
Abstract
Liver cancer is one of the most prevalent cancers, and the third most common cause of cancer-related mortality worldwide. The therapeutic options for the main types of primary liver cancer—hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA)—are very limited. HCC and CCA are immunogenic cancers, but effective immune-mediated tumour control is prevented by their immunosuppressive tumour microenvironment. Despite the critical involvement of key co-inhibitory immune checkpoint interactions in immunosuppression in liver cancer, only a minority of patients with HCC respond to monotherapy using approved checkpoint inhibitor antibodies. To develop effective (combinatorial) therapeutic immune checkpoint strategies for liver cancer, in-depth knowledge of the different mechanisms that contribute to intratumoral immunosuppression is needed. Here, we review the co-inhibitory pathways that are known to suppress intratumoral T cells in HCC and CCA. We provide a detailed description of insights from preclinical studies in cellular crosstalk within the tumour microenvironment that results in interactions between co-inhibitory receptors on different T-cell subsets and their ligands on other cell types, including tumour cells. We suggest alternative immune checkpoints as promising targets, and draw attention to the possibility of combined targeting of co-inhibitory and co-stimulatory pathways to abrogate immunosuppression.
Similar content being viewed by others


Background
Liver cancer is one of the most prevalent and aggressive cancers, and represents the third most common cause of cancer-related mortality worldwide.1 The most common primary liver cancer is hepatocellular carcinoma (HCC), an aggressive malignancy derived from hepatocytes. A second main type of primary liver cancer, cholangiocarcinoma (CCA), currently accounts for 10% of primary liver cancers, but its incidence is rising steadily.2,3 CCA is an aggressive hepatobiliary malignancy originating from the biliary tract epithelium with features of cholangiocyte differentiation.2,3 Potentially curative treatment options for both types of liver cancer, such as surgical resection and liver transplantation, are available for patients with early-stage disease; unfortunately, however, at the time of first presentation around 80% of patients are beyond this stage.4,5 Traditionally, multikinase inhibitors such as sorafenib and lenvatinib have been the only effective drugs for the treatment of advanced HCC.6 Sorafenib profoundly downregulates RAF/RAS/MAPK and STAT3/Akt signaling pathways, which are both crucial for proliferation and survival of HCC cells.7 The potent activity of lenvatinib against pro-angiogenic signalling pathways FGFRs 1-4 is a distinctive feature of lenvatinib.8 Chemotherapy is the only systemic treatment for advanced CCA. However, these treatments prolong patient survival by only a few months.4,5 Therefore, more effective (most likely combinatorial) approaches for the treatment of primary liver cancer and for preventing cancer recurrence are urgently needed.
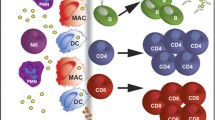
HCC and CCA have both been shown to be immunogenic,9,10 but effective immune-mediated tumour control is prevented by their immunosuppressive tumour microenvironment (TME). Monotherapy with antibodies that inhibit interactions between co-inhibitory receptors on T cells and their ligands on tumour cells and other cells in the tumour microenvironment, such as those mediated by cytotoxic T-lymphocyte-associated protein 4 (CTLA4), programmed cell death-1 (PD-1) and programmed cell death ligand-1 (PD-L1), has shown remarkable clinical efficacy in patients with different types of malignancy, including melanoma,11,12 non-small cell lung cancer13,14 and renal cell carcinoma.15 However, despite the critical involvement of key co-inhibitory immune checkpoint interactions in immunosuppression in liver cancer, checkpoint inhibitors have shown limited clinical effects in patients with HCC or CCA. To develop more effective therapeutic immune checkpoint strategies for the treatment of liver cancers, further knowledge of the different immune inhibitory checkpoint mechanisms that contribute to intratumoral T-cell suppression in liver cancer is needed. In this review, we provide an overview of insights from preclinical studies regarding the interactions between co-inhibitory receptors expressed on different T-cell subsets and their ligands expressed on other cell types within the liver TME (Fig. 1). We briefly summarise the results of clinical studies of agents designed to overcome the PD-1- or CTLA4-mediated inhibition of T cells in liver cancer, and suggest alternative immune checkpoints that could be targeted to abrogate intratumoral T-cell co-inhibition. Furthermore, we highlight the current gaps in our knowledge of T-cell co-inhibitory pathways in HCC and CCA. Although some co-inhibitory pathways can also suppress natural killer (NK) cells, we will focus on their effects on T-cell immunity.

a Potential interactions between co-inhibitory receptors on T cells and their corresponding ligands on tumour cells or antigen-presenting cells (dendritic cells, macrophages/monocytes) in HCC or CCA. b Expression of co-inhibitory ligands on diverse cell types and expression of co-inhibitory receptors on different T cells in HCC or CCA tumours, according to the data currently available.
Anti-tumour T-cell responses and their inhibition
HCC and CCA are both immunogenic—that is, they are both capable of soliciting an immune response by virtue of their expression of tumour-associated antigens (TAAs) and neoantigens that can be recognised as ‘foreign’ entities.
Immunogenicity in liver cancers
Tumour-associated antigens
Tumour-antigen-specific CD4+ T cells and CD8+ T cells, which recognise classic tumour-associated antigens (TAAs), have been detected in the circulation of HCC patients,16,17 and are also present in tumour tissue.10,18 Higher numbers of intratumoral CD8+ T cells in liver cancer are associated with better survival.19 Amongst the classic TAAs present in HCC cells and recognised by T cells in HCC patients are cancer-testis antigens, oncofetal antigens, and overexpressed antigens.10,16,17,20 Whether tumour-specific T cells that recognise classic TAAs exist in CCA patients, and whether those cells are functionally competent is still unclear. However, Löffler 21 reported that TAA-specific T cells could be induced in a patient with CAA by TAA-peptide vaccination, and that this response resulted in strong immune cell infiltration into the tumour lesions.
Neoantigens
Liver cancers also express neoantigens encoded by non-synonymous somatic mutations in protein-encoding DNA sequences that are only present in tumour cells.22,23,24 Their absence during fetal development makes these neoantigens highly specific targets for anti-tumour T-cell immunity,25,26 in which the neoantigenic peptides are presented by MHC molecules expressed on tumour cells and recognised by the patient’s T cells. MHC-binding prediction tools have been used to predict that such mutations in HCC give rise to potential neoantigenic peptides that can be presented by MHC class I molecules to CD8+ T cells.27,28 Moreover, HLA-immunopeptidome data have proven that neoantigenic epitopes are indeed presented by MHC class I in HCCs, albeit at relatively low levels. The same study provided the first experimental evidence for the presence of CD8+ T cells that recognise neoantigenic peptides in HCC patients.29 CD4+ tumour-infiltrating T cells (TILs) that recognise a mutated antigen have been identified in the tumour of a patient with CCA, and these TILs were effective in mediating tumour regression upon in vitro expansion followed by adoptive transfer.24 Neoantigen-specific CD8+ T cells have not yet been identified in CCA patients.
Loss of immune-mediated anti-tumour responses
Increasing evidence indicates that liver tumour cells can be recognised by T cells, and that T cells that recognise classical TAAs as well as those that recognise tumour-encoded neoantigens are present in HCC patients and CCA patients. Nevertheless, spontaneous T-cell immunity is apparently insufficient to clear the tumour. Several mechanisms are responsible for the loss of effective immune-mediated anti-tumour responses in the control of many types of cancer, including HCC and CCA.
Loss of neoantigens and MHC
Many types of tumour downregulate MHC class I molecules or have defective peptide processing or loading machinery; consequently, immunotherapy aimed at stimulating CD8+ T-cell responses will not be successful in such tumours.30 Notably, however, the expression of MHC class I molecules is actually upregulated in most HCC cells,29,31 although, in a minority of HCC patients, loss of heterozygosity of HLA alleles can hamper the ability of tumour cells to present antigens to CD8+ T cells.29 The possible mechanisms that regulate the expression of MHC class I on HCC cells remain controversial. MHC class I expression on HCC cells is related to underlying viral infections and other inflammatory liver diseases, and cytokines such as interferon (IFN)-γ released by T cells infiltrated in HCC tissue might contribute to the increased expression.31 Interestingly, loss of HLA heterozygosity was observed in HCCs in which tumour-infiltrating T cells showed a limited T-cell receptor (TCR) diversity, whereas tumours infiltrated by T cells with a high TCR diversity showed limited neoantigen expression.29 These data suggest that most HCCs try to escape immune pressure through the loss of neoantigens, but partial loss of HLA occurs in a minority. However, MHC class I expression in CCA shows wider differences between patients,32,33 ranging from complete negativity, to heterogeneous expression, to strong homogeneous expression in tumours,;32,33 therefore tumour cells can probably not be recognised by CD8+ T cells in a fraction of CCA patients.
T-cell dysfunction
The inability of T cells to eliminate tumours is probably at least partially due to the number of functional tumour-specific T cells being insufficient.16 Indeed, it has been demonstrated in a mouse liver cancer model that T cells become dysfunctional early during tumorigenesis at the premalignant phase.34 T-cell dysfunction is a cell-intrinsic status that is initially reversible but subsequently becomes irreversible owing to epigenetic changes. The process of a T cell becoming dysfunctional is largely due to an antigen-driven dynamic differentiation program in response to continuous exposure to tumour antigen(s) rather than microenvironmental factors,34 although the intratumoral expression of the immune co-inhibitory molecule B7-H4 by infiltrating myeloid cells might exacerbate T-cell exhaustion in HCC.35 This exhaustion program in mouse liver tumours is driven by the transcription factor TOX.36 In microsatellite stable colorectal cancer, vascular endothelial growth factor-A (VEGF-A) induces TOX expression, which increases the levels of the co-inhibitory immune checkpoint molecules PD-1, TIM3, LAG3 and TIGIT (see below) in CD8+ T cells and thereby drives T-cell exhaustion.37 Whether this occurs in liver cancer is as yet unknown, but the overexpression of VEGF in HCC is associated with disease progression.38
Inhibition of migration of immune effector cells into tumors
Additionally, effector T cells might be unable to enter the tumours. We have shown that both HCC and CCA tumors contain lower numbers of cytotoxic immune cells such as NK cells and CD8+ T cells compared to tumor-free liver tissues of the same patients.9,18 This suggests that tumors may inhibit immigration of cytotoxic immune cells, which is another way of immune evasion. For several other cancer types it has been demonstrated that collagen and endothelial barriers prevent infiltration of immune cells.39,40 The TME creates multiple defense mechanisms limiting T cells from migrating and reaching the tumor bed, the dysregulated extracellular matrix and tumor-associated macrophages make tumors a hostile environment for T-cell ability to contact and kill malignant cells.41,42 Chemoattractant molecules are also essential for regulating T-cell motile behavior such as T-cell trafficking into and within the tumors,41 chemokines and their receptors directly or indirectly shape the TME and regulate the biological behavior of tumor.43
Immunosuppressive mechanisms
A fourth important constraint for effective anti-tumour immunity is the presence of immunosuppressive mechanisms within the TME, including the intratumoral accumulation of immunosuppressive cells such as regulatory T (TREG) cells, anti-inflammatory macrophages and other myeloid cells;44,45 the intratumoral expression of enzymes, such as indoleamine 2,3-dioxygenase (IDO) and arginase, that catalyse the generation of immunosuppressive metabolites, such as kynurenine; and the intratumoral expression of immune inhibitory checkpoint molecules.
