
Abstract
Background
Individualising treatment in breast cancer requires effective predictive biomarkers. While relatively few genomic aberrations are clinically relevant, there is a need for characterising patients across different subtypes to identify actionable alterations.
Methods
We identified genomic alterations in 49 potentially actionable genes for which drugs are available either clinically or via clinical trials. We explored the landscape of mutations and copy number alterations (CNAs) in actionable genes in seven breast cancer subtypes utilising The Cancer Genome Atlas. To dissect the genomic complexity, we analysed the patterns of co-occurrence and mutual exclusivity in actionable genes.
Results
We found that >30% of tumours harboured putative actionable events that are targetable by currently available drugs. We identified genes that had multiple targetable alterations, representing candidate targets for combination therapy. Genes predicted to be drivers in primary breast tumours fell into five categories: mTOR pathway, immune checkpoints, oestrogen signalling, tumour suppression and DNA damage repair. Our analysis also revealed that CNAs in 34/49 (69%) and mutations in 13/49 (26%) genes were significantly associated with gene expression, validating copy number events as a dominant oncogenic mechanism in breast cancer.
Conclusion
These results may enable the acceleration of personalised therapy and improve clinical outcomes in breast cancer.
Similar content being viewed by others


Background
The comprehensive analysis of human cancer molecular data by The Cancer Genome Atlas (TCGA) and other studies supports the notion that treatment could be tailored to a tumour’s specific genetic signature (https://tcga-data.nci.nih.gov/tcga/) [1]. Although sequencing studies have provided a wide repertoire of information on the cancer genome, the implementation of genomic profiling in clinical decision-making remains underdeveloped. Genomic events are defined as any detectable alterations that results in a change in the gene or pathway. Actionable alterations are those with clinically actionable targets that are potentially responsive to targeted therapies available in clinical trials, or the Food and Drug Administration (FDA)-approved or experimental drugs. Detection of genomic aberrations in potentially clinically actionable genes enables the rational selection of existing or novel therapeutic agents and also helps in predicting tumour response to targeted therapy.
Breast cancer is a complex and heterogeneous disease, comprising multiple entities with a wide range of clinical behaviours, disparate treatment responses, and differences in survival rates [2, 3]. Invasive ductal carcinoma (IDC) accounts for 80% of invasive breast cancer cases, while invasive lobular cancer (ILC) represents ~10% of all breast malignancies [4]. These entities can be further divided into classes based on protein biomarkers or gene expression profiles that can be used to design therapeutic approaches [5]. Perou et al. [6] reported that hierarchical clustering analysis of gene expression profiles segregate breast cancer into five different molecular subtypes. The luminal A (LumA) subtype was defined as oestrogen receptor (ER) and/or progesterone receptor (PR) positive, human epidermal growth factor receptor 2 (HER2) negative; luminal B (LumB) as ER and/or PR-positive, HER2 positive; HER2 enriched as ER and PR-negative, HER2 positive; basal-like/ triple-negative breast cancer (TNBC) as ER, PR and HER2 negative, and a normal breast-like group as ER and/or PR-positive, HER2 negative. However, the American Society of Clinical Oncology (ASCO) clinical practice guidelines endorse that only the clinical-grade biomarkers ER, PR and HER2 receptor status should be used to guide the selection of adjuvant systemic therapy in breast cancer [7].
Mutational loads vary substantially in human cancers [8]. The systematic analysis of 21 cancer types from the TCGA ‘Pan-Cancer’ dataset has revealed a lower mutational burden in breast cancer as compared to many other malignancies [9]. The Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) has performed an integrated analysis by combining gene copy number and expression in primary breast tumours that may help to identify targets for therapy using ten integrative clusters [10]. A recent analysis of the genomic data from 12 cancer types from TCGA suggests that M-class (mutation-driven) and C-class (CN-driven) tumours are associated with different oncogenic signatures and provide insights into potentially actionable alterations [11]. In addition to these large-scale studies, the OncoKB database has provided detailed information on therapeutically actionable alterations with the level of evidence (levels 1–4 and level R1/R2) for targetability of driver alterations in more than 50 tumour types. Similarly, the European Society for Medical Oncology (ESMO) also developed a scale to rank genomic alterations into different tiers (I–V and X) based on clinical evidence of actionability in early breast cancer [12]. These large-scale genomic and transcriptomic studies enabled the identification of molecular alterations/signatures for tumour classification and uncovered molecular mechanisms necessary for tumour development. However, some of these genetic alterations that might create therapeutic vulnerabilities have not been extensively studied. Building on the strengths of the publicly available TCGA dataset, our study is based on the identification of genetic alterations in druggable or actionable genes that are predictors of response or resistance to existing cancer therapies in breast cancer.
The objective of our study was to evaluate the gene expression and genomic alterations in potentially clinically actionable genes present in a comprehensive genomic analysis of invasive breast carcinomas from the TCGA dataset, with the goal of identifying the most significant contributors to the variance in breast cancer subtypes. A further goal of this analysis was to evaluate whether RNA sequencing (RNA-Seq) or whole-exome sequencing (WES) is more suitable for providing evidence of these actionable alterations.
