
Abstract
Microtubules play an important role in regulating several vital cellular activities, including cell division and tissue organization, through their dynamic protofilament network. In addition to forming the cytoskeleton, microtubules regulate the intracellular trafficking of cytoplasmic components and various signaling molecules, depending on the presence of post-transitional modifications (PTMs) and binding proteins. Accumulating evidence indicates the significant role of microtubule PTMs on cancer behavior. The PTMs that frequently occur on microtubules include acetylation, detyrosination, tyrosination, polyglutamylation, and polyglycylation. Alterations in these PTMs cause global effects on intracellular signal transduction, strongly linked to cancer pathogenesis. This review provides an update on the role of microtubule PTMs in cancer aggressiveness, particularly regarding cell death, sensitivity to chemotherapy, cell migration, and invasion. Additionally, it provides a mechanistic explanation of the molecular signaling pathways involved. This information might prove useful for predictive or therapeutic purposes.
Similar content being viewed by others

Introduction
Microtubules, the major elements of the cytoskeleton, are assembled from highly-conserved tubulin heterodimers and required to construct several subcellular organelles. The essential biological functions of the network of microtubules include the control of both cell growth and cell movement; they represent a key signaling phenomenon that regulates several indispensable cellular activities [1, 2]. Microtubules consist of α- and β-tubulin heterodimers organized to form a hollow cylindrical structure [1, 3]. They are highly dynamic and persistently change in form (rapid shrinkage [microtubule catastrophe] to growth [microtubule rescue]) throughout the cell cycle [3]. Microtubules are nucleated, and they adhere to microtubule-organizing centers (MTOCs) such as centrosomes at the minus-end, while the plus-end is extended toward the cell periphery. Additionally, microtubules function closely with an array of microtubule-associated proteins (MAPs), which regulate their assembly and dynamic stability by specifically binding to the plus- or minus-ends and/or cooperating with the cellular trafficking of organelles and molecules along the microtubules [4, 5].
Microtubules’ other cellular regulatory functions are carried out through the spatial and temporal generation of specialized microtubule identities, particularly the post-translational modifications (PTMs) [6]. Most modifications are localized at the C-terminus of tubulin and accumulated on both long-lived and highly dynamic microtubules [6, 7]. Mechanistically, tubulin PTMs seem to mediate microtubule functions by directly altering their mechanical properties or controlling their interactions with other proteins [7]. Several groups of PTMs specifically preserved on microtubules, such as tyrosination, detyrosination, glutamylation, and glycylation, have been explored previously [8]. These modifications are known to contribute to the various functions of the microtubules. Accumulative studies report that microtubule PTMs participate in the tubulin code and act as key regulators of the functions of microtubules [9, 10]. Although these modifications affect the single-cell level, alterations in their functions could cause massive disruptions in tissue homeostasis and eventually result in disease development [11]. Although alterations in the tubulin code have been reported in many types of cancer, the presence of a functional association between tubulin PTMs and cancer aggressiveness remains largely unknown. In this study, we have reviewed and summarized the updated information about the role of tubulin PTMs in cancer cell behavior.
Microtubules and their dynamic structure
Microtubules have a highly dynamic filamentous structure. They play important roles in sustaining the cell’s shape and transporting vesicles or other components within eukaryotic cells. Microtubules consist of α- and β-tubulin heterodimers [1, 2]. Constructing the tubulin dimer from the α- and β-monomers requires adding a guanosine triphosphate (GTP) molecule to the β-tubulin subunit-conjugated to the α-tubulin [12]. The interaction between the α- and β-tubulins at the interface of growing protofilaments mediates the process of GTP hydrolysis, which contributes to the flexibility of the conformation and the catastrophe-rescue cycle (Fig. 1). Adding and removing the tubulin heterodimers at the plus-ends of the microtubules is a highly dynamic process [13]. The GTP molecules associated with β-tubulin are hydrolyzed to guanosine diphosphate (GDP) after polymerization. The GDP-bound β-tubulins are different from the previously-formed GTP-associated β-tubulins because they are strongly related to the depolymerized state of the microtubule. However, a GTP-tubulin cap (region at the tip of the polymerizing microtubule) may be lost. In that case, the microtubule switches to a shrinking state by a process termed catastrophe [14], which is a crucial determiner of the stability of the microtubules.

α- and β-tubulin dimers are incorporated into the growing lattice; GTP-β-tubulin (green) forms a GTP-cap and is shortly hydrolyzed to GDP-β-tubulin (pink). GTP exchange causes a dynamic switch between microtubule polymerization and depolymerization, resulting in a catastrophe (transition from growth to shrinkage) or rescue (transition from shortening to growth). The catastrophe occurs due to the loss of the GTP-cap. Tubulin post-translational modifications (PTMs; in gray) are localized in the tubulin carboxy-terminal tail. They determine the stability of the microtubule and potentially play specific roles in cancer behavior.
The minus-end of the microtubules bind to microtubule-organizing centers (MTOCs), such as the centrosome, Golgi complex, and microtubule-binding proteins, which help with microtubule nucleation, anchoring, and stabilization [15, 16]. These characteristics are involved in the transition from microtubule growth or polymerization to shrinkage or depolymerization, thus allowing for their interaction with other cytoskeletal and cytoplasmic contents, consequently altering cellular activities and morphologies [17]. Also, the tubulin isotypes and the dynamic variations in tubulin PTMs interact with a wide range of signaling systems and contribute to the various roles of the microtubules [18, 19].
In humans, microtubules consist of a combination of 7 α-tubulin and 8 β-tubulin isotypes [20]. The tubulin family members share a structural homology composed of the formation of a globular body from the N-terminal with particularly different sequences at the C-terminal tail [21]. The tail at the C-terminal consists of a specific site for the tubulin PTMs, which aid in regulating the protein-protein interactions and their functions, thereby contributing to the remarkable properties of each tubulin isotype [22] (Fig. 1). Despite their high degree of conservation, microtubules exhibit varying behaviors and molecular structures among species and cell types due to the convergence of the tubulin isotypes and their PTMs.
Microtubule PTMs
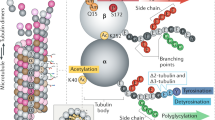
The therapeutic strategy of targeting microtubules has gained attention after reports on differences in microtubule PTMs between normal and cancer cells [23, 24]. Microtubule PTMs are thought to regulate protein-protein interactions within the microtubules, thereby affecting the signaling events within the cells. Most modifications (tubulin tyrosination, detyrosination, acetylation, glutamylation, and glycylation) are localized at the tubulin C-terminus, and each PTM is associated with a specific function. As shown in Fig. 2, the microtubule PTMs can markedly generate binary on and off signals by adding or eliminating single functional residues (acetylation and detyrosination) or moderately adding different numbers of residues (glutamylation, amination, and glycylation) [25].

