
Abstract
Hypoxia, via the activity of hypoxia-inducible factors (HIFs), plays a crucial role in fibrosis, inflammation, and oxidative injury, processes which are associated with progression of cardiovascular and kidney diseases. HIFs are key transcription heterodimers consisting of regulatory α-subunits (HIF-1α, HIF-2α, HIF-3α) and a constitutive β-subunit (HIF-β). The stability of HIFs is regulated by the prolyl hydroxylases (PHDs). Specific PHD inhibitors (PHD-i) are being investigated as a therapeutic approach to modulate the cellular signaling pathways and harness the native protective adaptive responses to hypoxia. Selective inhibition of PHD leads to the stabilization of the HIFs, which is the transcriptional gatekeeper of a multitude of genes involved in angiogenesis, energy metabolism, apoptosis, inflammation, and fibrosis. PHD-i downregulate hepcidin, improve iron absorption, and increase the endogenous production of erythropoietin. Furthermore, this pharmacological group has also been proven to ameliorate ischemic injuries in several organs, opening a new and promising field in cardiovascular research.. In this review, we present the basic and clinical potential of PHD-i treatment in different scenarios, such as ischemic heart disease, cardiac hypertrophy and heart failure, and their interplay with other pharmacological agents with proven cardiovascular benefits, such as sodium-glucose cotransporter 2 (SGLT2) inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
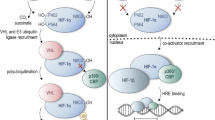
Hypoxia-inducible factors (HIFs) are transcription factors that respond to changes in the level of oxygen by binding to specific DNA sequences that control the transcription rate of genes involved in angiogenesis, metabolism, erythropoiesis, apoptosis, cell migration, and other tissue‐specific functions, including inflammation and fibrosis [1]. In its active form, HIF is a dimer composed of the HIF-α and HIF-β subunits. The HIF-α subunit accumulates under hypoxic conditions, whereas under normoxic conditions it is degraded by prolyl hydroxylase (PHD) domain proteins, which require oxygen to function (Fig. 1). The degradation of HIF-α by PHD proteins under normoxic conditions prevents its dimerization with HIF-β, leading to the inhibition of its transcriptional activity [2].

HIF signaling pathway: O2-dependent degradation of HIF-1α (left side of the picture) and transcription of hypoxia-response genes (right side). Under normoxic conditions, HIF-1α undergoes proteasomal degradation by a mechanism that involves hydroxylation of proline residues on HIF-1α by prolyl hydroxylases (PHDs) and subsequent ubiquitination by the VHL protein. Under hypoxic conditions, HIF-1α is stabilized and enters in the nucleus, where it binds to its dimerization partner HIF-1β to regulate the transcription of downstream genes
The HIF-PHD enzymes serve as gatekeepers of the coordinated transcriptional response to hypoxia and oxidative stress that plays key roles in the pathophysiology of several common disorders [1]. Thus, the selective inhibition of PHD activity—generating a pharmacologically induced hypoxia-like state favoring the dimerization of HIF—could be an effective therapeutic opportunity to reinforce the (protective) physiological adaptive response of cells when exposed to hypoxia. These interventions could have a significant impact in organs with high-energy demand and high oxygen requirements, such as kidney and heart.
Anemia in CKD Patients
Anemia is a frequent and serious complication of chronic kidney disease (CKD) that affects millions of patients worldwide [3, 4]. As many as 15.4% of patients with CKD experience anemia which is associated with reduced quality of life and increased morbidity and mortality [5,6,7,8]. Key factors responsible for anemia of CKD include relative deficiency in erythropoietin (EPO) production and decreased iron availability for Hemoglobin (Hb) synthesis associated with inflammation. Recently, it has become apparent that other factors are also involved in the etiology of anemia, most notably dysfunctional iron metabolism, mediated via increased hepcidin activity and reduced clearance.
Currently, anemia of CKD is managed by the administration of recombinant human EPO or erythropoiesis stimulating agents (ESAs), along with adjuvant iron supplementation [9]. However, some studies have reported that high doses of ESAs increase the risk of serious adverse events, including death, myocardial infarction, congestive heart failure, and stroke [10,11,12]. On the other hand, some patients are hypo-responsive to ESAs and require larger doses because of functional iron deficiency associated with inflammation [8, 13]. Thus, there is a need for safer and more effective interventions to address the impaired EPO production and functional iron deficiency affecting anemic CKD patients.
PHD-i are currently being developed for the treatment of anemia in CKD patients, and their benefits have been recently reported [14,15,16]. One of the major advantages of PHD-i is the ability to circumvent the generation of unstable and supra-physiological plasma levels of EPO. PHD-i have demonstrated the ability to improve iron absorption, downregulate hepcidin, and physiologically increase the endogenous production of EPO [17]. Recently, the PHD-i vadadustat was shown in two clinical trials to be non-inferior to ESA (darbepoetin alfa) in terms of cardiovascular safety and the correction and maintenance of Hb concentrations in CKD patients undergoing dialysis [15]. Nonetheless, growing evidence suggests that PHD-i could also have a role beyond CKD, with potentially significant and independent beneficial effects in cardiovascular disease [18,19,20].