Methods
TCGA data and potentially breast cancer-related genes
We analysed somatic mutations, copy number alterations (CNAs) and RNA expression data for clinically actionable genes in seven breast cancer subtypes (127 ILC, 490 IDC, 201 LumA, 122 PAM50 LumB, 51 PAM50 HER2 enriched, 107 PAM50 basal-like and 82 TNBC). The TCGA breast cancer study has followed the ASCO/College of American Pathologists (CAP) guidelines for hormone receptor status definition [13]. Tumours with HER2 IHC level 3+ and FISH ratio > = 2.2 were called HER2 positive and <10% ER/PR nuclear staining (10 for ER and 33 for PR) were ER/PR-positive cases. Some of the cases with higher staining levels were not called ER/PR-positive (six for ER and seven for PR). For <10% staining level, a different threshold was used to call ER/PR-positive or negative. The data used in this study are from the 2015 TCGA breast cancer publication [14]. The clinical information for breast cancer subtypes was retrieved from the TCGA data portal (https://tcga-data.nci.nih.gov/tcga/). We developed a list of 49 potential gene targets based on an extensive literature query for biomarker/drug pairs with evidence of preclinical and clinical effectiveness in breast cancer [15]. We considered genes as clinically relevant or actionable that are associated with FDA-approved or investigational drugs in use in the clinic or in clinical trials [16]. As our study was based on analysing specific genes of interest, we performed the analysis on the processed data for mutations, CNAs, and normalised gene expression data of RNA-Seq obtained from cBioportal. The study protocol 21–654 (Date of approval: June 2, 2021) was approved by the Cleveland Clinic Institutional Review Board (IRB) Committee. All breast cancer patient samples were de-identified and encoded with TCGA sample codes.
Analysis of DNA mutations from exome-sequencing data
The Kruskal–Wallis test was applied to assess the prevalence of mutation types and to characterise the variant allele fractions (VAFs) of somatic variants in each breast cancer subtype. Based on the method described by Chang et al. for defining the mutational hotspots [17], we retained the mutational hotspots affecting ≥2% of the tumours. The R package Maftools was used to calculate the frequency of mutation type, the most frequently mutated genes, the identification of mutually exclusive or co-occurring set of genes, and to perform a cancer-driver gene analysis [18]. Quantile–Quantile (QQ) plots were generated to compare the expected and the observed distribution of the FM bias P values and to identify the significantly mutated genes among actionable targets using the OncodriveFML approach [19].
Classification of actionable alterations
The OncoKB database, which comprehensively considered FDA and National Comprehensive Cancer Network (NCCN) guidelines, was used to identify and grade actionable alterations based on a four-level classification system (version 2.0) [20]. Level 1 includes predictive biomarkers that are responsive to FDA-approved drugs. Level 2 includes standard of care predictive biomarkers recommended by the NCCN and are responsive to FDA-approved treatments. Level 3A includes candidate predictive biomarkers of drug response based on compelling clinical evidence in reported tumour types and level 3B includes standard of care biomarkers of response to FDA-approved or investigational drugs. Level 4 includes alterations that are candidate predictive biomarkers of drug response based on compelling biological evidence.
CNA calling and interpretation
To measure the burden of copy number, we applied a re-segmentation approach using the CNApp tool [21]. For the identification of significant copy number events, the genomic identification of significant targets in cancer (GISTIC) was applied to the segmented data of each breast cancer subtype. CNA was characterised by measured copy number (expressed as a log2 ratio), and by the extent of change in the genome. The CNA thresholds were determined according to the set of discrete copy number calls provided by GISTIC: homozygous deletion (−2), hemizygous deletion (−1), low-level gain (1) and high-level amplification (2). Fisher’s exact test was used to determine the frequencies of high-level amplifications and homozygous deletions.
Analysis of RNA-Seq data
We downloaded the normalised RNA-Seq data processed by RSEM (RNA-Seq by Expectation Maximisation) method from cBioPortal for each breast cancer subtype. The z score cut-off was set at −2 and +2 and this represents the standard deviation of expression value above or below the mean in the reference sample [22]. We then transformed z score to P values using one-tailed hypothesis testing. Multiple hypothesis testing was performed on calculated P values to identify significant overexpressed/downregulated actionable genes. Heatmap and clustering analysis was performed in Morpheus (https://software.broadinstitute.org/morpheus/) using Euclidean distance and average linkage.
Comparison of RNA-Seq with mutation/CNA data
We evaluated the possible association of somatic mutations and CNAs with gene expression data. We selected only two classes of CNAs: high-level amplification and homozygous deletion as these are more biologically relevant and are known to be associated with cancer. The normalised expression of wild-type/mutant cases and amplification/homozygous loss versus diploid for each gene was evaluated using a Mann–Whitney test.
Ingenuity pathway analysis
Ingenuity pathways analysis (IPA) was used to identify key molecules and signalling pathways affected by actionable alterations in breast tumours. A list of genes with variants in 49 actionable genes was subjected to IPA application (Qiagen Inc., https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis). The variants that fit the American College of Medical Genetics (ACMG) criteria for classification as pathogenic or likely pathogenic were used to visualise the gene interactions [23].
Statistical analysis
Statistical analysis for comparing the mutations, CNAs, and gene expression in breast cancer subtypes were performed using GraphPad Prism 8.3.1 (GraphPad Software, San Diego, CA, USA). The two-stage linear step-up procedure of Benjamini, Kreiger and Yekutieli by setting the false discovery rate (FDR) (Q) to 5% was used to correct P values for multiple testing. The threshold for statistical significance was set at 0.05 for all statistical tests.
Results
Detection of breast cancer-associated mutations in actionable genes
We analysed whole-exome sequencing data in seven breast cancer subtypes in the TCGA dataset. Clinical features and patient characteristics are described in Table 1. The majority of patients included had early-stage breast cancer and 12 patients received neoadjuvant treatment.
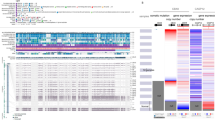
We first profiled the mutational landscape for 49 actionable genes in all seven breast cancer subtypes. A total of 1614 somatic variants comprising 1084 missense mutations, 489 truncating mutations and 41 inframe mutations were identified in all TCGA breast cancer patients. Among seven breast cancer subtypes, 100 (93%) basal, 47 (92%) HER2 enriched, 74 (90%) TNBC, 401 (82%) IDC, 99 (81%) LumB, 149 (74%) LumA and 84 (66%) ILC samples harboured mutations in 49 actionable targets. No statistically significant differences were observed in regards to mutational frequencies of these genes (Kruskal–Wallis test, P = 0.4) (Fig. 1a). However, a significant difference was observed in VAFs in hotspot mutations between the distinct subtypes (Kruskal–Wallis test, P < 0.0001) indicating a substantial degree of allelic heterogeneity in breast cancer (Fig. 1b). No substantial variation was observed in six classes of mutation types. The most frequent mutational signature, characterised by a high prevalence of C>T transition substitutions, was identified in all seven breast cancer subtypes (Fig. 1c). This signature was also found to be the most dominant mutation in 21 primary breast cancer genomes [24].