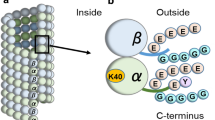
Tubulin PTMs are generally located on various tubulin dimer regions (α-tubulins, yellow; β-tubulins, pink). The acetylation occurs at lysine 40 (K40) on the luminal surface of the α-tubulin. Detyrosination/tyrosination and Δ2- and Δ3-tubulin are found at the carboxy-terminal tails of the α-tubulins. Polyglutamylation and polyglycylation involve the addition of glutamine- and glycine- side chains, respectively, on the carboxy-terminal tails of both α- and β-tubulins. Polyamination at glutamine 15 (Q15) and phosphorylation at serine 172 (S172) are mostly seen in the globular part of β-tubulin.
A range of enzymes catalyzes tubulin PTMs. The acetylation at lysine 40 found on the luminal surface of the α-tubulin requires the α-tubulin N-acetyl-transferase 1 (αTAT1), whereas the deacetylation process is dependent on tubulin deacetylating enzymes such as histone deacetylase 6 (HDAC6) and sirtuin 2 (SIRT2) [26,27,28]. Tubulin acetylation appears to increase the stability and longevity of microtubules. The addition of tyrosine (tyrosination) at the α-tubulin C-terminal is mediated by tubulin tyrosine ligase (TTL), whereas its removal requires the catalytic activity of carboxypeptidases [25]. Detyrosinated tubulin (referred to as Glu-tubulin) provides stability to the microtubules [25]. Following the detyrosination process, the α-tubulin carboxy-terminal tail can be modified further by removing the penultimate glutamate residue, which results in the generation of Δ2-and Δ3-tubulin [7]. Polyglutamylation and polyglycylation of tubulin are associated with the addition of glutamine- and glycine-side chains, respectively, which extend from glutamine residues on the tubulin C-terminal. The tubulin tyrosine ligase-like enzyme family controls this activity; removing the polyglutamylate reaction is catalyzed by the cytosolic carboxypeptidase (CCP) family [29,30,31]. During polyamination, the polyamines are added to the globular part of β-tubulin at glutamine (Q15) followed by phosphorylation at serine (S172), thus impacting the assembly rates and stability of the microtubules [32].
Microtubules and their biological roles in cancer
Microtubules play a significant role in regulating protein kinases, which are associated with the behaviors of several cancers [33]. They are known to govern intracellular trafficking and molecular signaling through their dynamic properties [34], linked with the behavior and morphology of the cells during both tissue development and pathological disease. For instance, the interactions between cytoskeletal elements and several signaling molecules contribute to activating the downstream effectors related to cancer cell movement and cell survival [35,36,37]. It has been reported that tubulin was strongly associated with the functions of the tumor suppressor protein p53; the translocation of p53 to the nucleus is dependent on the microtubule tracking protein dynein, in which its function was controlled by tubulin dynamics [38].
The microtubule can mediate cancer cells to accomplish intricate functions, such as cell migration and adhesion [39, 40]. The Rho guanosine-5’ triphosphatases (GTPases) signaling pathway is one of the major cellular signaling pathways associated with the cytoskeletal microtubules [39]. This pathway plays a powerful role as an indispensable regulator in various biochemical processes associated with cancer migration, such as cell-cell junction assembly and disassembly, directional sensing, integrin-matrix adhesion, and cytoskeletal dynamics [41]. A recent study demonstrated the role of microtubules in tumor motility. It reported that alterations in microtubule dynamics enhance the RhoA activity, which stimulates Rho-associated protein kinase (ROCK), a downstream signaling molecule in the Rho GTPase pathway. Consequently, the cytoskeleton is reorganized in gastric adenocarcinoma cells [42]. Microtubule dynamics influence cancer motility via the regulation of Rho GEF-H1, which functions on RhoA-regulating downstream myosin light chain phosphorylation [43]. Accumulative evidence also demonstrates that microtubules participate with Rho GTPases in regulating cancer metastasis [44, 45].
It is of note that microtubules coordinate with the extracellular environment in the regulation of cell morphology and function. The indispensable controller for cancer cell-mechanics dependent function is the mechanical cues such as substrate stiffness, cell stretch, and shear stress [46]. These mechanical cues are crucial for intrinsic tumor cell biology, which is governed by mechanical interactions between cancer cells and their extracellular matrix (ECM) stiffness. Consequently, these interactions change the balancing of mechanical force in the ECM and microtubule organization [47]. Some microtubule PTMs are defined as a necessary mediator for microtubule network reorganization under mechanical stress from the tumor microenvironment [48, 49]. Recent work reported that microtubule glutamylation plays a causative function in response to ECM remodeling, leading to breast cancer progression [48]. Low shear stress, a biomechanical force, promotes microtubule dynamics through downregulating tubulin acetylation, thereby increasing the integrin β1 trafficking and enhancing breast cancer cell migration [49]. It indicated that microtubules participate in a biomechanical cue contributed by the tumor microenvironment in the regulation of cancer cell behavior through their PTMs alteration. However, the effect of mechanical forces on other microtubule PTMs and cancer aggressiveness required further elucidation.
