
Abstract
Adoptive cell therapy using tumor-infiltrating lymphocytes (TIL) is arguably the most effective treatment for patients with metastatic melanoma. With higher response rates than ipilimumab or IL-2, and longer durations of response than vemurafenib, TIL therapy carries the potential to transform current outcomes in melanoma, while also defining the way cell-based immunotherapy gets incorporated into mainstream cancer treatment. This paper will review the current state of TIL therapy in melanoma, the strategies to improve its efficacy, the current obstacles, and future directions to expand the availability of TIL to the general patient population.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Melanoma has long been recognized as one of the most immunogenic of all cancers. Of all tumor types, melanoma has provided the most fertile ground for the development of immunotherapeutic strategies. It was in melanoma that we first learned it was possible to achieve tumor regression solely by activating the immune system via interleukin-2, a T-cell growth factor [1, 2]. More recently, it has been in melanoma that the unprecedented therapeutic potential of T-cell checkpoint blockade has been realized, with durable responses to ipilimumab now lasting 8 years and beyond [3].
At the other end of the immunotherapy spectrum from IL-2 and ipilimumab lies adoptive cell transfer (ACT), a more technical approach in which the patient’s autologous T cells are expanded, manipulated ex vivo, and then re-infused into the patient to exert an anti-tumor response. Over the past 10 to 15 years, investigators have pursued different modalities of ACT: tumor-infiltrating lymphocytes (TIL), antigen-specific autologous T-cell clones, donor anti-tumor lymphocytes, and more recently, genetically engineered human lymphocytes. Of these, TIL therapy has been most thoroughly studied and has demonstrated the most consistently favorable results, supporting its viability as a mainstream treatment for metastatic melanoma.
Although currently less-heralded and much less accessible than ipilimumab, TIL therapy is arguably more successful, with response rates of over 50 % and durable complete response rates of 20 % in patients with metastatic melanoma who have failed other therapies [4••]. By comparison, ipilimumab offers long-term disease control for approximately 20 % of patients, but complete responses are rare. High-dose IL-2 provides complete responses in only 6 %–7 % of patients [5]. Standard chemotherapy induces objective responses in less than 20 %, rarely leads to CR, and benefits last only months. Thus, the need is urgent to expand the accessibility of TIL therapy so that it becomes a treatment option available to all metastatic melanoma patients.
Background
Early investigation into the use of immune cells to inhibit cancer growth was inspired in the 1950s by studies showing solid transplant rejection was mediated by cellular immunity [6]. Animal studies over the next decade established that lymphocytes from immunized donors could be transferred to mediate tumor regression in syngeneic recipients, and that IL-2 could be used to expand these lymphocytes [7–9]. Donohue et al took this further by demonstrating that the concurrent administration of IL-2 in vivo enhanced the anti-tumor efficacy of lymphocytes in a murine model [10]. However, the need for an immunized syngeneic donor as a source for anti-tumor lymphocytes remained an inherent obstacle to the translation of this work in humans, who lack such a source.
In 1986, this barrier was surmounted when Rosenberg and colleagues, at the Surgery Branch of the NIH, pioneered the use of tumor-infiltrating lymphocytes in mice and demonstrated that the combination of autologous TIL and cyclophosphamide could induce regression of metastases [11]. This was followed quickly with their landmark publication in 1988 of the first human study that showed TIL could induce cancer regression when administered to patients with metastatic melanoma [12]. An analysis of 86 melanoma patients treated with TIL followed by high-dose IL-2 at the NIH from 1987 to 1992 demonstrated a 34 % overall response rate, with similar activity in patients with prior IL-2 exposure or no prior IL-2 [13]. Five patients (6 %) had a complete response but only 2 were durable at 21 and 46 months. Since then, there has been considerable effort at the NIH and other institutions towards improving these results through modifications to the TIL generation and selection methodologies, as well as changes in the preparative regimens given prior to TIL.