a Bar graph maps depicting the percentage of cases with mutations in actionable genes in different breast cancer subtypes. The Kruskal–Wallis test was applied to identify the differences in mutation frequencies. b Violin plot showing the distribution of variant allele fractions (VAFs) in hotspot mutations in breast cancer subtypes (Kruskal–Wallis test, P < 0.05). c Top panel: Oncoplot showing the ten most frequently mutated genes colour-coded by type of mutations in breast cancer subtypes. The percentage to the right of the oncoplot shows the percent of samples with variants for the corresponding gene. The x axis showed the total number of samples mutated and the y axis showed the frequently mutated genes. By default, samples are ordered by the most frequently mutated genes. Bottom panel: Distribution of base substitutions in seven breast cancer subtypes revealed a signature characterised predominantly by C>T transition substitutions. The percent mutations are shown on the x axis and substitution mutation types are on the y axis. d Mutation landscape in breast cancer subtypes with different types of mutations in each actionable gene. The right panel shows the percentage of cases with mutations in each actionable gene across breast cancer subtypes. OncodriveFM analysis identifies the drivers by computing the mutation rate of each gene in each subtype and is significantly greater than the background. Significant differences (P < 0.05) are shown in red.
We next utilised the Oncodrive function of Maftools to identify driver genes and the top ten most frequently mutated genes in each breast cancer subtype [25]. Missense mutations were found to be more prevalent compared to truncating and inframe mutations in all subtypes (Kruskal–Wallis test, P < 0.0001). Maftools analysis showed that TP53 was the most frequently mutated gene across IDC, LumA, HER2 enriched, basal and TNBC subtypes. PIK3CA was the second most commonly mutated gene across ILC subtypes (Fig. 1c). Interestingly, HER2-enriched subtype showed the highest rate of TP53 mutations (86%, P = 1E-7), and the ILC subtype showed the highest rate of PIK3CA mutations (48%, q = 1E-4). IDC subtype showed the significant enrichment of mutations in 7/49 (14%) actionable genes when compared with other subtypes. Notably, only in basal subtype 21/49 genes (43%) were mutated in ≥2% of cases when compared with other subtypes, suggesting that actionable changes predominantly differ among different subtypes (Fig. 1d). Oncodrive favours the detection of oncogenic events and is less efficient in identifying mutations in tumour suppressor genes. We also applied the OncodriveFML approach that identifies drivers by computing the functional impact (FI) score using the set of somatic mutations in a gene [19, 26]. Quantile–quantile plots and the most significant genes for each breast cancer subtype are presented in Supplementary Fig. S1a and Supplementary Table S1. A total of 9/49 genes displayed a significant P value (P < 0.05) and a q value <0.05 in all seven breast cancer subtypes. These nine genes fell into five categories of tumour drivers: mTOR pathway (PIK3CA, AKT1 and NF1), immune checkpoints (GATA3), oestrogen signalling (ESR1), tumour suppressor activators (TP53, RB1) and PARP (ATM, BRCA2).
The frequency of actionable mutations in 49 targets in ILC and IDC subtypes were also compared between early breast cancer (EBC) (TCGA cohort) and two metastatic breast cancer (MBC) cohorts: Razavi et al. [27] and the Genomics, Evidence, Neoplasia, Information, Exchange (GENIE) [28] cohort. We observed a higher frequency of mutations in metastatic cases than in primary breast cancer in six genes (ESR1, BRCA2, RB1, TP53, ERBB2 and AKT1) in ILC and seven genes (ESR1, GATA3, BARD1, CDKN2A, ERBB2, AKT1 and NF1) in IDC subtype (Supplementary Fig. S1b). For HER2-enriched cases, no significant differences were observed in the frequencies of mutations in the TCGA breast cohort versus Angus et al. [29] and Lefebvre et al. [30] metastatic cohorts. The frequency of mutations in TNBC TCGA cases was also compared with Angus et al. [29] study and no significant differences were observed between early and metastatic breast cases.
Mutual exclusivity and co-occurrence among actionable alterations and therapeutic actionability
We identified 22 pairs of mutually exclusive alterations and 72 pairs of co-occurring alterations using Maftools (Fig. 2a and Supplementary Table S2). TP53 was mutually exclusive with PIK3CA in ILC, IDC and LumA subtypes. We also found numerous mutually exclusive pairs of TP53 with GATA3, BRCA2 and NF1 in more than one breast cancer subtype. Interestingly, AKT1 mutations were also found mutually exclusive with PIK3CA mutations in 45% of LumA cases. Among co-occurrences, we found many alterations affecting genes in different pathways rather than in the same pathway (Supplementary Table S2). For instance, the co-occurrence of BRCA2 and FGFR2 in IDC and LumB tumours, NF1 with RPTOR, PIK3CB and ERBB3 in IDC and HER2-enriched tumours.

a Mutually exclusive (in pink) or co-occurrence (in green) of gene alterations in breast cancer subtypes. Asterisks indicate significant P values (bold <0.01, non-bold <0.05). Fisher’s exact test was used within Maftools to identify significant somatic interactions. b Left panel: Bar plot showing the percentage of samples with actionable alterations in each subtype, broken down by level of evidence (levels 1–4) using the OncoKB database. Right panel: Heatmap showing the fraction of samples with actionable alterations per gene across breast cancer subtypes. c Boxplots showing the gene expression level of 13 actionable genes in wild type (WT) (green), missense (MS) (blue), and truncating mutated (TM) (red) cases across breast cancer subtypes. The x axis showed the expression level (z score) of wild-type and mutated cases.