Co-inhibitory immune checkpoints
Mechanisms of suppressive function
In this review we will focus on the contribution of co-inhibitory immune checkpoint pathways to intratumoral T-cell inhibition in liver cancers. Co-inhibitory receptor-ligand interactions can mediate immune-suppressive functions at least by the following mechanisms (Fig. 2). (i) Upon binding by their corresponding ligand, co-inhibitory receptors can deliver suppressive signals within the T cells on which they are expressed; these signals suppress activatory receptor signalling (e.g. by the T-cell-receptor or CD28). (ii) Co-inhibitory receptor binding can deliver suppressive signals to the ligand-expressing cells. (iii) Co-inhibitory receptors can compete with co-stimulatory receptors for the same ligand. (iv) Co-inhibitory receptors can block homodimerization of co-stimulatory receptors, thereby preventing the co-stimulatory signal from being generated.46,47,48,49

Several mechanisms of immune-suppressive function can be mediated by co-inhibitory receptor-ligand interactions. (i) Upon binding by their corresponding ligand, co-inhibitory receptors can deliver suppressive signals within the T cells on which they are expressed; these signals suppress activatory receptor signalling (e.g. by the T-cell-receptor (TCR) or CD28). (ii) Co-inhibitory receptor binding can deliver suppressive signals to the ligand-expressing cells (e.g. PD-1 binding to PD-L1 induces functional changes in antigen-presenting cells). (iii) Co-inhibitory receptors can compete with co-stimulatory receptors for the same ligand (e.g. CTLA4 and CD28 compete for CD80 and CD86, TIGIT and CD226 compete for CD155). (iv) Co-inhibitory receptors can block homodimerization of co-stimulatory receptors, thereby preventing the co-stimulatory signal from being generated (TIGIT physically prevents CD226 from signaling). Purple arrows indicate stimulatory signals, red arrows indicate inhibitory signals.
Below we outline current knowledge of the interactions between co-inhibitory receptors expressed on different T-cell subsets and their ligands expressed on other cell types within the liver TME.
CTLA4 and CD80 (B7-1)/CD86 (B7-2)
CTLA4 is the target of the first therapeutic immune checkpoint inhibitor that was approved (ipilimumab for the treatment of melanoma).
Targeting CTLA4 in HCC
CTLA4 is highly expressed on tumour-infiltrating CD4+Foxp3+ TREG cells in patients with HCC,18,50 and anti-CTLA4 antibodies have been shown to partially alleviate the suppressive capacity of these cells in HCC patients.51 CTLA4 expression is also induced on effector T cells upon activation, and interaction with its ligands, CD80 or CD86, directly inhibits effector T-cell functions. We have demonstrated that blocking CTLA4 increases the responses of TILs from HCC patients against TAAs ex vivo, and that combined blockade of CTLA4 and PD-L1 further enhances these responses.10
Although these preclinical data show that CTLA4 blockade can reinvigorate the responses of T cells derived from HCCs, only one clinical study using anti-CTLA4 antibody monotherapy in HCC patients has been carried out (Table 1). A phase 2 clinical trial showed that treatment with the anti-CTLA4 agent tremelimumab resulted in a partial response in 17.6% of patients with advanced HCC associated with hepatitis C virus.52 In another clinical trial, 26.3% of evaluable advanced HCC patients achieved a partial response after treatment with tremelimumab in combination with subtotal radiofrequency ablation or chemoablation (Table 2).53 Activation of tumor-specific T cells and a decrease in T-cell clonality indicating broadening of the T-cell receptor repertoire, was seen in the peripheral blood of HCC patients upon tumour ablation combined with tremelimumab treatment, but it is unclear whether these changes were caused by tumour ablation or tremelimumab treatment.54 One potential reason for the limited efficacy of tremelimumab might be that this antibody belongs to the human IgG2 antibody subclass, which has limited capacity for antibody-dependent T-cell-mediated cytotoxicity; the efficacy of anti-CTLA4 therapy can be at least partly attributed to the intratumoral depletion of TREG cells by antibody-dependent cell-mediated cytotoxicity (ADCC).55,56,57 The anti-CTLA4 agent ipilimumab belongs to the IgG1 subclass, which can better mediate ADCC, and has shown robust anti-tumour effects in many cancer types; however, its clinical efficacy in HCC has not yet been investigated.
Targeting CTLA4 in CCA
Zhou et al.9 have shown that TILs from CCA patients express increased levels of CTLA4 compared with T cells from matched tumour-free liver tissue and blood, and that ipilimumab increases the proliferation of CD4+ and CD8+ TILs ex vivo, suggesting that anti-CTLA4 therapy might be able to enhance intratumoral T-cell reactivity.9 In a clinical trial of patients with biliary tract cancer, including CCA, 12.5% achieved a partial response upon treatment with tremelimumab and microwave ablation; however, the relative contribution of anti-CTLA4 treatment to this result is unclear.58 Ongoing clinical trials are investigating whether combination treatments of anti-CTLA4 with anti-PD-L1 or anti-PD-1 demonstrate clinical efficacy in HCC and CCA (Table 2).59 The results of the first few studies have been published and are discussed below.
PD-1 and PD-L1 (B7-H1)/PD-L2 (B7-DC)
Blockade of the interaction between the co-inhibitory receptor PD-1 and its ligand PD-L1 has shown enormous therapeutic success and has been approved for the treatment of several types of cancer over the past few years.60,61,62
PD-1 and PD-L1/PD-L2 expression in HCC
The PD-1–PD-L1 pathway has been relatively well-studied in HCC. In HCC patients, PD-1 is overexpressed on intratumoral CD4+ and CD8+ T cells compared with T cells in tumour-free liver tissue and blood; PD-L1 is expressed on intratumoral monocytes/macrophages, whereas the expression of PD-L1 on tumour cells is strongly variable between patients.10,63,64 Much less is known about PD-1–PD-L2 pathway in HCC. PD-L2 is expressed on the surface of tumour cells in HCC tissue,65 but very limited evidence is available for the involvement of the PD-1–PD-L2 co-inhibitory pathway in HCC.66
HCC patients with aggressive tumours have a discrete subset of CD8+ PD-1high T cells in their tumours that express multiple markers of T-cell exhaustion, including the co-inhibitory receptors TIM3 and LAG3,67 and high levels of TOX, which, as mentioned previously, is involved in T-cell exhaustion.68 CD8+ tissue-resident memory T cells and TREG cells from hepatitis B (HBV)-associated HCCs express more PD-1 and are functionally more exhausted and suppressive than their counterparts from non-virus-associated HCC.69 As the surface expression levels of PD-1 and exhaustion status of tumour-infiltrating CD8+ T cells from HCC patients are promoted by TOX, downregulating TOX expression exerts synergistic effects with anti-PD-1 therapy in improving the anti-tumour function of HCC patient-derived tumour-infiltrating CD8+ T cells in immunocompromised mice transplanted with tumour material derived from HCC patients.68 Data obtained from an orthotopic mouse liver cancer model indicate that hepatocyte growth factor (HGF), which has been associated with tumor initiation and progression through HGF/c-Met signaling pathways,70 might contribute to the enhanced expression of PD-1 on tumour-infiltrating T cells in HCC.71
The expression of PD-L1 on tumour cells is induced by IFN-γ produced by pre-existing, activated CD8+ T cells in the HCC milieu, and might represent an adaptive immune resistance mechanism in response to endogenous anti-tumour activity.72 PD-L1 expression on cancer cells and stromal cells is also promoted by hypoxia in HCC.73 Additionally, tumour-derived soluble factors including hyaluronan fragments enhance the levels of glycolysis in tumour-associated monocytes, which increases the expression of PD-L1 on these cells and subsequently attenuates cytotoxic T-cell responses in HCC.74 In orthotopic-grafted and induced murine models of HCC, VEGFR-2 was selectively expressed in tumor endothelial cells; and PD-L1 expression in murine HCC cells was found to be induced in a paracrine manner upon antibody-mediated VEGFR-2 blockade in endothelial cells and in part through IFN-γ expression by endothelial cells, particularly in hypoxic conditions which mimic the in vivo effects.75 Furthermore, the expression of the transcription factor myocyte enhancer factor 2D (MEF2D) by both human and mouse HCC cells increases PD-L1 expression.76 Depletion of the tumour-intrinsic cell-cycle related kinase (CCRK) oncogene upregulates PD-L1 expression and consequently improves the efficacy of anti-PD-L1 blockade to eradicate mouse HCC.77 By contrast, the epigenetic modifier EZH2, a histone methyltransferase, suppresses the expression of PD-L1 on human hepatoma cell lines.78 PD-L1 expression in HCCs is therefore regulated by different environmental factors, which might explain the differences in the levels of PD-L1 observed in tumours from individual patients.
Targeting the PD-1–PD-L1 interaction in HCC patients
In ex vivo assays, blockade of PD-1/PD-L1 restores the functionality of TILs of HCC patients,10,67 and PD-L1 blockade has also been shown to restore ex vivo TAA-specific responses of tumour-infiltrating CD4+ and CD8+ T cells from HCC patients.10 Moreover, the immunosuppression exerted by tumour-derived PD-1+ TREG cells is reversed with anti-PD-1 blockade.69 HCC tumour-infiltrating PD-1+ memory CD8+ T cells are the predominant T-cell subset to respond to anti-PD-1 treatment in in vitro studies.44
More data are available on the clinical efficacy of blockade of PD-1/PD-L1 in HCC patients compared with CTLA4 blockade (Table 1). In the first phase 1/2 clinical trial, the anti-PD-1 nivolumab resulted in an objective response rate of 20% in patients with advanced HCC regardless of etiology.79 The objective response rate of the intent-to-treat sorafenib-experienced population was 14%.80 The anti-PD-1 antibody pembrolizumab resulted in an objective response in 17% of patients with advanced HCC previously treated with sorafenib in a phase 2 trial.81 In a subsequent phase 3 trial, pembrolizumab induced objective responses in 18% of patients, but did not prolong patient survival.82 When studied in a phase 2 trial of previously treated patients with advanced HCC, the novel anti-PD-1 antibody camrelizumab achieved an objective response rate of 15%.83 The anti-PD-L1 antibody avelumab showed moderate efficacy, with a partial response rate of 10% in patients with advanced HCC.84 These studies show the potential of PD-1 blockade in HCC. However, similar to several other cancer types, only a subpopulation of patients with HCC responds to anti-PD-1 monotherapy, and most of these patients showed incomplete responses. Therefore, further research should aim to combine anti-PD-1 treatment with other therapeutic options to improve its efficacy.
