
Abstract
Background
CC-115, a dual mTORC1/2 and DNA-PK inhibitor, has promising antitumour activity when combined with androgen receptor (AR) inhibition in pre-clinical models.
Methods
Phase 1b multicentre trial evaluating enzalutamide with escalating doses of CC-115 in AR inhibitor-naive mCRPC patients (n = 41). Primary endpoints were safety and RP2D. Secondary endpoints included PSA response, time-to-PSA progression, and radiographic progression.
Results
Common adverse effects included rash (31.7% Grades 1–2 (Gr); 31.7% Gr 3), pruritis (43.9% Gr 1–2), diarrhoea (37% Gr 1–2), and hypertension (17% Gr 1–2; 9.8% Gr 3). CC-115 RP2D was 5 mg twice a day. In 40 evaluable patients, 80% achieved ≥50% reduction in PSA (PSA50), and 58% achieved ≥90% reduction in PSA (PSA90) by 12 weeks. Median time-to-PSA progression was 14.7 months and median rPFS was 22.1 months. Stratification by PI3K alterations demonstrated a non-statistically significant trend towards improved PSA50 response (PSA50 of 94% vs. 67%, p = 0.08). Exploratory pre-clinical analysis suggested CC-115 inhibited mTOR pathway strongly, but may be insufficient to inhibit DNA-PK at RP2D.
Conclusions
The combination of enzalutamide and CC-115 was well tolerated. A non-statistically significant trend towards improved PSA response was observed in patients harbouring PI3K pathway alterations, suggesting potential predictive biomarkers of response to a PI3K/AKT/mTOR pathway inhibitor.
Clinical trial registration
ClinicalTrials.gov identifier: NCT02833883.
Similar content being viewed by others


Introduction
Globally, over 375,000 men died of prostate cancer in 2020 [1, 2]. Although androgen-deprivation therapy (ADT) to lower the production of testosterone is initially effective, almost all cancers eventually stop responding, a stage termed castration-resistant prostate cancer (CRPC) with a median survival of about 3–5 years. Next-generation AR pathway targeted therapy, such as abiraterone, enzalutamide and apalutamide, has improved the survival of patients in both the metastatic castration-resistant and hormone-sensitive setting, but treatment resistance inevitably emerges, leading to a lethal outcome [3, 4].
Multiple mechanisms of resistance to AR pathway targeted therapy have been identified, and one major mechanism involves the activation of phosphoinositide-3 kinase/AKT/mammalian target of rapamycin (PI3K/AKT/mTOR) pathway [5, 6]. Reciprocal regulation between PI3K/AKT and AR pathways has been reported in prostate cancer, with inhibition of AR pathway leading to tumour cell survival via feedback activation of PI3K pathway [7, 8]. In addition, deletion or loss-of-function mutation in PTEN, a critical negative regulator of the PI3K/AKT/mTOR pathway, occurs in up to 50% of mCRPC, and the presence of such alterations can portend to attenuated response to AR-targeted therapies and worse clinical outcomes [9,10,11,12,13]. Thus, dual inhibition of AR and PI3K/AKT/mTOR pathways may enhance anti-tumour response and delay the development of drug resistance to AR-targeted therapy alone. Indeed, clinical support for this hypothesis emerged from a randomised Phase 3 trial where addition of ipatasertib, a AKT inhibitor, to abiraterone improved radiographic progression-free survival (rPFS) compared to abiraterone alone in mCRPC patients with PTEN loss [14].
DNA-dependent protein kinase (DNA-PK), a member of the PI3K-related kinase family, plays a significant role in DNA damage response, cell survival and proliferation, and has emerged as a potentially important therapeutic target in cancer [15, 16]. Clinically, DNA-PK dysregulation has been associated with aggressive disease, tumour progression and poor clinical outcomes in many cancers, including prostate cancer [17,18,19]. Furthermore, pre-clinical studies have identified a potential interaction between AR and DNA-PK pathways in DNA repair in prostate cancer, suggesting that dual inhibition of AR and DNA-PK pathways may provide synergistic killing of prostate cancer cells [20, 21].
CC-115 is a selective and potent dual inhibitor of mTORC1/2 and DNA-PK. It has demonstrated anti-tumour activity in a large panel of cancer cell lines in vitro [22]. In the first-in-human Phase I study in patients with advanced solid or haematologic malignancies, CC-115 had an acceptable safety and toxicity profile at 10 mg twice a day dosing, and an encouraging preliminary anti-tumour activity in several tumour types [23]. Recently, another dual mTOR and DNA-PK inhibitor, samotolisib, has demonstrated a tolerable safety profile and an improved PFS when combined with enzalutamide in a randomised, placebo-controlled Phase 1b/2 study in mCRPC patients after progression on abiraterone [24].
In this current study, we report the results of a Phase 1b multicentre trial evaluating the safety and preliminary efficacy of escalating doses of CC-115 (5–10 mg twice daily) in combination with enzalutamide in AR pathway inhibitor-naive mCRPC patients (n = 41) after progression on ADT. PK analysis, pre-clinical analysis on target pathway inhibition, and exploratory biomarker analysis with tumour genomic profiling and circulating tumour DNA (ctDNA) targeted sequencing are also reported here.