We also evaluated the level of clinical evidence of mutations in 49 genes in 7 breast cancer subtypes, using the OncoKB database. Overall, >30% of tumours in ILC, IDC, LumA, LumB and >10% of tumours in TNBC and basal subtypes harboured level 1 alterations (FDA-approved) in PIK3CA gene. In OncoKB database, genomic alterations in NF1 and ATM are classified as level 1 in neurofibroma and prostate cancer, respectively. Likewise, genomic alterations in BRCA1 and BRCA2 are classified as level 1 in the ovary and prostate cancer. In basal and TNBC subtypes, we found that <10% of the patients harbour same alterations in NF1, BRCA1, BRCA2 and ATM genes that are classified as level 1 alterations in other cancers. Among all breast cancer subtypes, very few cases (<10%) had mutations in BRCA1, BRCA2, ERBB2, AKT1, ESR1, KRAS, MAP2K1, ATM, PALB2, NF1, CDKN2A and FGFR2 genes that were annotated as levels 2–4 (Fig. 2b). According to the OncoKB classification, ESR1 oncogenic mutations are classified as level 3A in breast cancer [20]. Three missense ESR1 mutations (E380Q, Y537S and D538G) are ranked tier IIA in breast cancer according to ESMO classification. Both of these tools provide consistent variant annotations based on their levels of clinical and/or experimental evidence. Patients with ESR1 mutations gain less benefit from aromatase inhibitor therapy but are predicted to benefit from fulvestrant [31]. We also observed a significant association of gene expression levels with mutations in 13/49 genes in mutated breast cancer cases compared with wild-type cases (Fig. 2c).
Taken together, these results revealed that TP53 (34%) and PIK3CA (35%) were the most frequently mutated genes in primary breast cancer patients. PIK3CA mutations were enriched in ILC and luminal tumours, especially LumA subtype. In addition, >30% of the primary breast tumours in ILC, IDC, LumA and LumB harboured potentially actionable mutations. A total of nine genes (PIK3CA, AKT1, PIK3CB, NF1, RPTOR, TP53, GATA3, FGFR2 and BRCA2) formed mutually exclusive pairs (group of genes that are genetically altered, tend not to occur in the same patient and are less common than is expected by random chance). Proliferation, stem cell signalling and mismatch repair targets were frequently co-altered across breast cancer subtypes. The landscape of somatic mutations affecting potentially actionable genes varied markedly between different breast cancer subtypes, revealing genetic heterogeneity between individuals.
Detection of breast cancer-associated CNAs in actionable genes
The genome-wide landscape of CNAs indicates widespread alterations across different breast cancer subtypes (Fig. 3a). Consistent with previous studies [32,33,34], our analysis confirmed high-level amplifications (>50%) in chromosome 1q and 8q as well as deletions (>50%) in 8p, 16q and 17p in breast cancer patients irrespective of intrinsic subtypes. We identified subtype-specific CNAs such as amplification at 10p and deletions at 5q, 14q and 15p in basal and TNBC subtypes, 16p amplification in LumA, 16p, 17q, 20q amplification in LumB, and 17q in HER2-enriched subtypes (Fig. 3a).

a Genome-wide copy number landscape showing the widespread alterations in each breast cancer subtype. The y axis showed the percentage of the occurrence of somatic gains (red) and losses (blue) in sub-cytoband regions. b Significant amplified and deleted regions identified by GISTIC, which contains potentially actionable genes from our list.
To identify the most clinically relevant alterations in breast cancer subtypes, we focused on identifying CNAs in our list of 49 potentially actionable genes [15]. GISTIC analysis identified 17 loci with amplifications and 30 loci affected by deletions in breast cancer subtypes that were statistically significant (Fig. 3b and Supplementary Table S3). Significant copy number amplification in ESR1 and NF1 (q = 0.0001) was identified only in IDC patients. Similarly, JAK2 was significantly amplified only in IDC (q = 0.02) and basal (q = 0.03). Significant copy number deletions were identified in the tumour suppressor gene RB1 in IDC (q = 0.0001) and basal subtype (q = 0.01). Only IDC patients had significant copy number deletions in proliferative signalling targets (CDKN2A, CDKN2B and TP53). PIK3CA was most commonly altered in all 6 subtypes except ILC. Overall, we identified 28 actionable genes that were amplified and 4 genes deleted across the TCGA breast cancer dataset (Supplementary Table S3). A significant association of CNAs with RNA expression was observed in 34/49 actionable genes (Table 2).
These results indicate that copy number amplifications and deletions do not occur randomly but instead arise at specific genomic regions potentially affecting dosage of gene(s) and may confer a growth advantage to tumour cells in breast cancer. The subset of these alterations would result in the specific therapeutic vulnerabilities of these subtypes. Although breast cancer is complex and highly heterogeneous at the genomic level, these results highlight subtype-selective dependencies that can potentially guide treatment strategies for patients harbouring these actionable alterations.
Transcriptomic profiling of actionable genes
We next analysed and compared the transcriptomes of actionable genes in seven breast cancer subtypes. We considered candidate genes with a z score ≥2 or ≤−2 to be significantly expressed. The transcriptional landscape of actionable genes in seven breast cancer subtypes is presented in Supplementary Fig. S2. We found significantly upregulated expression of GRB7 a common target for IDC (P = 0.001) and HER2-enriched (P-1E-5) tumours. ERBB2 was overexpressed in these two subtypes. We also identified many genes that can distinguish breast cancer subtypes based on RNA expression of actionable genes. For example, cell cycle signalling target (CCNE1 (P = 0.00009)) characterises the basal subtype. Likewise, proliferative signalling targets (EGFR (P < 0.00001)), cell cycle signalling targets (CCNE1 (P = 0.005) and stem cell signalling target (MET(P = 0.006)) characterise TNBCs.