How TIL Are Generated
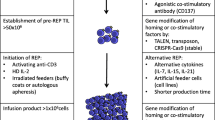
The generation of TIL was pioneered by the Surgery Branch at the NIH (Fig. 1) [14, 15]. A tumor ideally 2 cm in diameter is first harvested by excisional biopsy and then dissected into fragments (approximately 1–3 mm3) and placed in wells with media. IL-2 is added and over the following weeks, the TIL proliferate while the adherent tumor cells disappear as they die off, or are killed by the lymphocytes. After 2–4 weeks of growth, the TIL are tested for T-cell phenotype by FACS and for reactivity to autologous tumor cells. The wells containing cells with the highest cytokine-release after co-culture with autologous tumor cells are selected and expanded for 2 weeks using a rapid expansion protocol with agonistic concentrations of anti-CD3 antibody, IL-2 and irradiated, allogeneic feeder cells derived from normal donor pools of peripheral blood mononuclear cells. The total generation time from start to finish is approximately 5–6 weeks. Currently TIL can be generated successfully from 60 % to >90 % of melanoma tumor samples, at different institutions [15–18].

Generation of tumor-infiltrating lymphocytes. Methodologies for generating tumor-infiltrating lymphocytes were pioneered by the Surgery Branch of the NCI/NIH. Tumor is first harvested from the patient, dissected into fragments, and then cultured with IL-2 for 2–4 weeks until the lymphocytes reach 30–50 million in number. After lymphocytes are tested against tumor cells, they undergo a 2-week rapid expansion to achieve a goal of 50 billion cells, which are then infused into the patient
Host Factors Affecting TIL Generation
In 2011, Radvanyi and colleagues at MD Anderson performed an analysis of TIL growth in 226 consecutive patients with melanoma to determine what factors could predict successful TIL generation [16]. They found that younger patients and female patients carried a higher success rate for TIL generation. The TIL generation rate was 94 % for patients less than 30 years old compared with a 46 % success rate for patients over 60. Female patients had a significantly higher rate at 71 % vs 57 % for men. Patients who received systemic therapy less than 30 days before tumor harvest had a success rate of 47 % compared with a 66 % rate when the last therapy occurred greater than 90 days before harvest. Interestingly, exposure to prior immunotherapies such as ipilimumab or IL-2, even when given within 30 days of tumor harvest, did not increase the TIL yield.
Emerging Role of BRAF Inhibition in TIL Generation and Combination Treatment
It remains to be seen whether BRAF inhibition has an effect on TIL generation; the MD Anderson analysis described above did not have enough patients who received prior targeted therapy to reach statistical significance. This is a particularly interesting question because recently published in vitro work has shown that BRAF inhibition can increase the expression of melanoma differentiation antigens which, in turn, improves recognition by antigen-specific lymphocytes [19•]. This has fueled a great deal of excitement about the possibility of combination therapies with BRAF inhibition and immunotherapy.
In 2012, an examination of patient biopsies at the Melanoma Institute in Australia before and after BRAF inhibition revealed that the numbers of CD4+ and CD8+ lymphocytes infiltrating the tumors increased significantly 7 days after BRAF inhibition, and the increase in CD8+ lymphocytes correlated with tumor regression in patients [20]. Ribas and colleagues very recently published compelling murine data to support the superior efficacy of combination BRAF inhibition and adoptive cell therapy, using transgenic, antigen-specific T cell clones. This group found that BRAF inhibition increased cytotoxic activity and cytokine secretion by the transferred T cells, without affecting their expansion and trafficking to the tumor site [21]. The first protocol investigating the combination of BRAF inhibition and TIL was opened at the NIH this year using vemurafenib, the FDA-approved BRAF inhibitor, prior to TIL therapy (study identifier NCT01585415; clinicaltrials.gov).