Materials and methods
Patient selection
Patients 18 years of age and above with a life expectancy of at least 6 months and documented progressive mCRPC, serum testosterone <50 ng/dl, ECOG performance status of 0–1, willingness to provide a tumour sample via biopsy or adequate archival tissue from a metastatic site of disease to be collected at screening if safe and feasible per treating investigator discretion, and adequate haematologic, renal, and hepatic function were eligible to enrol. Patients with central nervous system metastases, and other major medical comorbidities were excluded from the trial. Prior exposure to enzalutamide, apalutamide, or other investigational AR-directed therapy, abiraterone acetate, ketoconazole or other specific CYP-17 inhibitors, agents specifically targeting mTOR complexes (dual TORC1 + TORC2 inhibitors), PI3K/AKT pathway, DNA repair pathway or chemotherapy for castration-resistant disease was not permitted.
Study design and treatment
This was a Phase 1b multicentre, open label trial testing enzalutamide (160 mg QD) with escalating doses (5 mg, 10 mg and amended to include 7.5 mg BID) of oral CC-115 using a standard 3 + 3 dose escalation design in AR pathway inhibitor-naive mCRPC patients. See schema for the study design and consort diagram in Fig. S1A, B, respectively. The starting dose for enzalutamide was 160 mg QD, the standard approved dose for enzalutamide monotherapy in prostate cancer. The starting dose for CC-115 was 5 mg BID, a lower dose than previously selected monotherapy dose in the first-in-human trial [23], due to concern for possible PK interaction and overlapping toxicity with enzalutamide. The trial was registered at clinicaltrials.gov as NCT02833883. A Data Monitoring Committee (DMC) was employed to determine whether the safety and PK profile seen in the dose escalation phase of the study was favourable to justify initiating the expansion cohort to further confirm safety and tolerability of the combination and assess preliminary evidence of anti-tumour activity. An interim analysis of 17 subjects in the Expansion Phase was conducted and evaluated by the DMC. Due to the unacceptable observed rate of grade 3 rash toxicities at the 7.5 and 10 mg BID expansion dose of CC-115 in the study, the protocol was amended to accrue an additional 15 patients in the expansion phase treated at 5 mg BID. Definition and evaluation of dose-limiting toxicities were provided in Supplemental Method section.
Study assessments
Safety and laboratory
All adverse events (AEs), defined as any untoward medical occurrence in a patient or clinical investigation subject administered a pharmaceutical product and which does not necessarily have to have a causal relationship with treatment (ICH E2A), were recorded by investigators and graded per NCI-CTCAE version 4.0. Laboratory tests, including serum PSA and testosterone, were monitored at screening and during the study.
Pharmacokinetics
Each subject in the dose escalation portion of the study and a minimum of six subjects treated at the 7.5 mg CC-115 BID expansion phase had pharmacokinetic (PK) specimens obtained to evaluate and compare CC-115 exposure in subjects administered CC-115 alone and in combination with enzalutamide to assess potential gross drug-drug interaction. PK parameters (Cmax, Tmax, AUC(INF), AUC(TAU), AI (Accumulation Index), Peak-to-trough fluctuation, Cmin and T½) were derived from blood or plasma concentration versus time data.
Clinical response analysis
PSA response was calculated based on percent change in serum PSA values (ng/ml) at 12-week of treatment (or earlier for those who discontinue therapy prior to 12 weeks) from baseline Cycle 1 Day 1 PSA values. The maximum decline in PSA that occurs at any point after treatment was reported for each subject. One patient was excluded from this analysis because he withdrew consent after signing, and did not have any treatment and PSA follow-up. Three patients discontinued the treatment and withdrew from the trial prior to 12 weeks, and their last PSA values (all at approximately 8 weeks) were used for PSA response calculation. This is consistent with Prostate Cancer Working Group 3 (PCWG3) recommendation. Time-to-PSA progression and radiographic progression-free survival were calculated according to PCWG3 [25].
Exploratory biomarkers associated with treatment response
The following specimens were obtained from each subject, when possible: (A) a paraffin-embedded (FFPE) tumour tissue sample from a metastatic site of disease (via biopsy) at screening; (B) blood for Epic Sciences to evaluate CTC count, CTC AR-V7 positivity, CTC chromosomal instability (measured by predicted large-scale transition number or pLST) and single-cell low-pass sequencing of CTCs as previously described [26,27,28,29,30]; (C) targeted tumour tissue sequencing assays with MSK-IMPACT (Integrated Mutation Profiling of Actionable Cancer Targets) or Foundation One genomic profiling to detect pan-cancer driver gene mutations and other critical genetic aberrations [31]; and (D) pre- and post-treatment plasma cell-free and leucocyte DNA, subjected to deep targeted sequencing to quantify ctDNA fraction and identify somatic mutations, copy number changes, and structural rearrangements in established prostate cancer genes, as previously described [32]. These biomarker analyses were exploratory and not pre-specified in the study protocol.
Statistical analysis
The phase I component of the study was designed to establish the RP2D of CC-115 combined with the standard dose of enzalutamide. Subjects were treated in cohorts of size three and six and the dosage were escalated if the clinical toxicity is acceptable. The maximum tolerated dose is defined as the highest dose level with an observed incidence of DLT in no more than one out of six subjects treated at a particular dose level. A DLT was defined by Cycle 1 toxicity, although all-cycle toxicity was recorded. The trial was designed to treat a minimum of two subjects and a maximum of 18 subjects. When considering the probability of escalating a dose level as a function of the true DLT rate, the probability of escalation was high when the risk of DLT is low.