Hypoxia-induced Signaling Pathways and Role of PHD-i
Oxygen homeostasis is central to the understanding of cell physiology. Oxygen serves as the major electron acceptor in oxidative phosphorylation, which is responsible for most of Adenosine triphosphate (ATP) production [21]. However, oxidative phosphorylation also generates harmful reactive oxygen species (ROS). The interactions between ROS and cellular macromolecules negatively impact their biochemical and physical properties, which ultimately lead to cell dysfunction and apoptosis. Hence, organisms have evolved metabolic and systemic physiological mechanisms to both maintain oxygen at desirable levels and also protect themselves against oxidative cellular damage [21].
HIF was first identified by Semenza et al. as the transcription factor responsible for the hypoxic induction of EPO [22]. The dimeric HIF form (HIF-α and HIF-β) binds to specific DNA sequences (hypoxia-responsive elements, HREs) and modulates expression of a diverse set of genes. This expression profile of genes includes glycolytic enzymes, vascular endothelial growth factors, EPO, and other factors in a tissue-specific manner. This transcriptional response also includes the downregulation of some genes, such as pyruvate dehydrogenase kinase 1 (PDK1) [23], which decreases mitochondrial oxygen consumption. In general, this broad range of transcriptional responses aims to assist cells in resisting the damaging effects of hypoxic environments, while simultaneously promoting the delivery of and reducing the requirement of O2.
The transcriptional activity and stability of HIF are tightly regulated by oxygen availability. HIF-α is constantly expressed in the cytoplasm of cells and, unless degraded, migrates towards the nucleus to bind with HIF-β and create a functional HIF dimer. Hydroxylation of specific proline residues within the HIF-α subunit results in its inactivation, and this process, carried out by PHDs, requires molecular oxygen as a co-substrate. Therefore, PHDs are sensitive to environmental oxygen, acting as oxygen sensors.
Under normal oxygen conditions, HIF-α is hydroxylated by PHDs. The Von Hippel-Lindau (VHL) protein attaches itself to the hydroxylated proline residue and ubiquitinates HIF-α. The ubiquitination of HIF-1α makes it identifiable by a protease enzyme, which ultimately leads to its destruction (Fig. 1, Left panel). On the other hand, under hypoxic conditions, PHD activity is reduced and hydroxylation, ubiquitination, and degradation do not occur, resulting in HIF-α accumulation in the cells (Fig. 1, right panel).
Three HIF-α isoforms have been identified, HIF-1α, HIF‐2α (also known as EPAS1), and HIF-3α. All 3 isoforms have the ability to bind to its dimerization partner HIF-1β in the nucleus and regulate the transcription of downstream genes. HIF-1α and HIF‐2α present a comparable amino acid sequence, protein structure, and domain arrangement [18]. However, although they are closely related and activate HRE-dependent gene expression, both isoforms have unique target genes and require distinct transcriptional cofactors [24], implying that they have different mechanisms of expression and cellular actions. HIF-1α is expressed in a wide range of cell types [18], whereas HIF‐2α expression is tissue specific and restricted primarily to the endothelium, liver, lungs, kidneys, heart, brain, and intestines [25].
In general, HIF-1α predominantly promotes the transcription of proteins that decrease oxygen use. For example, HIF-1α, but not HIF-2α, is responsible for the regulation of transcription of genes encoding enzymes involved in the glycolytic pathway [26]. On the other hand, HIF‐2α is the main isoform involved in upregulating EPO gene expression [27]. The gene encoding divalent metal transporter 1 (DMT1), the principal intestinal iron transporter, is only regulated by HIF‐2α [28]. Oct4, a crucial transcription factor regulating stem cell self-renewal, is a specific, direct target of HIF‐2α but not HIF-1α [29].
The specificity of their action suggests that both isoforms are non-redundant and play an important role in the hypoxia response. The interplay between HIF-1α and HIF-2α is thought to provide a sliding scale response, with different predominance depending on the intensity and duration of the hypoxic stimulus. For example, HIF-1α drives the initial response to hypoxia, but under chronic hypoxic exposure, it is HIF-2α that plays the major role. A disbalance between HIF-1α and HIF-2α could directly impact physiological and pathophysiological processes [18, 19, 30].
There are three HIF PHDs, termed PHD1, PHD2, and PHD3. They differ with respect to their extent, time course, and cellular localization [31]. Regulation of expression of PHD isoforms varies in a cell-type specific manner, as does the affinity of PHDs to HIF-1α or HIF-2α isoforms (Fig. 2):
-
PHD1 is mainly located in the nucleus. It is highly expressed in testis, with low levels of expression in the heart [32, 33].
-
PHD2 expression is induced by desferroxamine, CoCl2, and all known PHD inhibitors (PHD-i). It is predominantly localized in the cytoplasm. Inhibition of PHD2 acts primarily to boost the activity of HIF-1α, rather than HIF‐2α. Basal expression levels have been found to be high in the heart and testis [25], and moderate in brain, kidney, and liver [33].
-
PHD3 distributes equally in the cytoplasm and nucleus. It is highly expressed in the heart and liver. PHD3 acts preferentially on HIF-2α [34].