Some TIL Are Better Than Other TIL
TIL represent a heterogeneous population of lymphocytes that are growing within a tumor. This population as a whole is evidently ineffective in eradicating the tumor at that site, for complex reasons that likely include inadequate numbers of anti-tumor cells, anti-tumor cells that have become senescent or anergic, and high numbers of immunosuppressive cells, such as regulatory T cells. Local secreted factors like indole dioxygenase and arginase derived from immature dendritic cells and myeloid-derived suppressor cells in the tumor microenvironment also contribute [22]. The rationale of TIL therapy is that the anti-tumor immune response can be enhanced by removing cells with anti-tumor potential from the immunosuppressive tumor microenvironment to a setting where they can be expanded in vitro and then returned in high enough numbers that allow them to traffic to tumor sites and kill tumor targets and possibly other cell targets that sustain the tumor, such as vascular endothelial cells. Thus, an important goal of TIL investigation has been the identification of the ideal subtype within the initial mixed TIL outgrowth to expand preferentially and infuse back into the patient.
Over recent years, our understanding of T cell differentiation has led to a model in which, upon antigenic stimulation, T cells develop through progressive stages of differentiation from the naïve CD8+ T cells with high proliferative capacity to the terminally differentiated effector memory cell with very little proliferative or self-renewal capacity [23–25]. Animal and clinical studies over the last 5 years demonstrated that TIL at an earlier differentiation stage, with longer telomeres, and a younger central memory phenotype (CD62L+, CD27+, CD28+), are associated with longer in vivo persistence, and superior clinical response [26–28].
This led to the development by Dudley et al in 2010 of a streamlined “Young TIL” protocol that pools lymphocytes from multiple tumor fragments, without additional selection, to obtain the cell number needed prior to rapid expansion, shortening the time compared with that of growing cells from individual microcultures to sufficient numbers [15]. By minimizing the time in culture, this new method enriched for TIL with an earlier differentiation stage with higher expression of CD27 and CD28 and longer telomeres. Svane and colleagues in Denmark performed a side-by-side comparison of ‘standard’ TIL and ‘young’ TIL in 2011 and also found a higher expression of CD27 and longer telomeres [29]. Their average time to establishment of TIL culture, prior to the 14-day rapid expansion step, was approximately 25 days for ‘young’ TIL vs 45 days for standard TIL. This faster production time also carries significant clinical impact by decreasing the numbers of patients who become ineligible for treatment due to rapid disease progression and clinical decline during the period of TIL generation.
Weber and colleagues at Moffitt Cancer Center are currently investigating how TIL generation can be enhanced by targeting 4-1BB, a co-stimulatory molecule on activated T cells involved in T-cell proliferation and antigen-specific cytolytic activity [30]. They recently presented compelling in vitro data at the 2012 American Society of Clinical Oncology (ASCO) annual conference in which an agonistic 4-1BB antibody was added to the initial culture step of the melanoma tumor fragments, along with standard IL-2. 4-1BB agonism yielded a TIL product with a higher CD8+ population, lower T regulatory cell numbers, and higher markers of cytolytic function. Further studies will determine whether this modification results in the enrichment of the TIL product with a more terminally differentiated T cell proliferation that has less replicative potential.
Their most impressive finding was a dramatic enhancement of TIL proliferation that led to 4.3 × 107 cells generated under 4-1BB agonism vs 2.3 × 107 cells with isotype control. The kinetics of proliferation were increased as well and the 4-1BB treated fragments reached the threshold cell number of 3 × 107 required for rapid expansion in less than 11 days, which would allow an overall TIL generation time of 25 days only. This group is currently starting a clinical trial for melanoma patients using this method for TIL generation. If these favorable data are reproduced in a clinical trial that also confirms the safety and efficacy of the TIL product, this could dramatically increase the numbers of patients eligible for TIL, by making it a more viable option for patients who are experiencing disease progression and can’t wait months for treatment.