In the expansion phase, a 28 subject Simon two-stage design that differentiates between PSA response rates (≥90% by 12 weeks) of 0.50 and 0.75 was originally planned to assess treatment efficacy. Twelve subjects will be initially enrolled. If six or fewer subjects respond, the trial would have been terminated early and the treatment will be considered insufficiently active. Otherwise, the enrolment will be extended to a total of 28 subjects. If 18 or more of the 28 subjects respond, the treatment would be considered active. The probability of declaring the treatment sufficiently active is approximately 0.10 when the PSA response rate (≥90% at 12 weeks) in the population is 0.50 and increases to 0.90 when the population response rate is 0.75. Of note, due to unexpectedly high frequency of Grade 3 rash in the expansion phase, CC-115 was reduced from 10 mg to 7.5 mg and ultimately to 5 mg BID. This decision rule was not evaluated because the trial did not recruit to 28 patients at RP2D (CC-115 at 5 mg BID).
In order to monitor safety in the expansion phase, the design included a stopping boundary in the event of excessive DLTs. The boundary is derived using a repeated significance test. The boundary was based on an acceptable DLT rate of 0.15 and an unacceptable DLT rate of 0.35. The repeated significance test using this boundary has size 0.10 and power 0.90.
The association of PI3K/mTOR pathway activations with PSA response (≥50%, ≥90%) was evaluated using Fisher’s exact test. Time to PSA progression was estimated using the Kaplan–Meier method, and the association of PI3K/mTOR pathway activations with time to PSA progression was evaluated using the logrank test.
Preclinical in vitro cell growth and biochemistry assays
Human prostate cancer cell line (LNCaP) was treated with CC-115, NU-7441 (a specific DNA-PK inhibitor) and INK128 (a specific mTORC1/2 inhibitor) in a range of concentrations in vitro. Western blot analysis on phosphor-DNA-PKcs, phosphor-AKT (p473), phosphor-S6, phosphor-4EBP1, phosphor-H2-AX, phosphor-ATM and actin were performed to evaluate the effects of these drugs on the activation status of DNA-PK and PI3K/AKT/mTOR pathways. Two prostate cancer cell lines (LNCaP and CWR-22PC sgPTEN) were also treated with a vehicle control, CC-115, NU-7441, INK128 and a combination of these drugs over 7 days in vitro. Cell growth was evaluated by Celltiter-Glo assay.
Results
Patients
Overall, 41 patients were enrolled in both dose-escalation (n = 9) and expansion (n = 32) cohort in this Phase 1b study, including patients who received CC-115 at 10 mg (n = 13), 7.5 mg (n = 10), and 5 mg (n = 18) twice daily. One patient (7.5 mg cohort) was excluded from the clinical response analysis because he withdrew the consent after signing and did not receive any treatment. The demographic and baseline disease characteristics were listed in Table 1. Overall, median baseline PSA was 11.7 (range 0.7–627.4) ng/ml, and the majority (58.5%) of patients had Gleason 8–10 disease. 61% of the patients had bone only metastases, while 12.2% had visceral metastases (liver or lung) at the start of the trial. At the data cutoff date of July 15, 2021, 37 patients have discontinued the trial due to disease progression, side effects or withdrawal of consent with 4 patients remaining active on the trial.
Safety and tolerability
Treatment-emergent adverse events (TEAEs) are reported in Table 2. The most common toxicity included rash (63.4%), pruritus (43.9%), diarrhoea (36.6%), hypertension (26.8%) and fatigue (24.4%). Grade 3 toxicity included rash (31.7%), hypertension (9.8%) and increased lipase (7.3%). There was only 1 Grade 4 toxicity (hyperglycaemia) and no Grade 5 toxicity was observed. The most common Grade 3 toxicity was rash, which appeared to be related to the dose of CC-115. Rash was maculopapular with histology showing interface and spongiotic dermatitis with vasculopathic changes and associated eosinophils (Fig. S2). The rash occurred usually within the first cycle, which resolved with a brief course of topical and/or oral steroids and rarely recurred. The incidence of Grade 3 rash prompted dose reduction of CC-115 from 10 mg to 5 mg twice daily, leading to a corresponding reduction in Grade 3 rash from 46.2% to 16.7%. Hyperglycaemia, a common toxicity related to the class of drugs inhibiting PI3K/AKT/mTOR pathway, occurred in 19.5% of the patients, and was generally mild (Grade 1 or 2), with the exception of one Grade 4 toxicity that happened at 10 mg dose after more than 1 year on treatment prompting discontinuation. CC-115 at 5 mg twice daily was generally well tolerated and selected as the RP2D when combined with enzalutamide at 160 mg daily.
Pharmacokinetics (PK)
PK was measured in 6 patients after a single dose of CC-115 was administered 48 h prior to Cycle 1/Day 1 (C1D1), and after a combined dose of CC-115 and enzalutamide at 160 mg on C2D1 (Fig. S3). After a single dose of CC-115, 5 mg and 7.5 mg dosing achieved similar serum concentration at 3 h (peak, 30 ng/ml) and 8 h (13-14 ng/ml) after administration, while 10 mg dosing achieved about a twofold higher serum concentration at 3 h (61 ng/ml) and 8 h 31 (ng/ml) after administration. Serum concentrations of CC-115 was comparable when administered in combination with enzalutamide (Fig. S3). These results indicated that no significant PK interaction was observed between CC-115 and enzalutamide. CC-115 at 5 mg and 7.5 mg achieved comparable serum drug concentration, which was more than 50% lower than the serum concentration at 10 mg dosing.