Preferential inhibitory effects on HIF-1α and HIF-2α in hypoxia signaling pathway. PHD inhibitors (e.g., daprodustat, vadadustat, roxadustat) While they act on all PHD isoforms, these compounds have different affinities to each one. Roxadustat would primarily inhibit the isoform PHD3, although it also potentially suppresses other PHDs (depending on dose) The inhibition of PHD3 would ultimately boost HIF-2α. Conversely, PHD2 acts preferentially on HIF-1α. HIF-2α is also directly enhanced by SIRT1. SIRT1 sirtuin-1, SGLT2 sodium-glucose cotransporter 2 inhibitors, PDH prolyl hydroxylases
Role of HIF in Cardiovascular Diseases
Ischemic heart diseases, characterized by significant coronary occlusions resulting in disruptions in blood flow and oxygen supply, are major causes of mortality and disability in high‐income countries, followed by cerebrovascular disease [35]. However, a dysfunctional oxygen supply to the heart can also be attributed to more complex pathophysiology, such as coronary microvascular disease or a distorted and hypertrophic myocardium. Indeed, ischemia contributes, at least partially, to most cardiomyopathies [36].
HIF-α has a genuine pivotal role in the physiology and pathophysiology of the cardiovascular system. Several models of HIF deletion demonstrate the importance of HIF signaling in cardiac development and in embryonic development overall. The knockout of both HIF-1α and HIF-2α is lethal in mice. Hypoxia is an essential trigger to tissue differentiation, and HIF controls angiogenesis, glucose metabolism, and cellular proliferation [21].
In the adult heart, HIF-1α and HIF-2α are the major factors driving all mechanisms of endogenous cardiac protection against any ischemic stress. PHD-i provide the opportunity to harness these adaptive responses and reinforce the cardioprotective effects of HIF [37]. Two big groups of experimental models support the modulation of HIF as a potential therapeutic strategy. First, interventions which increase HIF activity have proved, in most cases, to improve ischemic outcomes (mainly, infarct size, and cardiac function) [38,39,40,41]. Second, experimental interventions that nullify the native HIF activity in cells significantly exacerbate the ischemic damage [42]. In general, activation of HIF would improve myocardial ischemia by multiple mechanisms: reprogramming of metabolism and induction of angiogenesis, effects on apoptosis, autophagy, cell survival, cell migration, and stem cell behavior.
Acute Myocardial Infarction
The reduced supply of oxygen to the cardiomyocytes during an acute MI causes nuclear accumulation of HIF-1α protein and enhancement of its transcriptional activity in target genes, including vascular endothelial growth factor (VEGF), angiopoietin-1 (Ang-1), angiopoietin-2 (Ang-2), platelet-derived growth factor beta, inducible nitric oxide synthase (iNOS), EPO, phosphoglycerate kinase, and stromal-derived factor-1 (SDF-1) [37]. Pharmacologically, PHD inhibition provides a novel way of utilizing body’s natural compensatory mechanisms in response to hypoxia, by increasing HIF transcriptional activity. Different studies suggest that PHD inhibition, and the subsequent HIF-1α overexpression, reduces ischemic injury [38,39,40,41].
Cardiomyocyte-specific deletion of PHD2 has been studied in a mice model of myocardial infarction. This intervention resulted in a significantly smaller area at risk and area of necrosis, which correlated with a decreased number of apoptotic cells in the infarcted myocardium and significantly improved cardiac function 3 weeks after myocardial infarction [38].
Similar results have been reported with direct activation of HIFα. Genetic overexpression of HIF-1α induced therapeutic angiogenesis, reduced infarct size, and improved myocardial function after acute coronary occlusion in murine models [40]. In other experiment, upregulation of HIF-1α by plasmid DNA improved myocardial perfusion, peri-infarct vascularization, and border zone survival in rats with acute myocardial infarction [39].
Intramyocardial transfection of HIF-1α has also been studied in mesenchymal stem cells (MSCs) models. Co-transplantation of MSCs and HIF-1α led to increased angiogenesis, reduced apoptosis, and increased survival and engraftment of these stem cells [41].
A clinical trial with the PHD-i Roxadustat (ROXAMI: Study of Roxadustat in the Treatment of Acute Myocardial Infarction) in acute ST-elevation myocardial infarction is currently ongoing. Roxadustat seems to act primarily on PHD3 although it also potentially suppresses other PHDs depending on the dose [18]. This preferential effect on PHD3 would stabilize and promote HIF-2α (Fig. 2). This trial aims to evaluate the efficacy and safety of early and short-term administration of a PHD-i in the treatment of acute myocardial infarction (NCT04803864, ClinicalTrials.gov).
Ischemia/Reperfusion
Ischemia–reperfusion (I/R) injury develops subsequent to the restoration (reperfusion) of blood supply to an organ that had been disrupted (ischemia). Although early reperfusion is the most effective way to salvage ischemic tissue, the restoration of blood flow is known to induce pathological events leading to a greater myocardial tissue injury in some cases than that caused by the original ischemic insult [43]. As ROS generation has been observed to be elevated when oxygen supply is restored following an ischemic event, oxidative stress is considered a hallmark of myocardial I/R pathophysiology [44].
Evidence suggests that HIF pathway activation, which promotes the production of oxygen-independent ATP production through enhanced anaerobic respiration during hypoxic conditions, confers protection against I/R injury [43].
