
Abstract
Prolonged T-cell immunodeficiency following HLA- incompatible hematopoietic stem cell transplantation (HSCT) represents a major obstacle hampering the more widespread use of this approach. Strategies to fasten T-cell reconstitution in this setting are highly warranted as opportunistic infections and an increased risk of relapse account for high rates of morbidity and mortality especially during early month following this type of HSCT. We have implemented a feeder free cell system based on the use of the notch ligand DL4 and cytokines allowing for the in vitro differentiation of human T-Lymphoid Progenitor cells (HTLPs) from various sources of CD34+ hematopoietic stem and precursor cells (HSPCs). Co- transplantion of human T-lymphoid progenitors (HTLPs) and non- manipulated HSPCs into immunodeficient mice successfully accelerated the reconstitution of a polyclonal T-cell repertoire. This review summarizes preclinical data on the use of T-cell progenitors for treatment of post- transplantation immunodeficiency and gives insights into the development of GMP based protocols for potential clinical applications including gene therapy approaches. Future clinical trials implementing this protocol will aim at the acceleration of immune reconstitution in different clinical settings such as SCID and leukemia patients undergoing allogeneic transplantation. Apart from pure cell-therapy approaches, the combination of DL-4 culture with gene transduction protocols will open new perspectives in terms of gene therapy applications for primary immunodeficiencies.
Similar content being viewed by others


Introduction
Allogeneic hematopoietic stem cell transplantation (HSCT) is a key treatment for a large number of acquired and inherited diseases of the hematopoietic system. Initially restricted to patients with HLA-identical siblings due to risks of T-cell mediated graft rejection and graft versus-host disease (GvHD), it is now widely used also for patients without HLA-identical donors thanks to the use of antithymocyte globulin (ATG), changes in graft handling (T-cell depletion, CD34+ hematopopoietic stem, and progenitor cell (HSPCs) selection) and, recently, the use of post-transplant cyclophosphamide (PTC) [1,2,3]. Although these modifications have greatly improved HSCT outcomes, T-cell immunodeficiency following transplantation remains a major obstacle in an HLA-mismatched setting and hampers the more widespread use of this approach.
Whereas innate immunity recovers quickly, reconstitution of adoptive immunity is a long process. In the case of T-cell replete haplo-identical HSCT, donor T cells present in the graft survive and expand, but this expansion concerns mainly a CD8+ memory T-cell population and leads to a contraction and skewing of the TCR repertoire, thus producing a partially ineffective immune response [4]. In CD34+-selected haplo-identical transplantation, this first wave of homeostatically expanded T cells is virtually absent. Consequently, in both settings, reconstitution of a functional, fully diverse and naive peripheral T-cell pool relies on de novo production of naive T cells from grafted CD34+ HSPCs [4,5,6]. Even in young patients (i.e., most of those transplanted for severe combined immunodeficiency (SCID)), donor-derived T-cells only appear in the blood after 3–6 months” [7, 8]. A period of 6–12 months is required to achieve CD4 + cell counts that provide protective immunity. More complete restoration of the overall T-cell compartment (i.e. naive T-cells exhibiting a polyclonal T-cell receptor (TCR) repertoire) is an even longer process and may require up to 2 years in adults [9,10,11].
In primary immunodeficiency patients transplanted in an HLA-incompatible setting the profound immunodeficiency following such a procedure leads to severe, opportunistic viral, bacterial, and fungal infections accounting for ~40% of the mortality. The importance of a functional T-cell compartment early after HSCT is further highlighted by the fact, that T-cell-depleted HSCT is associated with an increased risk of graft rejection and, in patients with malignant hematologic disorders, relapse [12].
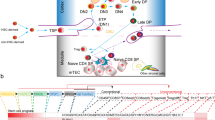
Gene therapy constitutes a valid alternative treatment for SCID-X1 patients without HLA-identical donor [13]. Indeed, the risk of vector-related leukemia has been greatly reduced by the introduction of new generations of safer vectors. Furthermore this treatment has been restricted to patients without HLA-identical donors (either matched related or matched unrelated). A restrospective analysis of haplo-identical transplantion versus gene therapy treatment for immune reconstitution of SCID-X1 patients showed that T-cell reconstitution was significantly faster in the gene therapy group (14 patients) as compared to the haplo-identical group (13 patients) [8]. However, this overall positive output veils some disparities (Fig. 1a). T-cell reconstitution was very slow and incomplete in some patients from both groups. All of them shared one common feature: the presence of active infection at time of treatment (disseminated BCGitis, adenovirus infection…) (Fig. 1b).