Independent of its role in adaptive immunity, PD-1 can promote the growth of HCC by binding and promoting the phosphorylation of eukaryotic initiation factor 4E and ribosomal protein S6, which function downstream of the mammalian target of rapamycin (mTOR). Combining anti-PD-1 antibody treatment with mTOR inhibition using MLN0128/INK128 resulted in more durable and synergistic tumour regression compared with either single agent alone in mice with HCC.85 Similarly, the dual blockade of PD-1 and VEGFR-2 overcame treatment resistance to either agent alone, inhibited primary tumour growth and doubled survival in HCC murine models.75 In a phase 3 trial involving patients with unresectable HCC, the anti-PD-L1 antibody atezolizumab combined with the anti-VEGF bevacizumab antibody indeed resulted in better overall and progression-free survival than did sorafenib treatment (Table 2).86 Nivolumab combined with ipilimumab induced an objective response in 27–32% of patients with advanced HCC,87 while the combination of nivolumab and the TREG cell-depleting anti-CCR4 antibody mogamulizumab induced a partial response rate of 27%.88 In patients with unresectable HCC treated with pembrolizumab plus lenvatinib, an objective response was achieved in 36% of patients.89 In summary, combining anti-PD-1/PD-L1 antibodies with an additional target for immunomodulation seems to be a promising way to proceed.
Targeting the PD-1–PD-L1 interaction in CCA patients
In patients with CCA, the expression of PD-1 and PD-L1 is associated with intratumoral immunosuppression, indicating that this inhibitory checkpoint might be a potential therapeutic target for this type of liver cancer as well.33,90,91,92,93,94,95,96,97 Whole-genome analysis and integrative clustering showed that fluke-negative CCA tumours exhibited high PD-1 and PD-L2 expression,98 while another integrative genomic analysis revealed that CCA tumours in which adaptive immune response genes were upregulated had overexpression of PD-1 and PD-L1 and molecular features associated with a better response to checkpoint inhibitors shown in other types of cancer.99 Increased PD-1 expression has been demonstrated on CD4+ and CD8+ TILs in CCA patients, and nivolumab was shown to enhance the production of effector cytokines in TILs cultured ex vivo.9
In a clinical trial investigating anti-PD-1 therapy in diverse types of cancer deficient in mismatch repair (MMR) genes (thus increasing their tumour mutational burden and rendering them more immunogenic), pembrolizumab resulted in tumour regression in a few of the MMR-deficient CCA patients that were included (Table 1).100 In another cohort of MMR-deficient cancers treated with pembrolizumab, an objective response rate of 40.9% was observed in those patients with CCA.101 In a separate multicentre phase 2 study, nivolumab resulted in an objective response in 24% of CCA patients and all responders had MMR-proficient tumours.102 Nivolumab in combination with ipilimumab induced an objective response in 31% of MMR-proficient patients with intrahepatic CCA but none in patients with extrahepatic CCA.103 Clinical trials of anti-PD-1/PD-L1 antibodies in combination with other treatments in CCA patients are currently ongoing (Table 2).59,104
CD276 (B7-H3)
B7-H3 is an immune inhibitory protein expressed on tumour cells and antigen-presenting cells, with an otherwise limited expression in healthy tissue. Its receptor is yet unknown.105,106 B7-H3 has been shown to suppress anti-tumour T-cell responses.107,108 HCCs express strongly enhanced levels of B7-H3 compared with healthy liver tissue,109 and this higher expression level on tumour cells is associated with aggressive tumour features, poor survival110,111,112 and increased recurrence after tumour resection.112,113 Moreover, B7-H3 expression on tumour cells correlates inversely with TIL proliferation, as measured by Ki-67 expression, suggesting that B7-H3 might inhibit intratumoral T-cell expansion in HCC (Table 3).112
Targeting B7-H3
Several antibodies against B7-H3 are being tested in clinical trials.114 However, because the receptor and exact function of B7-H3 are as yet unknown, these antibodies have been designed to induce ADCC against B7-H3-expressing tumours rather than to block B7-H3. Patients with intrahepatic CCA that were deficient in B7-H3 expression showed higher overall survival and cancer-specific survival rates than those with B7-H3 expression.115 Together, the available preliminary data suggest that B7-H3 might be a potential target for the induction of ADCC against liver cancers. However, it is as yet unclear whether B7-H3 serves as a T-cell co-inhibitory molecule in liver cancers.
VTCN1 (B7-H4)
V-set domain-containing T-cell activation inhibitor 1 (VTCN1/B7-H4) is another co-inhibitory ligand that suppresses T cells,116 but whose receptor is as yet unknown. Human single chain antibody fragments against B7-H4 can restore anti-tumour T-cell responses in vitro.117 The currently available antibodies against B7-H4 can induce ADCC and block inhibition of T cells. B7-H4 is not expressed in healthy liver, but 45% of HCC tumours express B7-H4 and its expression positively correlates with aggressive tumour features. The levels of B7-H4 expression are higher in HBV-positive HCCs than in HBV-negative HCCs.118 Interestingly, in a mouse HCC model, B7-H4 promoted T-cell exhaustion via upregulation of the transcription factor Eomes, and its increased expression on myeloid cells from human HCCs correlates with intratumoral CD8+ T-cell dysfunction. B7-H4 blockade synergised with PD-1 blockade in eliciting anti-tumour responses in mouse models of liver cancer (Table 3 and Table 4).35
In CCA patients, B7-H4 expression in tumour cells negatively correlates with the density of CD8+ T cells in the tumour stroma,119 suggesting that B7-H4 might inhibit T-cell infiltration and/or survival in CCA tumours. Knockdown of B7-H4 in CCA cell lines was able to restore cytotoxic T-cell function in co-culture experiments.119 Similar to B7-H3, extensive research including elucidation of the receptor of B7-H4 is required to clarify whether B7-H4 serves as a co-inhibitory ligand in liver cancers.
VISTA (B7-H5)
B7-H5, also known as V-domain immunoglobulin suppressor of T-cell activation (VISTA),120,121 is a co-inhibitory molecule that shares homology with PD-L1.120,121,122,123 VISTA is highly expressed on myeloid cells and TREG cells in the TME of murine cancer models.124 Preclinical studies have demonstrated a potential role for VISTA blockade in the anti-tumour T-cell response, leading to impeded tumour growth and improved survival.125 Combined treatment using monoclonal antibodies specific for VISTA and PD-L1 achieved synergistic therapeutic efficacy in murine tumour models.126 Similar to B7-H3 and B7-H4, the putative receptor for VISTA is unknown. In HCC patients, VISTA is expressed on both tumour cells and intratumoral immune cells; VISTA expression was associated with a high density of CD8+ TIL, a high pathological grading and absence of liver cirrhosis; and patients with dual positive VISTA+ cells and CD8+ cells in the TME showed a better overall survival (Table 3).127 Further preclinical investigation of the potential role of VISTA in the suppression of TILs in liver cancers is required.
HHLA2 (B7-H7)
B7-H7, also called HERV-H LTR-associating 2 (HHLA2), has both co-inhibitory and co-stimulatory effects on T cells, depending on its activation history. Its co-stimulatory receptor on T cells is CD28H (TMIGD2), which is expressed on naïve CD4+ and CD8+ T cells.128 The existence of an unknown co-inhibitory receptor that is expressed on activated memory T cells has been postulated to explain its co-inhibitory function.129 HHLA2 protein is widely expressed in human cancers, with preliminary data indicating that it is expressed by tumour cells in a subgroup of HCC patients.130 In CCA tumours, HHLA2 is more frequently expressed than PD-L1, and has been identified as an independent prognostic indicator for overall survival (Table 3).131 The expression and potential function of HHLA2 in liver cancer immunity awaits further research.
TIM3 and galectin-9
T-cell immunoglobulin and mucin domain-containing molecule 3 (TIM3) is a negative regulator of T-cell functions in viral infections and cancer.132,133,134,135 In HCC patients, intratumoral CD4+ and CD8+ T cells express high levels of TIM3, whereas its ligand galectin-9 is expressed on tumour cells in most patients, as well as on intratumoral Kupffer cells, dendritic cells and B cells.10,64,136 The long non-coding RNA Lnc-TIM3 promotes CD8+ T-cell exhaustion in HCC patients via binding to TIM3.137 Blocking the TIM3–galectin-9 interaction improved the ex vivo functionality of CD4+ and CD8+ TILs from HCC patients,10,136 and the combined blockade of TIM3 and PD-L1/PD-1 synergistically enhances ex vivo CD4+ and CD8+ TIL functions.10,67
Interestingly, in HCC patients, TIM3 is not only expressed on T cells but also on peripheral blood monocytes and tumour-associated macrophages. Downregulation or blockade of TIM3 on macrophages suppressed HCC cell growth in an experimental mouse model (Table 3 and Table 4).138 Together, the data suggest that TIM3 might be a promising target for blockade therapy in HCC. Clinical trials on several different anti-TIM3 antibodies, either alone or in combination with anti-PD-1, are ongoing for HCC patients (NCT03680508, NCT03652077).59 The expression and function of TIM3 and whether TIM3 is involved in suppression of the anti-tumour response in CCA are unknown.
LAG3
Lymphocyte activating 3 (LAG3) is a co-inhibitory receptor involved in the regulation of T-cell expansion and function.139,140
LAG3 expression on T cells and MHC class II molecules as LAG3 ligands
The interaction between LAG3 and its major ligand, MHC class II, is implicated in the regulation of dendritic cell function and in maintaining tolerance of CD8+ T cells.141,142 In several murine non-liver cancer models, LAG3 and PD-1 are co-expressed on tumour-infiltrating CD8+ and CD4+ T cells, and the combined blockade of LAG3 and PD-1 synergised to improve anti-tumour CD8+ T-cell responses.143 In humans, the co-expression of LAG3 and PD-1 was reported to mark dysfunctional CD8+ T cells in ovarian cancer, and the combined blockade of LAG3 and PD-1 improved the cytokine production and proliferation of TAA-specific CD8+ T cells derived from ovarian cancer patients.144 In patients with HCC, tumour-infiltrating TREG cells and tissue-resident memory CD8 + T cells express multiple markers for T-cell exhaustion, including LAG3 and PD-1.33,134 Our results indicate that LAG3 expression is increased on TAA-specific CD8 + TILs in HCC patients, and that LAG3 blockade increased the responses of CD4 + and CD8 + TILs ex vivo;8 the combined blockade of LAG3 and PD-L1 additively enhanced the effects.8,55 Many clinical trials studying LAG3 blockade as a monotherapy or in combination with anti-PD-1 antibodies are currently ongoing in patients with diverse types of cancer.59
LSECtin and FGL1 as additional LAG3 ligands
Experimental evidence indicates that LSECtin, a type II transmembrane protein of the C-type lectin receptor superfamily, can serve as an alternative ligand to MHC class II molecules for LAG3 and that the LAG3–LSECtin interaction inhibits anti-tumour T-cell responses in melanoma.145 As LSECtin is highly expressed on liver sinusoidal endothelial cells,146 this interaction might also be relevant for liver cancer. Fibrinogen-like protein 1 (FGL1), a liver-secreted protein, has also been demonstrated in mice to be a major ligand of LAG3, independent of MHC II. Blocking the FGL1–LAG3 interaction using monoclonal antibodies stimulated anti-tumour immunity and was therapeutically effective against mouse colon tumours, and preliminary data suggest that inhibiting this interaction also reduced tumour growth by a HCC cell line inoculated subcutaneously into syngeneic mice. FGL1 mRNA was also found to be expressed in liver cancer, although at lower levels compared to normal liver tissue (Table 3 and Table 4).147 Together, the preclinical data suggest that LAG3 might be a promising co-inhibitory target for immunotherapy in HCC patients. Currently, a clinical trial of LAG3 blockade that involves HCC patients (NCT03538028) and a clinical trial with both HCC and CCA patients combining LAG3, CTLA4 and PD-1 blockade (NCT03849469) are ongoing.59 In contrast to HCC, LAG3 expression is not increased on TILs in CCA patients,9 and therefore the suitability of LAG3 blockade as immunotherapy in CCA is questionable.