Pre-clinical in vitro analysis of target pathway inhibition
CC-115 at 5 or 7.5 mg twice daily oral dose reached a peak serum concentration of ~30 ng/ml (equivalent of 100 nM) based on PK studies (Fig. S3). We performed experiments to investigate the effects of CC-115 at this concentration on biochemical signalling and prostate cancer cell growth in vitro. While CC-115 effectively inhibited the downstream targets of PI3K/AKT/mTOR signalling biochemically, including phosphor-AKT (pAKT), phosphor-S6 (pS6) and phosphor 4EBP1 (p4EBP1), starting at 100 nM in a dose-dependent manner, it had no detectably inhibitory effect on DNA-PK pathway measured by phosphor-DNA-PKcs (pDNA-PKcs) and phosphor-H2-AX (pH2-AX) (Fig. S4A). Furthermore, CC-115 had a significant inhibitory effect on two human prostate cancer cell lines (LNCaP and CWR-22PC sgPTEN) prostate cancer cell growth in vitro, similarly to another specific mTORC1/2 inhibitor (INK128). However, a specific DNA-PK inhibitor, NU-7441, had only modest inhibitory effect on prostate cancer cell line growth (Fig. S4B, C). Taking together, these pre-clinical data suggested that CC-115 may predominantly inhibit mTORC1/2, but may be insufficient to block the DNA-PK pathway at 5 or 7.5 mg dose.
Clinical response
Percentage PSA reduction by 12 weeks from the baseline (Fig. 1), median time-to-PSA progression (TTPP) (Fig. S4), median radiographic progression-free survival (rPFS) (Fig. 2) and duration on treatment (Fig. 3) were used to evaluate the preliminary antitumour effects in this trial. In 40 evaluable patients, 80% achieved a ≥50% reduction in PSA (PSA50) and 58% achieved a ≥90% reduction in PSA (PSA90) by 12 weeks (Fig. 1). Median TTPP was 14.7 months and median rPFS was 22.1 months in all 40 evaluable patients (Figs. 2 and S5). Consistent with PK data, there was a numerically higher PSA50 response in patients receiving 10 mg (PSA50 of 92%) than patients receiving 5 mg and 7.5 mg (PSA50 of 78% and 67%, respectively) of CC-115. In addition, PSA50 response was observed in 12 out of 13 (92%) patients with Grade 3 rash and in 20 out of 27 (74%) without Grade 3 rash although the comparison was not statistically significant (Fig. 1a, b).

a Waterfall plot of PSA response at 12 weeks by CC-115 dose, PTEN mutation status, TSC1, TSC2 or PIK3CA mutation status and the presence of Grade 3 rash. b Summary table of PSA50 and PSA90 response by CC-115 dose and mutation status.

Kaplan–Meier curves of rPFS in all patients (a), separated by PTEN mutation status (b) and by PI3K pathway mutation status (including PTEN, PIK3CA, TSC1 and TSC2) (c). d Summary table of median rPFS by mutation status.

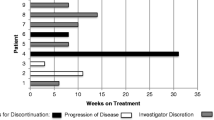
Swimmer’s plot was annotated with doses, PI3K pathway mutation status and Grade 3 rash. Annotation of potential mechanisms of resistance in two patients (Patients A and B) of early progression (<3 months after the beginning of treatment) who had both screening and end-of-treatment ctDNA analysis.
Exploratory biomarker analyses
Exploratory biomarkers from tumour tissue genomic analysis
To explore whether tumour genomic alterations, in particular mutations or copy number changes leading to activation of the PI3K/mTOR pathway, may be potential biomarkers to predict response to CC-115, for 33 patients we performed next-generation sequencing (NGS) of biopsy tumour tissue samples using MSK-IMPACT or Foundation One tests. Sample insufficiency resulted in technical failure in two patients. Among the remaining 31 patients, 16 patients (51.6%) harboured mutations predicted to lead to PI3K/AKT/mTOR pathway activation, including 11 (35%) with PTEN loss-of-function mutation or deletion, 4 (13%) with TSC1 or TSC2 loss-of-function mutations, and 1 (3%) with PIK3CA activation mutation. Interestingly, in this exploratory biomarker analysis, PSA50 was 94% in the 16 patients with any PTEN/PI3K/mTOR pathway activation mutations, including loss-of-function mutations in PTEN, TSC1 and TSC2, and activating mutation in PIK3CA, versus 67% in the 15 patients without such mutations (Fisher’s Exact test, p = 0.08) (Fig. 1b). Because of small sample sizes and low progression event rate at the time of data cutoff, in particularly for radiographic progression, there were no statistically significant differences on median TTPP and rPFS with this biomarker analysis (Figs. 2 and S5).
Exploratory biomarkers from liquid biopsy
Newer technologies have emerged to allow less invasive analysis of circulating tumour DNA (ctDNA) and circulating tumour cells (CTCs) overtime. We have taken a systematic approach in this trial to serially collect peripheral blood for ctDNA and CTC analysis to gain biological insight into the potential biomarker of response and the mechanism of drug resistance. Patients’ plasma was collected at baseline, C3D1 and end-of-treatment in selected patients for ctDNA analysis by NGS [31]. In addition, peripheral blood was also collected at baseline, C3D1 and end-of-treatment in selected patients for CTC analysis for cell enumeration, AR-V7 protein detection, cellular phenotypic heterogeneity (Shannon index) and chromosomal instability (pLST; large scale transition) analysis [26,27,28,29, 33]. However, because of the small sample size and 92% of the patients had 0 to less than 5 CTC count per ml of blood at screening, consistent with other first line mCRPC patients, no further analysis from CTCs would be presented here. Results from ctDNA analysis will be presented below.