Immune reconstitution in SCID-X1 patients after gene therapy and In vitro generation of human T-lymphoid progenitors (HTLPs). a Reconstitution of naïve CD4 T cells over time in patients from trial #1 (upper graph) and trial #2 (bottom graph). b Correlation of CD3 reconstitution over time in the absence (upper graph) or presence of infection (bottom graph). Data include patients of both trials (indicated by SCID#1 and SCID#2). c Descriptive scheme of human T-cell development. d Experimental protocol for the 7-day generation of CD7+ T-cell precursors (=HTLPs) from CD34+ hematopoietic stem and precursor cells (HSPCs) by co- culture with immobilized notch ligand DL4 and a cocktail of cytokines
Many well-established pre-transplant parameters influence immune reconstitution after HSCT: diagnosis, age of the recipient, degree of HLA mismatches, conditioning regimen, type of graft (origin and manipulation), but also the infectious status of donor and recipient. Immune reconstitution is further impacted by clinical complications including leukemia relapse, infections and GvHD, or treatments [14,15,16,17]. However, in the small comparative study published by our group [8] as well as in the study conducted by Clave and colleagues [12], GvHD did not significantly impact the immunological outcome in young patients.
Beside our own observations (Fig. 1a, b), the deleterious impact of bacterial and viral infections on immune reconstitution has been observed by several groups in murine models [18,19,20] as well as by clinical observations [21]. Some of these studies show a significant association between infections (viral and bacterial) and poor thymic output quantified by T-cell receptor excision circles (TREC) in the blood [4, 22].
As mentioned above, delayed T-cell reconstitution represents a major obstacle to the widespread use of HLA-mismatched HSCT. In order to shorten post-transplant immunodeficiency and thus improve clinical outcome, we chose to implement a new treatment strategy consisting of co- transplantion of human T-lymphoid progenitors (HTLPs) together with non-manipulated HSPCs. These progenitors are able to seed the thymus and generate a wave of mature and polyclonal T-cells significantly faster than it is usually observed [11, 13]. Herein we are going to describe this approach as well as its potential applications for both HLA-mismatched HSCT and gene therapy.
Ex vivo generation of human T-lymphoid progenitors
T-cell generation proceeds through three main stages: the first one consists in the production of lymphoid progenitors able to leave the bone marrow (where they are generated from HSCs) and seed the thymus; the second one is T-cell commitment accompanied by the loss of other lineage potentials and first T-cell rearrangements at the δ, γ, and β loci leading to the production of either γδ TCR+ T cells (<5% of circulating mature T cells) or immature single positive cells that (stage 3) further rearrange TCR α locus and undergo positive and negative selection to ultimately give rise to functional, non autoreactive αβ TCR+ T cells (95% of circulating T cells) (Fig. 1c). Of note, the first step of thymic T-cell differentiation takes half of the time required for the whole thymopoietic process. Finding a way to produce quickly ex vivo large numbers of HTLPs appeared thus as a promising approach to fasten T-cell reconstitution after HSCT.
The early stage of T-cell development is dependant on Notch signaling in mouse and human, in particular Delta-like ligand 4 (DL-4) [23,24,25], as well as on key cytokines implicated in the survival and proliferation of thymocytes [26,27,28]. Based on these findings, we implemented a feeder cell-free culture system relying on a recombinant fusion protein composed of the extracellular domain of human DL-4 and the Fc part of human IgG2 (Fig. 1d). The use of this modification allowed immobilisation of DL-4 on the culture surface and thus eliminated the necessity of using a stromal cell line similar to a previous study implementing DL-1 for the expansion of CB HSCPs [29].
Our DL4-Fc culture system allowed the generation large numbers of CD34+/-CD7+CD5-icCD3+ HTLPs from cord blood CD34+ HSPCs within 7 days (an average of 2.5 HTLPs per CD34+ HSPC,) [30]. HTLPs displayed the gene expression profile of early thymic precursors as demonstrated by significant levels of transcripts of PTA, IL7RA, RAG1, and BCL11B. T-cell differentiation in limiting dilution conditions on OP9/DL-1 cells revealed a significant increase in in vitro T-cell potential (from 1/350 at day 0 to 1/12 at day 7). Once transplanted into both, irradiated adult or non-irradiated neonate NOD/SCID/γc(-/-) (NSG) recipients, these T-cell precursors seeded the thymus and generated mature, polyclonal, and functional T cells [30]. Co-transplantation of HTLPs and untreated CD34+ HSCPs was used to mimick future clinical applications. In this setting reconstitution of the T-cell compartment from cultured HTLPs was robust and rapid, with other hematopoietic lineages being produced from the non-manipulated HSCPs.