PHD inhibition has been shown to markedly reduce I/R injury by pharmacologically improving hypoxic responses. In a murine I/R model, the PHD-i roxadustat showed a reduction in infarct size compared with placebo [45]. Cell death induced by hypoxia/reoxygenation was also significantly prevented with pretreatment in cultured cardiomyocyte. Furthermore, oxygen consumption rate ([OCR] representing aerobic respiration) was significantly suppressed and extracellular acidification rate ([ECAR] representing anaerobic respiration) significantly enhanced in cultured cardiomyocytes with roxadustat treatment. Conversely, silencing HIF-1α abolished the enhancement of ECAR, but not the reduction of OCR. These results suggested that roxadustat enhances anaerobic respiration through HIF-1α, having an additional effect reducing OCR independently of HIF-1α [45].
Genetic deletion of PHD-1 [PHD-1(− / −)] in mice facilitates HIF-1α-mediated cardioprotection in I/R, contributing to a smaller infarct size compared with control mice [46]. Other studies have examined the role of HIF-1 activation on proinflammatory chemokine and adhesion molecule expression during post-ischemic cardiac inflammation [47]. In murine cardiomyocytes in vitro and in intact murine hearts following in vivo I/R injury, HIF-1 activation, both pharmacologically and via small-interfering RNA, significantly attenuated tumor necrosis factor-alpha-induced chemokines and ICAM-1 (intracellular adhesion molecule) expression in cardiomyocytes [47].
Ischemic Preconditioning
Preconditioning has been extensively investigated, and HIF is thought to play a role in this cardioprotective phenomenon [6]. Eckle et al. observed that preconditioning induces a robust activation of cardiac HIF-1α in mice [42]. The cardioprotection with ischemic preconditioning was abolished by the experimental suppression of cardiac HIF-1α. In contrast, pretreatment with the HIF-1α was associated with cardioprotection similar to the benefit obtained with ischemic preconditioning itself [42]. Different studies have shown that increased HIF-1α transcriptional activity under normoxic conditions preconditions murine hearts to withstand subsequent events of ischemia [42, 48]. Selective inactivation of PHD2 by small interfering RNA has been shown to cause significant activation of HIF-1α and reduced myocardial infarct sizes in murine endothelial cells in vitro and in murine cardiac tissue. Experiments with iNOS knockout mice suggest that NO plays a key role in mediating the cardioprotection observed following HIF-1α activation [48].
The specific implication of HIF-2α in cardiac preconditioning has also been the subject of some studies. The role of HIF-2α would come into play especially with long-lasting or sustained hypoxic stimulus (hours) [49]. Its expression is increased in remote myocardial areas and in the peri-infarct zone after infarction [49]. Furthermore, HIF-2α has been suggested to contribute to the protective adaptive responses of kidney tissue [50] and neurons [51].
Although significant progress has been made in this field, the mechanisms, signaling pathways, and effectors that mediate protection in preconditioning are not yet completely defined [44]. Mitochondrial ROS production has been linked to preconditioning [44]. Hypoxia itself leads to an increased ROS production, and elevated levels of ROS cause peroxidation of proteins, DNA, and lipids and trigger mitochondria-induced cell death pathways. However, in contrast to the detrimental effects of massive ROS production, sub-lethal amounts of ROS could serve as a trigger for cardioprotection in ischemia preconditioning [52].
Low levels of ROS can act as signaling molecules that induce HIF pathway activation, which then triggers the metabolic program that provides cellular protection against oxygen deprivation [53].
Regardless of the mechanism of action, what seems clear is that HIF is associated with increased myocardial resistance to hypoxia, resulting in a profound reduction in myocardial infarct sizes. Accordingly, different HIF-1α loss-of-function models are consistent in showing the reversal of this protection, and as a result, mice present bigger infarct size after ischemia [42].
Cardiac Hypertrophy
In response to different haemodynamic stresses, such as hypertension or ischemia, cardiac performance is initially maintained by a beneficial compensatory increase in myocyte size (hypertrophy) [54]. Cardiac myocytes activate intracellular hypertrophic signaling pathways to re-use embryonic transcription factors and to increase the synthesis of various proteins, such as structural and contractile proteins [54]. The increase in myocardial mass increases oxygen demand and promotes myocardial angiogenesis to ameliorate the hypoxic situation. In these circumstances, angiogenesis is a key to maintaining sufficient cardiac perfusion to support beneficial hypertrophy and avoid the transition to heart failure and adverse cardiac remodeling. Angiogenesis requires secreted angiogenic growth factors, most of which are regulated by HIF-1α [21].
Preclinical pressure-overload heart failure models have been used to study the role of HIF-1α activation in myocardial hypertrophy. Pressure overload is experimentally induced with severe transverse aorta constriction (TAC), and cardiac hypertrophy gradually develops to maintain heart function. Expression of HIF-1α, but not HIF‐2α, is increased by TAC from day 3 [55]. In these models, the number of microvessels per cardiomyocyte usually increases in parallel with cardiomyocyte hypertrophy until day 14 when it reaches its peak. However, if the hemodynamic stress becomes chronic, hypertrophy eventually progresses to adverse ventricular dilatation. Myocyte apoptosis, interstitial fibrosis, and detrimental matrix remodeling contribute to this process, leading to heart failure.
Several cellular pathways have been implicated in this beneficial-to-adverse hypertrophic transition. Chronic hemodynamic pressure and stress on the ventricular wall lead to accumulation of p53, an inhibitor of HIF-1α, causing inactivation of HIF-1α’s downstream angiogenic targets. Accumulation of p53 restrains neovascularization and precipitates a detrimental stage of ventricular dilatation and eventual heart failure [55]. Mice deficient in p53 in TAC models show higher cardiac HIF-1α activity and VEGF levels, with a significant increase in the number of microvessels. After chronic pressure overload, p53-deficient mice have more marked cardiac hypertrophy, better systolic function, and lower atrial natriuretic factor levels [55].