ctDNA analysis
A total of 45 baseline and 30 end-of-treatment plasma samples were subjected to cell-free DNA (cfDNA) sequencing using a custom panel that captures coding and non-coding regions from 73 prostate cancer driver genes [34, 35]. After using patient-matched leucocyte DNA to filter variants related to clonal haematopoiesis, 57% (43 out of 75 samples) had detectable ctDNA levels above the detection threshold (1%), which is comparable to previously published results in a similar patient population [35]. Twenty-four patients at baseline had ctDNA detected in their plasma at baseline. For these 24 patients, the most frequent mutations or copy deletions were TP53 (33.3%), PTEN (25.0%), APC (25.0%), FOXA1 (16.7%), SPOP (16.7%) and AR (8.3%; two patients with ligand-binding domain mutations) (Fig. 4a). In addition, amplification/copy-number gain of AR and MYC were detected in 50.0% and 20.8% of the 24 patients at baseline, respectively (Fig. 4a). Further analysis of AR gene locus revealed that 10/12 patients with AR amplification/copy-number gain had co-amplification of both the gene body and upstream AR enhancer element (Fig. S6). Six of the 12 patients had a high level of AR copy amplification (≥8 copies, after normalising by ctDNA fraction), which has been shown to be associated with poor response to AR-targeted therapies [35].

a ctDNA analysis from 24 patients at screening. b ctDNA analysis from 16 patients with matched screening and end-of-treatment samples.
In addition, matched ctDNA samples at baseline and end-of-treatment from 16 patients were also evaluable (Fig. 4b). Tracking genomic alterations in plasma ctDNA allows minimally invasive monitoring of the emergence or persistence of resistant tumour cells overtime. Overall, the patients’ genomic profiles were similar between baseline and end-of-treatment (EOT), suggesting that enzalutamide/CC-115 treatment did not result in a dramatic shift in clonal selection (Fig. 4b). Furthermore, ctDNA analysis revealed the persistence and/or emergence of aggressive clones, including TP53 and PTEN mutation/deletion (Patient A), and CDK12 mutations and MDM2 amplification (Patient B), in two patients with early progressive disease within 3-4 months of enzalutamide and CC-115 treatment (Figs. 3 and 4b).
Discussion
This Phase 1b trial investigated the combination of enzalutamide with a selective dual mTORC1/2 and DNA-PK inhibitor, CC-115, in the first line patients with mCRPC that have progressed after ADT. Overall, this trial demonstrated an acceptable safety profile of CC-115 (5 mg BID) in combination with enzalutamide and a preliminary efficacy signal.
Maculopapular rash was the most common treatment-emergent adverse event, and was dose-dependent. Grade 3 rash was observed in 46% of the patients taking 10 mg twice a day dosing, which necessitated dose-reduction ultimately to 5 mg twice a day. At 5 mg dosing, Grade 3 rash was reduced to 16.7%. Most of the rashes were successfully managed with a short course of topical or oral corticosteroids, as has been reported previously for the AR inhibitor apalutamide [36]. The rate of rash observed in this trial appeared to be higher than that reported in the first-in-human CC-115 monotherapy study [23]. This is likely due to some overlapping skin toxicity between CC-115 and enzalutamide. Although less common than apalutamide, enzalutamide can cause skin toxicity, including pruritis (~3.8%) and rash (~2.6%) [37, 38].
Interestingly, 92.3% (12 out of 13) patients who developed a Grade 3 rash had a PSA reduction of at least 50%. This is reminiscent of EGFR tyrosine kinase inhibitors used in NSCLC, where skin rash is predictive of clinical response [39]. In contrast, another dual mTOR and DNA-PK inhibitor, samotolisib in clinical development had a very different side effect profile with fatigue and gastrointestinal side effects being the most common adverse effects, while rash was not reported [24]. This is possibly due to different chemical structures and differential kinase target inhibition between CC-115 and samotolisib. Overall, CC-115 at 5 mg twice a day was well tolerated with acceptable safety profile when combined with enzalutamide. There was no PK interaction observed with the combination.