Pathway analysis
We performed IPA analysis to identify the pathways that were altered by gene mutations in each breast cancer subtype. We identified 281 signalling pathways in IDC, 253 in HER2 enriched, 246 in LumA, 243 in basal, 242 in TNBC, 236 in LumB and 221 in ILC that have a −log(P value) greater than 1.3. Of note, we found 298 overlapping pathways in all 7 breast cancer subtypes primarily involved in DNA damage repair pathway, ERBB signalling, hereditary breast cancer signalling, immunoregulatory pathways, NOTCH signalling, PIK3/AKT signalling and several others. The top 15 signalling pathways in each subtype are presented in Fig. 4. The Fisher’s exact test was used to identify significant pathways (P < 0.05).

Bar chart showing the enriched canonical pathways related to actionable genes in each subtype. The y axis on the left shows the percentage of genes overlapping in each pathway having pathogenic (pink) and likely pathogenic (blue) variants. The y axis on the right shows the significance level. The number on the top of each stacked bar indicates the total number of genes present in each pathway. The orange line represents the threshold value (0.05) for the significance level for −log(P value). The graph is displaying only those entities that have a −log (P value) greater than 1.3.
Discussion
The emerging field of precision oncology implies that comprehensive tumour profiling is required for the identification of actionable alterations for targeted therapy. The FDA-approved FoundationOne assay, MSK-IMPACT actionable gene panel and Caris comprehensive tumour profiling offer a targeted approach for the identification of actionable alterations. However, the major challenge remains in the identification of truly actionable variants, particularly when there is variation in their methodology, sequencing platforms and data analysis pipelines. Moreover, this variation also creates an additional level of complexity for clinicians for diagnosis and/or guiding treatment. Recently, Tsimberidou and Kurzrock [35] pointed out serious weaknesses in the SHIVA trial by stating that monotherapies given to patients with advanced cancer are less effective than combinations of drugs. Thus, a deeper understanding of the detailed mechanism and co-occurrence of these actionable alterations is critical for the rational development of new therapeutic approaches that can improve patient care.
In this study, our approach is primarily based on three steps: (1) reducing the genomic complexity by selecting alterations of clinical relevance; (2) to identify which actionable alterations are associated with gene expression and if this association varies by breast cancer subtype and (3) to identify biological networks affected by these alterations by deciphering the mutations, copy number and gene expression status of potentially actionable genes. We applied this framework to analyse the TCGA breast cancer dataset to perform a comprehensive characterisation of 49 potentially clinically actionable targets relevant to breast cancer treatment [15]. The analysis revealed similarities and differences in frequencies of alteration of actionable genes in seven breast cancer subtypes. More generally, we identified that there are relatively few genes that are associated with high-frequency mutations in comparison to CNAs. In our analysis, the nine most frequently altered genes correlated with five key pathways of tumorigenesis: mTOR pathway (PIK3CA, AKT1 and NF1), immune checkpoints (GATA3), oestrogen signalling (ESR1), tumour suppressor activators (TP53, RB1) and PARP (ATM, BRCA2). Both of the TCGA and METABRIC studies have also shown that CNAs are the dominant feature in breast cancer [10, 14]. The first notable finding in our study is that CNAs in actionable genes 34/49 (69%) in the TCGA dataset displayed a significant association with gene expression. For all seven breast cancer subtypes, we found variability in the frequencies of amplifications and deletions in actionable genes. Copy number amplifications were strikingly more common than deletions in proliferative, cell cycle, stem cell, and mismatch repair target genes in all breast cancer subtypes. This indicated that amplification events could be the principal mechanism for the deregulated gene expression in breast cancer. Interestingly, we also identified some actionable genes such as GRB7, CCND1, FGFR1, AKT3, RPTOR, ERBB2 and PALB2 whose expression level is tightly controlled by copy number amplification only (Table 2), not by mutations, in breast cancer subtypes. These results suggest that the majority of the breast cancer genetic drivers are, in fact, the result of CNAs and that perturbations of these actionable genes with altered expression levels could therefore be potential therapeutic targets. These findings provide a strong rationale for exploring copy number changes for potentially actionable genes that are linked to FDA-approved or investigational therapeutics.
Another notable finding in this study is that mutations in 13/49 (26%) genes were significantly associated with gene expression (Fig. 2c), indicating that not all mutations are expressed and actionable. Furthermore, 17/49 (35%) genes from our actionable gene list were found to be significantly mutated in a significant proportion of early-stage breast cancer patients (Supplementary Table S1). These 17 genes are predicted to be the targets of drug inhibitors including mTOR, RAS, CDKs, NOTCH, immune checkpoints, oestrogen receptor and PARP. Consistent with the TCGA study [14], we found that only three of these driver genes were altered in ≥20% of cases (PIK3CA in ILC, IDC, LumA, LumB and basal; TP53 in IDC, LumB, HER2 enriched, basal and TNBC; GATA3 in LumA). Other actionable genes were altered in 0–20% of breast cancer cases. Importantly, we found that only basal subtype (≥2% of cases) had mutations in 21/49 actionable genes. These results suggest high variability of mutations in clinically actionable genes and a lack of recognisable subtype-specific pattern of genomic alterations, thus reflecting substantial heterogeneity.
