
Abstract
Tumour immunotherapy has achieved remarkable clinical success in many different types of cancer in the past two decades. The outcome of immune checkpoint inhibitors in cancer patients has been linked to the quality and magnitude of T cell, NK cell, and more recently, B cell within the tumour microenvironment, suggesting that the immune landscape of a tumour is highly connected to patient response and prognosis. It is critical to understanding tumour immune microenvironments for identifying immune modifiers of cancer progression and developing cancer immunotherapies. The infiltration of solid tumours by immune cells with anti-tumour activity is both a strong prognostic factor and a therapeutic goal. Recent approaches and applications of new technologies, especially single-cell mRNA analysis in dissecting tumour microenvironments have brought important insights into the biology of tumour-infiltrating immune cells, revealed a remarkable degree of cellular heterogeneity and distinct patterns of immune response. In this review, we will discuss recent advances in the understanding of tumour infiltrated lymphocytes, their prognostic benefit, and predictive value for immunotherapy.
Similar content being viewed by others

Background
In recent years, multiple strategies for eliciting anti-tumour immunity have been developed in different clinical studies. Currently, immunotherapy was clinically validated as an effective treatment option for many tumours such as melanoma, non-small cell lung cancer (NSCLC) and renal cell carcinoma (RCC) [1]. Base on the negative T cell regulatory pathways are often overactive in tumour microenvironment (TME), the overall therapeutic aims of a large variety of immunotherapy drugs in clinical trials are to disrupt or counteract tumour-mediated immunosuppression [2]. Among these, immune checkpoint blockade (ICB) has proven to be an effective strategy for enhancing the effector activity and clinical impact of anti-tumour T cells. Checkpoint inhibitors targeting CTLA-4, PD-1, and PD-L1 have yielded unprecedented and durable responses in a significant percentage of cancer patients [3]. Since the majority of people with cancer do not respond to these antibodies, effective immunotherapy requires the knowledge of TME, especially the interplay among the immune system, the tumour and treatment.
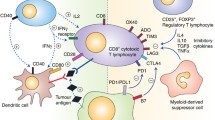
The TME is heterogeneous and complex in its molecular and immunological features. Multiple immune subsets and factors in the TME greatly influence the outcome of immunotherapy and disease prognosis. Over the last decade, it has become increasingly clear that the immune cells in the TME play a critical role in controlling or promoting tumour growth [4, 5]. The exact composition of the infiltrate immune cells can vary widely within and between tumours and clearly modulate the effectiveness of the anti-tumour therapy. CD8+ cytotoxic T cells, T-helper 1 cells producing interferon-γ, and natural killer cells (NK) are generally associated with favourable responses, along with macrophages polarised to an M1 phenotype and dendritic cells showing a DC1 phenotype. Immunosuppressive effects are seen with T-helper 2 cells, M2 macrophages, DC2 dendritic cells, myeloid-derived suppressor cells and FOXP3+ regulatory T cells producing IL-10 and TGFβ [6]. B cells and plasma cells can also adopt either effector or regulatory phenotypes, and hence can carry positive or negative anti-tumour associations depending on contextual factors in TME. The function of T lymphocytes in ICB-mediated anti-tumour responses has been well characterised, but the function of B lymphocytes and others are less clear. The presence of tertiary lymphoid structures in TME is correlated with better prognosis in many solid tumours [7, 8]. The numbers, localisation and phenotypes of tumour-infiltrating lymphocytes (TILs) are not only predictive of response to immunotherapy but also key modulators of disease progression [9].
The programmed death ligand 1 (PD-L1) expression level, tumour mutation burden (TMB) and microsatellite instability (MSI) are biomarkers currently used to predict response to immune checkpoint inhibitors, but more comprehensive characterisation of TME will likely provide deeper insights for patient stratification and drug development [9]. The assessment of immune infiltrates in tumour, most commonly referred to as TILs, is gaining importance in the current quest for optimal biomarkers to select patients who will get benefits. Several strategies to measure the interaction between tumour and immune system have been published, including analysis of haematoxylin/eosin (H&E) staining, evaluation of specific subgroups of immune cells by immunohistochemistry (IHC) [10], immunofluorescence or flow cytometry as well as measuring expression of immune-related genes [9]. In contrast to conventional bulk mRNA-seq which process mixtures of all cells to average out potential differences in cell type-specific transcriptomes, single-cell RNA-seq profiles the gene expression pattern of each individual cell and decodes its intercellular signalling networks, could provide clear and unbiased insights into the entire tumour ecosystem, such as mechanisms of intra- and inter-tumoural heterogeneity, as well as cell–cell interactions through ligand-receptor signalling [11]. In this paper, we explore the sate-of-art prognostic relevance of TILs pattern, profiling and enumerating various lymphocyte subsets, also the new strategies to measure the immune status in TME and discuss their application in clinical.
Tumour-infiltrating lymphocytes
TILs are immune cells that have migrated from the blood stream into tumour tissue with the purpose of tumour elimination [6]. In most cases, TILs are comprised primarily of CD3 + T cells, and a smaller proportion of B and NK cells. The driven force of tumour infiltrating derived from TILs’ tumour specificity which is always assimilated and suppressed by TME. Curative effect of TILs was verified by Professor Seven Rosenberg firstly and applied in patients with melanoma in 1986, 60% (9/15) of enrolled patients with metastatic melanoma had an objective response [12]. Compared with other cellular immunotherapies, autologous tumour tissue derived TILs without genetic modification are potent in tumour killing and meanwhile safe from immune-related adverse events (irAEs).
TILs, as one of the most intricate immune subgroups within TME, the overall existence of TILs would serve as the rough indicator of patients’ survival and therapeutic effect of ICB, even no further clustering and function details are provided. According to the results of certain clinical trials as KEYNOTE-086, which shows curative response of Keytruda is positively related with HE staining defined TILs abundance, and as KEYNOTE-173, which reported the homodromous consistency between patients’ objective response rate (ORR) and complete response (CR) improvement and biopsy defined enhancive TILs expression in neoadjuvant ICB treatment in triple-negative breast cancer (TNBC). TME is classified as four different subtypes by scientists based on the presence or absence of TILs and expression of PD-L1: type I (PD-L1 and TILs positive), type II (PD-L1 and TILs negative), type III (PD-L1 positive with TILs negative), and type IV (PD-L1 negative with TIL positive) [13]. Patients of type I subgroup are most likely to benefit from ICB due to the evidence of pre-existing intratumour T cells that are turned off by PD-L1 engagement even the risk of further activation of PD-1+Treg may also exist. Around 41% melanoma patients present with a type II TME which indicated poor prognosis based on the lack of detectable immune reaction. Administration of single agent ICB would most likely not to be successful given the lack of pre-existing TILs within this subgroup. At least three strategies should be considered in this scenario: (1) Combinational therapies, such as the combination of anti-CTLA-4 and anti-PD-1, which would enhance tumour tissue trafficking and infiltrating also referred as “tumour penetrating” of immune potent T cells. The 45–50% response rates and 70% of 2-year overall survival rate were reported by Wolchok et al. in advanced melanoma patients and increased infiltrating of TILs in situ were also confirmed by IHC in a combining trial of ipilimumab and nivolumab [14]. (2) Establishment of type I IFN response would be helpful to changeover the PD-L1-TILs- immunosuppressive TME [15]. (3) Adoptive cell transfer or tumour vaccination would be other choices to attract tumour-specific T cells into the hostile TME. The former would passively remodelling the host anti-tumour immunity by infusing abundant specific immune potent cells such as ex vivo expanded TILs, CAR-T or TCR-T cells, while the later would mobilise host anti-tumour immunity in the proactive way [16]. Another potential strategy which deserved to be mentioned here is immunogenic cell death which induced by certain chemo/radio-therapy or small molecules would favour the process of type II tumours remoulding [17]. Patients within type III group would not benefit from ICB owing to lack of TILs as downstream executor of tumour killing which means same strategies should be employed to recruit lymphocytes into tumour lesions. Type IV tumour microenvironment refers to the other suppressive pathways such as LAG3, TIM3, TIGIT, IDO etc. induced immune tolerated state. In this condition, other immune checkpoint inhibitors should be considered rather than PD-1/PD-L1 mAb and TILs abundance could not be simply interpreted as one monodirectional variate due to its promiscuous state [18].