CC-115 plus enzalutamide showed encouraging preliminary efficacy in patients with PTEN mutation/deletion or other PI3K pathway alteration, such as PIK3CA activating mutation and TSC1/2 loss-of-function mutations. In 40 evaluable patients (irrespective of mutation status), PSA50 and PSA90 response was 80% and 58%, respectively. This is in line with the enzalutamide arm of the PREVAIL trial, which showed PSA50 and PSA90 response of 78% and 47% respectively in a similar mCRPC population post ADT [3]. However, when stratified by PI3K pathway mutation status, patients with PI3K pathway alteration had a PSA50 response reaching 94% (15 out of 16) and a PSA90 response of 63% (10 out of 16), compared to PSA50 and PSA90 of 67% and 47% respectively in patients without such mutations. It is important to note that this comparison did not reach statistical significance and the biomarker analysis was not a pre-specified subgroup analysis. Given the limitation of such exploratory analysis, PI3K pathway mutations as a predictive biomarker for PI3K/AKT/mTOR pathway inhibitor in mCRPC patients require further validation. The finding in this study is consistent with a randomised double-blinded Phase 3 trial (IPATential150) that showed ipatasertib, an AKT inhibitor, plus abiraterone improved rPFS in mCRPC patients with PTEN-loss tumours, but not in the all-comer intention-to-treat population [14]. Interestingly, in a recently published randomised Phase 1b/2 trial in mCRPC, the combination of samotolisib and enzalutamide showed an improved PFS compared to placebo plus enzalutamide, but PTEN protein loss by IHC was not found to be a biomarker for response. The patient populations were different between these two studies. The current study and the Phase 3 IPATential150 enrolled first line mCRPC patients, while the study with samotolisib enrolled patients who had already progressed on abiraterone. In addition, the authors postulated several other reasons and suggested that the overall response in PTEN-intact patients was likely driven by samotolisib’s DNA-PK inhibitor activity [24]. In contrast, CC-115 at the lower dosing level, such as 5 or 7.5 mg, was likely primarily acting as an mTORC1/2 inhibitor. This is partly supported by in vitro Western blot analysis of LNCaP prostate cancer cell line treated with CC-115, which showed robust biochemical inhibition of AKT/mTOR pathway but no evidence of DNA-PKcs inhibition by CC-115 at the concentration (0.1 μM) that is roughly equivalent to peak serum concentration of ~30 ng/ml (Fig. S4A). Furthermore, CC-115 had a significant growth inhibitory effect on two human prostate cancer cell lines (LNCaP and CWR-22PC sgPTEN) in vitro, similarly to INK128, a selective mTORC1/2 inhibitor without DNA-PK inhibitor activity (Fig. S4B, C). Taken together, these data suggested that CC-115 at the clinically tolerated dose in combination with enzalutamide predominantly inhibits mTORC1/2, but may be insufficient to block the DNA-PK pathway. However, this cannot be confirmed in clinical samples as post-treatment biopsy and assay to measure DNA-PK inhibition in real time in patients were not available.
The ability to understand the mechanism of therapeutic resistance and monitor the emergence of resistant mutations/ genomic alterations non-invasively in patients has significant scientific and clinical values. This study incorporated several biomarker analyses, including tumour tissue mutation profiling by NGS and ctDNA mutation analysis. In particularly, serial analyses of ctDNAs before, during and after treatment provided insight into the potential mechanisms of drug resistance to enzalutamide plus CC-115. In two patients who progressed early within 3 months of beginning treatment, ctDNA analysis revealed the persistence and emergence of PTEN deletion and TP53 mutation in Patient A, and CDK12 mutations and multiple additional gene amplification, including MDM2, in Patient B at the time of disease progression (Fig. 4). The presence of CDK12 mutation [40, 41] and TP53/PTEN double mutations [42,43,44] have been shown in the literature to confer resistance to targeted therapies in prostate cancer.
Although CC-115 is no longer being developed for prostate cancer, this study is an important proof of concept supporting AR and PI3K/AKT/mTOR co-inhibition in mCRPC and suggests that PI3K pathway mutations may be a promising biomarker for further studies involving targeted therapy against the PI3K/AKT/mTOR pathway. Trials are now underway evaluating dual inhibition of AR and PI3K/AKT/mTOR pathways in earlier hormone sensitive states which have the potential to show even greater response particularly if selecting for patients with an appropriate biomarker [43, 44].
Data availability
All relevant data generated or analysed during this study are included in this published article and its Supplementary Information files.
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. https://doi.org/10.3322/caac.21660.
American Cancer Society. Cancer Statistics Center. 2022. http://cancerstatisticscenter.cancer.org.
Beer TM, Armstrong AJ, Rathkopf DE, Loriot Y, Sternberg CN, Higano CS, et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N Engl J Med. 2014;371:424–33. https://doi.org/10.1056/NEJMoa1405095.
de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. https://doi.org/10.1056/NEJMoa1014618.
Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat Rev Cancer. 2015;15:701–11. https://doi.org/10.1038/nrc4016.
Sarker D, Reid AH, Yap TA, de Bono JS. Targeting the PI3K/AKT pathway for the treatment of prostate cancer. Clin Cancer Res. 2009;15:4799–805. https://doi.org/10.1158/1078-0432.CCR-08-0125.
Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011;19:575–86. https://doi.org/10.1016/j.ccr.2011.04.008.
Mulholland DJ, Tran LM, Li Y, Cai H, Morim A, Wang S, et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell. 2011;19:792–804. https://doi.org/10.1016/j.ccr.2011.05.006.
Jamaspishvili T, Berman DM, Ross AE, Scher HI, De Marzo AM, Squire JA, et al. Clinical implications of PTEN loss in prostate cancer. Nat Rev Urol. 2018;15:222–34. https://doi.org/10.1038/nrurol.2018.9.
Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28. https://doi.org/10.1016/j.cell.2015.05.001.
Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, et al. Integrative genomic profiling of human prostate cancer. Cancer Cell. 2010;18:11–22. https://doi.org/10.1016/j.ccr.2010.05.026.
Rescigno P, Lorente D, Dolling D, Ferraldeschi R, Rodrigues DN, Riisnaes R, et al. Docetaxel treatment in PTEN- and ERG-aberrant metastatic prostate cancers. Eur Urol Oncol. 2018;1:71–7. https://doi.org/10.1016/j.euo.2018.02.006.
Ferraldeschi R, Nava Rodrigues D, Riisnaes R, Miranda S, Figueiredo I, Rescigno P, et al. PTEN protein loss and clinical outcome from castration-resistant prostate cancer treated with abiraterone acetate. Eur Urol. 2015;67:795–802. https://doi.org/10.1016/j.eururo.2014.10.027.
Sweeney C, Bracarda S, Sternberg CN, Chi KN, Olmos D, Sandhu S, et al. Ipatasertib plus abiraterone and prednisolone in metastatic castration-resistant prostate cancer (IPATential150): a multicentre, randomised, double-blind, phase 3 trial. Lancet. 2021;398:131–42. https://doi.org/10.1016/S0140-6736(21)00580-8.