Mutually exclusive or co-occurring alterations have been frequently observed within several tumour types [11, 36,37,38,39,40]. The co-occurrence of genomic alterations contributes to the heterogeneity of oncogenic drivers [41] and can generate vulnerabilities that confer therapeutic resistance [42]. Our third notable finding is that we identified a complex interplay of co-occurring and mutually exclusive actionable alterations between individual tumours and within each subtype, representing attractive candidate targets for combination therapies. We found a high prevalence of significant co-occurrence alterations and many mutually exclusive alterations within proliferative signalling, cell cycle, immunoregulatory, and mismatch repair targets. For instance, TP53 mutations were found to be mutually exclusive with mutations affecting GATA3, BRCA2, RPTOR, NF1 and PIK3CA in more than one breast cancer subtype. BRCA2 and TP53 mutations were mutually exclusive in HER2-enriched, basal and TNBC tumours. These results suggest that one alteration is sufficient to perturb the activity of the pathway associated with these genes.
Our correlation analysis suggested that both genetic alterations and RNA expression are important in predicting actionability even if they may not be associated with each other. Several ongoing trials are evaluating the synthetic lethality of PARP inhibitors with DNA-damaging agents (e.g., platinum-based therapies) in BRCA-mutated tumours, few have translated to clinical success (https://clinicaltrials.gov/ct2/show/NCT02000622?term=nct02000622&rank=1; https://clinicaltrials.gov/ct2/show/NCT02032823?term=nct02032823&rank=1; https://clinicaltrials.gov/ct2/show/NCT02163694?term=nct02163694&rank=1; https://clinicaltrials.gov/ct2/show/NCT02032277?term=nct02032277&rank=1) [43,44,45]. Therefore, targeting the co-occurrence of mutual exclusive alterations might be more effective in eliminating cancer cell population [46, 47]. We identified many alterations among mutually exclusive or co-occurrence pairs, such as TP53, which are still under investigation as potential therapies (levels 3 or 4).
Pathway analysis showed alterations in many breast cancer-related pathways such as hereditary breast cancer signalling, HER2 signalling, TP53, PI3K/AKT and cell cycle regulation. We also observed co-occurrence patterns in these genes indicating a potential for combinational therapies in these breast cancer subtypes. Many preclinical studies have shown that PIK3CA mutations are the possible cause for therapeutic resistance in HER2-positive breast cancer patients [48]. There is increasing preclinical evidence indicating that the therapeutic effects of certain drug combinations are superior to standard care and monotherapy [49]. As an example, promising results were present by the combination of PI3K inhibitors and HER2 inhibitors in HER2-positive/PIK3CA mutant breast cancer [50].
There are several limitations to our approach. We focused on a curated list of highly relevant genes that has no claim on being complete. Few of these 49 biomarkers/drug targets are currently available for clinical use outside of early phase clinical trials and are usually reserved for metastatic breast cancer. Some exceptions to this are HER2 (trastuzumab, neratinib, T-DM1, pertuzumab). For hormone receptor-positive, HER2-negative disease, endocrine-based therapy is a nice example of how utilising combination therapy to target multiple pathways results in improved clinical outcomes. For example, patients with an identified PIK3CA mutation are eligible to receive fulvestrant (an oestrogen receptor degrader) in combination with alpelisib (a PIK3CA targeted agent) [51]. Similarly, many patients receive combination aromatase inhibitor with CDK4/6 inhibitors in the metastatic setting. Alterations in the ESR1 gene have been shown to predict resistance to aromatase inhibitors but the benefit to fulvestrant [31]. Furthermore, sample size varies between different breast cancer subtypes in the TCGA dataset (51–490) potentially skewing the analysis in favour of larger sample groups. The current analysis requires further exploration of certain actionable alterations that occur at very low frequencies. In addition, the selection of candidate functional events depends on the integrative analysis of mutation, CNAs, gene expression and protein translation which has not been addressed in this analysis in a systems biology approach.
The current landscape of precision medicine is characterised by the availability of many targeted agents [52], yet very little is known about potentially actionable therapeutic targets in different subtypes of breast cancer. Other databases also provide insight into specific potentially actionable molecular alteration—although, these have varying degrees of curation. Thus, more caution should be exercised when interpreting these genetic alterations. There is some functional evidence where these inhibitors seem to be effective in in vivo models, but the final picture seems to be complex as these agents are often not successful in clinical trials [53,54,55].
Through integrated genomic analysis, we have identified clinically relevant genomic alterations across 49 genes in different subtypes of breast cancer using one of the largest genomic datasets. A key contribution of our study is that our results expand the search for potentially targetable alterations combining transcriptomic and genomic data that goes beyond a single-gene approach by analysing co-occurrence gene-interaction network that could also help for the identification of effective personalised cancer treatment. Our strategy is also based on identifying the molecular pathways affected by clinically relevant alterations in breast cancer subtypes. Breast cancer clinical trials have frequently shown that rational drug combination regimens are more effective than monotherapy. Our results highlight an opportunity to enhance single-agent therapeutic strategies currently under investigation in clinical trials, as patients may have more than one driver mutation or CNA. Such information, if appropriately implemented, could help in the design of rational clinical trials and eventually more effective therapies.
Data availability
The data analysed in this study are available in the TCGA GDC data portal and cBioportal platform.
References
Chin L, Andersen JN, Futreal PA. Cancer genomics: from discovery science to personalized medicine. Nat Med. 2011;17:297–303. https://doi.org/10.1038/nm.2323
Spitale A, Mazzola P, Soldini D, Mazzucchelli L, Bordoni A. Breast cancer classification according to immunohistochemical markers: clinicopathologic features and short-term survival analysis in a population-based study from the South of Switzerland. Ann Oncol. 2009;20:628–35. https://doi.org/10.1093/annonc/mdn675
Tang P, Wang J, Bourne P. Molecular classifications of breast carcinoma with similar terminology and different definitions: are they the same? Hum Pathol. 2008;39:506–13. https://doi.org/10.1016/j.humpath.2007.09.005
Li CI, Anderson BO, Daling JR, Moe RE. Trends in incidence rates of invasive lobular and ductal breast carcinoma. J Am Med Assoc. 2003;289:1421–4.