Phenotype of TILs within TME is intricate and erratic. It is difficult to define certain subset of TILs as indicator of prognosis only based on unstable surface markers. So researchers turn to set up profile of TILs related immune-predictive signature genes with equal or distinct weight within system such as IFN-γ–related mRNA profile(T cell-inflamed GEP) [19] or IFN stimulated genes(ISGs) profile combined with ISG resistance signature(ISG.RS) profile [20]. T cell-inflamed GEP including IFN-γ–responsive genes related to chemokine expression, antigen presenting, T cell cytotoxicity, and immune tolerance is designed as pan-tumour indicator derived from 220 patients with 9 cancers which are necessary but not always sufficient. IFN stimulated genes profile refer to around 200 genes such as HLA-A, GZMA, ICAM, etc., which are tightly related IFN signalling pathway in immune cells especially in TILs while ISGs resistance signature profile refers to 38 genes such as HLA-G, TIM3, LAMP3, etc., which are related with immunosuppressive or immune tolerance mechanism of cancer cells, tumour associated fibroblast, type 2 macrophage and so on. Putting ISGs together with ISG.RS would reinforce the Signal to Noise Ratio(SNR) of the predictive system.
According to the theory of clonal selection [21], tumour-specific TILs clone would be collectively expanded within TME under stimulation of tumour antigen before cancer induced clonal suppression and deletion, which means the intratumour richness and clonality of TILs and diversity TILs repertoires may correlate with ICB responses, while it is far more complicated in reality [22, 23]. TILs clonality may related with ICB response in some studies but not in others which may indicate that some restrict low density TILs clones are also functional in ICB treatment. The Immune Repertoire (IR)-Index is further induced by researchers to highlight the proportion of potent neoantigen stimulated TCRs in ICB background, while the predictive efficacy of IR-Index is only confirmed in two distinct cohorts and its prognosis value in pan-tumour should be further validated [24].
TIL subsets and tumour-specific TILs (TSTs) within TME
CD8 + TILs
Cytotoxic CD8 + TILs are definite focus of host anti-tumour immunity from basic research in tumour immunology to clinical application of adoptive cell therapy (ACT). We could highlight the characters of CD8 + TILs by sketching the fate of intratumour cytotoxic T lymphocytes (CTLs) with cellular and functional features [25]. Naive T cells activated by antigens via transcription factors of TCR pathway as BATF, IRF-4, and NFAT-AP1 and differentiated into KLRG-1loCD127hi memory precursor T cells (MPECs). Depending cytokine crosstalk and antigen stimulation, CD8 + TILs would further differentiate to final stage along different trajectories: (1) Effector T (Teff) cells are characterised as KLRG-1hiT-bet + +Blimp-1 + CD8 + T cells which are functional in cytokine production and antigen elimination. (2) Memory T cells are characterised as EomeshiTcf-1+ self-renewing CD8 + T cells. (3) Resident memory T (Trm) cells are characterised as CD103 + CD69 + CD127 lo/–KLF-2lo Hobit+ Eomes lo CD8 + T cells which escape from circulation and retain in tumour tissue. (4) Exhausted T (Tex) cells subjectively divide into early and terminal subgroups according to distinct master transcription or surface markers as PD-1IntTcf-1+Eomeslo early exhausted CD8 + TILs and PD-1hiEomeshiTbetloTcf-1- terminal exhausted CTLs. Repeated continuous antigen stimulation is the driven force of T cell exhaustion.
According to the study published by Lee et al. on JAMA Oncology, among the 36 enrolled potent risk factors as PD-L1 expression, intratumour heterogeneity, TMB, etc., estimated CD8 + TILs abundance within TME is the most independent predictive marker (P < 2.3 × 10−4) of anti–PD-1/PD-L1 therapy across 21 cancer types (7187 patients) [26]. Similar conclusion is also reported by Bruni et al. on data of 18,700 patients across 17 solid cancer types but totally opposite observation is presented meanwhile in patients with clear cell RCC (ccRCC) and prostate cancer, which shows abundance of CD8+ TILs within TME correlate with shorter progression-free survival (PFS) and overall survival (OS) [27,28,29,30]. Exhausted state of infiltrated CTLs in the case of ccRCC is one of the potential explanations which is not yet validated in the case of prostate cancer [31]. Immunoscore, reported by Galon et al., is established on algorithm of CD3/CD8 expression at the core and edge of tumour lesion in colorectal cancer (CRC) [32]. By quantifying CD3+ and CD8+ TILs densities in both locations, potential anti-tumour immunity which tightly correlates with disease-free survival (DFS), disease-specific survival (DSS) and OS is translated into measurable certain score ranging from 0 to 4 [33, 34]. In this predictive system, high scores refer to low recurrence and better survival in spite of TNM stage, tumour differentiation, sex, mucinous tumour type and MSI status. Even further, the prognostic value CD3/CD8 based Immunoscore is confirmed by studies not only in CRC but also in other cancers as breast cancer and ovarian cancer. Up to now, Immunoscore is approved for clinical use and industrialised successfully as the predictive platform. Beside the entire CD8 + TILs group, selective expression of transcription factor 7 (TCF7) on memory-like CD8 + TILs in melanoma strongly correlate with clinical benefit and OS in patients treated with anti-PD-1 mAb [35]. Defined by high expression of exhaustion markers as PD-1 and CTLA-4, phenotypically exhausted CD8 + TILs could be functional meanwhile in certain degree to produce cytokine as IFN-γ rather than IL-2 or TNF-α and serve as marker of clinical response of anti-PD-1 blockade in metastatic melanoma [36]. One recent study verified the fact that chemotactic reprogramming inducing via upregulation of chemokine receptor CXCR6 in TME would convert stem-like CD8 + TILs into effector-like CTLs and provide survival signals together with IL-15 trans-presentation which means CXCR6 is critical for CTL-mediated tumour control [37].