Mohiuddin IS, Kang MH. DNA-PK as an emerging therapeutic target in cancer. Front Oncol. 2019;9:635 https://doi.org/10.3389/fonc.2019.00635.
Yue X, Bai C, Xie D, Ma T, Zhou PK. DNA-PKcs: a multi-faceted player in DNA damage response. Front Genet. 2020;11:607428 https://doi.org/10.3389/fgene.2020.607428.
Dylgjeri E, McNair C, Goodwin JF, Raymon HK, McCue PA, Shafi AA, et al. Pleiotropic impact of DNA-PK in cancer and implications for therapeutic strategies. Clin Cancer Res. 2019;25:5623–37. https://doi.org/10.1158/1078-0432.CCR-18-2207.
Goodwin JF, Kothari V, Drake JM, Zhao S, Dylgjeri E, Dean JL, et al. DNA-PKcs-mediated transcriptional regulation drives prostate cancer progression and metastasis. Cancer Cell. 2015;28:97–113. https://doi.org/10.1016/j.ccell.2015.06.004.
Bouchaert P, Guerif S, Debiais C, Irani J, Fromont G. DNA-PKcs expression predicts response to radiotherapy in prostate cancer. Int J Radiat Oncol Biol Phys. 2012;84:1179–85. https://doi.org/10.1016/j.ijrobp.2012.02.014.
Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer Discov. 2013;3:1254–71. https://doi.org/10.1158/2159-8290.CD-13-0108.
Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013;3:1245–53. https://doi.org/10.1158/2159-8290.CD-13-0172.
Tsuji T, Sapinoso LM, Tran T, Gaffney B, Wong L, Sankar S, et al. CC-115, a dual inhibitor of mTOR kinase and DNA-PK, blocks DNA damage repair pathways and selectively inhibits ATM-deficient cell growth in vitro. Oncotarget. 2017;8:74688–702. https://doi.org/10.18632/oncotarget.20342.
Munster P, Mita M, Mahipal A, Nemunaitis J, Massard C, Mikkelsen T, et al. First-in-human phase I study of a dual mTOR kinase and DNA-PK inhibitor (CC-115) in advanced malignancy. Cancer Manag Res. 2019;11:10463–76. https://doi.org/10.2147/CMAR.S208720.
Sweeney CJ, Percent IJ, Babu S, Cultrera JL, Mehlhaff BA, Goodman OB, et al. Phase 1b/2 study of enzalutamide with samotolisib (LY3023414) or placebo in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2022 https://doi.org/10.1158/1078-0432.CCR-21-2326.
Scher HI, Morris MJ, Stadler WM, Higano C, Basch E, Fizazi K, et al. Trial design and objectives for castration-resistant prostate cancer: updated recommendations from the Prostate Cancer Clinical Trials Working Group 3. J Clin Oncol. 2016;34:1402–18. https://doi.org/10.1200/JCO.2015.64.2702.
Schonhoft JD, Zhao JL, Jendrisak A, Carbone EA, Barnett ES, Hullings MA, et al. Morphology-predicted large-scale transition number in circulating tumor cells identifies a chromosomal instability biomarker associated with poor outcome in castration-resistant prostate cancer. Cancer Res. 2020;80:4892–903. https://doi.org/10.1158/0008-5472.CAN-20-1216.
Brown LC, Halabi S, Schonhoft JD, Yang Q, Luo J, Nanus DM, et al. Circulating tumor cell chromosomal instability and neuroendocrine phenotype by immunomorphology and poor outcomes in men with mCRPC treated with abiraterone or enzalutamide. Clin Cancer Res. 2021;27:4077–88. https://doi.org/10.1158/1078-0432.CCR-20-3471.
Scher HI, Armstrong AJ, Schonhoft JD, Gill A, Zhao JL, Barnett E, et al. Development and validation of circulating tumour cell enumeration (Epic Sciences) as a prognostic biomarker in men with metastatic castration-resistant prostate cancer. Eur J Cancer. 2021;150:83–94. https://doi.org/10.1016/j.ejca.2021.02.042.
Scher HI, Lu D, Schreiber NA, Louw J, Graf RP, Vargas HA, et al. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. JAMA Oncol. 2016;2:1441–9. https://doi.org/10.1001/jamaoncol.2016.1828.
Greene SB, Dago AE, Leitz LJ, Wang Y, Lee J, Werner SL, et al. Chromosomal instability estimation based on next generation sequencing and single cell genome wide copy number variation analysis. PLoS ONE. 2016;11:e0165089 https://doi.org/10.1371/journal.pone.0165089.
Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med. 2017;23:703–13. https://doi.org/10.1038/nm.4333.
Annala M, Taavitsainen S, Khalaf DJ, Vandekerkhove G, Beja K, Sipola J, et al. Evolution of castration-resistant prostate cancer in ctDNA during sequential androgen receptor pathway inhibition. Clin Cancer Res. 2021;27:4610–23. https://doi.org/10.1158/1078-0432.CCR-21-1625.
Scher HI, Graf RP, Schreiber NA, McLaughlin B, Jendrisak A, Wang Y, et al. Phenotypic heterogeneity of circulating tumor cells informs clinical decisions between AR signaling inhibitors and taxanes in metastatic prostate cancer. Cancer Res. 2017;77:5687–98. https://doi.org/10.1158/0008-5472.CAN-17-1353.