Mobilized peripheral blood (mPB) is currently the main source of HSPCs in allogenic HSCT as such adult HSPCs are available in large quantity and exhibit several advantages over cord blood grafts in the clinical setting. However, the lymphoid potential of adult HSCPs, especially T-lymphoid potential, is diminished as compared to the one of CB [31]. We recently demonstrated the capacity to generate HTLPs from adult mPB CD34+ cells in DL-4 culture conditions in a very similar way as for cord blood cells [32]. Within 7 days, adult HSPCs produced CD7+ HTLPs expressing T lineage master genes (TCF7, IL7Ra and BCL11B, GATA3, and CD3E) and exhibited an in vitro T-cell differentiation potential similar to the one of CB-derived HTLPs (around 1/19). HTLPs derived from adult HSPCs expressed chemokine receptors implicated in thymus homing (CCR7, CCR9, and CXCR4) and efficiently produced polyclonal T cells upon transplantation in NOD/SCID/γc−/− (NSG) mice. However, due to intrinsic differences in terms of T-cell potential, survival, and proliferation, the yields of HTLPs recovered after 7 days were lower (around 0.45 HTLP/CD34+ HSPC). The lower efficiency of adult HSPCs to produce HTLPs in DL-4 culture conditions was accompanied by a higher rate of apoptosis and a lower rate of proliferation. To overcome this hurdle, we tested several molecules aiming at either increasing Notch signaling or enhancing T-cell differentiation and HTLPs survival and proliferation. The addition of a human fragment of fibronectin and Tumor Necrosis Factor alpha (TNFα) improved the yields of production of adult HTLPs by 10 fold (patents WO2016/055396, WO2018/146297). Our improved results demonstrate that, like CB HSPCs, adult HSPCs provide an effective and available source of in vitro cultured HTLPs in the context of future clinical applications directed to shorten T-cell recovery after HSCT.
Cell therapy based on HTLPs
Based on these preclinical data, the entire platform, including all reagents, DL-4, medium, serum, cytokines, was translated into a GMP grade process. Of note, results obtained in GMP conditions in the clinical runs were identical to the ones obtained following the research grade protocol. Subsequently, we carefully monitored the composition of the day-7-cellular product, with a special focus on cellular contaminants. Apart from HTLPs, DL-4 cultures contain no or very rare T-cells (undetectable upon analysis of TCR rearrangements by robust and highly sensitive techniques used to detect minimal residual disease in hematologic malignancies, coll. with Hematology Lab directed by E. McIntyre, Necker Hospital) and mostly myeloid precursors (around 12%).
As a third step, we tested the toxicity of the DL-4 protein and the day 7-cellular product of DL-4 cultures by injection of a 10 fold higher dose than used in any future clinical trial into the mice.
Of note, quantification of residual DL-4 at the end of the culture performed by a home-made ELISA using antibodies specific for either the DL-4 extracellular domain or the Fc part of DL-4 yielded negative results in all experiments but one the dose of which was defined as reference for injection into mice. We did not observe any toxicity or tumorigenicity upon injection of the DL-4 protein and the cellular product in mice. Furthermore, we also analyzed the caryotype of the T-cell precursors and performed CGH array, both of which were normal (data not shown).
Based on these results, we are currently implementing two phase 1/2 clinical trials using human in vitro generated HTLPs, with the financial support of the French Ministry of Health and the sponsorship of Assistance-Publique Hopitaux de Paris. The first clinical trial will include 12 SCID patients undergoing haplo-identical HSCT. In this trial, patients will receive a first transplant of CD34+-selected cells, followed 8 days later by the injection of HTLPs generated from the residual CD34+ fraction at doses (0.1 up to 1.5 × 10e6 CD7 + cells/kg, one single dose of HTLPs per patient). The protocol has been approved by the French Drug Agency and is open to inclusion. The primary objective will be to assess the procedure’s safety: dose-limiting toxicity (DLT), including grade III-IV graft versus host disease (GVHD) and grade III or higher CTCAE adverse events (AEs). Secondary objectives will include: graft failure, the presence of naïve CD4+ T cells at 6 months, kinetics of T-cell reconstitution, incidence of infections, relapse rate, and overall survival.