Overexpression of angiogenic factors also attenuates the transition from compensatory hypertrophy to dilated heart failure. Jian Guo et al. demonstrated that transgenic mice that overexpress the angiogenic factor CNPY2 (Canopy 2) presented a better vascularized myocardium and better cardiac performance during development of chronic heart failure [56]. Activation of p53 was also decreased in hearts after injury and the expression of HIF-1α better maintained, thereby potentially retaining a most beneficial pro-angiogenic program [56].
These results suggest the benefit of HIF-1α and angiogenic activity in injured hearts, helping to preserve cardiac function and preventing the switch from beneficial compensatory hypertrophy to ventricular dilatation and heart failure.
Induction of Cardiomyopathy
The role of HIF-1α signaling in chronic heart failure might be controversial. Although the HIF-mediated adaptations are beneficial to withstand acute ischemia, sustained overexpression of either HIF-1α or HIF-2α has been associated with the spontaneous development of cardiomyopathy [57].
Bekeredjian et al. showed that enhanced HIF activity is sufficient to cause contractile dysfunction in the adult mice heart [58]. In another study, the inactivation of PHD2, combined with the inactivation of PHD2/PHD3, and inactivation of VHL resulted in progressively more intense HIF activation and severe phenotypes of cardiac dysfunction [59].
The mechanisms underlying HIF-associated cardiomyopathy are unclear. Adaptations induced by HIF include changes in vascular tone, anaerobic metabolism, and calcium handling, which could explain the positive findings in HIF-1α overexpressing hearts, in the setting of a myocardial infarction or ischemia preconditioning. Regarding metabolism changes, HIF induces the expression of glycolytic genes favoring glucose consumption and anaerobic metabolism during ischemia. Although this energy-saving metabolism is beneficial for withstanding oxygen-deprived conditions, the same pathways are indicative of a defective cardiac homeostasis in the heart [57].
More studies are needed to determine the implications of long-lasting stabilization of HIF or determine its optimal level of activity to gain the maximum cardiovascular benefits without the unwanted risks.
Sodium-Glucose Cotransporter 2 Inhibitors (SGLT2i) and PDH Inhibitors
Several studies have demonstrated a significant cardioprotective and renoprotective benefit with SGLT2-i treatment, in both diabetic and nondiabetic patients [60,61,62]. These benefits cannot be readily explained by lowering blood glucose, blood pressure, or body weight. SGLT2-i have been shown to reduce serious heart failure and adverse renal events within the first few months of treatment initiation. A potential effect on non-hemodynamic pathogenic mechanism has been suggested for the beneficial actions of SGLT2-i [19, 63].
Chronic kidney failure, heart failure, obesity, and type 2 diabetes mellitus share a deranged cell signaling setup with downregulated HIF-2α, AMPK, Sirtuin-1 (SIRT1), and an impaired autophagic flux [19]. SIRT1, a deacetylase protein, acts as a redox sensor and augments HIF-2α signaling during hypoxia. Type 2 diabetes is characterized by renal hypoxia, oxidative stress, and defective nutrient deprivation signaling, which might induce activation of HIF-1α and suppression of HIF-2α. This shift in the balance of HIF-1α/HIF-2α activities promotes proinflammatory and profibrotic pathways in glomerular and renal tubular cells [19].
Increased and chronic HIF-1α expression in tubular epithelial cells leads to tubulointerstitial fibrosis. One of the major HIF-1α gene targets, the plasminogen activator inhibitor-1 (PAI-1), is a key effector in progression of kidney fibrosis. Indeed, previous studies have shown that genetically silencing PAI-1 improves fibrosis in Diabetic Kidney Disease (DKD) in mice [64, 65]. The pharmacological inhibition of HIF-1α also improves clinical manifestations of diabetic nephropathy [66].
Inhibiting SGLT2 seems to be efficient in rectifying this metabolic imbalance, reducing HIF-1α and promoting SIRT1 [67] and AMPK in DKD. It has been hypothesized that SGLT2 inhibition would also act to enhance HIF-2α signaling [20]. By promoting a selective upregulation of HIF‐2α over HIF-1α, these pharmacological agents would switch the balance between the two isoforms toward a more beneficial renal and cardiovascular profile [18]. Luseogliflozin has been demonstrated to inhibit HIF-1α expression and subsequent renal fibrosis in diabetic kidneys [64]. Another SGLT2 inhibitor, Dapagliflozin, nearly nullifies the increased level of HIF-1α in renal proximal tubule. This was observed in samples from both diabetic patients and mice with experimental streptozocin (STZ)-induced diabetes [68]. The tubulointerstitial damage, a common feature in diabetic kidney, was partially reversed by Dapagliflozin, with less macrophage infiltration and fibrosis. Another common feature in diabetic nephropathy, an increased intraglomerular filtration pressure, was also ameliorated by SGLT2 inhibition [19].