Microtubules play several roles in response to cellular stress in cancer by regulating the signaling pathways, particularly those involved with signaling trafficking throughout the cells [33, 35]. The cytoskeleton network plays an important role in cellular homeostasis by acting as a scaffold for sequestration or co-localizing the stress response-related proteins [38, 50]. A previous study reported that mitogen-activated protein kinase (MAPK) interacts with the microtubule network via kinesin motor proteins [51]. The coordination between microtubules and the MAPK superfamily, including extracellular regulated kinases 1/2 (ERK1/2) and c-Jun N-terminal protein kinase (JNK), extensively mediates several stress signaling pathways in cancer cells. An emerging study revealed that ERK binds to the kinesin family member 26B (KIF26B) on microtubules and causes an increase in the chemosensitivity of osteosarcoma cells [52]. Moreover, microtubule-associated protein 4 (MAP4), a member of the MAP family, can interact with and activate ERK1/2 leading to an upregulation in vascular endothelial growth factor A (VEGFA) expression, which enhances cell invasion and migration in esophageal cancer [53]. MAP7 has been associated with cancer metastasis, wherein it activates the MAPK signaling and promotes both cell growth and motility in cervical cancer [54]. JNK is another signaling pathway governed by microtubule dynamics in response to environmental stresses; it has been reported that tubulin-binding agents trigger JNK-mediated apoptosis in ovarian cancer cells [55]. The JNK pathway is necessary for apoptosis and autophagy activation after endoplasmic reticulum (ER) stress [56]. Moreover, the activation states of several signaling molecules, including protein kinase B (Akt), were induced by their interactions with tubulin, particularly the acetylated form. The phosphorylation at serine 473 and threonine 308 by phosphatidylinositol 3-kinase (PI3K) initiated its activity; however, the extension of the activated state has shed light on the regulation by microtubules [57]. The acetylated tubulin plays an important role in sustaining the Akt activity in a dynactin-dynein motor complex-dependent mechanism in response to decelerated cancer cell death [57]. Heat shock protein 90 (Hsp90)-mediated Akt stabilization is regulated by the localization of the Hsp90-Akt complex in the microtubule network [58]. In addition, microtubules interacting with kinesins KIF2A and KIF1B between perinuclear lysosomes regulate mammalian target of rapamycin (mTOR) signaling leading to autophagic initiation [59]. The activating molecule of BECN1-regulated autophagy 1 (AMBRA1) plays a vital role as a linker protein among microtubules and the PI3K signaling complex, which is responsible for the induction of the autophagy process [60]. Thus, due to their involvement in cellular trafficking, microtubules are required as a scaffold for the sequestration of signal transduction in response to cellular stress.
Microtubule dynamics also play roles in response to hypoxic stress through several signaling pathways such as p38/MAPK and hypoxia-inducible factor-1α/vascular endothelial growth factor (HIF-1α/VEGF) [61,62,63]. Previous work reported the coordination of microtubules and p38/MAPK that this signaling interacting to MAP4 on microtubules was activated under hypoxic condition, consequently facilitating microtubule depolymerization and attenuating cervical cancer cell death [61]. Additionally, HIF-1α protein, one of the main regulators of cellular hypoxia adaptation, was associated with polymerized microtubules and was transported into the nucleus by the microtubule-associated motor dynein protein [62]. Upon an interference to microtubule dynamics occurred, the nuclear translocation of HIF-1α was disrupted which inhibits VEGF transcription and thus attenuates in vitro and in vivo angiogenesis in breast cancer cells [63]. Evidence indicated that microtubule dynamics participated in several signalings of hypoxic adaptation, and an alteration of their behaviors might affect these signalings and cellular response.
Types of microtubule PTMs that affect cancer behavior
Aberrant levels of microtubule PTMs have been reported in cancers and linked with cancer aggressiveness. Alterations in tubulin PTM diversity are likely to disturb cellular homeostasis and affect the behavior of cancer.
Acetylation
Tubulin acetylation is a marker of microtubule stability. The α-tubulin K40 acetylation site is found on the microtubule’s luminal surface [26]. Upregulated levels of tubulin PTMs following acetylation have been reported in breast and head and neck squamous cell carcinomas in association with both cell growth and apoptotic cell death [64, 65]. The downregulation of tubulin acetylation retarded the growth of cells in lung cancer (A549 cells) and cervical cancer (HeLa cells) [66]. Alternatively, the tubulin deacetylase HDAC6, which decreases tubulin acetylation, promoted cell proliferation in several cancers [67, 68], suggesting that its role is possibly a cell-type-specific and/or signaling participated.
Tubulin acetylation is known to be involved in the chemosensitivity of lung cancer. The upregulation of tubulin acetylation in paclitaxel-resistant cells, which stabilizes and prevents the anti-apoptotic degradation of Mcl-1 has been reported previously [69]. Disruption of the microtubule resulted in Akt deactivation leading to accelerate cell death [57]. In contrast, increased acetylation of tubulin under nutrient deprivation can regulate kinesin-1-dependent JNK expression and stimulate autophagosome formation in HeLa cells [56]. Previous studies have demonstrated that a histone deacetylase inhibitor, which upregulated the level of acetylated tubulin, induced pancreatic cancer cell death [70] and enhanced the anticancer effect of paclitaxel [71, 72]. These findings suggest that the level of tubulin acetylation could predict the chemosensitivity of paclitaxel in naïve cancer cells; however, after chemotherapy, the cancer cells might develop resistance and exhibit hyperacetylation of the tubulin. Although, the role of tubulin acetylation on cell death regulation remains unclear, and extensive studies are required to confirm its role as a determiner of the cell’s sensitivity to death.
Apart from the regulation of cell death, elevated tubulin acetylation was associated with metastasis; a recent study reported that tubulin acetylation was upregulated in cases with lymph node metastases [73]. Increased α-tubulin acetylation enhanced cell migration and invasion, and a mutation at the acetylation site lysine 40 could inhibit these activities. The circulatory cancer cells exhibit a high level of stable tubulin, suggesting that the tubulin dynamics promote the detachment of these cells and potentiates metastasis [74]. Our recent study showed that acetylated tubulin promotes epithelial to mesenchymal transition in lung cancer via the CAMSAP3/Akt machinery [75]. Interestingly, suppression of tubulin acetylation via the αTAT1 knockout approach could suppress cell invasion and migration through Wnt/β-catenin and matrix metalloproteinase (MMP) 9 [76, 77], thus indicating that tubulin acetylation potentiates the ability to metastasize.
Tubulin acetylation participates in other cancer behaviors, including ER stress [78], intracellular trafficking [79], and microtubule dynamics regulation in cancer [80]. For instance, the disruption of microtubule acetylation inhibits breast cancer progression via upregulation of ER stress and alterations in the morphology of the ER [78]. Gao et al. showed that deficiency of HDAC6 could enhance tubulin acetylation and induce the microtubule-directed transport of epidermal growth factor receptor (EGFR)-loaded vesicles in lung cancer [79]. Moreover, increased microtubule stability caused by the pharmacological inhibition of HDAC6 was observed against cold- and nocodazole-induced microtubule depolymerization in breast cancer MCF-7 cells [80]. Thus, the effect of tubulin acetylation on cellular functions is considered to be highly significant in terms of controlling cellular homeostasis.
Detyrosination/Tyrosination
Detyrosination, observed in highly stabilized microtubules, typically regulates the C-terminal tails of α-tubulin via TTL and carboxypeptidases [25]. Alterations in the levels of detyrosination and the enzymes responsible for these changes have been seen in a wide range of cancers, particularly the aggressiveness of the tumor [24, 37, 81]. Increased level of detyrosinated tubulin was reported in breast cancer associated with poor prognosis [81]. Furthermore, detyrosinated tubulin reportedly regulates Rho/GEF-mediated actin stress fiber formation, which promotes in vitro cell motility, thereby indicating microtubules’ significant role in cancer metastasis [37]. Increased tubulin detyrosination facilitates tumor development by enhancing the production of a tubulin-based membrane protrusion called microtentacle, which interacts with the ECM [82, 83]. Low TTL levels might be frequently observed in cancer cells and could be involved in promoting cancer cell escape and invasion into the surrounding stroma. Transient alterations in microtubule stability at the invasive tumor front could remarkably affect metastatic efficiency. These assumptions are supported by studies, which show that microtubules are required for the attachment of circulating cancer cells to the blood vessel walls [84].