Another area of interest is whether the use of other T-cell-supporting cytokines such as IL-15 and IL-21 with or without IL-2 during the TIL generation would lead to a more effective T cell population than IL-2 alone in this step. Unlike IL-2, which promotes the differentiation of effector T cells and the proliferation of Treg cells as well as supporting T cell activation-induced cell death (AICD), IL-15 and IL-21 seem to induce a younger, less differentiated, central memory phenotype, and do not promote AICD [31, 32].
The Importance of Lymphodepletion
Perhaps the most significant advancement in the field of TIL therapy since its inception has been the addition of a lymphodepleting conditioning regimen. The creation of a lymphopenic environment prior to TIL infusion is believed to enhance TIL proliferation and activity by reducing the numbers of immunosuppressive regulatory T cells and myeloid derived suppressor cells that are otherwise promoted by many factors associated with tumor growth, such as TGF-β and apoptosis-inducing receptor-ligand interactions [33, 34]. It is also widely believed that the elimination of other lymphocytes decreases the competition for homeostatic cytokines IL-7 and IL-15 [35], providing both physical and biologic “space” for TIL and other potential effectors such as NK cells to proliferate, and survive. Total body irradiation contributes to lymphodepletion but also appears to increase the function of antigen-presenting cells by activating the innate immune system, in part due to bacterial translocation from gut mucosal damage, which provides activation signals to antigen-presenting cells through their toll-like receptors [36].
In a compelling 2008 analysis, Dudley et al presented the results of 3 sequential trials with increasing intensities of myeloablation prior to TIL infusion [37]. In the first trial, a nonmyeloablative regimen was administered using cyclophosphamide (60 mg/kg for 2 days) and fludarabine (25 mg/m2 for 5 days) followed by TIL infusion and high-dose IL-2. The two subsequent TIL trials gave cyclophosphamide and fludarabine at the same doses as before, but with the addition of 2 Gy TBI for the second trial and 12 Gy TBI for the third trial. Although the trials were conducted sequentially, limiting the reliability of a comparison, they reported that the overall response rates and complete response rates were progressively higher with the greater intensity of lympho- and myeloablation. For the no-TBI, 2 Gy-TBI, and 12 Gy-TBI trials, the objective response rate was 49 %, 52 % and 72 %, respectively, and complete response rate was 12 %, 20 %, and 40 %, respectively [4••]. All but 1 of the CRs have continued beyond 3 years.
While the response rates are impressive, the myeloablative regimens carried significant toxicity. The latter 2 trials required hematopoietic rescue with autologous stem cells after TIL infusion due to the intensity of the myeloablative regimen. There was 1 treatment-related death out of 93 patients and 1 patient with prolonged pulmonary hypertension; both were on the 2 Gy-TBI trial. Five patients on the 12 Gy-TBI arm developed long-standing microangiopathic nephropathy, but interestingly, all 5 patients also achieved CRs and recovered from their renal failure. Because the apparent superiority of the 12 Gy-TBI regimen may be biased by differences in patient population and other factors, a prospective randomized trial is ongoing in which patients will receive identical TIL cell and conditioning regimens, but half will also receive TBI with autologous stem cell support (study identifier NCT01319565, clinicaltrials.gov).
Uncertain Role of Adjuvant Interleukin-2
The inclusion of adjuvant high-dose IL-2 in the first TIL trial was based on animal studies that demonstrated the in vivo administration of IL-2 enhanced the anti-tumor efficacy of transferred lymphocytes [10]. The use of IL-2 is problematic because IL-2 also exerts very potent proliferative effects on regulatory T cells, which may contribute to the poor response from TIL experienced by many patients [38]. Whether high-dose i.v. IL-2 has an advantage over low-dose subcutaneous IL-2 for TIL cell support has not been evaluated. Yee et al examined the effect of low-dose, subcutaneous IL-2 on the in vivo survival of transferred T cells clones, and found the administration of low-dose IL-2 for 2 weeks following cell infusion increased the median duration of in vivo persistence of the T cell clones from 7 days without IL-2 to 17 days with IL-2 [39]. TIL may prove to be less dependent on IL-2 than CD8+ T cell clones, since TIL are comprised of a heterogenous population that includes CD4+ cells, which are the primary cellular source of IL-2. Also under investigation is whether IL-2 could eventually be replaced by other homeostatic cytokines such as IL-7 or IL-15, which demonstrate a greater selectivity for expanding CD8+ T cell populations over regulatory T cells [40].