In the synthesis and regulation of EPO, HIF2α is the main responsible factor [69]. The deficiency of EPO and the efficacy of PHD-i in CKD suggest that HIF-2α is downregulated in renal patients, which has been confirmed in preclinical studies [70]. In this regard, SGLT2 inhibitors, a group of drugs which promote EPO production and erythropoiesis in kidneys [71], would potentially act through HIF-2α signaling.
The interplay between HIF-1α/HIF-2α and SGLT2 inhibition could be a contributing factor in their actions towards alleviating renal hypoxia and cellular stress, and improving renal and cardiovascular outcomes.
Conclusions
Inhibition of PHD may provide the capability to modulate the endogenous adaptive programs that are triggered in response to hypoxia in cells. This has important implications in ischemic organ injuries, fibrosis, inflammation, and oxidative stress, all processes that have been implicated in the progression of cardiovascular and renal disease.
Several PHD-i (daprodustat, roxadustat, vadadustat, or molidustat) are being actively investigated for the treatment of anemia in CKD, in both non-dialysis-dependent and dialysis-dependent adult patients, as well as chemotherapy-induced and anemia associated with myelodysplastic syndromes. These compounds might also be useful for treating ischemic heart disease. As we have highlighted in this review, numerous studies show the potential of PHD-i to reduce the severity of acute myocardial infarction, ameliorate the ischemia–reperfusion injury, and improve function and vascularization in cardiac hypertrophy.
However, a more complete understanding of HIF signaling is still needed to determine its specificity of action and the exact therapeutic window. The possibility also exists that long-term drug-induced HIF elevation could be deleterious, and associated with increased rather than decreased cardiac dysfunction. Furthermore, the pleiotropic effects of HIF and the activation of a wide range of genetic responses imply the risk of unwanted or unexpected side effects.
The current challenge is the development of tissue-specific and PHD-selective inhibition, which could allow independently targeting each isoform of HIF to promote a favorable HIF-1α/HIF-2α balance. To know when, how much and, most importantly, for how long PDH inhibitors should be used would be key to avoiding its potential detrimental effects and obtaining the maximum benefit of hydroxylase inhibition.
Improving understanding of the pathophysiology of hypoxic tissue damage and the endogenous adaptive processes that are triggered by hypoxia remains a major challenge for the development of new therapies for myocardial infarction. Unraveling the mechanisms underlying hypoxia will also help us to define the full clinical value of novel pharmacological agents in cardiovascular and renal disease, such as PHD-i or SGLT2 inhibitors.
Data Availability
Not applicable.
References
Lee JW, Ko J, Ju C, Eltzschig HK. Hypoxia signaling in human diseases and therapeutic targets. Exp Mol Med. 2019;51(6):1–13.
Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. 2018;27(2):281–98.
Stauffer ME, Fan T. Prevalence of anemia in chronic kidney disease in the United States. PLoS One. 2014;9(1):e84943.
Kassebaum NJ, Jasrasaria R, Naghavi M, Wulf SK, Johns N, Lozano R, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood. 2014;123(5):615–24.
Akizawa T, Okumura H, Alexandre AF, Fukushima A, Kiyabu G, Dorey J. Burden of anemia in chronic kidney disease patients in Japan: a literature review. Ther Apher Dial. 2018;22(5):444–56.
Locatelli F, Fishbane S, Block GA, Macdougall IC. Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol. 2017;45(3):187–99.
Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23(10):1631–4.
Yilmaz MI, Solak Y, Covic A, Goldsmith D, Kanbay M. Renal anemia of inflammation: The name is self-explanatory. Blood Purif. 2011;32(3):220–5.
Biggar P, Kim GH. Treatment of renal anemia: Erythropoiesis stimulating agents and beyond. Kidney Res Clin Pract. 2017;36(3):209–23.
Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355(20):2085–98.
Szczech LA, Barnhart HX, Inrig JK, Reddan DN, Sapp S, Califf RM, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int. 2008;74(6):791–8.
Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361(21):2019–32.
Macdougall IC, Cooper AC. Hyporesponsiveness to erythropoietic therapy due to chronic inflammation. Eur J Clin Invest. 2005;35(Suppl 3):32–5.
Li ZL, Tu Y, Liu BC. Treatment of renal anemia with roxadustat: Advantages and achievement. Kidney Dis (Basel). 2020;6(2):65–73.
Pergola PE, Spinowitz BS, Hartman CS, Maroni BJ, Haase VH. Vadadustat, a novel oral HIF stabilizer, provides effective anemia treatment in nondialysis-dependent chronic kidney disease. Kidney Int. 2016;90(5):1115–22.
Yan Z, Xu G. A Novel choice to correct inflammation-induced anemia in CKD: Oral hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat. Front Med (Lausanne). 2020;7:393.
Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: A potential new treatment for anemia in patients with CKD. Am J Kidney Dis. 2017;69(6):815–26.
Packer M. Mutual antagonism of hypoxia-inducible factor isoforms in cardiac, vascular, and renal disorders. JACC Basic Transl Sci. 2020;5(9):961–8.
Packer M. Mechanisms leading to differential hypoxia-inducible factor signaling in the diabetic kidney: Modulation by SGLT2 inhibitors and hypoxia mimetics. Am J Kidney Dis. 2021;77(2):280–6.
Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: Implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol. 2020;31(5):907–19.
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, et al. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev. 1998;12(2):149–62.
Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12(12):5447–54.
Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3(3):177–85.