BTLA, CD160, CD244; and HVEM and CD48
Other co-inhibitory receptors expressed on T cells include B and T-lymphocyte attenuator (BTLA), CD160 and CD244 (2B4). Interaction with their ligands HVEM, HVEM and CD48, respectively, might also suppress anti-tumour T-cell responses.140,148
HVEM is expressed on tumour cells in almost all HCC patients,64 and the proportion of HVEM+ tumour cells is inversely associated with the number of tumour-infiltrating CD8+, CD4+ and CD45RO+ lymphocytes, as well as the expression of granzyme B, perforin and IFN-γ in HCC tissue,149 suggesting that HVEM expression might suppress TILs number and function. However, we found that TILs from HCC and CCA patients express only low levels of BTLA.,9,10 and our unpublished and published data show reduced CD160 expression on TILs in HCC and CCA tumours compared with paired tumour-free liver tissue. Nevertheless, a minor subset of BTLA+ PD-1+ CD4+ TILs in HCC was shown to be highly dysfunctional, indicating that BTLA signals might participate in suppressing CD4+ TILs in HCC.150
CD48 is expressed on monocytes/macrophages in HCC tissue,151 but our unpublished and published data demonstrate that CD244 is expressed at similar levels on CD8+ T cells in tumours and paired tumour-free liver tissue from HCC and CCA patients (Table 3).9 Blockade of CD244 could therefore result in liver immunotoxicity due to unwanted targeting of highly expressed CD244 on CD8+ T cells in the tumour-free liver. Together, these data suggest that CD160–HVEM and CD244–CD48 co-inhibitory pathways are probably not good targets for checkpoint inhibitors in liver cancer, whereas the potential involvement of the BTLA–HVEM interaction in the regulation of tumour-specific immunity in liver cancers requires further investigation.
TIGIT and CD155 (PVR)
T-cell immunoreceptor with Ig and ITIM domains (TIGIT) is a co-inhibitory receptor that limits anti-tumour and other CD8+ T-cell-dependent chronic immune responses.152,153 The high affinity ligand of TIGIT, poliovirus receptor (PVR/CD155), is expressed on dendritic cells and endothelial cells, and overexpressed on tumour cells in many types of cancer.154 TIGIT shares CD155 with the co-stimulatory receptor CD226 and reportedly counterbalances CD226 signalling.155 TIGIT is highly expressed on TILs in several types of solid cancer, and blockade of both TIGIT and PD-L1 specifically and synergistically enhanced CD8+ T-cell function in models of both cancer and chronic viral infection, resulting in tumour and viral clearance.156 In melanoma patients, TIGIT expression is elevated on TAA-specific CD8+ T cells, which often co-express PD-1.157 Accordingly, blocking TIGIT and PD-1 enhanced the functions of TAA-specific CD8+ T cells in the presence of TIGIT-ligand-expressing cells ex vivo.157
TIGIT and CD155 expression in HCC
Murine HCC cells upregulate the expression of poliovirus-receptor-related 1 (PVRL1), which stabilises the cell-surface expression of CD155 and suppresses the CD8+ T-cell response via TIGIT; accordingly, blocking either PVRL1 or TIGIT overcame tumour resistance to PD-1 blockade in mice.158 The expression levels of TIGIT and CD155 are higher in more undifferentiated cancerous tissue than in highly differentiated cancerous tissue from HCC patients, suggesting that the TIGIT–CD155 pathway might be involved in the pathogenesis of HCC.159 CD155 expression in HCCs is increased compared to surrounding non-cancerous liver tissue, and higher CD155 expression levels in tumours are associated with poor survival after tumour resection,160 supporting an immunosuppressive function of CD155. Recently, in ex vivo experiments using TILs from human HCC, we demonstrated that co-blockade of TIGIT and PD-1 improves functionality of CD8+ TILs that do not respond to single PD-1 blockade. Therefore co-blockade of TIGIT and PD-1 could be a promising immune therapeutic strategy for HCC patients (Table 3 and Table 4).161
TIGIT and CD155 expression in CCA
Similar to HCC, the mRNA and protein expression of CD155 is increased in tumour tissue from CCA patients compared with corresponding para-cancerous tissue. Increased levels of CD155 were associated with aggressive clinicopathologic characteristics, angiogenesis and shorter survival after surgical resection in CCA patients.162 No data on the expression or function of TIGIT are yet available for CCA. Based on the limited preclinical data that are available, blocking the TIGIT–CD155 interaction might be an interesting strategy for patients with liver cancer. Whereas clinical trials that involve co-blocking TIGIT and PD-1/PD-L1 in multiple solid tumours are ongoing, no drugs targeting TIGIT have yet been evaluated in liver cancer patients, to our knowledge.
Combined targeting of T-cell co-inhibitory and co-stimulatory pathways
T-cell dysfunction in cancer patients can be overcome not only by the use of antagonistic antibodies that block co-inhibitory pathways, but also by using agonistic antibodies that bind to co-stimulatory receptors to stimulate T cells. Most T-cell stimulatory antibodies so far developed for cancer immunotherapy are directed against co-stimulatory molecules of the tumour necrosis factor receptor superfamily (TNFRSF), such as CD134 (OX40), CD137 (4-1BB) and the glucocorticoid-induced tumour necrosis factor receptor (GITR).163,164 The expression of OX40 on T cells is higher in tumours of HCC patients than in adjacent liver tissue.165 Treatment with an Fc-engineered anti-OX40 antibody (anti-OX40_v12) with selectively enhanced FcγRIIB affinity stimulated the expansion in vitro of TILs from HCC patients, as well as their secretion of cytokines and chemokines.166 An adenovirus expressing a human soluble fusion protein comprising the extracellular domains of PD-1 and the CD137 ligand CD137L suppressed tumour growth in a CD8+ T-cell-dependent way by activating the CD137 pathway and blocking the PD-L1–PD-1 pathway and improved survival in a humanised mouse HCC model.167 CD137 expression was indicated to mark a distinct activation state among highly exhausted PD-1high CD8+ TILs from HCC patients, and a CD137 agonistic antibody enhanced the ex vivo functions of CD8+ TILs and showed additive effects on CD8+ TIL responses in combination with anti-PD-1 therapy.168 Ex vivo agonistic GITR engagement partially reduces the suppression exerted by tumour-infiltrating TREG cells derived from HCC patients.18 Increased GITR expression has been reported on tumour-infiltrating effector T cells in both HCC and CCA patients, and the agonistic ligation of GITR enhances the ex vivo functions of CD4+ and CD8+ TILs from HCC patients as well as from CCA patients.9,169
A promising new development for cancer immunotherapy is combined treatment with antibodies that target co-inhibitory molecules and antibodies that target co-stimulatory molecules. We demonstrated that a combination of low doses of CTLA4 blocking antibody and GITR agonistic ligand completely abrogated the ex vivo immunosuppression mediated by tumour-infiltrating TREG cells from HCC patients.51 Moreover, PD-1 blockade in combination with GITR ligation further invigorated TAA-specific responses of TILs from some patients with HCC.169 No data on the combined targeting of co-inhibitory and co-stimulatory molecules in CAA are available. The combination of a GITR agonistic antibody and PD-1 and/or CTLA4 antagonistic antibodies (NCT04021043, NCT03126110) is currently being tested in clinical trials involving patients with liver cancer.59
Conclusions and future perspectives
The preclinical data summarised in this article support that, in addition to the PD-1 and CTLA4 pathways, the TIM3, LAG3 and TIGIT co-inhibitory pathways are also involved in suppression of T cells in the TME of HCC. Therefore, these co-inhibitory receptors and their ligands should be considered as promising targets for immune checkpoint therapy in HCC. Clinical trials of antibody blockade of TIM3 and LAG3, but not TIGIT blockade, are currently ongoing in HCC patients. Whether these pathways contribute to intratumoral immunosuppression in CCA is as yet unknown, and requires further investigation. These novel co-inhibitory interactions should not only be considered as targets for single antibody blockade, but also for combined treatment in conjunction with anti-PD-1/PD-L1 antibodies. Combinatorial treatments of anti-CTLA4 and anti-PD-L1/PD-1 have demonstrated additive clinical efficacy in several types of cancer, and clinical studies investigating these combinations, as well as combinations with anti-TIM3 or anti-LAG3 antibodies, are ongoing in patients with HCC and CCA.
There is an urgent need to expand our understanding of liver cancer immunology in order to develop more effective immunotherapeutic treatments for patients with primary liver cancer. Our knowledge of the immunological TME of CCA, in particular, is very limited. We need to deepen our understanding of the expression and function of alternative promising immune inhibitory checkpoint molecules, such as PD-L2 and BTLA, in the liver cancer microenvironment. In addition, we need to identify the missing receptors of various immune checkpoint ligands, such as B7-H3, B7-H4, VISTA and HHLA2, and to study their impact on different immune cells in the TME of liver cancer. We also need to understand the mechanistic basis that underlies the synergistic effects observed by targeting different immune checkpoint molecules. Moreover, as tumours can shield the influx of cytotoxic immune cells—for example, through collagen or endothelial barriers39,40,170—ways to break down such barriers should be studied with a view to boosting the infiltration of immune cells into liver tumours.
Evidence exists that the gut microbiome impacts the response to checkpoint inhibitors in cancer treatment,171,172,173,174 which is a gap of knowledge in HCC and CCA and is worth investigating. Preliminary data suggest that systemic antibiotic treatment might indeed be associated with a worse outcome of anti-PD-1 therapy in patients with HCC.175 The safety and effectiveness of various strategies that combine differential treatments need to be tested, including immune checkpoint antibodies with adoptive cell transfer, cytokines or vaccines, and immunotherapy with molecular targeted therapy or gene therapy. Identifying and validating biomarkers to facilitate patient stratification to be able to individualise and tailor treatments is of imminent importance. For example, in HCC, the intratumoral expression of PD-L1 does not accurately predict the response to anti-PD-1 therapy.79,81 Interestingly, somatic mutations that lead to β-catenin activation have been suggested to predict the resistance of HCC patients to immune checkpoint inhibitor therapy.176,177 Comprehensive molecular profiling of tissue from patients with primary liver cancer treated with different types of immunotherapy might therefore enable the identification of molecular biomarkers that can assist patient selection for optimal personalised immunotherapy.
References
Bray, F., Ferlay, J., Soerjomataram, I., Siegel, R. L., Torre, L. A. & Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
Patel, T. Cholangiocarcinoma-controversies and challenges. Nat. Rev. Gastroenterol. Hepatol. 8, 189–200 (2011).