Tyanova S, Albrechtsen R, Kronqvist P, Cox J, Mann M, Geiger T. Proteomic maps of breast cancer subtypes. Nat Commun. 2016;7:10259 https://doi.org/10.1038/ncomms10259
Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. https://doi.org/10.1038/35021093
Harris LN, Ismaila N, McShane LM, Andre F, Collyar DE, Gonzalez-Angulo AM, et al. Use of biomarkers to guide decisions on adjuvant systemic therapy for women with early-stage invasive breast cancer: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2016;34:1134–50. https://doi.org/10.1200/jco.2015.65.2289
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. https://doi.org/10.1038/nature12477
Aran D, Sirota M, Butte AJ. Systematic pan-cancer analysis of tumour purity. Nat Commun. 2015;6:8971 https://doi.org/10.1038/ncomms9971
Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. https://doi.org/10.1038/nature10983
Ciriello G, Miller ML, Aksoy BA, Senbabaoglu Y, Schultz N, Sander C. Emerging landscape of oncogenic signatures across human cancers. Nat Genet. 2013;45:1127–33. https://doi.org/10.1038/ng.2762
Cardoso F, Kyriakides S, Ohno S, Penault-Llorca F, Poortmans P, Rubio IT, et al. Early breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up†. Ann Oncol. 2019;30:1194–220. https://doi.org/10.1093/annonc/mdz173
Wolff AC, Hammond ME, Schwartz JN, Hagerty KL, Allred DC, Cote RJ, et al. American Society of Clinical Oncology/College of American Pathologists guideline recommendations for human epidermal growth factor receptor 2 testing in breast cancer. Arch Pathol Lab Med. 2007;131:18–43. https://doi.org/10.1043/1543-2165(2007)131[18:Asocco]2.0.Co;2
Ciriello G, Gatza ML, Beck AH, Wilkerson MD, Rhie SK, Pastore A, et al. Comprehensive molecular portraits of invasive lobular breast cancer. Cell. 2015;163:506–19. https://doi.org/10.1016/j.cell.2015.09.033
Kaur P, Porras TB, Ring A, Carpten JD, Lang JE. Comparison of TCGA and GENIE genomic datasets for the detection of clinically actionable alterations in breast cancer. Sci Rep. 2019;9:1482 https://doi.org/10.1038/s41598-018-37574-8
Van Allen EM, Wagle N, Stojanov P, Perrin DL, Cibulskis K, Marlow S, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nat Med. 2014;20:682–8. https://doi.org/10.1038/nm.3559
Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol. 2016;34:155–63. https://doi.org/10.1038/nbt.3391
Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018;28:1747–56. https://doi.org/10.1101/gr.239244.118
Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. OncodriveFML: a general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 2016;17:128 https://doi.org/10.1186/s13059-016-0994-0
Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, et al. OncoKB: a precision oncology knowledge base. JCO Precision Oncol. 2017. https://doi.org/10.1200/po.17.00011.
Franch-Expósito S, Bassaganyas L, Vila-Casadesús M, Hernández-Illán E, Esteban-Fabró R, Díaz-Gay M, et al. CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications. eLife 2020;9. https://doi.org/10.7554/eLife.50267.
Deng M, Bragelmann J, Schultze JL, Perner S. Web-TCGA: an online platform for integrated analysis of molecular cancer data sets. BMC Bioinforma. 2016;17:72 https://doi.org/10.1186/s12859-016-0917-9
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. https://doi.org/10.1038/gim.2015.30
Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. https://doi.org/10.1016/j.cell.2012.04.024
Mayakonda A, Koeffler HP. Maftools: efficient analysis, visualization and summarization of MAF files from large-scale cohort based cancer studies. bioRxiv:052662,2016 [Preprint]. 2016. Available from: https://doi.org/10.1101/052662.
Tamborero D, Gonzalez-Perez A, Lopez-Bigas N. OncodriveCLUST: exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics. 2013;29:2238–44. https://doi.org/10.1093/bioinformatics/btt395
Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34:427–.e426. https://doi.org/10.1016/j.ccell.2018.08.008
AACR Project GENIE: powering precision medicine through an International Consortium. Cancer Discov. 2017; 7:818, https://doi.org/10.1158/2159-8290.CD-17-0151.