A recent study identified vasohibins (VASH1 or VASH2) as enzymes that encode the tubulin carboxypeptidase activity necessary for detyrosination [85]. VASHs were initially recognized as secretory proteins stimulated by VEGF during angiogenesis [86]. VASH1 is reported to inhibit tumor lymphangiogenesis and metastasis [87] and suppress tumor growth in an in vivo xenograft model of lung cancer [88]. On the contrary, VASH2 functions as a mediator of proliferation [89]. It promotes tumor growth and angiogenesis [90,91,92] and decreases the chemosensitivity to paclitaxel in various types of cancer [93]. These findings demonstrate the contradictory roles of VASH1 and VASH2 in cancers. The distinctive functions of VASH1 and VASH2 might be depended heavily on the overall homology at the amino acid level, accounted only for 52% similarity, which contributes to their difference in a three-dimensional structure and the molecular interaction [86]. However, the regulatory role of these VASHs in cancers is remaining largely unknown, particularly in association with microtubule PTMs. Nonetheless, the extensive range of human carcinoma patients had VASHs mutation that impacts their tubulin detyrosination activity [94]. Previous evidence also reported the relation of VASHs and microtubule detyrosination that co-depletion of VASH1 and VASH2 was able to reduce microtubule detyrosination level along with the mitotic spindles [95]. This information suggested that VASHs-mediated microtubule detyrosination regulates the cell cycle by regulation of chromosome segregation. Moreover, a recent study revealed that VASH2 markedly increased α-tubulin detyrosination that is required for cancer cell migration [96], which tubulin detyrosination is related to metastasis through the formation of cellular protrusive microtentacles in breast cancer cells [83]. According to this evidence, VASH2 has a fundamental role in the regulation of detyrosinated tubulin in such circumstances. The understanding of VASHs/microtubule detyrosination could be of great importance for achieving benefits for cancer therapy.
Furthermore, dysregulation of tubulin detyrosination altered the transportation of the centromere-associated protein E (CENP-E)-mediated chromosomes to the spindle equator [97]. During metaphase, chromosomes are aligned at the center in preparation for their segregation into daughter cells; CENP-E/kinesin-7 is required to guide the proximal chromosomes to the metaphase plate along the microtubules at the equator [98]. Microtubule detyrosination enhanced this process. The suppression of detyrosinated spindle microtubules led to a delayed mitotic process [97]. Their highly proliferative activity characterizes cancer cells, and the inhibition of CENP-E was found to inhibit this activity [99]. In cooperation with microtubules, the suppression of tubulin detyrosination might contribute to CENP-E dysfunction and delay cell division.
Tyrosination of α-tubulin is known to play a vital role in chemotherapy resistance, cell division, and migratory ability, in which the alteration of tyrosinated tubulin level is occurred in some cancers and is associated with tumor aggression [100,101,102]. Upregulation of tyrosinated α-tubulin levels has been correlated with paclitaxel resistance in MCF-7 breast cancer cells [100]. Abundant tyrosinated tubulin was observed along with the cytoplasmic extensions and in the mitotic spindle of dividing glioblastoma cells [101]. Recently, an elevation in the level of tyrosinated tubulin due to overexpression of low-density lipoprotein receptor-related protein-6 (LRP6) was associated with metastasis in colorectal cancer [102]. The effects of these PTMs on cancer behavior remain unclear and have some contradictory effects on cancer aggressiveness with tubulin detyrosination/tyrosination status. As above mention, an increasing tubulin detyrosination level facilitates tumor development, particularly migration and metastasis activities in breast cancer [82, 83]. Notwithstanding, it has reported that an upregulation of tubulin tyrosination influences varying cancer behaviors in several cancer cell types [100,101,102]. According to all this evidence, tubulin detyrosination/tyrosination may have disparate mechanisms of dominant pathways participated for regulating cancer activity and have dependability on cancer cell-type specificity. Further investigations on the molecular mechanisms involved are required to fully understand the potential implications of these tubulin PTMs in tumorigenesis.
Polyglutamylation/Polyglycylation
Polyglutamylation and polyglycylation occur at the C-termini of the α-and β-tubulins [29, 30]. Tubulin polyglutamylation is commonly observed in several organisms. They enhance the negative charge at the C-terminal tails of the α- and β-tubulins and alter the surface conformation of the microtubule, thus impacting the MAPs and the motor protein interactions [103]. Tubulin polyglycylation has been reported to play an important role in cell motility and cytokinesis [104]. Mechanistically, polyglutamylation is carried out by a tubulin tyrosine ligase-like (TTLL) enzyme and can be reversed by CCP enzymes [29,30,31]. However, the enzyme responsible for removing polyglycylation remains unknown [105]. Members of the TTLL enzyme family catalyze diverse tubulin PTMs; for example, TTLL-1, -4, -5, -6, -7, -9, -11, -12, and -13 are specific to polyglutamylate α-tubulin or β-tubulin [106]. TTLL-3 and TTLL-10 have been reported as tubulin glycine ligases [105].
Downregulation of polyglutamylated α-tubulin after increased polyglutamylation enzyme TTLL-12 inhibited prostate cancer cell growth [107]. TTLL-12 also plays an important role as a molecular marker of invasion and progression in ovarian cancer [108]. On the contrary, increased polyglutamylation of β-tubulin following an increased TTLL-4 expression level was associated with a shorter recurrence-free survival period, brain metastasis, and increased extracellular vesicles (EV) trafficking in breast cancer [109], indicating that polyglutamylation of microtubules directly modulates cancer behavior and provides a metastatic niche by altering the EV signature. Additionally, a high level of polyglutamylated tubulin has been observed in prostate cancer [24]. Microtubule glutamylation was upregulated in breast cancer and associated with tumor cell proliferation and increased ECM remodeling [48]. In contrast, a lack of tubulin glycine ligase TTLL-3 resulting in low tubulin glycylation levels strongly mediated tumorigenesis in colon cancer in vivo, thereby indicating the suppressive role of tubulin glycylation [110].