Feasibility of TIL as a Mainstream Treatment Option
The clinical benefits of TIL in metastatic melanoma are fairly well established. The clinical limitations that will characterize how it is used as a treatment option include the 5–6 week period currently required for TIL generation, a wait time that is excessive for many patients with aggressive metastatic melanoma. Also, the intense myeloablative conditioning regimen associated with the highest response rates would exclude a significant number of patients who are not fit enough to tolerate the regimen-related toxicities and intense support required to complete the therapy safely [4••]. Thus, as TIL therapy becomes more available to the general unselected melanoma patient population, the question of how much clinical benefit is gained from each level of lymphodepletion becomes increasingly important.
A societal limitation of this treatment is the expense of TIL generation and associated costs of inpatient care for the preparative lymphodepletion and high-dose IL-2. However, TIL generation will likely become less expensive if production is scaled for higher numbers of patients than the small groups typically treated on clinical trials. Also, the high cost of TIL production, estimated in the tens of thousands of dollars, needs to be viewed in the light of other therapeutic options for advanced melanoma, including recombinant fully human antibodies that currently cost in excess of $100,000 for a course of therapy as approved by the US FDA. The reported higher complete response rates from TIL over all other treatment options also suggest that TIL from a tumor harvested prior to some other cytoreductive therapy followed by infusion of the TIL could lead to cures in a substantial fraction of melanoma patients. Given in this fashion, the schedule and “dose” of consolidative TIL therapy would be limited, further lowering the costs and dramatically increasing the benefit to cost ratio.
The logistical barrier to TIL therapy becoming a widely available treatment option is the technical requirements of TIL generation, which is currently only performed in a limited number of academic institutions. One possibility is for TIL therapy to follow the path of hematopoietic stem cell transplantation, which can be performed at multiple institutions in major U.S. cities, and at sites around the world; patients can travel to these sites for treatment, and then return home for follow-up care. This model to obtain specialized treatment is feasible for many patients because, like TIL therapy, the transplant is a one-time treatment package with curative intent, making the need for travel and housing temporary. Another proposal by the CTEP subcommittee on adoptive cell therapy is the development of a centralized TIL growth facility that would receive the tumor, grow the TIL and ship the expanded cells out to the collaborating institutions [41].
Conclusion
Metastatic melanoma is generally regarded to have a bleak prognosis. It is notorious for affecting young men and women in their 30s and 40s, and thus, the loss of productive years is one of the most significant among cancers [42]. Its incidence is also rising at a rate greater than almost any other cancer [43]. Both ipilimumab and vemurafenib represent important advances in the treatment of melanoma, yet the limited benefit of CTLA4 blockade, and the short-lived response duration of BRAF inhibitors, whose use is limited to patients whose tumors carry a BRAF V600 mutation, argue that neither will change the clinical outcome for the majority of melanoma patients in a meaningful way.
In contrast, TIL therapy has consistently demonstrated response rates 50 % and higher and impressive durable complete response rates of greater than 20 %, but has been available to only a small, selected group of patients in clinical trials at a handful of institutions. At a time when cancer treatment is becoming more personalized and personalized treatments are becoming more cost-effective for the general public, the feasibility of translating TIL therapy into an accessible, mainstream cancer treatment has never been greater. TIL therapy carries the potential to transform the state of melanoma care, while leading the way for future adoptive cellular therapy in many other cancers as well.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE, et al. Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med. 1985;313:1485–92.
Lotze MT, Chang AE, Seipp CA, Simpson C, Vetto JT, Rosenberg SA. High-dose recombinant interleukin 2 in the treatment of patients with disseminated cancer. Responses, treatment-related morbidity, and histologic findings. JAMA. 1986;256:3117–24.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23.