Along with the development of single-cell sequencing technologies, properties of immune cells in TME could be described and exhibited on a more elaborate level. According to data of single-cell sequencing, CD8 + TILs could be labelled in a subtle way based on the otherness and specificity of genetic fingerprints. Exhausted CTLs which annotated as LAYN+Tex in TME are usually dysfunctional by increased expression of inhibitory receptors such as PD-1, CTLA-4, LAG3, TIM3, TIGI, etc. and low/absent expression of T-box transcription factor (TBX21), IL-2 and TNF-α, along with reactively elevated inflammatory and cytotoxic associated genes as TOX, TNFRSF9, CXCL13, IFNG, GZMB, and PRF1 [35, 38,39,40]. In certain cancer types as melanoma, CRC and liver, lung, breast, bladder cancer, proliferative CTLs(Mki67+) are defined as close but distinct subsets of Tex and shared TCR between the two subgroups suggesting the same origin [41,42,43]. CD45RO+GZMK+ T effector memory (Tem) is another common group revealed by scRNA-seq by expression of GZMK, GZMA, CCL5, IFNG, RUNX3, EOMES, CXCR3, CXCR4, CD44 instead of exhaustion gene profile but TCR overlaps are still widely detectable between Tem and Tex which indicate the existence of crosstalk and interaction rather than isolation [44]. The ratio of Tem/Tex could serve as predictive marker of OS in ICB treated NSCLC and melanoma and it also varies within different cancer types and stages [43]. Pre-exhausted Trm cells which annotated as ZNF683+ Trm in scRNA-seq analysis are highlighted by ZNF683, HOPX and ITGAE expression in TME and share TCR frequently with Tex cells [45]. Comparably, another Trm subgroup in peri-tumour site is annotated as XCL1+ Trm which is characterised by expression of XCL1, XCL2, MYADM, CAPG, CD6, NR4A1/2/3, CD69, ITGAE in CRC [41].
CD4 + TILs
T helper cells in TME represent one complicated subgroup of miscellaneous TILs including T helper 1 (Th1) cells, T helper 2 (Th2) cells, T helper 17 (Th17) cells, regulatory T cells, T follicular helper (Tfh) cells, etc., which are phenotypically and functionally distinct. Th1 cells produce cytokines like IFN-γ and TNF-α would help to establish type 1 inflammation and strongly associated with better clinical outcome in almost all cancer types, while Th2 cells produce cytokines like IL-4, IL-5 and IL-6 would favour type 2 inflammation and associated with poor survival [27]. One exception case should be mentioned that Th1 abundance are associated with worse prognosis in NSCLC after surgery. The exception case for positive prognostic value of Th2 is in Hodgkin lymphoma. In glioblastoma model, ratio of Th1/Th2 is applied as predictive marker in ICB treatment [46]. The indicative role of Th17 is also controversy: better survival correlations are observed in gastric, ovarian and cervical cancers [47,48,49] while worse survival correlations are reported in NSCLC and CRC [50, 51]. IL-17 produced by Th17 would aid process of oncogenesis while CCL5 and CCL20 produced by Th17 would induce enrichment of CCR5+CCR6+CTLs in situ for tumour elimination in CRC [52, 53]. Tfh cells are frequently reported as the central component of tertiary lymphoid structures which are correlated with establishment of anti-tumour immunity and clinical benefit after ICB treatment [54]. CXCR5 expressed on Tfh cells would induce chemotaxis migration of tumour specific CXCL13+ T cells in in CRC and breast cancer [55, 56].
Treg cell is the typical CD4+ T cell subset and characterise by its high expression of IL-2Rα (CD25) and the transcription factor forkhead box protein P3 (FOXP3), meanwhile low or negative expression of IL-7Rα (CD127) [57]. Physiologic suppressive function of Treg cells (Tregs) are executed by production of inhibitive cytokines as IL-10, IL-35 and TGF-β, induction of adenosine or IDO luxuriant microenvironment and metabolic hostile milieu, IL-2 competition, negative regulation of DC development, maturation and function via LAG3 and CTLA-4 expressing and CTLs elimination by direct contact and GZMB production. Treg cells are assimilated and domesticated in the procedure of oncogenesis and work as the core of pathologic immunotolerance. CCR8 would be helpful to discriminate the highly suppressive intratumour Tregs as function marker, so as to CD39 and CD73 [58]. Based on sustained Foxp3 expression, hypomethylation at the CNS2 locus and maintained suppressive function, Tregs are defined as 3 main subgroups [59]: (1) Stable Tregs are hypomethylated at CNS2 and express stabilising markers like NRP1 and Helios. Stable Tregs are Foxp3 positive and function in immune suppression. (2) Fragile Tregs are Foxp3 positive while with low IL-10 and TGF-β production and are incapable in inhibitory function. Anti-tumour activity of fragile Tregs is reported owing to their tumour antigen specific IFN-γ secretion. (3) Unstable Tregs are Foxp3 negative and disable in effector suppression [60, 61]. Abundance of Tregs within TME is related with poor prognosis in cervical and renal cancer [62, 63], but no confirmed correlation with prognosis in SCC and glioblastoma [64, 65], or even positively related with clinical benefit in CRC, bladder, head and neck cancer [66, 67]. One key mechanism of hyperprogressive disease in Patients with NSCLC treated with anti-PD-1/PD-L1 Abs is due to the PD-1 expression on Tregs [68]. This point is circumstantially evidenced by the study published by Kumagai et al. concerning the PD-1 expression balance between Teff and Tregs would predict the clinical efficacy of PD-1 blockade [69]. Another explanation for the contradictory predictive value of Tregs in ICB treatment is its nonuniformity and non-pureness, since Foxp3 could not be the unique key marker of Tregs as we mentioned above and also in other publications. Existence of two functionally distinct Foxp3+ populations are reported by Saito et al., within which “classical” Foxp3hi Tregs are immunosuppressive while CD45RA-Foxp3lo Tregs (non-Tregs) would produce inflammatory cytokines, as the fragile Tregs mentioned before [35].
From the aspect of scRNA-seq, CD4 + T cells within tumour adjacent site are specified as TCF7+ naive T (Tn) and follicular helper T cells, respectively. Distinctive markers for the former are CCR7, TCF7, RGS1, CD69, CXCR5 and for the later are CXCR5, BCL6, ICA1, TOX, TOX2, IL6ST, MAGEH1, BTLA, ICOS, PDCD1, CD200 [70, 71]. As the mirror group in CD4+ TILs, CD4+GZMK+Tem cells share similar profile of signature genes with GZMK+ CTLs. Corresponding matchup of CD8+LAYN+ Tex cells in T helper are CXCL13+ Th1-like cells whose signature genes are CXCL13, IFNG, CXCR3, BHLHE40, GZMB, PDCD1, HAVCR2, ICOS, IGFLR1, ITGAE. Subgroups which are CD4+ TILs specific are CXCR6+ Trm cells (CXCR6, CD69, KLRB1, PTGER4, IL7R, NR4A1/2/3, MYADM) and Th17 cells (IL17A, IL23R, RORC, FURIN, CTSH, CCR6, KLRB1,CAPG, ITGAE). Tregs within tumour lesion are usually functionally active with high expression of CCR8 OXP3, CTLA4, LAYN, TNFRSF9, TNFRSF18, IKZF2, RTKN2, BATF and annotated as tumour Tregs reported in melanoma, breast cancer, NSCLC, HNSCC, TNBC and CRC [70, 72].