The role of tubulin PTMs in chemotherapeutic resistance has been specifically implicated in cancer. Increased levels of polyglutamylated tubulin have been related to paclitaxel resistance in breast cancer cells [111, 112]. An extensive investigation of the roles of tubulin polyglutamylation and polyglycylation in tumorigenesis, and the clarification of their diverse enzyme activities that affect cancer progression are necessary to establish the PTMs as potential diagnostic and/or prognostic biomarkers.
Perspectives
Overall evidence provides the complex model of tubulin PTMs that emerged the mechanistic details of their different biological functions in cancers. Many recent works demonstrated correlations between alterations in tubulin PTMs and cancer aggressiveness. However, in-depth knowledge about the functions of these PTMs in many cancer types is largely unknown. Further systematic and methodical studies on the functions of tubulin PTMs are required to establish their roles in cancer and their use as diagnostic and prognostic markers. Additional knowledge regarding these PTMs will elucidate their prospective therapeutic value for the treatment of human cancers.
References
Nogales E. Structural insights into microtubule function. Annu Rev Biophys Biomol Struct. 2001;30:397–420.
Vicente JJ, Wordeman L. The quantification and regulation of microtubule dynamics in the mitotic spindle. Curr Opin Cell Biol. 2019;60:36–43.
Valiron O, Caudron N, Job D. Microtubule dynamics. Cell Mol Life Sci. 2001;58:2069–84.
Bodakuntla S, Jijumon AS, Villablanca C, Gonzalez-Billault C, Janke C. Microtubule-associated proteins: structuring the cytoskeleton. Trends Cell Biol. 2019;29:804–19.
Akhmanova A, Steinmetz MO. Control of microtubule organization and dynamics: two ends in the limelight. Nat Rev Mol Cell Biol. 2015;16:711–26.
Gadadhar S, Bodakuntla S, Natarajan K, Janke C. The tubulin code at a glance. J Cell Sci. 2017;130:1347–53.
Magiera MM, Janke C. Post-translational modifications of tubulin. Curr Biol. 2014;24:R351–R354.
Janke C. The tubulin code: molecular components, readout mechanisms, and functions. J Cell Biol. 2014;206:461–72.
Eshun-Wilson L, Zhang R, Portran D, Nachury MV, Toso DB, Löhr T, et al. Effects of α-tubulin acetylation on microtubule structure and stability. Proc Natl Acad Sci USA. 2019;116:10366–71.
Portran D, Schaedel L, Xu Z, Thery M, Nachury MV. Tubulin acetylation protects long-lived microtubules against mechanical ageing. Nat Cell Biol. 2017;19:391–8.
Magiera MM, Singh P, Gadadhar S, Janke C. Tubulin posttranslational modifications and emerging links to human disease. Cell 2018;173:1323–7.
Sept D. Microtubule polymerization: one step at a time. Curr Biol. 2007;17:R764–R766.
Aranson IS, Tsimring LS. Pattern formation of microtubules and motors: Inelastic interaction of polar rods. Phys Rev E. 2005;71:050901.
Burbank KS, Mitchison TJ. Microtubule dynamic instability. Curr Biol. 2006;16:R516–R517.
Etienne-Manneville S. Microtubules in cell migration. Annu Rev Cell Dev Biol. 2013;29:471–99.
Jiang K, Hua S, Mohan R, Grigoriev I, Yau KW, Liu Q, et al. Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev Cell. 2014;28:295–309.
Jiang K, Akhmanova A. Microtubule tip-interacting proteins: a view from both ends. Curr Opin Cell Biol. 2011;23:94–101.
Sharma N, Kosan ZA, Stallworth JE, Berbari NF, Yoder BK. Soluble levels of cytosolic tubulin regulate ciliary length control. Mol Biol Cell. 2011;22:806–16.
Schappi JM, Krbanjevic A, Rasenick MM. Tubulin, actin and heterotrimeric G proteins: coordination of signaling and structure. Biochim Biophys Acta. 2014;1838:674–81.
Ludueña RF, Banerjee A. The isotypes of tubulin. In The role of microtubules in cell biology, neurobiology, and oncology. Humana Press. 2008;123–75.
Mariani M, Karki R, Spennato M, Pandya D, He S, Andreoli M, et al. Class III β-tubulin in normal and cancer tissues. Gene 2015;563:109–14.
Janke C, Bulinski JC. Post-translational regulation of the microtubule cytoskeleton: mechanisms and functions. Nat Rev Mol Cell Biol. 2011;12:773–86.
Borys F, Joachimiak E, Krawczyk H, Fabczak H. Intrinsic and extrinsic factors affecting microtubule dynamics in normal and cancer cells. Molecules 2020;25:3705.
Souček K, Kamaid A, Phung AD, Kubala L, Bulinski JC, Harper RW, et al. Normal and prostate cancer cells display distinct molecular profiles of α‐tubulin posttranslational modifications. Prostate. 2006;66:954–65.
Wloga D, Joachimiak E, Fabczak H. Tubulin post-translational modifications and microtubule dynamics. Int J Mol Sci. 2017;18:2207.
Friedmann DR, Aguilar A, Fan J, Nachury MV, Marmorstein R. Structure of the α-tubulin acetyltransferase, αTAT1, and implications for tubulin-specific acetylation. Proc Natl Acad Sci USA. 2012;109:19655–60.
Hubbert C, Guardiola A, Shao R, Kawaguchi Y, Ito A, Nixon A, et al. HDAC6 is a microtubule-associated deacetylase. Nature 2002;417:455–8.
North BJ, Marshall BL, Borra MT, Denu JM, Verdin E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol Cell. 2003;11:437–44.
Wloga D, Webster DM, Rogowski K, Bré MH, Levilliers N, Jerka-Dziadosz M, et al. TTLL3 is a tubulin glycine ligase that regulates the assembly of cilia. Dev Cell. 2009;16:867–76.
Janke C, Rogowski K, Wloga D, Regnard C, Kajava AV, Strub JM, et al. Tubulin polyglutamylase enzymes are members of the TTL domain protein family. Science 2005;308:1758–62.
Kimura Y, Kurabe N, Ikegami K, Tsutsumi K, Konishi Y, Kaplan OI, et al. Identification of tubulin deglutamylase among Caenorhabditis elegans and mammalian cytosolic carboxypeptidases (CCPs). J Biol Chem. 2010;285:22936–41.
Song Y, Kirkpatrick LL, Schilling AB, Helseth DL, Chabot N, Keillor JW, et al. Transglutaminase and polyamination of tubulin: posttranslational modification for stabilizing axonal microtubules. Neuron 2013;78:109–23.
Parker AL, Kavallaris M, McCarroll JA. Microtubules and their role in cellular stress in cancer. Front Oncol. 2014;4:153.