Rizvi, S. & Gores, G. J. Pathogenesis, diagnosis, and management of cholangiocarcinoma. Gastroenterology 145, 1215–1229 (2013).
Razumilava, N. & Gores, G. J. Cholangiocarcinoma. Lancet 383, 2168–2179 (2014).
Forner, A., Reig, M. & Bruix, J. Hepatocellular carcinoma. Lancet 391, 1301–1314 (2018).
Kudo, M., Finn, R. S., Qin, S., Han, K. H., Ikeda, K., Piscaglia, F. et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 391, 1163–1173 (2018).
Matsuda, Y. & Fukumoto, M. Sorafenib: complexities of Raf-dependent and Raf-independent signaling are now unveiled. Med. Mol. Morphol. 44, 183–189 (2011).
Al-Salama, Z. T., Syed, Y. Y. & Scott, L. J. Lenvatinib: a review in hepatocellular carcinoma. Drugs 79, 665–674 (2019).
Zhou, G., Sprengers, D., Mancham, S., Erkens, R., Boor, P. P. C., van Beek, A. A. et al. Reduction of immunosuppressive tumor microenvironment in cholangiocarcinoma by ex vivo targeting immune checkpoint molecules. J. Hepatol. 71, 753–762 (2019).
Zhou, G., Sprengers, D., Boor, P. P. C., Doukas, M., Schutz, H., Mancham, S. et al. Antibodies against immune checkpoint molecules restore functions of tumor-infiltrating T cells in hepatocellular carcinomas. Gastroenterology 153, 1107–19 e10 (2017).
Khair, D. O., Bax, H. J., Mele, S., Crescioli, S., Pellizzari, G., Khiabany, A. et al. Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front. Immunol. 10, 453 (2019).
Schadendorf, D., van Akkooi, A. C. J., Berking, C., Griewank, K. G., Gutzmer, R., Hauschild, A. et al. Melanoma. Lancet 392, 971–984 (2018).
Hirsch, F. R., Suda, K., Wiens, J. & Bunn, P. A. Jr. New and emerging targeted treatments in advanced non-small-cell lung cancer. Lancet 388, 1012–1024 (2016).
Vansteenkiste, J., Wauters, E., Reymen, B., Ackermann, C. J., Peters, S. & De Ruysscher, D. Current status of immune checkpoint inhibition in early-stage NSCLC. Ann. Oncol. 30, 1244–1253 (2019).
Atkins, M. B., Clark, J. I. & Quinn, D. I. Immune checkpoint inhibitors in advanced renal cell carcinoma: experience to date and future directions. Ann. Oncol. 28, 1484–1494 (2017).
Flecken, T., Schmidt, N., Hild, S., Gostick, E., Drognitz, O., Zeiser, R. et al. Immunodominance and functional alterations of tumor-associated antigen-specific CD8+ T-cell responses in hepatocellular carcinoma. Hepatology 59, 1415–1426 (2014).
Mizukoshi, E., Nakamoto, Y., Arai, K., Yamashita, T., Sakai, A., Sakai, Y. et al. Comparative analysis of various tumor-associated antigen-specific t-cell responses in patients with hepatocellular carcinoma. Hepatology 53, 1206–1216 (2011).
Pedroza-Gonzalez, A., Verhoef, C., Ijzermans, J. N., Peppelenbosch, M. P., Kwekkeboom, J., Verheij, J. et al. Activated tumor-infiltrating CD4+ regulatory T cells restrain antitumor immunity in patients with primary or metastatic liver cancer. Hepatology 57, 183–194 (2013).
Gabrielson, A., Wu, Y., Wang, H., Jiang, J., Kallakury, B., Gatalica, Z. et al. Intratumoral CD3 and CD8 T-cell densities associated with relapse-free survival in HCC. Cancer Immunol. Res. 4, 419–430 (2016).
Sideras, K., Bots, S. J., Biermann, K., Sprengers, D., Polak, W. G., J. N., I. J. et al. Tumour antigen expression in hepatocellular carcinoma in a low-endemic western area. Br. J. Cancer. 112, 1911–1920 (2015).
Loffler, M. W., Chandran, P. A., Laske, K., Schroeder, C., Bonzheim, I., Walzer, M. et al. Personalized peptide vaccine-induced immune response associated with long-term survival of a metastatic cholangiocarcinoma patient. J Hepatol. 65, 849–855 (2016).
Lee, J. S. The mutational landscape of hepatocellular carcinoma. Clin. Mol. Hepatol. 21, 220–229 (2015).
Zou, S., Li, J., Zhou, H., Frech, C., Jiang, X., Chu, J. S. et al. Mutational landscape of intrahepatic cholangiocarcinoma. Nat. Commun. 5, 5696 (2014).
Tran, E., Turcotte, S., Gros, A., Robbins, P. F., Lu, Y. C., Dudley, M. E. et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014).
Schumacher, T. N. & Schreiber, R. D. Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015).
Pritchard, A. L., Burel, J. G., Neller, M. A., Hayward, N. K., Lopez, J. A., Fatho, M. et al. Exome sequencing to predict neoantigens in melanoma. Cancer Immunol. Res. 3, 992–998 (2015).
Gao, Q., Zhu, H., Dong, L., Shi, W., Chen, R., Song, Z. et al. Integrated proteogenomic characterization of HBV-related hepatocellular carcinoma. Cell 179, 561–77 e22 (2019).
Losic, B., Craig, A. J., Villacorta-Martin, C., Martins-Filho, S. N., Akers, N., Chen, X. et al. Intratumoral heterogeneity and clonal evolution in liver cancer. Nat. Commun. 11, 291 (2020).
Dong, L. Q., Peng, L. H., Ma, L. J., Liu, D. B., Zhang, S., Luo, S. Z. et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. J. Hepatol. 72, 896–908 (2020).
Garrido, F., Aptsiauri, N., Doorduijn, E. M., Garcia Lora, A. M. & van Hall, T. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr. Opin. Immunol. 39, 44–51 (2016).
Deng, X. L., Chen, W., Cai, M. Y. & Wei, D. P. Expression of class I MHC molecule, HSP70 and TAP in human hepatocellular carcinoma. World J. Gastroenterol. 9, 1853–1855 (2003).
Goeppert, B., Frauenschuh, L., Zucknick, M., Roessler, S., Mehrabi, A., Hafezi, M. et al. Major histocompatibility complex class I expression impacts on patient survival and type and density of immune cells in biliary tract cancer. Br. J. Cancer 113, 1343–1349 (2015).
Sabbatino, F., Villani, V., Yearley, J. H., Deshpande, V., Cai, L., Konstantinidis, I. T. et al. PD-L1 and HLA class I antigen expression and clinical course of the disease in intrahepatic cholangiocarcinoma. Clin. Cancer Res. 22, 470–478 (2016).
Schietinger, A., Philip, M., Krisnawan, V. E., Chiu, E. Y., Delrow, J. J., Basom, R. S. et al. Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401 (2016).
Li, J., Lee, Y., Li, Y., Jiang, Y., Lu, H., Zang, W. et al. Co-inhibitory molecule B7 superfamily member 1 expressed by tumor-infiltrating myeloid cells induces dysfunction of anti-tumor CD8(+) T cells. Immunity 48, 773–786 e5 (2018).
Scott, A. C., Dundar, F., Zumbo, P., Chandran, S. S., Klebanoff, C. A., Shakiba, M. et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571, 270–274 (2019).
Kim, C. G., Jang, M., Kim, Y., Leem, G., Kim, K. H., Lee, H. et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci. Immunol. 4, eaay0555 (2019).
Morse, M. A., Sun, W., Kim, R., He, A. R., Abada, P. B., Mynderse, M. et al. The role of angiogenesis in hepatocellular carcinoma. Clin. Cancer Res. 25, 912–920 (2019).
Guzman, A., Ziperstein, M. J. & Kaufman, L. J. The effect of fibrillar matrix architecture on tumor cell invasion of physically challenging environments. Biomaterials 35, 6954–6963 (2014).
Baker, E. L., Srivastava, J., Yu, D., Bonnecaze, R. T. & Zaman, M. H. Cancer cell migration: integrated roles of matrix mechanics and transforming potential. PLoS ONE 6, e20355 (2011).
Nicolas-Boluda, A. & Donnadieu, E. Obstacles to T cell migration in the tumor microenvironment. Comp. Immunol. Microbiol. Infect. Dis. 63, 22–30 (2019).
Lanitis, E., Dangaj, D., Irving, M. & Coukos, G. Mechanisms regulating T-cell infiltration and activity in solid tumors. Ann. Oncol. 28, xii18–xii32 (2017).
Xue, D., Zheng, Y., Wen, J., Han, J., Tuo, H., Liu, Y. et al. Role of chemokines in hepatocellular carcinoma (Review). Oncol. Rep. 45, 809–823 (2021).
Chew, V., Lai, L., Pan, L., Lim, C. J., Li, J., Ong, R. et al. Delineation of an immunosuppressive gradient in hepatocellular carcinoma using high-dimensional proteomic and transcriptomic analyses. Proc. Natl Acad. Sci. USA 114, E5900–E5909 (2017).
Zheng, C., Zheng, L., Yoo, J. K., Guo, H., Zhang, Y., Guo, X. et al. Landscape of infiltrating T cells in liver cancer revealed by single-cell sequencing. Cell 169, 1342–1356 e16 (2017).
Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 12, 252–264 (2012).
Gotwals, P., Cameron, S., Cipolletta, D., Cremasco, V., Crystal, A., Hewes, B. et al. Prospects for combining targeted and conventional cancer therapy with immunotherapy. Nat. Rev. Cancer 17, 286–301 (2017).
Salama, A. K. & Moschos, S. J. Next steps in immuno-oncology: enhancing antitumor effects through appropriate patient selection and rationally designed combination strategies. Ann. Oncol. 28, 57–74 (2017).
Rotte, A., Jin, J. Y. & Lemaire, V. Mechanistic overview of immune checkpoints to support the rational design of their combinations in cancer immunotherapy. Ann. Oncol. 29, 71–83 (2018).
Han, Y., Chen, Z., Yang, Y., Jiang, Z., Gu, Y., Liu, Y. et al. Human CD14+ CTLA-4+ regulatory dendritic cells suppress T-cell response by cytotoxic T-lymphocyte antigen-4-dependent IL-10 and indoleamine-2,3-dioxygenase production in hepatocellular carcinoma. Hepatology 59, 567–579 (2014).
Pedroza-Gonzalez, A., Zhou, G., Singh, S. P., Boor, P. P., Pan, Q., Grunhagen, D. et al. GITR engagement in combination with CTLA-4 blockade completely abrogates immunosuppression mediated by human liver tumor-derived regulatory T cells. Oncoimmunology 4, e1051297 (2015).
Sangro, B., Gomez-Martin, C., de la Mata, M., Inarrairaegui, M., Garralda, E., Barrera, P. et al. A clinical trial of CTLA-4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J. Hepatol. 59, 81–88 (2013).
Duffy, A. G., Ulahannan, S. V., Makorova-Rusher, O., Rahma, O., Wedemeyer, H., Pratt, D. et al. Tremelimumab in combination with ablation in patients with advanced hepatocellular carcinoma. J. Hepatol. 66, 545–551 (2017).