B cells
As an integral component of the TME, B cells exist in all stages of cancer, including all major B cell subsets, from naive B cells to plasma cell (PC). Compared to the significant attention to T cells, the roles of B cells are not well characterised and studies examining their prognostic potential are limited [73]. However, B cells also display important functions apart from producing antibodies, evidence has shown that B cells can both restrain and promote anti-tumour immune responses [74]. And new findings suggest that some aspects of the humoral immune response may improve clinical outcomes through B cell tumour infiltration and antibody expression in lesions or circulation [9]. Briefly, B cells can impede tumour development by promoting cytotoxicity to tumour cells, producing tumour-specific antibodies, and acting as APC, especially where DC function may be impaired or DC may be present [75]. In the last 5 years, multiple studies have demonstrated an association between the presence of B cells in the TME and improved clinical outcome [74, 76].
Researchers from Lund University discovered that when B cell structures were present in the tumour, the patient had a better prognosis and responded better to immunotherapy in melanoma [77]. Improved survival of patients with high CD20 gene expression and with high numbers of CD20+ B cells in the majority of patient cohorts were reported, strongly supports an anti-tumour role for conventional B cells [78, 79]. In TNBC, tumour-infiltrating B lymphocytes assemble in clusters, undergoing B cell receptor–driven activation, proliferation, isotype switching, and the clonally expanded, IgG isotype-biased humoral immunity primarily associates with favourable prognosis [80]. These suggests that humoral immune responses may contribute to clinical outcomes, especially in more immunogenic subtypes. Patients with the enrichment of B cell and Tertiary lymphoid structure (TLS) were demonstrated to obtain significant therapeutic advantages in CRC [81]. However, the characteristics of B cells and their clinical significance remain unclear. It was found that the proportion of IgG plasma cells in the tumour site was higher than that in the adjacent normal mucosa. In addition, B cells and the CCL28-CCR10 axis is pivotal for IgG plasma cell migration from the periphery of TLS to the tumour stroma [82, 83].
Recent studies indicate that proliferating B cells are observed in 35% of lung cancer patients [84]. And can be observed in all stages, and their presence differs between stage and histological subtypes, suggesting a critical role for B cells during lung tumour progression [75]. B cells and TLSs in NSCLC correlate positively with prognosis and are associated with increased CD4+ T cell repertoire clonality and a reduction in the frequency of Treg [85]. In a recent study, B cells were remarkably enriched in major pathological response (MPR) tumour lesions, suggesting that B cells may be a key determinant of therapeutic response to neoadjuvant pembrolizumab and chemotherapy in NSCLC patients [81]. IGHA1, IGHA2, and JCHAIN were significantly decreased in neoadjuvant MPR tumour lesions than in non-MPR tumour lesions, while IGHG1 and IGHG3 were significantly upregulated in MPR tumour lesions, indicating the B cells class switched to two dominant antibody isotype IgG1 and IgG3 during the therapy [86]. The multiplex fluorescent immunohistochemistry (mIHC) results demonstrated that neoadjuvant chemoimmunotherapy promoted more intratumoural CD20+ B cell infiltration (mean 7.59% versus 14.25%). And there are more CD4+IL21+ and IgG-positive cells in MPR tumour tissues, while no difference in nearest non-cancer tissues, and LNs. Taken all together, the analysis suggested that B cells class switched to IgG subclass mediated favourable anti-tumour immune response during neoadjuvant chemoimmunotherapy, and that increased IL-21 secreted by CD4+ T cells was the driving force in tumour lesion [87]. Another study also shows that B cell density represents a new prognostic biomarker for the survival of NSCLC patients, and makes the link between TLS and a protective B cell–mediated immunity [88].
Regulatory B (Breg) cells are immunosuppressive cells that support immunological tolerance through the production of interleukin-10 (IL-10), IL-35, and transforming growth factor β (TGF-β) or intercellular contact [89], evidences shows that Bregs play a pivotal role in regulating immune responses involved in inflammation, autoimmunity and cancer [90]. In accordance with these findings, patients with ovarian cancer also express elevated frequencies of IL-10-producing B cells that correlate with higher frequencies of FOXP3+CD4+ Tregs and exhibit suppressed IFN-γ production by CD8+ T cells, which is mediated through both IL-10 secretion and cell-to-cell contact via CD80/CD86 expression [91]. Bregs were also reported to correlate with increased frequencies of CD14+ HLA-DRlo myeloid-derived suppressor cells as well as increased progression of carcinoma and reduced patient survival. IL-10 secretion by Bregs were reported to induce the conversion of CD4+ T cells into Tregs, which could be one of the mechanisms that modulate cancer progression [92]. Taken together, Bregs attenuate anti-tumour immune responses by suppressing diverse cell subtypes, including T cells, through the secretion of anti-inflammatory mediators and can facilitate the conversion of T cells to regulatory T cells [93].
As discussed above, B cells play widely varied roles in the context of tumour immunity, and in different cancers contribute to either tumour growth or anti-tumour immunity. Correlative studies on human tumour-infiltrating B cells are somewhat limited by their inability to differentiate between functional B cells present in tumours and bystander cells simply recruited as a result of the local cytokine milieu [75]. These observations suggest that both the functional diversity as well as the spatial localisation of B cells and plasma cells are important considerations when determining their prognostic and biological significance in the TME [76]. The prognostic and mechanistic significance of the localisation of B-TIL and other leucocytes within tertiary lymphoid structure is a vital avenue of investigation in tumour immunology [94].
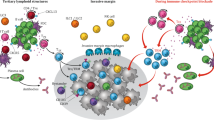
Tertiary lymphoid structures
Tertiary lymphoid structure is transient ectopic lymphoid tissues which develop in the context of chronic inflammatory responses such as in chronic viral infections, autoimmune diseases, allograft rejection and cancer [54]. TLSs are characterised by an inner zone of CD20+ B cells that is surrounded by CD3+ T cells, and can vary in complexity from lymphocyte clusters to highly organised in the tumour, spatially segregated structures bearing a strong resemblance to secondary lymphoid organs, with the exception that TLSs are not surrounded by fibrous capsules like lymph node. And TLSs are also observed to contain high endothelial venules (HEV), distinct T cell zones with mature DC and B cell follicles with a germinal centre, and evidence of antibody class-switching [94]. Histologically, TLSs are found in a proportion of cancers of various types such as NSCLC, melanoma, sarcoma and colorectal cancer [95], and correlated with a good prognosis and prolonged patient’s survival in 12 different types of cancer [96], and usually located in the invasive margin or in the stroma rather than the tumour core, and HEV are detected in close proximity to TLSs. Using H&E, mature TLS (mTLS) can be identified either as well differentiated lymphoid follicles which include structures resembling germinal centres, which can be assessed by multiplex immunofluorescence assay combining markers with CD4, CD8, CD20, CD21 and CD23 [54, 97].
In patients with soft-tissue sarcoma, high expression of a transcriptomic B cell signature correlated with longer OS, regardless of other immune factors such as T cell abundance. This B cell signature was positively correlated with expression of CXCL13 and the presence of TLS detected by IHC, mostly in peritumoural areas [95]. A study on a cohort of 328 patients treated with anti-PD1 or anti-PD-L1 monoclonal antibodies shows that the presence of mTLS was the most significant predictive factor for objective response, and had an independent predictive value for both PFS and OS, independent of PD-L1 TPS score and the level of CD8 + T cell infiltration [97]. In another validation cohort included 81 patients from MATCH-R study, mTLS-positive patients had a significantly better outcome than patients with no mTLSs, with an ORR of 38.4% versus 11.8% (P = 0.02), a median PFS of 10.9 versus 2.1 months (P = 0.079) and a median overall survival of 24.6 versus 8.1 months (P = 0.036).