Ganguly A, Yang H, Sharma R, Patel KD, Cabral F. The role of microtubules and their dynamics in cell migration. J Biol Chem. 2012;287:43359–69.
Giannakakou P, Nakano M, Nicolaou KC, O’Brate A, Yu J, Blagosklonny MV, et al. Enhanced microtubule-dependent trafficking and p53 nuclear accumulation by suppression of microtubule dynamics. Proc Natl Acad Sci USA. 2002;99:10855–60.
Stewart ZA, Tang LJ, Pietenpol JA. Increased p53 phosphorylation after microtubule disruption is mediated in a microtubule inhibitor- and cell-specific manner. Oncogene 2001;20:113–24.
Nagae S, Meng W, Takeichi M. Non-centrosomal microtubules regulate F-actin organization through the suppression of GEF-H1 activity. Genes Cells. 2013;18:387–96.
Galigniana MD, Harrell JM, O’Hagen HM, Ljungman M, Pratt WB. Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J Biol Chem. 2004;279:22483–9.
Fife CM, McCarroll JA, Kavallaris M. Movers and shakers: cell cytoskeleton in cancer metastasis. Br J Pharm. 2014;171:5507–23.
Stehbens S, Wittmann T. Targeting and transport: how microtubules control focal adhesion dynamics. J Cell Biol. 2012;198:481–9.
Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–8.
Eitaki M, Yamamori T, Meike S, Yasui H, Inanami O. Vincristine enhances amoeboid-like motility via GEF-H1/RhoA/ROCK/Myosin light chain signaling in MKN45 cells. BMC Cancer. 2012;12:469.
Nalbant P, Chang YC, Birkenfeld J, Chang ZF, Bokoch GM. Guanine nucleotide exchange factor-H1 regulates cell migration via localized activation of RhoA at the leading edge. Mol Biol Cell 2009;20:4070–82.
Amin E, Dubey BN, Zhang SC, Gremer L, Dvorsky R, Moll JM, et al. Rho-kinase: regulation, (dys)function, and inhibition. Biol Chem. 2013;394:1399–410.
Fortin Ensign SP, Mathews IT, Symons MH, Berens ME, Tran NL. Implications of Rho GTPase signaling in glioma cell invasion and tumor progression. Front Oncol. 2013;3:241.
Shieh AC. Biomechanical forces shape the tumor microenvironment. Ann Biomed Eng. 2011;39:1379–89.
Malik R, Lelkes PI, Cukierman E. Biomechanical and biochemical remodeling of stromal extracellular matrix in cancer. Trends Biotechnol. 2015;33:230–6.
Torrino S, Grasset EM, Audebert S, Belhadj I, Lacoux C, Haynes M, et al. Mechano-induced cell metabolism promotes microtubule glutamylation to force metastasis. Cell Metab. 2021;33:1342–57.
Tang K, Li S, Li P, Xia Q, Yang R, Li T, et al. Shear stress stimulates integrin β1 trafficking and increases directional migration of cancer cells via promoting deacetylation of microtubules. Biochim Biophys Acta Mol Cell Res. 2020;1867:118676.
Kevenaar JT, Bianchi S, Van Spronsen M, Olieric N, Lipka J, Frias CP, et al. Kinesin-binding protein controls microtubule dynamics and cargo trafficking by regulating kinesin motor activity. Curr Biol. 2016;26:849–61.
Liang YJ, Yang WX. Kinesins in MAPK cascade: how kinesin motors are involved in the MAPK pathway? Gene 2019;684:1–9.
Pu Y, Yi Q, Zhao F, Wang H, Cai W, Cai S. MiR-20a-5p represses multi-drug resistance in osteosarcoma by targeting the KIF26B gene. Cancer Cell Int. 2016;16:64.
Jiang YY, Shang L, Shi ZZ, Zhang TT, Ma S, Lu CC, et al. Microtubule-associated protein 4 is an important regulator of cell invasion/migration and a potential therapeutic target in esophageal squamous cell carcinoma. Oncogene 2016;35:4846–56.
Tang N, Lyu D, Chang JF, Liu ZT, Zhang Y, Liu HP. Enhanced expression of microtubule-associated protein 7 functioned as a contributor to cervical cancer cell migration and is predictive of adverse prognosis. Cancer Cell Int. 2020;20:1–16.
Zhou J, Gupta K, Yao J, Ye KQ, Panda D, Giannakakou P, et al. Paclitaxel-resistant human ovarian cancer cells undergo c-Jun NH2-terminal kinase-mediated apoptosis in response to noscapine. J Biol Chem. 2002;277:39777–85.
Geeraert C, Ratier A, Pfisterer SG, Perdiz D, Cantaloube I, Rouault A, et al. Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J Biol Chem. 2010;285:24184–94.
Jo H, Loison F, Luo HR. Microtubule dynamics regulates Akt signaling via dynactin p150. Cell Signal. 2014;26:1707–16.
Giustiniani J, Daire V, Cantaloube I, Durand G, Poüs C, Perdiz D, et al. Tubulin acetylation favors Hsp90 recruitment to microtubules and stimulates the signaling function of the Hsp90 clients Akt/PKB and p53. Cell Signal. 2009;21:529–39.
Korolchuk VI, Saiki S, Lichtenberg M, Siddiqi FH, Roberts EA, Imarisio S, et al. Lysosomal positioning coordinates cellular nutrient responses. Nat Cell Biol. 2011;13:453–60.
Di Bartolomeo S, Corazzari M, Nazio F, Oliverio S, Lisi G, Antonioli M, et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J Cell Biol. 2010;191:155–68.
Hu JY, Chu ZG, Han J, Dang YM, Yan H, Zhang Q, et al. The p38/MAPK pathway regulates microtubule polymerization through phosphorylation of MAP4 and Op18 in hypoxic cells. Cell Mol Life Sci. 2010;67:321–33.
Carbonaro M, Escuin D, O’Brate A, Thadani-Mulero M, Giannakakou P. Microtubules regulate hypoxia-inducible factor-1α protein trafficking and activity: implications for taxane therapy. J Biol Chem. 2012;287:11859–69.
Mabjeesh NJ, Escuin D, LaVallee TM, Pribluda VS, Swartz GM, Johnson MS, et al. 2ME2 inhibits tumor growth and angiogenesis by disrupting microtubules and dysregulating HIF. Cancer Cell. 2003;3:363–75.
Oba T, Ono M, Matoba H, Uehara T, Hasegawa Y, Ito KI. HDAC6 inhibition enhances the anti-tumor effect of eribulin through tubulin acetylation in triple-negative breast cancer cells. Breast Cancer Res Treat. 2021;186:37–51.