Agdashian, D., ElGindi, M., Xie, C., Sandhu, M., Pratt, D., Kleiner, D. E. et al. The effect of anti-CTLA4 treatment on peripheral and intra-tumoral T cells in patients with hepatocellular carcinoma. Cancer Immunol. Immunother. 68, 599–608 (2019).
Simpson, T. R., Li, F., Montalvo-Ortiz, W., Sepulveda, M. A., Bergerhoff, K., Arce, F. et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J. Exp. Med. 210, 1695–1710 (2013).
Selby, M. J., Engelhardt, J. J., Quigley, M., Henning, K. A., Chen, T., Srinivasan, M. et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol. Res. 1, 32–42 (2013).
Romano, E., Kusio-Kobialka, M., Foukas, P. G., Baumgaertner, P., Meyer, C., Ballabeni, P. et al. Ipilimumab-dependent cell-mediated cytotoxicity of regulatory T cells ex vivo by nonclassical monocytes in melanoma patients. Proc. Natl Acad. Sci. USA 112, 6140–6145 (2015).
Xie, C., Duffy, A. G., Mabry-Hrones, D., Wood, B., Levy, E., Krishnasamy, V. et al. Tremelimumab in combination with microwave ablation in patients with refractory biliary tract cancer. Hepatology 69, 2048–2060 (2019).
U.S. National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/home.
Wolchok, J. D., Chiarion-Sileni, V., Gonzalez, R., Rutkowski, P., Grob, J. J., Cowey, C. L. et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 377, 1345–1356 (2017).
Ribas, A., Hamid, O., Daud, A., Hodi, F. S., Wolchok, J. D., Kefford, R. et al. Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 315, 1600–1609 (2016).
Goldberg, S. B., Gettinger, S. N., Mahajan, A., Chiang, A. C., Herbst, R. S., Sznol, M. et al. Pembrolizumab for patients with melanoma or non-small-cell lung cancer and untreated brain metastases: early analysis of a non-randomised, open-label, phase 2 trial. Lancet Oncol. 17, 976–983 (2016).
Calderaro, J., Rousseau, B., Amaddeo, G., Mercey, M., Charpy, C., Costentin, C. et al. Programmed death ligand 1 expression in hepatocellular carcinoma: relationship with clinical and pathological features. Hepatology 64, 2038–2046 (2016).
Sideras, K., Biermann, K., Verheij, J., Takkenberg, B. R., Mancham, S., Hansen, B. E. et al. PD-L1, Galectin-9 and CD8+ tumor-infiltrating lymphocytes are associated with survival in hepatocellular carcinoma. Oncoimmunology 6, e1273309 (2017).
Jung, H. I., Jeong, D., Ji, S., Ahn, T. S., Bae, S. H., Chin, S. et al. Overexpression of PD-L1 and PD-L2 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Res. Treat. 49, 246–254 (2017).
Xu, P., Sun, Z., Wang, Y. & Miao, C. Long-term use of indomethacin leads to poor prognoses through promoting the expression of PD-1 and PD-L2 via TRIF/NF-kappaB pathway and JAK/STAT3 pathway to inhibit TNF-alpha and IFN-gamma in hepatocellular carcinoma. Exp Cell Res. 337, 53–60 (2015).
Kim, H. D., Song, G. W., Park, S., Jung, M. K., Kim, M. H., Kang, H. J. et al. Association between expression level of PD1 by tumor-infiltrating CD8(+) T cells and features of hepatocellular carcinoma. Gastroenterology 155, 1936–1950 e17 (2018).
Wang, X., He, Q., Shen, H., Xia, A., Tian, W., Yu, W. et al. TOX promotes the exhaustion of antitumor CD8(+) T cells by preventing PD1 degradation in hepatocellular carcinoma. J. Hepatol. 71, 731–741 (2019).
Lim, C. J., Lee, Y. H., Pan, L., Lai, L., Chua, C., Wasser, M. et al. Multidimensional analyses reveal distinct immune microenvironment in hepatitis B virus-related hepatocellular carcinoma. Gut 68, 916–927 (2019).
Wang, H., Rao, B., Lou, J., Li, J., Liu, Z., Li, A. et al. The function of the HGF/c-Met axis in hepatocellular carcinoma. Front. Cell Dev. Biol. 8, 55 (2020).
Qi, X., Yang, M., Ma, L., Sauer, M., Avella, D., Kaifi, J. T. et al. Synergizing sunitinib and radiofrequency ablation to treat hepatocellular cancer by triggering the antitumor immune response. J. Immunother. Cancer. 8, e001038 (2020).
Xie, Q. K., Zhao, Y. J., Pan, T., Lyu, N., Mu, L. W., Li, S. L. et al. Programmed death ligand 1 as an indicator of pre-existing adaptive immune responses in human hepatocellular carcinoma. Oncoimmunology 5, e1181252 (2016).
Chen, Y., Ramjiawan, R. R., Reiberger, T., Ng, M. R., Hato, T., Huang, Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology 61, 1591–1602 (2015).
Chen, D. P., Ning, W. R., Jiang, Z. Z., Peng, Z. P., Zhu, L. Y., Zhuang, S. M. et al. Glycolytic activation of peritumoral monocytes fosters immune privilege via the PFKFB3-PD-L1 axis in human hepatocellular carcinoma. J Hepatol. 71, 333–343 (2019).
Shigeta, K., Datta, M., Hato, T., Kitahara, S., Chen, I. X., Matsui, A. et al. Dual programmed death receptor-1 and vascular endothelial growth factor receptor-2 blockade promotes vascular normalization and enhances antitumor immune responses in hepatocellular carcinoma. Hepatology 71, 1247–1261 (2020).
Xiang, J., Zhang, N., Sun, H., Su, L., Zhang, C., Xu, H. et al. Disruption of SIRT7 increases the efficacy of checkpoint inhibitor via MEF2D regulation of programmed cell death 1 ligand 1 in hepatocellular carcinoma cells. Gastroenterology 158, 664–78 e24 (2020).
Zhou, J., Liu, M., Sun, H., Feng, Y., Xu, L., Chan, A. W. H. et al. Hepatoma-intrinsic CCRK inhibition diminishes myeloid-derived suppressor cell immunosuppression and enhances immune-checkpoint blockade efficacy. Gut 67, 931–944 (2018).
Xiao, G., Jin, L. L., Liu, C. Q., Wang, Y. C., Meng, Y. M., Zhou, Z. G. et al. EZH2 negatively regulates PD-L1 expression in hepatocellular carcinoma. J. Immunother. Cancer. 7, 300 (2019).
El-Khoueiry, A. B., Sangro, B., Yau, T., Crocenzi, T. S., Kudo, M., Hsu, C. et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 389, 2492–2502 (2017).
Yau, T., Hsu, C., Kim, T. Y., Choo, S. P., Kang, Y. K., Hou, M. M. et al. Nivolumab in advanced hepatocellular carcinoma: sorafenib-experienced Asian cohort analysis. J. Hepatol. 71, 543–552 (2019).
Zhu, A. X., Finn, R. S., Edeline, J., Cattan, S., Ogasawara, S., Palmer, D. et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. Lancet Oncol. 19, 940–952 (2018).
Finn, R. S., Ryoo, B. Y., Merle, P., Kudo, M., Bouattour, M., Lim, H. Y. et al. Pembrolizumab as second-line therapy in patients with advanced hepatocellular carcinoma in KEYNOTE-240: a randomized, double-blind, Phase III Trial. J. Clin. Oncol. 38, 193–202 (2020).
Qin, S., Ren, Z., Meng, Z., Chen, Z., Chai, X., Xiong, J. et al. Camrelizumab in patients with previously treated advanced hepatocellular carcinoma: a multicentre, open-label, parallel-group, randomised, phase 2 trial. Lancet Oncol. 21, 571–580 (2020).
Lee, D. W., Cho, E. J., Lee, J. H., Yu, S. J., Kim, Y. J., Yoon, J. H. et al. Phase II study of avelumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib. Clin. Cancer Res. 27, 713–718 (2021).
Li, H., Li, X., Liu, S., Guo, L., Zhang, B., Zhang, J. et al. PD-1 Checkpoint blockade in combination with an mTOR inhibitor restrains hepatocellular carcinoma growth induced by hepatoma cell-intrinsic PD-1. Hepatology (2017).
Finn, R. S., Qin, S., Ikeda, M., Galle, P. R., Ducreux, M., Kim, T. Y. et al. Atezolizumab plus bevacizumab in unresectable hepatocellular carcinoma. N. Engl. J. Med. 382, 1894–1905 (2020).
Yau, T., Kang, Y. K., Kim, T. Y., El-Khoueiry, A. B., Santoro, A., Sangro, B. et al. Efficacy and safety of nivolumab plus ipilimumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib: The CheckMate 040 Randomized Clinical Trial. JAMA Oncol. 6, e204564 (2020).
Doi, T., Muro, K., Ishii, H., Kato, T., Tsushima, T., Takenoyama, M. et al. A Phase I study of the anti-CC chemokine receptor 4 antibody, mogamulizumab, in combination with nivolumab in patients with advanced or metastatic solid tumors. Clin. Cancer Res. 25, 6614–6622 (2019).
Finn, R. S., Ikeda, M., Zhu, A. X., Sung, M. W., Baron, A. D., Kudo, M. et al. Phase Ib study of lenvatinib plus pembrolizumab in patients with unresectable hepatocellular carcinoma. J. Clin. Oncol. 38, 2960–2970 (2020).
Gani, F., Nagarajan, N., Kim, Y., Zhu, Q., Luan, L., Bhaijjee, F. et al. Program death 1 immune checkpoint and tumor microenvironment: implications for patients with intrahepatic cholangiocarcinoma. Ann. Surg. Oncol. 23, 2610–2617 (2016).
Ma, K., Wei, X., Dong, D., Wu, Y., Geng, Q. & Li, E. PD-L1 and PD-1 expression correlate with prognosis in extrahepatic cholangiocarcinoma. Oncol. Lett. 14, 250–256 (2017).
Wang, L., Dong, H., Ni, S., Huang, D., Tan, C., Chang, B. et al. Programmed death-ligand 1 is upregulated in intrahepatic lymphoepithelioma-like cholangiocarcinoma. Oncotarget 7, 69749–69759 (2016).
Fontugne, J., Augustin, J., Pujals, A., Compagnon, P., Rousseau, B., Luciani, A. et al. PD-L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget 8, 24644–24651 (2017).
Walter, D., Herrmann, E., Schnitzbauer, A. A., Zeuzem, S., Hansmann, M. L., Peveling-Oberhag, J. et al. PD-L1 expression in extrahepatic cholangiocarcinoma. Histopathology 71, 383–392 (2017).
Sato, Y., Kinoshita, M., Takemura, S., Tanaka, S., Hamano, G., Nakamori, S. et al. The PD-1/PD-L1 axis may be aberrantly activated in occupational cholangiocarcinoma. Pathol. Int. 67, 163–170 (2017).
