka M, Tanaka A, Tsunemi A, Yamamoto H. Predominance of M2-polarized macrophages in bladder cancer affects angiogenesis, tumor grade and invasiveness. Oncol Lett. 2016;11:3403–8.
Komohara Y, Jinushi M, Takeya M. Clinical significance of macrophage heterogeneity in human malignant tumors. Cancer Sci. 2014;105:1–8.
Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265–77.
Hindupur SV, Schmid SC, Koch JA, Youssef A, Baur EM, Wang D, et al. STAT3/5 Inhibitors Suppress Proliferation in Bladder Cancer and Enhance Oncolytic Adenovirus Therapy. Int J Mol Sci. 2020;21:1106.
Reinert T, Borre M, Christiansen A, Hermann GG, Orntoft TF, Dyrskjot L. Diagnosis of bladder cancer recurrence based on urinary levels of EOMES, HOXA9, POU4F2, TWIST1, VIM, and ZNF154 hypermethylation. PLoS One. 2012;7:e46297.
Guan B, Xing Y, Xiong G, Cao Z, Fang D, Li Y, et al. Predictive value of gene methylation for second recurrence following surgical treatment of first bladder recurrence of a primary upper-tract urothelial carcinoma. Oncol Lett. 2018;15:9397–405.
Costa VL, Henrique R, Danielsen SA, Duarte-Pereira S, Eknaes M, Skotheim RI, et al. Three epigenetic biomarkers, GDF15, TMEFF2, and VIM, accurately predict bladder cancer from DNA-based analyses of urine samples. Clin Cancer Res. 2010;16:5842–51.
Guo RQ, Xiong GY, Yang KW, Zhang L, He SM, Gong YQ, et al. Detection of urothelial carcinoma, upper tract urothelial carcinoma, bladder carcinoma, and urothelial carcinoma with gross hematuria using selected urine-DNA methylation biomarkers: A prospective, single-center study. Urol Oncol. 2018;36:342 e15–342.e23.
Hu J, Zhou L, Song Z, Xiong M, Zhang Y, Yang Y, et al. The identification of new biomarkers for bladder cancer: A study based on TCGA and GEO datasets. J Cell Physiol. 2019;13:5931.
Rouas-Freiss N, Moreau P, LeMaoult J, Carosella ED. The dual role of HLA-G in cancer. J Immunol Res. 2014;2014:359748.
Gan LH, Huang LF, Zhang X, Lin A, Xu DP, Wang Q, et al. Tumor-specific upregulation of human leukocyte antigen-G expression in bladder transitional cell carcinoma. Hum Immunol. 2010;71:899–904.
Carosella ED, Rouas-Freiss N, Tronik-Le Roux D, Moreau P, LeMaoult J. HLA-G: An Immune Checkpoint Molecule. Adv Immunol. 2015;127:33–144.
Desgrandchamps F, LeMaoult J, Goujon A, Riviere A, Rivero-Juarez A, Djouadou M, et al. Prediction of non-muscle-invasive bladder cancer recurrence by measurement of checkpoint HLAG’s receptor ILT2 on peripheral CD8+ T cells. Oncotarget. 2018;9:33160–9.
Pauty J, Usuba R, Cheng IG, Hespel L, Takahashi H, Kato K, et al. A Vascular Endothelial Growth Factor-Dependent Sprouting Angiogenesis Assay Based on an In Vitro Human Blood Vessel Model for the Study of Anti-Angiogenic Drugs. EBioMedicine. 2018;27:225–36.
Ghosh S, Sullivan CA, Zerkowski MP, Molinaro AM, Rimm DL, Camp RL, et al. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol. 2008;39:1835–43.
Pignot G, Bieche I, Vacher S, Guet C, Vieillefond A, Debre B, et al. Large-scale real-time reverse transcription-PCR approach of angiogenic pathways in human transitional cell carcinoma of the bladder: identification of VEGFA as a major independent prognostic marker. Eur Urol. 2009;56:678–88.