•• Rosenberg SA, Yang JC, Sherry RM, Kammula US, Hughes MS, Phan GQ, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–7. This paper presents the most comprehensive long-term data on clinical responses in patients treated with TIL.
Rosenberg SA, Yang JC, White DE, Steinberg SM. Durability of complete responses in patients with metastatic cancer treated with high-dose interleukin-2: identification of the antigens mediating response. Ann Surg. 1998;228:307–19.
Mitchison NA. Studies on the immunological response to foreign tumor transplants in the mouse. I. The role of lymph node cells in conferring immunity by adoptive transfer. J Exp Med. 1955;102:157–77.
Fefer A. Immunotherapy and chemotherapy of Moloney sarcoma virus-induced tumors in mice. Cancer Res. 1969;29:2177–83.
Eberlein TJ, Rosenstein M, Rosenberg SA. Regression of a disseminated syngeneic solid tumor by systemic transfer of lymphoid cells expanded in interleukin 2. J Exp Med. 1982;156:385–97.
Cheever MA, Kempf RA, Fefer A. Tumor neutralization, immunotherapy, and chemoimmmunotherapy of a Friend leukemia with cells secondarily sensitized in vitro. J Immunol. 1977;119:714–8.
Donohue JH, Rosenstein M, Chang AE, Lotze MT, Robb RJ, Rosenberg SA. The systemic administration of purified interleukin 2 enhances the ability of sensitized murine lymphocytes to cure a disseminated syngeneic lymphoma. J Immunol. 1984;132:2123–8.
Rosenberg SA, Spiess P, Lafreniere R. A new approach to the adoptive immunotherapy of cancer with tumor-infiltrating lymphocytes. Science. 1986;19(233):1318–21.
Rosenberg SA, Packard BS, Aebersold PM, Solomon D, Topalian SL, Toy ST, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–80.
Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, et al. Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst. 1994;86:1159–66.
Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA. Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother. 2003;26:332–42.
Dudley ME, Gross CA, Langhan MM, Garcia MR, Sherry RM, Yang JC, et al. CD8+ enriched "young" tumor infiltrating lymphocytes can mediate regression of metastatic melanoma. Clin Cancer Res. 2010;16:6122–31.
Joseph RW, Peddareddigari VR, Liu P, Miller PW, Overwijk WW, Bekele NB, et al. Impact of clinical and pathologic features on tumor-infiltrating lymphocyte expansion from surgically excised melanoma metastases for adoptive T-cell therapy. Clin Cancer Res. 2011;17:4882–91.
Goff SL, Smith FO, Klapper JA, Sherry R, Wunderlich JR, Steinberg SM, et al. Tumor infiltrating lymphocyte therapy for metastatic melanoma: analysis of tumors resected for TIL. J Immunother. 2010;33:840–7.
Besser MJ, Shapira-Frommer R, Treves AJ, Zippel D, Itzhaki O, Schallmach E, et al. Minimally cultured or selected autologous tumor-infiltrating lymphocytes after a lympho-depleting chemotherapy regimen in metastatic melanoma patients. J Immunother. 2009;32:415–23.
• Boni A, Cogdill AP, Dang P, Udayakumar D, Njauw CN, Sloss CM, et al. Selective BRAFV600E inhibition enhances T-cell recognition of melanoma without affecting lymphocyte function. Cancer Res. 2010;70:5213–9. An important paper that demonstrates BRAF inhibition increases tumor antigen expression, providing the scientific basis for a potential synergy between targeted therapy and immunotherapy.
Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF, et al. Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma. Clin Cancer Res. 2012;18:1386–94.
Koya RC, Mok S, Otte N, Blacketor KJ, Comin-Anduix B, Tumeh PC, et al. BRAF inhibitor vemurafenib improves the antitumor activity of adoptive cell immunotherapy. Cancer Res. 2012.