Sangkhamanon, S., Jongpairat, P., Sookprasert, A., Wirasorn, K., Titapun, A., Pugkhem, A. et al. Programmed Death-Ligand 1 (PD-L1) expression associated with a high neutrophil/lymphocyte ratio in cholangiocarcinoma. Asian Pac. J. Cancer Prev. 18, 1671–1674 (2017).
Lu, J. C., Zeng, H. Y., Sun, Q. M., Meng, Q. N., Huang, X. Y., Zhang, P. F. et al. Distinct PD-L1/PD1 profiles and clinical implications in intrahepatic cholangiocarcinoma patients with different risk factors. Theranostics 9, 4678–4687 (2019).
Jusakul, A., Cutcutache, I., Yong, C. H., Lim, J. Q., Huang, M. N., Padmanabhan, N. et al. Whole-genome and epigenomic landscapes of etiologically distinct subtypes of cholangiocarcinoma. Cancer Discov. 7, 1116–1135 (2017).
Montal, R., Sia, D., Montironi, C., Leow, W. Q., Esteban-Fabro, R., Pinyol, R. et al. Molecular classification and therapeutic targets in extrahepatic cholangiocarcinoma. J. Hepatol. 73, 315–327 (2020).
Le, D. T., Durham, J. N., Smith, K. N., Wang, H., Bartlett, B. R., Aulakh, L. K. et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017).
Marabelle, A., Le, D. T., Ascierto, P. A., Di Giacomo, A. M., De Jesus-Acosta, A., Delord, J. P. et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the Phase II KEYNOTE-158 Study. J. Clin. Oncol. 38, 1–10 (2020).
Kim, R. D., Chung, V., Alese, O. B., El-Rayes, B. F., Li, D., Al-Toubah, T. E. et al. A Phase 2 multi-institutional study of nivolumab for patients with advanced refractory biliary tract cancer. JAMA Oncol. 6, 888–894 (2020).
Klein, O., Kee, D., Nagrial, A., Markman, B., Underhill, C., Michael, M. et al. Evaluation of combination nivolumab and ipilimumab immunotherapy in patients with advanced biliary tract cancers: subgroup analysis of a Phase 2 nonrandomized clinical trial. JAMA Oncol. 6, 1405–1409 (2020).
Oh, D. Y. Lee, K. H., Lee, D. W., Kim, T. Y., Bang, J. H., Nam, A. R., Lee, Y., Zhang, Q., Rebelatto, M., Li, W. & Kim, J. W. Phase II study assessing tolerability, efficacy, and biomarkers for durvalumab (D) ± tremelimumab (T) and gemcitabine/cisplatin (GemCis) in chemo-naïve advanced biliary tract cancer (aBTC). J. Clin. Oncol 38, 4520–4520 (2020).
Suh, W. K., Gajewska, B. U., Okada, H., Gronski, M. A., Bertram, E. M., Dawicki, W. et al. The B7 family member B7-H3 preferentially down-regulates T helper type 1-mediated immune responses. Nat. Immunol. 4, 899–906 (2003).
Leitner, J., Klauser, C., Pickl, W. F., Stockl, J., Majdic, O., Bardet, A. F. et al. B7-H3 is a potent inhibitor of human T-cell activation: no evidence for B7-H3 and TREML2 interaction. Eur. J. Immunol. 39, 1754–1764 (2009).
Chen, C., Shen, Y., Qu, Q. X., Chen, X. Q., Zhang, X. G. & Huang, J. A. Induced expression of B7-H3 on the lung cancer cells and macrophages suppresses T-cell mediating anti-tumor immune response. Exp. Cell Res. 319, 96–102 (2013).
Vigdorovich, V., Ramagopal, U. A., Lazar-Molnar, E., Sylvestre, E., Lee, J. S., Hofmeyer, K. A. et al. Structure and T cell inhibition properties of B7 family member, B7-H3. Structure 21, 707–717 (2013).
Seaman, S., Zhu, Z., Saha, S., Zhang, X. M., Yang, M. Y., Hilton, M. B. et al. Eradication of tumors through simultaneous ablation of CD276/B7-H3-positive tumor cells and tumor vasculature. Cancer Cell 31, 501–15 e8 (2017).
Kang, F. B., Wang, L., Li, D., Zhang, Y. G. & Sun, D. X. Hepatocellular carcinomas promote tumor-associated macrophage M2-polarization via increased B7-H3 expression. Oncol. Rep. 33, 274–282 (2015).
Kang, F. B., Wang, L., Jia, H. C., Li, D., Li, H. J., Zhang, Y. G. et al. B7-H3 promotes aggression and invasion of hepatocellular carcinoma by targeting epithelial-to-mesenchymal transition via JAK2/STAT3/Slug signaling pathway. Cancer Cell Int. 15, 45 (2015).
Sun, T. W., Gao, Q., Qiu, S. J., Zhou, J., Wang, X. Y., Yi, Y. et al. B7-H3 is expressed in human hepatocellular carcinoma and is associated with tumor aggressiveness and postoperative recurrence. Cancer Immunol. Immunother. 61, 2171–2182 (2012).
Zheng, Y., Liao, N., Wu, Y., Gao, J., Li, Z., Liu, W. et al. High expression of B7H2 or B7H3 is associated with poor prognosis in hepatocellular carcinoma. Mol. Med. Rep. 19, 4315–4325 (2019).
Ni, L. & Dong, C. New checkpoints in cancer immunotherapy. Immunol. Rev. 276, 52–65 (2017).
Cheng, R., Chen, Y., Zhou, H., Wang, B., Du, Q. & Chen, Y. B7-H3 expression and its correlation with clinicopathologic features, angiogenesis, and prognosis in intrahepatic cholangiocarcinoma. APMIS 126, 396–402 (2018).
Wang, X., Hao, J., Metzger, D. L., Ao, Z., Chen, L., Ou, D. et al. B7-H4 treatment of T cells inhibits ERK, JNK, p38, and AKT activation. PLoS ONE 7, e28232 (2012).
Dangaj, D., Lanitis, E., Zhao, A., Joshi, S., Cheng, Y., Sandaltzopoulos, R. et al. Novel recombinant human b7-h4 antibodies overcome tumoral immune escape to potentiate T-cell antitumor responses. Cancer Res. 73, 4820–4829 (2013).
Hong, B., Qian, Y., Zhang, H., Sang, Y. W., Cheng, L. F., Wang, Q. et al. Expression of B7-H4 and hepatitis B virus X in hepatitis B virus-related hepatocellular carcinoma. World J. Gastroenterol. 22, 4538–4546 (2016).
Zhao, X., Guo, F., Li, Z., Jiang, P., Deng, X., Tian, F. et al. Aberrant expression of B7-H4 correlates with poor prognosis and suppresses tumor-infiltration of CD8+ T lymphocytes in human cholangiocarcinoma. Oncol. Rep. 36, 419–427 (2016).
Lines, J. L., Pantazi, E., Mak, J., Sempere, L. F., Wang, L., O’Connell, S. et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 74, 1924–1932 (2014).
Wang, L., Le Mercier, I., Putra, J., Chen, W., Liu, J., Schenk, A. D. et al. Disruption of the immune-checkpoint VISTA gene imparts a proinflammatory phenotype with predisposition to the development of autoimmunity. Proc. Natl Acad. Sci. USA 111, 14846–14851 (2014).
Wang, L., Rubinstein, R., Lines, J. L., Wasiuk, A., Ahonen, C., Guo, Y. et al. VISTA, a novel mouse Ig superfamily ligand that negatively regulates T cell responses. J. Exp. Med. 208, 577–592 (2011).
Flies, D. B., Han, X., Higuchi, T., Zheng, L., Sun, J., Ye, J. J. et al. Coinhibitory receptor PD-1H preferentially suppresses CD4(+) T cell-mediated immunity. J. Clin. Invest. 124, 1966–1975 (2014).
Le Mercier, I., Chen, W., Lines, J. L., Day, M., Li, J., Sergent, P. et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 74, 1933–1944 (2014).
Lines, J. L., Sempere, L. F., Broughton, T., Wang, L. & Noelle, R. VISTA is a novel broad-spectrum negative checkpoint regulator for cancer immunotherapy. Cancer Immunol. Res. 2, 510–517 (2014).
Liu, J., Yuan, Y., Chen, W., Putra, J., Suriawinata, A. A., Schenk, A. D. et al. Immune-checkpoint proteins VISTA and PD-1 nonredundantly regulate murine T-cell responses. Proc. Natl Acad. Sci. USA 112, 6682–6687 (2015).
Zhang, M., Pang, H. J., Zhao, W., Li, Y. F., Yan, L. X., Dong, Z. Y. et al. VISTA expression associated with CD8 confers a favorable immune microenvironment and better overall survival in hepatocellular carcinoma. BMC Cancer 18, 511 (2018).
Zhu, Y., Yao, S., Iliopoulou, B. P., Han, X., Augustine, M. M., Xu, H. et al. B7-H5 costimulates human T cells via CD28H. Nat. Commun. 4, 2043 (2013).
Xiao, Y. & Freeman, G. J. A new B7:CD28 family checkpoint target for cancer immunotherapy: HHLA2. Clin. Cancer Res. 21, 2201–2203 (2015).
Janakiram, M., Chinai, J. M., Fineberg, S., Fiser, A., Montagna, C., Medavarapu, R. et al. Expression, clinical significance, and receptor identification of the newest B7 family member HHLA2 protein. Clin. Cancer Res. 21, 2359–2366 (2015).
Jing, C. Y., Fu, Y. P., Yi, Y., Zhang, M. X., Zheng, S. S., Huang, J. L. et al. HHLA2 in intrahepatic cholangiocarcinoma: an immune checkpoint with prognostic significance and wider expression compared with PD-L1. J. Immunother. Cancer. 7, 77 (2019).
Huang, Y. H., Zhu, C., Kondo, Y., Anderson, A. C., Gandhi, A., Russell, A. et al. Corrigendum: CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature 536, 359 (2016).
Das, M., Zhu, C. & Kuchroo, V. K. Tim-3 and its role in regulating anti-tumor immunity. Immunol. Rev. 276, 97–111 (2017).
Liu, F., Liu, Y. & Chen, Z. Tim-3 expression and its role in hepatocellular carcinoma. J. Hematol. Oncol. 11, 126 (2018).
Barathan, M., Gopal, K., Mohamed, R., Ellegard, R., Saeidi, A., Vadivelu, J. et al. Chronic hepatitis C virus infection triggers spontaneous differential expression of biosignatures associated with T cell exhaustion and apoptosis signaling in peripheral blood mononucleocytes. Apoptosis 20, 466–480 (2015).
Li, H., Wu, K., Tao, K., Chen, L., Zheng, Q., Lu, X. et al. Tim-3/galectin-9 signaling pathway mediates T-cell dysfunction and predicts poor prognosis in patients with hepatitis B virus-associated hepatocellular carcinoma. Hepatology 56, 1342–1351 (2012).
Ji, J., Yin, Y., Ju, H., Xu, X., Liu, W., Fu, Q. et al. Long non-coding RNA Lnc-Tim3 exacerbates CD8 T cell exhaustion via binding to Tim-3 and inducing nuclear translocation of Bat3 in HCC. Cell Death Dis. 9, 478 (2018).