The second trial will include 10 adult patients treated by double cord blood HSCT for leukemia. One cord blood will be injected without manipulation. The other one will be exposed 7 days to DL-4 culture conditions before infusion into the patient.
Both trials will allow not only evaluation of the procedure’s safety, but also the ability of adult versus CB-derived T-cell precursors to accelerate immune reconstitution in a different clinical context.
The first clinical trial for SCID patients will start in Q2 2019, followed by the second one in Q1 2020.
Gene therapy based on HTLPs
Our DL-4 culture system provides a unique plateform not only for cellular but also for gene therapy applications. We also explored the possibility to genetically modify HTLPs by combining gene transduction by SIN-retroviral (SIN-RV) and lentiviral (LV) vectors with DL-4 based culture. Several conditions were tested, transduction followed by DL-4 culture, DL-4 culture followed by transduction and a combination of both using either a VSVG SIN-retroviral vector and a BaEv –pseudotyped LV expressing either IL2RG gene or mCherry. The presence of gene-corrected HTLPs was evaluated at day 4 and day 7 of culture by the frequency and number of mCherry+ HTLPs at day 4 and day 7 and the quantification of T-cell potential in limiting dilution assay as described elsewhere [27]. Healthy donor HSPCs and SCID-X1 patients’ HSPCs were used, the results were compared to the ones obtained in classical gene transduction protocols. As a reminder, SCID-X1 patients exhibit a severe T-cell defect due to an early-stage blockade of T-cell differentiation caused by a loss-of-function mutation in the γC gene. SCID-X1 γC deficiency can be safely and efficiently corrected by the ex vivo transduction of HSPCs, as demonstrated in patients included in gene therapy protocols [33]. For both types of vectors (SIN-RV and BaEv-LV) and samples (healthy or SCID-X1 HSPCs), the combination of transduction and DL-4 culture conditions led to the appearance of a population of gene-corrected HTLPs from day 4 on (>30% [34]). Furthermore, we demonstrated that upon co-culture on OP9/DL1 cells, CD4+CD8+ double positive cells and TCR+ T cells appeared earlier and in higher numbers when gene transduction was combined to the DL-4 plateform. Our data indicated the correction of γC deficiency in BM SCID-X1 CD34+ HSPCs under both conventional and DL-4 culture conditions. However, T-cell differentiation was faster and more efficient under the DL-4 condition.
The possibility to obtain gene-corrected HTLPs able to accelerate T-cell reconstitution following gene therapy in infected SCID patients, but also for other clinical indications, needs to be further evaluated. We therefor plan to confirm both our in vitro and in vivo results in order to develop a GMP compatible protocol combining DL-4 culture with gene therapy.
A gene therapy protocol for infected SCID X1 patients including both, the infection of gene-corrected HSPCs and gene-corrected HTLPs, remains the ultimate goal. This proof of concept will be of key importance for the extension of the DL-4 protocol to other clinical applications.
Methods
Search strategy and selection criteria The first three references correspond to three major breakthroughs in HLA-partially incompatible HSCT. We searched PubMed between 1 Jan 2007, and 31 August 2018, with the terms « allogeneic hematopoietic stem cell transplantation », « haplo-identical HSCT » in combination with « thymus », « thymopoiesis », « immune reconstitution », « viral complications », « T-cell receptor excision circles », « SCID », and « lymphoid progenitors », « T-cell development » in combination with « Notch », « Delta-like-4 ligand » and « Delta-like-1 ligand ». We restricted our search to English publications. We selected reports from the past 5 years but did not exclude important and highly cited older publications. We searched the reference lists of articles identified by this search strategy and selected the 33 that we judged relevant. Review articles are also cited to provide more detail.
Conclusions
We have demonstrated the possibility to generate large amounts of HTLPs exhibiting the phenotypic and molecular signatures of their thymic counterparts from any source of CD34+ HSPCs.