In immature TLSs, B cells may produce molecules released in the TME or expressed on their membrane that impair an efficient anti-tumour immune response. In contrast, in germinal centres within mature TLS, B cell clones are selectively activated and amplified, and undergo antibody class switching and somatic hypermutation. Subsequently, these B cell clones differentiate into plasma cells that can produce IgG or IgA antibodies targeting tumour-associated antigens. Meanwhile, these B cells may instruct CD8 + T cells by presenting them with tumour-derived antigens. Although the precise mechanism by which mTLSs control tumour growth and predict the response to ICB is not fully understood, it is established that plasma cells are generated in TLS germinal centres [98].
Response to ICB immunotherapy and survival were better predicted in multiple cancer types by the presence of B cells, plasma cells, and a mature TLS. In order to better understand the mechanism behind, Fridman group performed spatial transcriptomic profiling on 24 human ccRCC samples [99]. In TLS+ tumours, B cell lineage genes representing all stages of development were found preferentially expressed in “hot spots” within the TLS, indicating that the maturation of B cells and PCs occurs in the TLS. Additionally, they saw the higher number of different B cell clonotypes in TLS+ tumours, further data from spatial B cell receptor repertoire profiling using Visium, reveals that the diversification, selection, and expansion of B cell clones occurred in the TLS. Importantly, expression of a PC-associated gene, MZB1, was high in TLS, low in the tumour area, but found in many spots at a distance from the TLS, and B cell repertoire analysis revealed the presence of fully mature clonotypes at distance which disseminated into the tumour beds along fibroblastic tracks.
Interestingly, the presence of mTLSs was found significantly associated with improved outcomes in tumours with a high infiltration of CD8 + T cells, whereas poor for patients with low infiltrated CD8+ T cells, regardless of the TLS status. Moreover, in ovarian cancer, antigen-experienced CD20+ B cells colocalised with activated CD8+ T cells, and the presence of both populations correlated with increased patient survival compared with the presence of CD8+ T cells alone [100]. This suggests that the presence CD8+ T cells is necessary but not sufficient to induce a sustained anti-tumour immune response, which indeed requires crucial cooperation with B cells [97]. TLSs presence in the TME is generally thought to reflect the capacity of a patient’s immune response to recognise and respond immunologically to their tumour as an anomaly. Together, these findings open new avenues for improving patient selection for immunotherapy given that the identification of mature TLS is readily feasible for standard pathology laboratories.
NK cells
Natural killer cells are important innate cytotoxic lymphocytes with a rapid and efficient capacity to recognise and kill tumour cells without prior sensitisation. According to the principle of “missing self” recognition, NK cells can eliminate targets that do not express sufficiently large numbers of MHC-I molecules [101]. The expression of these molecules is often lost during viral infection and neoplastic transformation, enabling the cells to escape CD8+ T cell immunosurveillance [102]. NK cells participate in immune responses to tumours by killing target cells and producing chemokines and cytokines, which also greatly impact the adaptive anti-tumour immune response. However, different tumour-related soluble factors (IL-10, IDO, PGE2, TGF-β1, etc.) produced by different tumour-infiltrating immune cells (M2 macrophages, MDSC, DC, Treg), may negatively affect NK cell activity [103].
Beside the direct killing, there exists another anti-tumour role of NK cells by modulating the immune response, the activation of the NK natural cytotoxic receptor 1 induces IFNγ production which, in turn modulates fibronectin 1 expression on tumour cells, preventing metastatic spread. The presence of NK cells within the TME may be associated with a good prognosis. Indeed, low NK-cell numbers are observed in primary head and neck squamous cell carcinoma tumour sections [104] and were associated with the insufficient tumour rejection. In a study of resectable oral squamous cell carcinoma, a higher intratumour infiltration with CD56+ cells was significantly correlated with locoregional disease control and improved survival (OR = 3.669, CI 95% 1.09–15.37, p = 0.035) [105]. Higher levels of NCR1 expression at the tumour site are associated with better survival. There is also a positive correlation between the infiltration of NK cells and CD8 + T cells into the TME and prolonged survival in patients with colorectal carcinoma [106]. In gastric and oesophageal cancers [107], the proportion of CD56dim NK cells infiltrating tumours gradually decreases with disease progression in patients. A NK gene expression signature shows strong survival effects for patients from a skin cutaneous melanoma cohort with either a high or moderate NK score. These survival effects are largely recapitulated across a selection of marker genes associated with: T cell infiltration (CD3D), cytokine signalling, which promotes NK cell and T cell infiltration (IL15), a shared component of the IL2 and IL15 receptors, interferon-gamma responsive checkpoint markers (CD274), NK secreted chemokines/cytokines associated with dendritic cell recruitment and stimulation (CCL5, XCL1) and cytotoxic effector molecules (GZMB, FASLG) [108].
The finding above indicating that NK cells play a role in the regulation of human tumours and highlight potential survival effects associated with increased NK cell activity. But the total number of NK cells infiltrating solid tumours including those considered “highly” infiltrated was relatively low compared with other immune populations, and several studies asserted that the number of NK cells was too low to pursue prognostication. Although the exact roles of infiltrating NK cells remain to be determined, that NK cells are competent “serial killers”, capable of polarising the TME, recruiting and activating additional effector cells suggests that their low frequency should not be interpreted as a lack of power or importance.
Conclusions and perspectives
The TME is heterogeneous, complex in molecular and immunological features, many immune subsets and factors greatly influence the outcome of immunotherapy and disease prognosis. As described above, the balance between immune evasion and anti-tumour activity, as well as a patient’s response, is greatly affected by the recruitment or accumulation of specific immune cells, which in turn influence the immune response. Increasing evidence shows that infiltrating immune cells such as T cells, B cells, macrophages, dendritic cells, monocytes, neutrophils, and mast cells can regulate cancer development and progression [12]. Tumour cells in the TME can invade surrounding tissues or metastasise through lymphatic or blood vessels, the infiltrated tumour cells can stimulate the host’s immune response, including releasing chemokines, cytokines and anibodies, regulating the expression of realted receptors, and other factors (Fig. 1), which directly or indirectly promote or inhibit tumour cell proliferation [4].

The lymphocytes play different roles by releasing specific molecules in TME, could form the organized lymphoid aggregates - Tertiary lymphoid structures.
The anti-tumourigenic function in the TME is not only regulated by lymphocytes such as T cells, B cells and NK cells discussed above; others like dendritic cells, macrophages and normal fibroblasts also play important roles. The pro-tumourigenic function is also mediated by other immune cells such as myeloid-derived suppressor cells, M2-tumour-associated macrophages (TAMs) and regulatory T cells, as well as carcinoma-associated fibroblasts (CAFs), adipocytes and endothelial cells [5]. In ovarian and endometrial cancer patients, the immunosuppressive factors specifically TGF-β, CD47 and TAMs play a significant role in predicting low survival outcome [109].
Effective immunotherapy requires the knowledge of the tumour microenvironment, especially the interplay among the various compartments in TME. However, measurement of immune heterogeneity and its impact on outcomes has been limited by the lack of standardised methods and suitable metrics. The complex interaction between tumour cells, immune cells and their longitudinal temporal evolution can give rise to spatial intratumour heterogeneity. In the attempt to address the need to capture cellular heterogeneity and detect rare immune subsets, approaches have advanced toward single-cell capabilities and high-dimensional analyses with new technologies such as single-cell RNA-Seq (scRNA-Seq), cytometry by time of flight (CyTOF) [110] and multiplex IHC (mIHC). Fluorescent in situ hybridisation (FISH) allows measurement of gene expression using fluorescent probes recognising RNAs linking transcription patterns with spatial organisation of single cell cells within the tissue.