Yan, W., Liu, X., Ma, H., Zhang, H., Song, X., Gao, L. et al. Tim-3 fosters HCC development by enhancing TGF-beta-mediated alternative activation of macrophages. Gut 64, 1593–1604 (2015).
Anderson, A. C., Joller, N. & Kuchroo, V. K. Lag-3, Tim-3, and TIGIT: Co-inhibitory receptors with specialized functions in immune regulation. Immunity 44, 989–1004 (2016).
Nguyen, L. T. & Ohashi, P. S. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat. Rev. Immunol. 15, 45–56 (2015).
Andrews, L. P., Marciscano, A. E., Drake, C. G. & Vignali, D. A. LAG3 (CD223) as a cancer immunotherapy target. Immunol. Rev. 276, 80–96 (2017).
Goldberg, M. V. & Drake, C. G. LAG-3 in cancer immunotherapy. Curr. Top. Microbiol. Immunol. 344, 269–278 (2011).
Woo, S. R., Turnis, M. E., Goldberg, M. V., Bankoti, J., Selby, M., Nirschl, C. J. et al. Immune inhibitory molecules LAG-3 and PD-1 synergistically regulate T-cell function to promote tumoral immune escape. Cancer Res. 72, 917–927 (2012).
Matsuzaki, J., Gnjatic, S., Mhawech-Fauceglia, P., Beck, A., Miller, A., Tsuji, T. et al. Tumor-infiltrating NY-ESO-1-specific CD8+ T cells are negatively regulated by LAG-3 and PD-1 in human ovarian cancer. Proc. Natl Acad. Sci. USA 107, 7875–7880 (2010).
Xu, F., Liu, J., Liu, D., Liu, B., Wang, M., Hu, Z. et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T-cell responses. Cancer Res. 74, 3418–3428 (2014).
Liu, W., Tang, L., Zhang, G., Wei, H., Cui, Y., Guo, L. et al. Characterization of a novel C-type lectin-like gene, LSECtin: demonstration of carbohydrate binding and expression in sinusoidal endothelial cells of liver and lymph node. J Biol. Chem. 279, 18748–18758 (2004).
Wang, J., Sanmamed, M. F., Datar, I., Su, T. T., Ji, L., Sun, J. et al. Fibrinogen-like protein 1 Is a major immune inhibitory ligand of LAG-3. Cell 176, 334–47 e12 (2019).
Le Mercier, I., Lines, J. L. & Noelle, R. J. Beyond CTLA-4 and PD-1, the generation Z of negative checkpoint regulators. Front. Immunol. 6, 418 (2015).
Hokuto, D., Sho, M., Yamato, I., Yasuda, S., Obara, S., Nomi, T. et al. Clinical impact of herpesvirus entry mediator expression in human hepatocellular carcinoma. Eur. J. Cancer. 51, 157–165 (2015).
Zhao, Q., Huang, Z. L., He, M., Gao, Z. & Kuang, D. M. BTLA identifies dysfunctional PD-1-expressing CD4(+) T cells in human hepatocellular carcinoma. Oncoimmunology 5, e1254855 (2016).
Wu, Y., Kuang, D. M., Pan, W. D., Wan, Y. L., Lao, X. M., Wang, D. et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology 57, 1107–1116 (2013).
Joller, N., Lozano, E., Burkett, P. R., Patel, B., Xiao, S., Zhu, C. et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity 40, 569–581 (2014).
Gur, C., Ibrahim, Y., Isaacson, B., Yamin, R., Abed, J., Gamliel, M. et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity 42, 344–355 (2015).
Zhu, Y., Paniccia, A., Schulick, A. C., Chen, W., Koenig, M. R., Byers, J. T. et al. Identification of CD112R as a novel checkpoint for human T cells. J. Exp. Med. 213, 167–176 (2016).
Martinet, L. & Smyth, M. J. Balancing natural killer cell activation through paired receptors. Nat. Rev. Immunol. 15, 243–254 (2015).
Johnston, R. J., Comps-Agrar, L., Hackney, J., Yu, X., Huseni, M., Yang, Y. et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell 26, 923–937 (2014).
Chauvin, J. M., Pagliano, O., Fourcade, J., Sun, Z., Wang, H., Sander, C. et al. TIGIT and PD-1 impair tumor antigen-specific CD8(+) T cells in melanoma patients. J. Clin. Invest. 125, 2046–2058 (2015).
Kung-Chun Chiu, D., Wai-Hin Yuen, V., Wing-Sum Cheu, J., Wei, L. L., Ting, V., Fehlings, M. et al. Hepatocellular carcinoma cells upregulate PVRL1, stabilizing PVR and inhibiting the cytotoxic T-cell response via TIGIT to mediate tumor resistance to PD1 inhibitors in mice. Gastroenterology. 159, 609–623 (2020).
Duan, X., Liu, J., Cui, J., Ma, B., Zhou, Q., Yang, X. et al. Expression of TIGIT/CD155 and correlations with clinical pathological features in human hepatocellular carcinoma. Mol. Med. Rep. 20, 3773–3781 (2019).
Sun, H., Huang, Q., Huang, M., Wen, H., Lin, R., Zheng, M. et al. Human CD96 correlates to natural killer cell exhaustion and predicts the prognosis of human hepatocellular carcinoma. Hepatology 70, 168–183 (2019).
Ge, Z., Zhou, G., Carrascosa, L. C., Gausvik, E., Boor, P. P. C., Noordam, L. et al. TIGIT and PD1 Co-blockade restores ex vivo functions of human tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Cell Mol. Gastroenterol. Hepatol. S2352-345X(21)00055-2. https://doi.org/10.1016/j.jcmgh.2021.03.003 (2021).
Huang, D. W., Huang, M., Lin, X. S. & Huang, Q. CD155 expression and its correlation with clinicopathologic characteristics, angiogenesis, and prognosis in human cholangiocarcinoma. Onco Targets Ther. 10, 3817–3825 (2017).
Sanmamed, M. F., Pastor, F., Rodriguez, A., Perez-Gracia, J. L., Rodriguez-Ruiz, M. E., Jure-Kunkel, M. et al. Agonists of co-stimulation in cancer immunotherapy directed against CD137, OX40, GITR, CD27, CD28, and ICOS. Semin. Oncol. 42, 640–655 (2015).
Chen, L. & Flies, D. B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 13, 227–242 (2013).
Xie, K., Xu, L., Wu, H., Liao, H., Luo, L., Liao, M. et al. OX40 expression in hepatocellular carcinoma is associated with a distinct immune microenvironment, specific mutation signature, and poor prognosis. Oncoimmunology 7, e1404214 (2018).
Campos Carrascosa, L., van Beek, A. A., de Ruiter, V., Doukas, M., Wei, J., Fisher, T. S. et al. FcgammaRIIB engagement drives agonistic activity of Fc-engineered alphaOX40 antibody to stimulate human tumor-infiltrating T cells. J. Immunother. Cancer. 8, 2 (2020).
Zhang, Y., Zhang, H., Wei, M., Mou, T., Shi, T., Ma, Y. et al. Recombinant adenovirus expressing a soluble fusion protein PD-1/CD137L subverts the suppression of CD8(+) T cells in HCC. Mol. Ther. 27, 1906–1918 (2019).
Kim, H. D., Park, S., Jeong, S., Lee, Y. J., Lee, H., Kim, C. G. et al. 4-1BB delineates distinct activation status of exhausted tumor-infiltrating CD8(+) T cells in hepatocellular carcinoma. Hepatology 71, 955–971 (2020).
van Beek, A. A., Zhou, G., Doukas, M., Boor, P. P. C., Noordam, L., Mancham, S. et al. GITR ligation enhances functionality of tumor-infiltrating T cells in hepatocellular carcinoma. Int. J. Cancer. 145, 1111–1124 (2019).
Salmon, H., Franciszkiewicz, K., Damotte, D., Dieu-Nosjean, M. C., Validire, P., Trautmann, A. et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Invest. 122, 899–910 (2012).
Shaikh, F. Y., Gills, J. J. & Sears, C. L. Impact of the microbiome on checkpoint inhibitor treatment in patients with non-small cell lung cancer and melanoma. EBioMedicine 48, 642–647 (2019).
Nagasaka, M., Sexton, R., Alhasan, R., Rahman, S., Azmi, A. S. & Sukari, A. Gut microbiome and response to checkpoint inhibitors in non-small cell lung cancer—a review. Crit. Rev. Oncol. Hematol. 145, 102841 (2020).
Fidelle, M., Yonekura, S., Picard, M., Cogdill, A., Hollebecque, A., Roberti, M. P. et al. Resolving the paradox of colon cancer through the integration of genetics, immunology, and the microbiota. Front. Immunol. 11, 600886 (2020).
Koulouridi, A., Messaritakis, I., Gouvas, N., Tsiaoussis, J. & Souglakos, J. Immunotherapy in solid tumors and gut microbiota: the correlation—a special reference to colorectal cancer. Cancers (Basel) 13, 43 (2020).
Spahn, S., Roessler, D., Pompilia, R., Gabernet, G., Gladstone, B. P., Horger, M. et al. Clinical and genetic tumor characteristics of responding and non-responding patients to PD-1 inhibition in hepatocellular carcinoma. Cancers (Basel) 12, 383 (2020).
Ruiz de Galarreta, M., Bresnahan, E., Molina-Sanchez, P., Lindblad, K. E., Maier, B., Sia, D. et al. beta-catenin activation promotes immune escape and resistance to anti-PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 9, 1124–1141 (2019).
Harding, J. J., Nandakumar, S., Armenia, J., Khalil, D. N., Albano, M., Ly, M. et al. Prospective genotyping of hepatocellular carcinoma: clinical implications of next-generation sequencing for matching patients to targeted and immune therapies. Clin. Cancer Res. 25, 2116–2126 (2019).
Lee, M. S., Ryoo, B. Y., Hsu, C. H., Numata, K., Stein, S., Verret, W. et al. Atezolizumab with or without bevacizumab in unresectable hepatocellular carcinoma (GO30140): an open-label, multicentre, phase 1b study. Lancet Oncol. 21, 808–820 (2020).
Yarchoan, M., Xing, D., Luan, L., Xu, H., Sharma, R., Popovic, A. et al. Characterization of the immune microenvironment in hepatocellular carcinoma (HCC). Clin. Cancer Res. 23, 7333–7339 (2017).
Acknowledgements
Not applicable.
Author information
Authors and Affiliations
Contributions
G.Z. contributed to drafting and revising the manuscript, making figures and tables. P.B. contributed to drafting the manuscript. M.B. and D.S. contributed to revising the manuscript. J.K. contributed to drafting and revising the manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent to publish
Yes.
Data availability
Not applicable.
Competing interests
The authors declare no competing interests.
Funding information
No.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhou, G., Boor, P.P.C., Bruno, M.J. et al. Immune suppressive checkpoint interactions in the tumour microenvironment of primary liver cancers. Br J Cancer 126, 10–23 (2022). https://doi.org/10.1038/s41416-021-01453-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41416-021-01453-3
- Springer Nature Limited