Blaveri E, Simko JP, Korkola JE, Brewer JL, Baehner F, Mehta K, et al. Bladder cancer outcome and subtype classification by gene expression. Clin Cancer Res. 2005;11:4044–55. Jun 1
Fristrup N, Ulhoi BP, Birkenkamp-Demtroder K, Mansilla F, Sanchez-Carbayo M, Segersten U, et al. Cathepsin E, maspin, Plk1, and survivin are promising prognostic protein markers for progression in non-muscle invasive bladder cancer. Am J Pathol. 2012;180:1824–34.
Kähler CM, Schratzberger P, Kaufmann G, Hochleitner B, Bechter O, Götsch C, et al. Transendothelial migration of leukocytes and signalling mechanisms in response to the neuropeptide secretoneurin. Regul Pept. 2002;105:35–46.
Yon L, Guillemot J, Montero-Hadjadje M, Grumolato L, Leprince J, Lefebvre H, et al. Identification of the secretogranin II-derived peptide EM66 in pheochromocytomas as a potential marker for discriminating benign versus malignant tumors. J Clin Endocrinol Metab. 2003;88:2579–85.
Luo Y, Chen L, Zhou Q, Xiong Y, Wang G, Liu X, et al. Identification of a prognostic gene signature based on an immunogenomic landscape analysis of bladder cancer. J Cell Mol Med. 2020;24:13370–82.
Wang C, Lu Y, Chen L, Gao T, Yang Q, Zhu C, et al. Th9 cells are subjected to PD-1/PD-L1-mediated inhibition and are capable of promoting CD8 T cell expansion through IL-9R in colorectal cancer. Int Immunopharmacol. 2020;78:106019.
Li P, Hao S, Ye Y, Wei J, Tang Y, Tan L, et al. Identification of an Immune-Related Risk Signature Correlates With Immunophenotype and Predicts Anti-PD-L1 Efficacy of Urothelial Cancer. Front Cell Dev Biol. 2021;9:646982.
Saiki Y, Horii A. Multiple functions of S100A10, an important cancer promoter. Pathol Int. 2019;69:629–36.
Sparrow E, Bodman-Smith MD. Granulysin: The attractive side of a natural born killer. Immunol Lett. 2020;217:126–32.
Zhao H, Bo Q, Wang W, Wang R, Li Y, Chen S, et al. CCL17-CCR4 axis promotes metastasis via ERK/MMP13 pathway in bladder cancer. J Cell Biochem. 2018;120:1979–89.
Feng J, He W, Song Y, Wang Y, Simpson RJ, Zhang X, et al. Platelet-derived growth factor receptor beta: a novel urinary biomarker for recurrence of non-muscle-invasive bladder cancer. PLoS One. 2014;9:e96671.
Romero D. TMB is linked with prognosis. Nat Rev Clin Oncol. 2019;16:336.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, et al. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56.
Kamat AM, Sylvester RJ, Bohle A, Palou J, Lamm DL, Brausi M, et al. Definitions, End Points, and Clinical Trial Designs for Non-Muscle-Invasive Bladder Cancer: Recommendations From the International Bladder Cancer Group. J Clin Oncol 2016;34:1935–44.
Author information
Authors and Affiliations
Contributions
ZW, LP, and LZ designed and supervised the study. ZW, LP, ZW, LL, and LZ analyzed the data and wrote the original draft. ZW, LP, and JS edited the draft. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of interest
JS’ conflicts can be found at https://www.nature.com/onc/editors. None are relevant here. The author declares no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

{kind=link}
Cite this article
Wang, Z., Zhu, L., Li, L. et al. Identification of an immune gene-associated prognostic signature in patients with bladder cancer. Cancer Gene Ther 29, 494–504 (2022). https://doi.org/10.1038/s41417-022-00438-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-022-00438-5
- Springer Nature America, Inc.
This article is cited by
-
Comprehensive analysis and prognostic assessment of senescence-associated genes in bladder cancer
Discover Oncology (2024)