Recent advances in computational power and the advent of whole-slide digitisation have led to several studies investigating quantitative approaches to interrogate the TME on high-resolution histology images. Research has focused on leveraging artificial intelligence and digital pathology approaches toward answering clinically relevant questions related to detection, diagnosis, prognosis, and treatment response. Researchers used an image analysis algorithm to capture morphologic attributes relating to the spatial interaction and architecture of tumour cells and TILs from digitised H&E images and presented a classifier called HistoTIL [111]. Deep learning approaches like handcrafted approaches have been shown to be able to predict underlying molecular signatures of the tumour and have also been shown to be associated with disease outcome and treatment response. Translating these novel technologies have the potential to revolutionise tumour immunopathology leading to altering our current understanding of cancer immunology and dramatically improving outcomes for patients.
Data availability
Not applicable.
References
Waldman AD, Fritz JM, Lenardo MJ. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020;20:651–68.
Esfahani K, Roudaia L, Buhlaiga N, Del Rincon SV, Papneja N, Miller WH Jr. A review of cancer immunotherapy: from the past, to the present, to the future. Curr Oncol. 2020;27:S87–S97.
Lim SM, Hong MH, Kim HR. Immunotherapy for non-small cell lung cancer: current landscape and future perspectives. Immune Netw. 2020;20:e10.
Anderson NM, Simon MC. The tumor microenvironment. Curr Biol. 2020;30:R921–R5.
Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther. 2021;6:72.
Lee S, Margolin K. Tumor-infiltrating lymphocytes in melanoma. Curr Oncol Rep. 2012;14:468–74.
Dieu-Nosjean MC, Goc J, Giraldo NA, Sautes-Fridman C, Fridman WH. Tertiary lymphoid structures in cancer and beyond. Trends Immunol. 2014;35:571–80.
Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends Cancer. 2016;2:747–57.
Bruno TC. New predictors for immunotherapy responses sharpen our view of the tumour microenvironment. Nature. 2020;577:474–6.
Loi S, Michiels S, Adams S, Loibl S, Budczies J, Denkert C, et al. The journey of tumor-infiltrating lymphocytes as a biomarker in breast cancer: clinical utility in an era of checkpoint inhibition. Ann Oncol. 2021;32:1236–44.
Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17:557–69.
Lin B, Du L, Li H, Zhu X, Cui L, Li X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed Pharmacother. 2020;132:110873.
Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139–45.
Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–33.
Bald T, Landsberg J, Lopez-Ramos D, Renn M, Glodde N, Jansen P, et al. Immune cell-poor melanomas benefit from PD-1 blockade after targeted type I IFN activation. Cancer Discov. 2014;4:674–87.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72.
Kalbasi A, June CH, Haas N, Vapiwala N. Radiation and immunotherapy: a synergistic combination. J Clin Investig. 2013;123:2756–63.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Investig. 2017;127:2930–40.
Benci JL, Johnson LR, Choa R, Xu Y, Qiu J, Zhou Z, et al. Opposing functions of interferon coordinate adaptive and innate immune responses to cancer immune checkpoint blockade. Cell. 2019;178:933.e14–48.e14.
Burnet FM. A modification of Jerne’s theory of antibody production using the concept of clonal selection. Aust J Sci. 1957;20:67–9.
Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171:934.e16–49.e16.
Bai R, Lv Z, Xu D, Cui J. Predictive biomarkers for cancer immunotherapy with immune checkpoint inhibitors. Biomark Res. 2020;8:34.
Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, et al. TCR repertoire diversity of peripheral PD-1(+)CD8(+) T cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol Res. 2020;8:146–54.
McLane LM, Abdel-Hakeem MS, Wherry EJ. CD8 T cell exhaustion during chronic viral infection and cancer. Annu Rev Immunol. 2019;37:457–95.
Lee JS, Ruppin E. Multiomics prediction of response rates to therapies to inhibit programmed cell death 1 and programmed cell death 1 ligand 1. JAMA Oncol. 2019;5:1614–8.
Bruni D, Angell HK, Galon J. The immune contexture and Immunoscore in cancer prognosis and therapeutic efficacy. Nat Rev Cancer. 2020;20:662–80.
Giraldo NA, Becht E, Pages F, Skliris G, Verkarre V, Vano Y, et al. Orchestration and prognostic significance of immune checkpoints in the microenvironment of primary and metastatic renal cell cancer. Clin Cancer Res. 2015;21:3031–40.
Remark R, Alifano M, Cremer I, Lupo A, Dieu-Nosjean MC, Riquet M, et al. Characteristics and clinical impacts of the immune environments in colorectal and renal cell carcinoma lung metastases: influence of tumor origin. Clin Cancer Res. 2013;19:4079–91.
Petitprez F, Fossati N, Vano Y, Freschi M, Becht E, Luciano R, et al. PD-L1 expression and CD8(+) T-cell infiltrate are associated with clinical progression in patients with node-positive prostate cancer. Eur Urol Focus. 2019;5:192–6.
Nakano O, Sato M, Naito Y, Suzuki K, Orikasa S, Aizawa M, et al. Proliferative activity of intratumoral CD8(+) T-lymphocytes as a prognostic factor in human renal cell carcinoma: clinicopathologic demonstration of antitumor immunity. Cancer Res. 2001;61:5132–6.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4.
Pages F, Mlecnik B, Marliot F, Bindea G, Ou FS, Bifulco C, et al. International validation of the consensus Immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128–39.
Mlecnik B, Tosolini M, Kirilovsky A, Berger A, Bindea G, Meatchi T, et al. Histopathologic-based prognostic factors of colorectal cancers are associated with the state of the local immune reaction. J Clin Oncol. 2011;29:610–8.
Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T cell states associated with response to checkpoint immunotherapy in melanoma. Cell. 2019;176:404.
Song Y, Li Z, Xue W, Zhang M. Predictive biomarkers for PD-1 and PD-L1 immune checkpoint blockade therapy. Immunotherapy. 2019;11:515–29.
Di Pilato M, Kfuri-Rubens R, Pruessmann JN, Ozga AJ, Messemaker M, Cadilha BL, et al. CXCR6 positions cytotoxic T cells to receive critical survival signals in the tumor microenvironment. Cell. 2021;184:4512.e22–30.e22.
Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–96.
Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard-Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775.e18–89.e18.
Puram SV, Tirosh I, Parikh AS, Patel AP, Yizhak K, Gillespie S, et al. Single-cell transcriptomic analysis of primary and metastatic tumor ecosystems in head and neck. Cancer Cell. 2017;171:1611.e24–24.e24.
Zhang L, Yu X, Zheng L, Zhang Y, Li Y, Fang Q, et al. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature. 2018;564:268–72.
Zheng C, Zheng L, Yoo JK, Guo H, Zhang Y, Guo X, et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell. 2017;169:1342.e16–56.e16.