Loboda A, Jozkowicz A, Dulak J. HIF-1 and HIF-2 transcription factors—Similar but not identical. Mol Cells. 2010;29(5):435–42.
Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W, et al. HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol. 2006;41(1):68–77.
Hu CJ, Wang LY, Chodosh LA, Keith B, Simon MC. Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol Cell Biol. 2003;23(24):9361–74.
Tanaka T, Nangaku M. Recent advances and clinical application of erythropoietin and erythropoiesis-stimulating agents. Exp Cell Res. 2012;318(9):1068–73.
Ingrassia R, Garavaglia B, Memo M. DMT1 Expression and iron levels at the crossroads between aging and neurodegeneration. Front Neurosci. 2019;13:575.
Covello KL, Kehler J, Yu H, Gordan JD, Arsham AM, Hu CJ, et al. HIF-2alpha regulates Oct-4: Effects of hypoxia on stem cell function, embryonic development, and tumor growth. Genes Dev. 2006;20(5):557–70.
Koh MY, Powis G. Passing the baton: The HIF switch. Trends Biochem Sci. 2012;37(9):364–72.
Czibik G. Complex role of the HIF system in cardiovascular biology. J Mol Med (Berl). 2010;88(11):1101–11.
Siddiq A, Aminova LR, Ratan RR. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: Center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem Res. 2007;32(4–5):931–46.
Lieb ME, Menzies K, Moschella MC, Ni R, Taubman MB. Mammalian EGLN genes have distinct patterns of mRNA expression and regulation. Biochem Cell Biol. 2002;80(4):421–6.
Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–65.
Moran AE, Forouzanfar MH, Roth GA, Mensah GA, Ezzati M, Murray CJ, et al. Temporal trends in ischemic heart disease mortality in 21 world regions, 1980 to 2010: The Global Burden of Disease 2010 study. Circulation. 2014;129(14):1483–92.
Severino P, D’Amato A, Pucci M, Infusino F, Adamo F, Birtolo LI, et al. Ischemic heart disease pathophysiology paradigms overview: From plaque activation to microvascular dysfunction. Int J Mol Sci. 2020;21(21).
Schreiber T, Salhofer L, Quinting T, Fandrey J. Things get broken: The hypoxia-inducible factor prolyl hydroxylases in ischemic heart disease. Basic Res Cardiol. 2019;114(3):16.
Holscher M, Silter M, Krull S, von Ahlen M, Hesse A, Schwartz P, et al. Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J Biol Chem. 2011;286(13):11185–94.
Shyu KG, Wang MT, Wang BW, Chang CC, Leu JG, Kuan P, et al. Intramyocardial injection of naked DNA encoding HIF-1alpha/VP16 hybrid to enhance angiogenesis in an acute myocardial infarction model in the rat. Cardiovasc Res. 2002;54(3):576–83.
Kido M, Du L, Sullivan CC, Li X, Deutsch R, Jamieson SW, et al. Hypoxia-inducible factor 1-alpha reduces infarction and attenuates progression of cardiac dysfunction after myocardial infarction in the mouse. J Am Coll Cardiol. 2005;46(11):2116–24.
Huang B, Qian J, Ma J, Huang Z, Shen Y, Chen X, et al. Myocardial transfection of hypoxia-inducible factor-1alpha and co-transplantation of mesenchymal stem cells enhance cardiac repair in rats with experimental myocardial infarction. Stem Cell Res Ther. 2014;5(1):22.
Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation. 2008;118(2):166–75.
Zheng J, Chen P, Zhong J, Cheng Y, Chen H, He Y, et al. HIF1alpha in myocardial ischemiareperfusion injury (Review). Mol Med Rep. 2021;23(5).
Cadenas S. ROS and redox signaling in myocardial ischemia-reperfusion injury and cardioprotection. Free Radic Biol Med. 2018;117:76–89.
Deguchi H, Ikeda M, Ide T, Tadokoro T, Ikeda S, Okabe K, et al. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice. Circ J. 2020;84(6):1028–33.
Adluri RS, Thirunavukkarasu M, Dunna NR, Zhan L, Oriowo B, Takeda K, et al. Disruption of hypoxia-inducible transcription factor-prolyl hydroxylase domain-1 (PHD-1-/-) attenuates ex vivo myocardial ischemia/reperfusion injury through hypoxia-inducible factor-1alpha transcription factor and its target genes in mice. Antioxid Redox Signal. 2011;15(7):1789–97.
Natarajan R, Salloum FN, Fisher BJ, Ownby ED, Kukreja RC, Fowler AA 3rd. Activation of hypoxia-inducible factor-1 via prolyl-4 hydoxylase-2 gene silencing attenuates acute inflammatory responses in postischemic myocardium. Am J Physiol Heart Circ Physiol. 2007;293(3):H1571–80.
Natarajan R, Salloum FN, Fisher BJ, Kukreja RC, Fowler AA 3rd. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ Res. 2006;98(1):133–40.
Bautista L, Castro MJ, Lopez-Barneo J, Castellano A. Hypoxia inducible factor-2alpha stabilization and maxi-K+ channel beta1-subunit gene repression by hypoxia in cardiac myocytes: role in preconditioning. Circ Res. 2009;104(12):1364–72.
Kojima I, Tanaka T, Inagi R, Kato H, Yamashita T, Sakiyama A, et al. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol. 2007;18(4):1218–26.