Muller AJ, Scherle PA. Targeting the mechanisms of tumoral immune tolerance with small-molecule inhibitors. Nat Rev Cancer. 2006;6:613–25.
Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–24.
Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, et al. A human memory T cell subset with stem cell-like properties. Nat Med. 2011;17:1290–7.
Hinrichs CS, Borman ZA, Gattinoni L, Yu Z, Burns WR, Huang J, et al. Human effector CD8+ T cells derived from naive rather than memory subsets possess superior traits for adoptive immunotherapy. Blood. 2011;117:808–14.
Zhou J, Shen X, Huang J, Hodes RJ, Rosenberg SA, Robbins PF. Telomere length of transferred lymphocytes correlates with in vivo persistence and tumor regression in melanoma patients receiving cell transfer therapy. J Immunol. 2005;175:7046–52.
Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305.
Robbins PF, Dudley ME, Wunderlich J, El-Gamil M, Li YF, Zhou J, et al. Cutting edge: persistence of transferred lymphocyte clonotypes correlates with cancer regression in patients receiving cell transfer therapy. J Immunol. 2004;173:7125–30.
Donia M, Junker N, Ellebaek E, Andersen MH, Straten PT, Svane IM. Characterization and comparison of "Standard" and "Young" tumor infiltrating lymphocytes for adoptive cell therapy at a Danish Translational Research Institution. Scand J Immunol. 2011.
Sarnaik A. Costimulatory effect of agonistic 4-1BB antibody on proliferation and effector phenotype of tumor-infiltrating lymphocytes in melanoma. 2012 ASCO Annual Meeting; 2012; Chicago. 2012.
Li Y, Bleakley M, Yee C. IL-21 influences the frequency, phenotype, and affinity of the antigen-specific CD8 T cell response. J Immunol. 2005;175:2261–9.
Hinrichs CS, Spolski R, Paulos CM, Gattinoni L, Kerstann KW, Palmer DC, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–33.
Seung LP, Rowley DA, Dubey P, Schreiber H. Synergy between T-cell immunity and inhibition of paracrine stimulation causes tumor rejection. Proc Natl Acad Sci U S A. 1995;92:6254–8.
Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR, et al. CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol. 2005;174:2591–601.
Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12.
Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L, et al. Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8+ T cells via TLR4 signaling. J Clin Invest. 2007;117:2197–204.
Dudley ME, Yang JC, Sherry R, Hughes MS, Royal R, Kammula U, et al. Adoptive cell therapy for patients with metastatic melanoma: evaluation of intensive myeloablative chemoradiation preparative regimens. J Clin Oncol. 2008;26:5233–9.
Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6:1142–51.
Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U S A. 2002;99:16168–73.
Rosenberg SA, Sportes C, Ahmadzadeh M, Fry TJ, Ngo LT, Schwarz SL, et al. IL-7 administration to humans leads to expansion of CD8+ and CD4+ cells but a relative decrease of CD4+ T-regulatory cells. J Immunother. 2006;29:313–9.
Weber J, Atkins M, Hwu P, Radvanyi L, Sznol M, Yee C. White paper on adoptive cell therapy for cancer with tumor-infiltrating lymphocytes: a report of the CTEP subcommittee on adoptive cell therapy. Clin Cancer Res. 2011;17:1664–73.
Cancer Epidemiology in Older Adolescents & Young Adults. SEER AYA Monograph; 2007. p. 53–7.
National Cancer Institute. [cited]; Available at http://seer.cancer.gov/csr/1975_2008/ ; based on November 2010 SEER data submission, posted to the SEER web site, 2011.
Acknowledgments
This work was supported by the NCI K12 Career Development in Pediatric and Medical Oncology Award (K12CA076930). The authors thank Mark Dudley for discussions about TIL generation.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lee, S., Margolin, K. Tumor-Infiltrating Lymphocytes in Melanoma. Curr Oncol Rep 14, 468–474 (2012). https://doi.org/10.1007/s11912-012-0257-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-012-0257-5