Guo X, Zhang Y, Zheng L, Zheng C, Song J, Zhang Q, et al. Global characterization of T cells in non-small-cell lung cancer by single-cell sequencing. Nat Med. 2018;24:978–85.
van der Leun AM, Thommen DS, Schumacher TN. CD8(+) T cell states in human cancer: insights from single-cell analysis. Nat Rev Cancer. 2020;20:218–32.
Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med. 2019;216:2128–49.
Schreck S, Friebel D, Buettner M, Distel L, Grabenbauer G, Young LS, et al. Prognostic impact of tumour-infiltrating Th2 and regulatory T cells in classical Hodgkin lymphoma. Hematol Oncol. 2009;27:31–9.
Wang JT, Li H, Zhang H, Chen YF, Cao YF, Li RC, et al. Intratumoral IL17-producing cells infiltration correlate with antitumor immune contexture and improved response to adjuvant chemotherapy in gastric cancer. Ann Oncol. 2019;30:266–73.
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, et al. Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood. 2009;114:1141–9.
Punt S, Fleuren GJ, Kritikou E, Lubberts E, Trimbos JB, Jordanova ES, et al. Angels and demons: Th17 cells represent a beneficial response, while neutrophil IL-17 is associated with poor prognosis in squamous cervical cancer. Oncoimmunology. 2015;4:e984539.
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–71.
Chen X, Wan J, Liu J, Xie W, Diao X, Xu J, et al. Increased IL-17-producing cells correlate with poor survival and lymphangiogenesis in NSCLC patients. Lung Cancer. 2010;69:348–54.
Fabre J, Giustiniani J, Garbar C, Antonicelli F, Merrouche Y, Bensussan A, et al. Targeting the tumor microenvironment: the protumor effects of IL-17 related to cancer type. Int J Mol Sci. 2016;17:1433.
Amicarella F, Muraro MG, Hirt C, Cremonesi E, Padovan E, Mele V, et al. Dual role of tumour-infiltrating T helper 17 cells in human colorectal cancer. Gut. 2017;66:692–704.
Sautes-Fridman C, Petitprez F, Calderaro J, Fridman WH. Tertiary lymphoid structures in the era of cancer immunotherapy. Nat Rev Cancer. 2019;19:307–25.
Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–95.
Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Investig. 2013;123:2873–92.
Schmetterer KG, Neunkirchner A, Pickl WF. Naturally occurring regulatory T cells: markers, mechanisms, and manipulation. FASEB J. 2012;26:2253–76.
Whiteside SK, Grant FM, Gyori DS, Conti AG, Imianowski CJ, Kuo P, et al. CCR8 marks highly suppressive Treg cells within tumours but is dispensable for their accumulation and suppressive function. Immunology. 2021;163:512–20.
Overacre-Delgoffe AE, Vignali DAA. Treg fragility: a prerequisite for effective antitumor immunity? Cancer Immunol Res. 2018;6:882–7.
Onizuka S, Tawara I, Shimizu J, Sakaguchi S, Fujita T, Nakayama E. Tumor rejection by in vivo administration of anti-CD25 (interleukin-2 receptor alpha) monoclonal antibody. Cancer Res. 1999;59:3128–33.
Dannull J, Su Z, Rizzieri D, Yang BK, Coleman D, Yancey D, et al. Enhancement of vaccine-mediated antitumor immunity in cancer patients after depletion of regulatory T cells. J Clin Investig. 2005;115:3623–33.
Li JF, Chu YW, Wang GM, Zhu TY, Rong RM, Hou J, et al. The prognostic value of peritumoral regulatory T cells and its correlation with intratumoral cyclooxygenase-2 expression in clear cell renal cell carcinoma. BJU Int. 2009;103:399–405.
Shah W, Yan X, Jing L, Zhou Y, Chen H, Wang Y. A reversed CD4/CD8 ratio of tumor-infiltrating lymphocytes and a high percentage of CD4(+)FOXP3(+) regulatory T cells are significantly associated with clinical outcome in squamous cell carcinoma of the cervix. Cell Mol Immunol. 2011;8:59–66.
Grabenbauer GG, Lahmer G, Distel L, Niedobitek G. Tumor-infiltrating cytotoxic T cells but not regulatory T cells predict outcome in anal squamous cell carcinoma. Clin Cancer Res. 2006;12:3355–60.
Heimberger AB, Abou-Ghazal M, Reina-Ortiz C, Yang DS, Sun W, Qiao W, et al. Incidence and prognostic impact of FoxP3+ regulatory T cells in human gliomas. Clin Cancer Res. 2008;14:5166–72.
Badoual C, Hans S, Rodriguez J, Peyrard S, Klein C, Agueznay Nel H, et al. Prognostic value of tumor-infiltrating CD4+ T-cell subpopulations in head and neck cancers. Clin Cancer Res. 2006;12:465–72.
Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High frequency of tumor-infiltrating FOXP3(+) regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer. 2010;126:2635–43.
Kim KH, Hur JY, Koh J, Cho J, Ku BM, Koh JY, et al. Immunological characteristics of hyperprogressive disease in patients with non-small cell lung cancer treated with anti-PD-1/PD-L1 Abs. Immune Netw. 2020;20:e48.
Kumagai S, Togashi Y, Kamada T, Sugiyama E, Nishinakamura H, Takeuchi Y, et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat Immunol. 2020;21:1346–58.
Savas P, Virassamy B, Ye C, Salim A, Mintoff CP, Caramia F, et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat Med. 2018;24:986–93.
Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade. Nat Med. 2019;25:1251–9.
Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. 2018;24:1277–89.
Sarvaria A, Madrigal JA, Saudemont A. B cell regulation in cancer and anti-tumor immunity. Cell Mol Immunol. 2017;14:662–74.
Largeot A, Pagano G, Gonder S, Moussay E, Paggetti J. The B-side of cancer immunity: the underrated tune. Cells. 2019;8:449.
Leong TL, Bryant VL. B cells in lung cancer-not just a bystander cell: a literature review. Transl Lung Cancer Res. 2021;10:2830–41.
Kinker GS, Vitiello GAF, Ferreira WAS, Chaves AS, Cordeiro de Lima VC, Medina TDS. B cell orchestration of anti-tumor immune responses: a matter of cell localization and communication. Front Cell Dev Biol. 2021;9:678127.
Cabrita R, Lauss M, Sanna A, Donia M, Skaarup Larsen M, Mitra S, et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature. 2020;577:561–5.
Griss J, Bauer W, Wagner C, Simon M, Chen M, Grabmeier-Pfistershammer K, et al. B cells sustain inflammation and predict response to immune checkpoint blockade in human melanoma. Nat Commun. 2019;10:4186.
Selitsky SR, Mose LE, Smith CC, Chai S, Hoadley KA, Dittmer DP, et al. Prognostic value of B cells in cutaneous melanoma. Genome Med. 2019;11:36.
Harris RJ, Cheung A, Ng JCF, Laddach R, Chenoweth AM, Crescioli S, et al. Tumor-infiltrating B lymphocyte profiling identifies IgG-biased, clonally expanded prognostic phenotypes in triple-negative breast cancer. Cancer Res. 2021;81:4290–304.
Edin S, Kaprio T, Hagstrom J, Larsson P, Mustonen H, Bockelman C, et al. The prognostic importance of CD20(+) B lymphocytes in colorectal cancer and the relation to other immune cell subsets. Sci Rep. 2019;9:19997.