Ralph GS, Parham S, Lee SR, Beard GL, Craigon MH, Ward N, et al. Identification of potential stroke targets by lentiviral vector mediated overexpression of HIF-1 alpha and HIF-2 alpha in a primary neuronal model of hypoxia. J Cereb Blood Flow Metab. 2004;24(2):245–58.
Zhou T, Chuang CC, Zuo L. Molecular Characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. Biomed Res Int. 2015;2015:864946.
Zhou T, Prather ER, Garrison DE, Zuo L. Interplay between ROS and antioxidants during ischemia-reperfusion injuries in cardiac and skeletal muscle. Int J Mol Sci. 2018;19(2).
Oka T, Akazawa H, Naito AT, Komuro I. Angiogenesis and cardiac hypertrophy: Maintenance of cardiac function and causative roles in heart failure. Circ Res. 2014;114(3):565–71.
Sano M, Minamino T, Toko H, Miyauchi H, Orimo M, Qin Y, et al. p53-induced inhibition of Hif-1 causes cardiac dysfunction during pressure overload. Nature. 2007;446(7134):444–8.
Guo J, Mihic A, Wu J, Zhang Y, Singh K, Dhingra S, et al. Canopy 2 attenuates the transition from compensatory hypertrophy to dilated heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2015;36(37):2530–40.
Holscher M, Schafer K, Krull S, Farhat K, Hesse A, Silter M, et al. Unfavourable consequences of chronic cardiac HIF-1alpha stabilization. Cardiovasc Res. 2012;94(1):77–86.
Bekeredjian R, Walton CB, MacCannell KA, Ecker J, Kruse F, Outten JT, et al. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS One. 2010;5(7):e11693.
Lei L, Mason S, Liu D, Huang Y, Marks C, Hickey R, et al. Hypoxia-inducible factor-dependent degeneration, failure, and malignant transformation of the heart in the absence of the von Hippel-Lindau protein. Mol Cell Biol. 2008;28(11):3790–803.
Zinman B, Lachin JM, Inzucchi SE. Empagliflozin, Cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. 2016;374(11):1094.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380(4):347–57.
Neal B, Perkovic V, Matthews DR. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377(21):2099.
Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Garcia-Ropero A, Ishikawa K, Watanabe S, et al. Empagliflozin ameliorates diastolic dysfunction and left ventricular fibrosis/stiffness in nondiabetic heart failure: A multimodality study. JACC Cardiovasc Imaging. 2021;14(2):393–407.
Bessho R, Takiyama Y, Takiyama T, Kitsunai H, Takeda Y, Sakagami H, et al. Hypoxia-inducible factor-1alpha is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep. 2019;9(1):14754.
Lassila M, Fukami K, Jandeleit-Dahm K, Semple T, Carmeliet P, Cooper ME, et al. Plasminogen activator inhibitor-1 production is pathogenetic in experimental murine diabetic renal disease. Diabetologia. 2007;50(6):1315–26.
Nayak BK, Shanmugasundaram K, Friedrichs WE, Cavaglierii RC, Patel M, Barnes J, et al. HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes. 2016;65(5):1387–97.
Umino H, Hasegawa K, Minakuchi H, Muraoka H, Kawaguchi T, Kanda T, et al. High basolateral glucose increases sodium-glucose cotransporter 2 and reduces sirtuin-1 in renal tubules through glucose transporter-2 detection. Sci Rep. 2018;8(1):6791.
Cai T, Ke Q, Fang Y, Wen P, Chen H, Yuan Q, et al. Sodium-glucose cotransporter 2 inhibition suppresses HIF-1alpha-mediated metabolic switch from lipid oxidation to glycolysis in kidney tubule cells of diabetic mice. Cell Death Dis. 2020;11(5):390.
Eckardt KU, Kurtz A. Regulation of erythropoietin production. Eur J Clin Invest. 2005;35(Suppl 3):13–9.
Olmos G, Munoz-Felix JM, Mora I, Muller AG, Ruiz-Torres MP, Lopez-Novoa JM, et al. Impaired erythropoietin synthesis in chronic kidney disease is caused by alterations in extracellular matrix composition. J Cell Mol Med. 2018;22(1):302–14.
Mazer CD, Hare GMT, Connelly PW, Gilbert RE, Shehata N, Quan A, et al. Effect of empagliflozin on erythropoietin levels, iron stores, and red blood cell morphology in patients with type 2 diabetes mellitus and coronary artery disease. Circulation. 2020;141(8):704–7.
Funding
Juan Antonio Requena-Ibáñez is the recipient of a Fellowship from the Alfonso Martin Escudero Foundation, Madrid (Spain).
Author information
Authors and Affiliations
Contributions
All authors have read, approved, and contributed equally to the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
All authors have read, approved, and contributed equally to the final version of the manuscript.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Juan Antonio Requena-Ibáñez is the recipient of a Fellowship from the Alfonso Martin Escudero Foundation, Madrid (Spain).
Rights and permissions
About this article

Cite this article
Requena-Ibáñez, J.A., Santos-Gallego, C.G., Rodriguez-Cordero, A. et al. Prolyl Hydroxylase Inhibitors: a New Opportunity in Renal and Myocardial Protection. Cardiovasc Drugs Ther 36, 1187–1196 (2022). https://doi.org/10.1007/s10557-021-07257-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-021-07257-0