Saba NF, Magliocca KR, Kim S, Muller S, Chen Z, Owonikoko TK, et al. Acetylated tubulin (AT) as a prognostic marker in squamous cell carcinoma of the head and neck. Head Neck Pathol. 2014;8:66–72.
Chien JY, Tsen SD, Chien CC, Liu HW, Tung CY, Lin CH. α-TAT1 downregulation induces mitotic catastrophe in HeLa and A549 cells. Cell Death Disco. 2016;2:1–9.
Wang Z, Hu P, Tang F, Lian H, Chen X, Zhang Y, et al. Hdac6 promotes cell proliferation and confers resistance to temozolomide in glioblastoma. Cancer Lett. 2016;379:134–42.
Gradilone SA, Radtke BN, Bogert PS, Huang BQ, Gajdos GB, LaRusso NF. Hdac6 inhibition restores ciliary expression and decreases tumor growth. Cancer Res. 2013;73:2259–70.
Wattanathamsan O, Thararattanobon R, Rodsiri R, Chanvorachote P, Vinayanuwattikun C, Pongrakhananon V. Tubulin acetylation enhances lung cancer resistance to paclitaxel-induced cell death through Mcl-1 stabilization. Cell Death Disco. 2021;7:1–13.
Wang G, He J, Zhao J, Yun W, Xie C, Taub JW, et al. Class I and class II histone deacetylases are potential therapeutic targets for treating pancreatic cancer. PloS one. 2012;7:e52095.
Zhang QC, Jiang SJ, Zhang S, Ma XB. Histone deacetylase inhibitor trichostatin A enhances antitumor effects of docetaxel or erlotinib in A549 cell line. Asian Pacific. J Cancer Prev. 2012;13:3471–6.
Cang S, Ma Y, Chiao J, Liu D. Phenethyl isothiocyanate and paclitaxel synergistically enhanced apoptosis and alpha-tubulin hyperacetylation in breast cancer cells. Exp Hematol Oncol. 2014;3:5.
Boggs AE, Vitolo MI, Whipple RA, Charpentier MS, Goloubeva OG, Ioffe OB, et al. α-Tubulin acetylation elevated in metastatic and basal-like breast cancer cells promotes microtentacle formation, adhesion, and invasive migration. Cancer Res. 2015;75:203–15.
Chakrabarti KR, Whipple RA, Boggs AE, Hessler LK, Bhandary L, Vitolo MI, et al. Pharmacologic regulation of AMPK in breast cancer affects cytoskeletal properties involved with microtentacle formation and re-attachment. Oncotarget 2015;6:36292–307.
Pongrakhananon V, Wattanathamsan O, Takeichi M, Chetprayoon P, Chanvorachote P. Loss of CAMSAP3 promotes EMT via the modification of microtubule-Akt machinery. J Cell Sci. 2018;131:jcs216168.
Oh S, You E, Ko P, Jeong J, Keum S, Rhee S. Genetic disruption of tubulin acetyltransferase, αTAT1, inhibits proliferation and invasion of colon cancer cells through decreases in Wnt1/β-catenin signaling. Biochem Biophys Res Commun. 2017;482:8–14.
Castro-Castro A, Janke C, Montagnac G, Paul-Gilloteaux P, Chavrier P. ATAT1/MEC-17 acetyltransferase and HDAC6 deacetylase control a balance of acetylation of alpha-tubulin and cortactin and regulate MT1-MMP trafficking and breast tumor cell invasion. Eur J Cell Biol. 2012;91:950–60.
Ko P, Choi JH, Song S, Keum S, Jeong J, Hwang YE, et al. Microtubule acetylation controls MDA-MB-231 breast cancer cell invasion through the modulation of endoplasmic reticulum stress. Int J Mol Sci. 2021;22:6018.
Gao YS, Hubbert CC, Yao TP. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J Biol Chem. 2010;285:11219–26.
Asthana J, Kapoor S, Mohan R, Panda D. Inhibition of HDAC6 deacetylase activity increases its binding with microtubules and suppresses microtubule dynamic instability in MCF-7 cells. J Biol Chem. 2013;288:22516–26.
Mialhe A, Lafanechèere L, Treilleux I, Peloux N, Dumontet C, Brémond A, et al. Tubulin detyrosination is a frequent occurrence in breast cancers of poor prognosis. Cancer Res. 2001;61:5024–7.
Whipple RA, Balzer EM, Cho EH, Matrone MA, Yoon JR, Martin S. Vimentin filaments support extension of tubulin-based microtentacles in detached breast tumor cells. Cancer Res. 2008;68:5678–88.
Whipple RA, Vitolo MI, Boggs AE, Charpentier MS, Thompson K, Martin SS. Parthenolide and costunolide reduce microtentacles and tumor cell attachment by selectively targeting detyrosinated tubulin independent from NF-κB inhibition. Breast Cancer Res. 2013;15:1–12.
Balzer EM, Whipple RA, Cho EH, Matrone MA, Martin SS. Antimitotic chemotherapeutics promote adhesive responses in detached and circulating tumor cells. Breast Cancer Res Treat. 2010;121:65–78.
Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, Mazouzi A, Stickel E, Celie P, et al. Vasohibins encode tubulin detyrosinating activity. Science 2017;358:1453–6.
Sato Y. The vasohibin family: a novel family for angiogenesis regulation. J Biochem. 2013;153:5–11.
Heishi T, Hosaka T, Suzuki Y, Miyashita H, Oike Y, Takahashi T, et al. Endogenous angiogenesis inhibitor vasohibin1 exhibits broad-spectrum antilymphangiogenic activity and suppresses lymph node metastasis. Am J Pathol. 2010;176:1950–8.
Hosaka T, Kimura H, Heishi T, Suzuki Y, Miyashita H, Ohta H, et al. Vasohibin-1 expression in endothelium of tumor blood vessels regulates angiogenesis. Am J Pathol. 2009;175:430–9.
Tu M, Liu X, Han B, Ge Q, Li Z, Lu Z, et al. Vasohibin-2 promotes proliferation in human breast cancer cells via upregulation of fibroblast growth factor-2 and growth/differentiation factor-15 expression. Mol Med Rep. 2014;10:663–9.
Kitahara S, Suzuki Y, Morishima M, Yoshii A, Kikuta S, Shimizu K, et al. Vasohibin-2 modulates tumor onset in the gastrointestinal tract by normalizing tumor angiogenesis. Mol Cancer. 2014;13:99.