Angus L, Smid M, Wilting SM, van Riet J, Van Hoeck A, Nguyen L, et al. The genomic landscape of metastatic breast cancer highlights changes in mutation and signature frequencies. Nat Genet. 2019;51:1450–8. https://doi.org/10.1038/s41588-019-0507-7
Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria J-C, et al. Mutational profile of metastatic breast cancers: a retrospective analysis. PLoS Med. 2016;13:e1002201 https://doi.org/10.1371/journal.pmed.1002201
Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 2016;7:11579 https://doi.org/10.1038/ncomms11579
Bergamaschi A, Kim YH, Wang P, Sørlie T, Hernandez-Boussard T, Lonning PE, et al. Distinct patterns of DNA copy number alteration are associated with different clinicopathological features and gene-expression subtypes of breast cancer. Genes, Chromosomes Cancer. 2006;45:1033–40. https://doi.org/10.1002/gcc.20366
Koboldt DC, Fulton RS, McLellan MD, Schmidt H, Kalicki-Veizer J, McMichael JF, et al. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. https://doi.org/10.1038/nature11412
Weigman VJ, Chao HH, Shabalin AA, He X, Parker JS, Nordgard SH, et al. Basal-likebreast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res Treat. 2012;133:865–80. https://doi.org/10.1007/s10549-011-1846-y
Tsimberidou AM, Kurzrock R. Precision medicine: lessons learned from the SHIVA trial. Lancet Oncol. 2015;16:e579–580. https://doi.org/10.1016/s1470-2045(15)00397-6
Boca SM, Kinzler KW, Velculescu VE, Vogelstein B, Parmigiani G. Patient-oriented gene set analysis for cancer mutation data. Genome Biol. 2010;11:R112 https://doi.org/10.1186/gb-2010-11-11-r112
Ciriello G, Cerami E, Sander C, Schultz N. Mutual exclusivity analysis identifies oncogenic network modules. Genome Res. 2012;22:398–406. https://doi.org/10.1101/gr.125567.111
Kim JW, Botvinnik OB, Abudayyeh O, Birger C, Rosenbluh J, Shrestha Y, et al. Characterizing genomic alterations in cancer by complementary functional associations. Nat Biotechnol. 2016;34:539–46. https://doi.org/10.1038/nbt.3527
Park S, Lehner B. Cancer type-dependent genetic interactions between cancer driver alterations indicate plasticity of epistasis across cell types. Mol Syst Biol. 2015;11:824–824. https://doi.org/10.15252/msb.20156102
Vandin F, Upfal E, Raphael BJ. De novo discovery of mutated driver pathways in cancer. Genome Res. 2012;22:375–85. https://doi.org/10.1101/gr.120477.111
Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509. https://doi.org/10.1038/s41568-019-0179-8
Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–42. https://doi.org/10.1038/nature11331
O’Shaughnessy J, Osborne C, Pippen J, Yoffe M, Patt D, Monaghan G, et al. Efficacy of BSI-201, a poly (ADP-ribose) polymerase-1 (PARP1) inhibitor, in combination with gemcitabine/carboplatin (G/C) in patients with metastatic triple-negative breast cancer (TNBC): results of a randomized phase II trial. J Clin Oncol. 2009;27:3–3. https://doi.org/10.1200/jco.2009.27.18_suppl.3
Tutt A, Robson M, Garber JE, Domchek S, Audeh MW, Weitzel JN, et al. Phase II trial of the oral PARP inhibitor olaparib in BRCA-deficient advanced breast cancer. J Clin Oncol. 2009;27:CRA501–CRA501. https://doi.org/10.1200/jco.2009.27.18_suppl.cra501
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009;361:123–34. https://doi.org/10.1056/NEJMoa0900212
Cisowski J, Sayin VI, Liu M, Karlsson C, Bergo MO. Oncogene-induced senescence underlies the mutual exclusive nature of oncogenic KRAS and BRAF. Oncogene. 2016;35:1328–33. https://doi.org/10.1038/onc.2015.186
Etemadmoghadam D, Weir BA, Au-Yeung G, Alsop K, Mitchell G, George J, et al. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc Natl Acad Sci USA. 2013;110:19489–94. https://doi.org/10.1073/pnas.1314302110
Loibl S, von Minckwitz G, Schneeweiss A, Paepke S, Lehmann A, Rezai M, et al. PIK3CA mutations are associated with lower rates of pathologic complete response to anti-human epidermal growth factor receptor 2 (her2) therapy in primary HER2-overexpressing breast cancer. J Clin Oncol. 2014;32:3212–20. https://doi.org/10.1200/jco.2014.55.7876
Levinson AD. Cancer therapy reform. Science. 2010;328:137 https://doi.org/10.1126/science.1189749
Fujimoto Y, Morita TY, Ohashi A, Haeno H, Hakozaki Y, Fujii M, et al. Combination treatment with a PI3K/Akt/mTOR pathway inhibitor overcomes resistance to anti-HER2 therapy in PIK3CA-mutant HER2-positive breast cancer cells. Sci Rep. 2020;10:21762 https://doi.org/10.1038/s41598-020-78646-y
André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med. 2019;380:1929–40. https://doi.org/10.1056/NEJMoa1813904
Tan DS, Thomas GV, Garrett MD, Banerji U, de Bono JS, Kaye SB, et al. Biomarker-driven early clinical trials in oncology: a paradigm shift in drug development. Cancer J. 2009;15:406–20. https://doi.org/10.1097/PPO.0b013e3181bd0445
Chow L, Sun Y, Jassem J, Baselga, J, Hayes D, Wolff A, et al. Phase 3 study of temsirolimus with letrozole or letrozole alone in postmenopausal women with locally advanced or metastatic breast cancer. Proc Breast Cancer Res Treat. 2006;S286.
Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46. https://doi.org/10.1038/nrd4504
Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES. Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene. 2010;29:4018–32. https://doi.org/10.1038/onc.2010.154
Acknowledgements
The authors wish to acknowledge the TCGA Research Network for sharing the TCGA breast cancer genomic datasets. The results presented here are in whole or part based upon data generated by TCGA managed by the NCI and NHGRI. We thank the USC Libraries Bioinformatics Service for assisting with data analysis. The bioinformatics software and computing resources used in the analysis are funded by the USC Office of Research and the Norris Medical Library.
Funding
The project was supported in part by award number P30CA014089 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institute of Health. The project was also supported by the Woodbury Foundation.
Author information
Authors and Affiliations
Contributions
Study design: PK and JEL; data acquisition: PK and JEL; data analysis: PK, JEL, AC, TP and AR; manuscript drafting: PK and JEL; critical revisions: PK, JEL, AR, TP, AC, JL and IK; funding: JEL.
Corresponding author
Ethics declarations
Competing interests
Dr. Kang is a member of Puma Biotechnology speaker bureau. The remaining authors declare no competing interests.
Ethics approval and consent to participate
The study was performed in strict accordance with the recommendations of data access guidelines of TCGA datasets. We received administrative permission for downloading the restricted-access data for breast cancer patients from the TCGA Data Access Committee (Project # 28198). The study was performed in accordance with the Declaration of Helsinki.
Consent to publish
Not applicable.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kaur, P., Porras, T.B., Colombo, A. et al. Identification of putative actionable alterations in clinically relevant genes in breast cancer. Br J Cancer 125, 1270–1284 (2021). https://doi.org/10.1038/s41416-021-01522-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01522-7
- Springer Nature Limited
This article is cited by
-
Identification of modules and key genes associated with breast cancer subtypes through network analysis
Scientific Reports (2024)