Wyatt AW, Annala M, Aggarwal R, Beja K, Feng F, Youngren J, et al. Concordance of circulating tumor DNA and matched metastatic tissue biopsy in prostate cancer. J Natl Cancer Inst. 2017. https://doi.org/10.1093/jnci/djx118.
Annala M, Vandekerkhove G, Khalaf D, Taavitsainen S, Beja K, Warner EW, et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018;8:444–57. https://doi.org/10.1158/2159-8290.CD-17-0937.
Pan A, Reingold RE, Zhao JL, Moy A, Kraehenbuehl L, Dranitsaris G, et al. Dermatological adverse events in prostate cancer patients treated with the androgen receptor inhibitor apalutamide. J Urol. 2022;207:1010–9. https://doi.org/10.1097/JU.0000000000002425.
Deng M, Chai H, Yang M, Wei X, Zhang W, Wang X, et al. Stevens-Johnson syndrome caused by enzalutamide: a case report and literature review. Front Oncol. 2021;11:736975 https://doi.org/10.3389/fonc.2021.736975.
FDA. Xtandi (enzalutamide) prescribing information. 2020. https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/203415lbl.pdf.
Liu HB, Wu Y, Lv TF, Yao YW, Xiao YY, Yuan DM, et al. Skin rash could predict the response to EGFR tyrosine kinase inhibitor and the prognosis for patients with non-small cell lung cancer: a systematic review and meta-analysis. PLoS ONE. 2013;8:e55128 https://doi.org/10.1371/journal.pone.0055128.
Schweizer MT, Ha G, Gulati R, Brown LC, McKay RR, Dorff T, et al. CDK12-mutated prostate cancer: clinical outcomes with standard therapies and immune checkpoint blockade. JCO Precis Oncol. 2020;4:382–92. https://doi.org/10.1200/po.19.00383.
Antonarakis ES, Isaacsson Velho P, Fu W, Wang H, Agarwal N, Sacristan, et al. CDK12-altered prostate cancer: clinical features and therapeutic outcomes to standard systemic therapies, poly (ADP-ribose) polymerase inhibitors, and PD-1 inhibitors. JCO Precis Oncol. 2020;4:370–81. https://doi.org/10.1200/po.19.00399.
Hamid AA, Gray KP, Shaw G, MacConaill LE, Evan C, Bernard B, et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol. 2019;76:89–97. https://doi.org/10.1016/j.eururo.2018.11.045.
Zou M, Toivanen R, Mitrofanova A, Floch N, Hayati S, Sun Y, et al. Transdifferentiation as a mechanism of treatment resistance in a mouse model of castration-resistant prostate cancer. Cancer Discov. 2017;7:736–49. https://doi.org/10.1158/2159-8290.CD-16-1174.
Wang Y, Wang Y, Ci X, Choi SYC, Crea F, Lin D, et al. Molecular events in neuroendocrine prostate cancer development. Nat Rev Urol. 2021;18:581–96. https://doi.org/10.1038/s41585-021-00490-0.
Acknowledgements
We thank all the patients, their families, and the study site investigators and staff who participated in the study.
Funding
The study was supported and funded by Celgene/BMS and Gateway for Cancer Research. The trial was managed by Prostate Cancer Clinical Trials Consortium (PCCTC). JLZ was supported by a K12 Paul Calabresi Career Development Award for Clinical Oncology and a Prostate Cancer Foundation Young Investigator Award.
Author information
Authors and Affiliations
Contributions
JLZ and ESA designed the study, acquired the data, interpreted the results, and drafted and revised the manuscript. HHC, DJG, RA, WA, FYF, KK, MEL and AWW designed the study, acquired the data, interpreted the results, and revised the manuscript. ER designed statistical methods, interpreted the results and drafted and revised the manuscript. TS designed the ctDNA analysis, acquired the data, interpreted the results and revised the manuscript. JDS and AA designed the CTC analysis, acquired the data, interpreted the results and revised the manuscript. NM, SH and BC designed the pre-clinical experiment, acquired the data, interpreted the results and revised the manuscript. BD and TC acquired the data, interpreted the results, and revised the manuscript. DR corresponding author, supervised the study, designed the study, acquired the data, interpreted the results, and drafted and revised the manuscript.
Corresponding author
Ethics declarations
Competing interests
JLZ is currently a full-time employee at AstraZeneca, which is not involved in the funding and conduct of the trial. The authors declare no competing interests.
Ethics approval and consent to participate
IRB committee approval was obtained from all participating sites and written consent was obtained by all participating patients. The study was performed in accordance with the Declaration of Helsinki.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
41416_2023_2487_MOESM2_ESM.pdf
Table S1. Summary table of PI3K, TP53 and DNA repair pathway mutations by next-generation sequencing (NGS) and PSA response by 12 weeks
41416_2023_2487_MOESM6_ESM.pdf
Figure S4. Pre-clinical biochemical analysis of target pathway inhibition (A) and in vitro cell proliferation (B and C).
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhao, J.L., Antonarakis, E.S., Cheng, H.H. et al. Phase 1b study of enzalutamide plus CC-115, a dual mTORC1/2 and DNA-PK inhibitor, in men with metastatic castration-resistant prostate cancer (mCRPC). Br J Cancer 130, 53–62 (2024). https://doi.org/10.1038/s41416-023-02487-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02487-5
- Springer Nature Limited
This article is cited by
-
Enzalutamide inhibits PEX10 function and sensitizes prostate cancer cells to ROS activators
Cell Death & Disease (2024)