Koyanagi T, Suzuki Y, Saga Y, Machida S, Takei Y, Fujiwara H, et al. In vivo delivery of sirna targeting vasohibin-2 decreases tumor angiogenesis and suppresses tumor growth in ovarian cancer. Cancer Sci. 2013;104:1705–10.
Takahashi Y, Koyanagi T, Suzuki Y, Saga Y, Kanomata N, Moriya T, et al. Vasohibin-2 expressed in human serous ovarian adenocarcinoma accelerates tumor growth by promoting angiogenesis. Mol Cancer Res. 2012;10:1135–46.
Koyanagi T, Saga Y, Takahashi Y, Tamura K, Yoshiba T, Takahashi S, et al. Knockout of vasohibin‐2 reduces tubulin carboxypeptidase activity and increases paclitaxel sensitivity in ovarian cancer. Cancer Med. 2021;10:2732–9.
Wang N, Bosc C, Ryul Choi S, Boulan B, Peris L, Olieric N, et al. Structural basis of tubulin detyrosination by the vasohibin-svbp enzyme complex. Nat Struct Mol Biol. 2019;26:571–82.
Liao S, Rajendraprasad G, Wang N, Eibes S, Gao J, Yu H, et al. Molecular basis of vasohibins-mediated detyrosination and its impact on spindle function and mitosis. Cell Res. 2019;29:533–47.
Iida‐Norita R, Kawamura M, Suzuki Y, Hamada S, Masamune A, Furukawa T, et al. Vasohibin‐2 plays an essential role in metastasis of pancreatic ductal adenocarcinoma. Cancer Sci. 2019;110:2296–308.
Barisic M, e Sousa RS, Tripathy SK, Magiera MM, Zaytsev AV, Pereira AL, et al. Microtubule detyrosination guides chromosomes during mitosis. Science 2015;348:799–803.
Owa M, Dynlacht B. A non-canonical function for centromere-associated protein-E controls centrosome integrity and orientation of cell division. Commun Biol. 2021;4:358.
Ohashi A, Ohori M, Iwai K, Nambu T, Miyamoto M, Kawamoto T, et al. A novel time-dependent CENP-E inhibitor with potent antitumor activity. PLoS One. 2015;10:e0144675.
Banerjee A. Increased levels of tyrosinated α-, βIII-, and βIV-tubulin isotypes in paclitaxel-resistant MCF-7 breast cancer cells. Biochem Biophys Res Commun. 2002;293:598–601.
Gadau SD. Morphological and quantitative analysis on α-tubulin modifications in glioblastoma cells. Neurosci Lett. 2018;687:111–8.
Yao Q, An Y, Hou W, Cao YN, Yao MF, Ma NN, et al. LRP6 promotes invasion and metastasis of colorectal cancer through cytoskeleton dynamics. Oncotarget 2017;8:109632–45.
Yu I, Garnham CP, Roll-Mecak A. Writing and reading the tubulin code. J Biol Chem. 2015;290:17163–72.
Thazhath R, Liu C, Gaertig J. Polyglycylation domain of β-tubulin maintains axonemal architecture and affects cytokinesis in Tetrahymena. Nat Cell Biol. 2002;4:256–9.
Rogowski K, Juge F, Van Dijk J, Wloga D, Strub JM, Levilliers N, et al. Evolutionary divergence of enzymatic mechanisms for posttranslational polyglycylation. Cell 2009;137:1076–87.
Van Dijk J, Rogowski K, Miro J, Lacroix B, Edde B, Janke C. A targeted multienzyme mechanism for selective microtubule polyglutamylation. Mol Cell. 2007;26:437–48.
Wasylyk C, Zambrano A, Zhao C, Brants J, Abecassis J, Schalken JA, et al. Tubulin tyrosine ligase like 12 links to prostate cancer through tubulin posttranslational modification and chromosome ploidy. Int J Cancer. 2010;127:2542–53.
Yang S, Liang Y, Qian H, Li Q. TTLL12 expression in ovarian cancer correlates with a poor outcome. Int J Clin Exp Pathol. 2020;13:239–47.
Arnold J, Schattschneider J, Blechner C, Krisp C, Schlüter H, Schweizer M, et al. Tubulin tyrosine ligase like 4 (TTLL4) overexpression in breast cancer cells is associated with brain metastasis and alters exosome biogenesis. J Exp Clin Cancer Res. 2020;39:1–15.
Rocha C, Papon L, Cacheux W, Marques Sousa P, Lascano V, Tort O, et al. Tubulin glycylases are required for primary cilia, control of cell proliferation and tumor development in colon. EMBO J. 2014;33:2247–60.
Froidevaux-Klipfel L, Targa B, Cantaloube I, Ahmed-Zaïd H, Poüs C, Baillet A. Septin cooperation with tubulin polyglutamylation contributes to cancer cell adaptation to taxanes. Oncotarget 2015;6:36063–80.
Targa B, Klipfel L, Cantaloube I, Salameh J, Benoit B, Poüs C, et al. Septin filament coalignment with microtubules depends on SEPT9_i1 and tubulin polyglutamylation, and is an early feature of acquired cell resistance to paclitaxel. Cell Death Dis. 2019;10:1–14.
Funding
This work was supported by the National Research Council of Thailand (NRCT; N41A640133 to VP) and the Second Century Fund (C2F to OW), Chulalongkorn University.
Author information
Authors and Affiliations
Contributions
All authors researched data, performed an extensive literature survey, and provided substantial contribution to the content of this article equally. All authors reviewed, edited, and approved the manuscript before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wattanathamsan, O., Pongrakhananon, V. Post-translational modifications of tubulin: their role in cancers and the regulation of signaling molecules. Cancer Gene Ther 30, 521–528 (2023). https://doi.org/10.1038/s41417-021-00396-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41417-021-00396-4
- Springer Nature America, Inc.
This article is cited by
-
Targeting AGTPBP1 inhibits pancreatic cancer progression via regulating microtubules and ERK signaling pathway
Molecular Medicine (2024)
-
Physiological roles of chloride ions in bodily and cellular functions
The Journal of Physiological Sciences (2023)
-
Inhibition of histone deacetylase 6 destabilizes ERK phosphorylation and suppresses cancer proliferation via modulation of the tubulin acetylation-GRP78 interaction
Journal of Biomedical Science (2023)
-
Dynamics of TUBB protein with five majorly occurring natural variants: a risk of cortical dysplasia
Journal of Molecular Modeling (2023)
-
Understanding the impact of structural modifications at the NNAT gene’s post-translational acetylation site: in silico approach for predicting its drug-interaction role in anorexia nervosa
Eating and Weight Disorders - Studies on Anorexia, Bulimia and Obesity (2023)