Xia J, Xie Z, Niu G, Lu Z, Wang Z, Xing Y, et al. Single-cell landscape and clinical outcomes of infiltrating B cells in colorectal cancer. Immunology. 2022;168:135–51.
Berntsson J, Nodin B, Eberhard J, Jirstrom K. Prognostic impact of tumor-associated B cells and plasma cells in colorectal cancer. J Clin Oncol. 2016;34:587.
Gottlin EB, Bentley RC, Campa MJ, Pisetsky DS, Herndon JE 2nd, Patz EF Jr. The association of intratumoral germinal centers with early-stage non-small cell lung cancer. J Thorac Oncol. 2011;6:1687–90.
Germain C, Devi-Marulkar P, Knockaert S, Biton J, Kaplon H, Letaief L, et al. Tertiary lymphoid structure-B cells narrow regulatory T cells impact in lung cancer patients. Front Immunol. 2021;12:626776.
Hui Z, Zhang J, Ren Y, Li X, Yan C, Yu W, et al. Single-cell profiling of immune cells after neoadjuvant pembrolizumab and chemotherapy in IIIA non-small cell lung cancer (NSCLC). Cell Death Dis. 2022;13:607.
Hao D, Han G, Sinjab A, Gomez-Bolanos LI, Lazcano R, Serrano A, et al. The single-cell immunogenomic landscape of B and plasma cells in early-stage lung adenocarcinoma. Cancer Discov. 2022;12:2626–45.
Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol. 2019;16:6–18.
Rosser EC, Mauri C. Regulatory B cells: origin, phenotype, and function. Immunity. 2015;42:607–12.
Zhang Y, Gallastegui N, Rosenblatt JD. Regulatory B cells in anti-tumor immunity. Int Immunol. 2015;27:521–30.
Wei X, Jin Y, Tian Y, Zhang H, Wu J, Lu W, et al. Regulatory B cells contribute to the impaired antitumor immunity in ovarian cancer patients. Tumour Biol. 2016;37:6581–8.
Zhou X, Su YX, Lao XM, Liang YJ, Liao GQ. CD19(+)IL-10(+) regulatory B cells affect survival of tongue squamous cell carcinoma patients and induce resting CD4(+) T cells to CD4(+)Foxp3(+) regulatory T cells. Oral Oncol. 2016;53:27–35.
Zhang C, Xin H, Zhang W, Yazaki PJ, Zhang Z, Le K, et al. CD5 binds to interleukin-6 and induces a feed-forward loop with the transcription factor STAT3 in B cells to promote cancer. Immunity. 2016;44:913–23.
Engelhard VH, Rodriguez AB, Mauldin IS, Woods AN, Peske JD, Slingluff CL Jr. Immune cell infiltration and tertiary lymphoid structures as determinants of antitumor immunity. J Immunol. 2018;200:432–42.
Petitprez F, de Reynies A, Keung EZ, Chen TW, Sun CM, Calderaro J, et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature. 2020;577:556–60.
Schumacher TN, Thommen DS. Tertiary lymphoid structures in cancer. Science. 2022;375:eabf9419.
Vanhersecke L, Brunet M, Guegan JP, Rey C, Bougouin A, Cousin S, et al. Mature tertiary lymphoid structures predict immune checkpoint inhibitor efficacy in solid tumors independently of PD-L1 expression. Nat Cancer. 2021;2:794–802.
Vaghjiani RG, Skitzki JJ. Tertiary lymphoid structures as mediators of immunotherapy response. Cancers. 2022;14:3748.
Meylan M, Petitprez F, Becht E, Bougouin A, Pupier G, Calvez A, et al. Tertiary lymphoid structures generate and propagate anti-tumor antibody-producing plasma cells in renal cell cancer. Immunity. 2022;55:527.e5–41.e5.
Nielsen JS, Sahota RA, Milne K, Kost SE, Nesslinger NJ, Watson PH, et al. CD20+ tumor-infiltrating lymphocytes have an atypical CD27- memory phenotype and together with CD8+ T cells promote favorable prognosis in ovarian cancer. Clin Cancer Res. 2012;18:3281–92.
Ljunggren HG, Karre K. In search of the ‘missing self’: MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–44.
Garrido F. MHC/HLA class I loss in cancer cells. Adv Exp Med Biol. 2019;1151:15–78.
Cozar B, Greppi M, Carpentier S, Narni-Mancinelli E, Chiossone L, Vivier E. Tumor-infiltrating natural killer cells. Cancer Discov. 2021;11:34–44.
Weil S, Memmer S, Lechner A, Huppert V, Giannattasio A, Becker T, et al. Natural killer group 2D ligand depletion reconstitutes natural killer cell immunosurveillance of head and neck squamous cell carcinoma. Front Immunol. 2017;8:387.
Caruntu A, Moraru L, Lupu M, Vasilescu F, Dumitrescu M, Cioplea M, et al. Prognostic potential of tumor-infiltrating immune cells in resectable oral squamous cell carcinoma. Cancers. 2021;13:2268.
Sconocchia G, Eppenberger S, Spagnoli GC, Tornillo L, Droeser R, Caratelli S, et al. NK cells and T cells cooperate during the clinical course of colorectal cancer. Oncoimmunology. 2014;3:e952197.
Izawa S, Kono K, Mimura K, Kawaguchi Y, Watanabe M, Maruyama T, et al. H(2)O(2) production within tumor microenvironment inversely correlated with infiltration of CD56(dim) NK cells in gastric and esophageal cancer: possible mechanisms of NK cell dysfunction. Cancer Immunol Immunother. 2011;60:1801–10.
Cursons J, Souza-Fonseca-Guimaraes F, Foroutan M, Anderson A, Hollande F, Hediyeh-Zadeh S, et al. A gene signature predicting natural killer cell infiltration and improved survival in melanoma patients. Cancer Immunol Res. 2019;7:1162–74.
Ni Y, Soliman A, Joehlin-Price A, Abdul-Karim F, Rose PG, Mahdi H. Immune cells and signatures characterize tumor microenvironment and predict outcome in ovarian and endometrial cancers. Immunotherapy. 2021;13:1179–92.
Giesen C, Wang HA, Schapiro D, Zivanovic N, Jacobs A, Hattendorf B, et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat Methods. 2014;11:417–22.
Wang X, Barrera C, Bera K, Viswanathan VS, Azarianpour-Esfahani S, Koyuncu C, et al. Spatial interplay patterns of cancer nuclei and tumor-infiltrating lymphocytes (TILs) predict clinical benefit for immune checkpoint inhibitors. Sci Adv. 2022;8:eabn3966.
Funding
Not applicable.
Author information
Authors and Affiliations
Contributions
Yarong Liu, Yanbin Liu and Zhenjiang Liu conceptualised the review. Yanbin Liu and Zhenjiang Liu contributed to writing early drafts of the paper. Jun Cui, Jingwei Sun and Yixiao Yang made substantial contributions to discussion of content of the review and edited the manuscript before submission. Yarong Liu, Jun Cui, Jingwei Sun, Yanbin Liu, Zhenjiang Liu and Yixiao Yang revised and approved the final version.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Y., Liu, Z., Yang, Y. et al. The prognostic and biology of tumour-infiltrating lymphocytes in the immunotherapy of cancer. Br J Cancer 129, 1041–1049 (2023). https://doi.org/10.1038/s41416-023-02321-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-023-02321-y
- Springer Nature Limited