Ex vivo generated HTLPs were able to colonize the thymus of NSG mice and produce polyclonal mature T cells in vitro and in vivo. The whole plateform has been moved to Necker GMP facility to implement two clinical trials including SCID and leukemic patients undergoing allogeneic transplantation in different settings. If we successfully shorten the severe immunodeficiency period following transplantation, the benefit for the patients and public health will be immense:
First of all, the reduction of incidence and severity of opportunistic infections (e.g., adenovirus, cytomegalovirus, enterovirus, and fungi infections) would significantly shorten the length of hospitalization for each patient. As a consequence the use of partially incompatible HSC donors could be extended to all patients requiring this procedure (even in transplantation units that are not familiar with this high-risk procedure), with a significant decrease in mortality and morbidity.
Apart from pure cell-therapy approaches, the combination of DL-4 culture with gene transduction protocols will open new perspectives in terms of gene therapy applications.
Change history
16 March 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41409-022-01629-2
References
Reisner Y, Itzicovitch LEA, Meshorer A, Sharon N. Hemopoietic stem cell transplant using mouse bone marrow spleen cells fractionated lectins. Proc Natl Acad Sci USA. 1978;75:2933–6.
Urbano-Ispizua A, Rozman C, Martínez C, Marín P, Briones J, Rovira M, et al. Rapid engraftment without significant graft-versus-host disease after allogeneic transplantation of CD34 + selected cells from peripheral blood. Blood. 1997;89:3967–73.
Robinson TM, O’Donnell PV, Fuchs EJ, Luznik L. Haploidentical bone marrow and stem cell transplantation: experience with post-transplantation cyclophosphamide. Semin Hematol 2016;53:90–7.
Toubert A, Glauzy S, Douay C, Clave E. Thymus and immune reconstitution after allogeneic hematopoietic stem cell transplantation in humans: Never say never again. Tissue Antigens. 2012;79:83–9.
Williams KM, Hakim FT, Gress RE. T cell immune reconstitution following lymphodepletion. Semin Immunol 2007;19:318–30.
Krenger W, Blazar BR, Hollander GA. Review article Thymic T-cell development in allogeneic stem cell transplant. Blood 2011;117:6768–76.
Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: A PIDTC natural history study. Blood. 2017;130:2718–27.
Touzot F, Moshous D, Creidy R, Neven B, Frange P, Cros G, et al. Faster T-cell development following gene therapy compared to haplo-identical hematopoietic stem cell transplantation in the treatment of SCID-X1. Blood. 2015;125:1–8.
Storek J, Geddes M, Khan F, Huard B, Helg C, Chalandon Y, et al. Reconstitution of the immune system after hematopoietic stem cell transplantation in humans. Semin Immunopathol 2008;30:425–37.
Bosch M, Khan FM, Storek J. Immune reconstitution after hematopoietic cell transplantation. Curr Opin Hematol 2012;19:324–55.
Reimann C, Dal Cortivo L, Hacein-Bey-Abina S, Fischer A, André-Schmutz I, Cavazzana-Calvo M. Advances in adoptive immunotherapy to accelerate T-cellular immune reconstitution after HLA-incompatible hematopoietic stem cell transplantation. Immunotherapy. 2010;2:481–96.
Clave E, Lisini D, Douay C, Giorgiani G, Busson M, Zecca M, et al. Thymic function recovery after unrelated donor cord blood or T-cell depleted HLA-haploidentical stem cell transplantation correlates with leukemia relapse. Front Immunol 2013;4:1–8.
Cavazzana M, Six E, Lagresle-Peyrou C, André-Schmutz I, Hacein-Bey-Abina S. Gene therapy for X-linked severe combined immunodeficiency: where do we stand? Hum Gene Ther 2016;27:108–16.
Hazenberg MD1, Otto SA, de Pauw ES, Roelofs H, Fibbe WE, Hamann D, et al. T-cell receptor excision circle and T-cell dynamics after allogeneic stem cell transplantation are related to clinical events. 2002;99:3449–53.
Clave E, Busson M, Douay C, Peffault de Latour R, Berrou J, Rabian C, et al. Acute graft-versus-host disease transiently impairs thymic output in young patients after allogeneic hematopoietic stem cell transplantation. Blood. 2011;113:6477–84.
Chaudhry MS, Velardi E, Malard F, van den Brink MR. Immune reconstitution after allogeneic hematopoietic stem cell transplantation: time to T up the thymus. J Immunol 2017;198:40–6.
Krenger W, Rossi S, Holländer GA. Apoptosis of thymocytes during acute graft-versus-host disease is independent of glucocorticoids. Transplantation. 2000;69:2190–3.
Wang SD, Huang KJ, Lin YS, Lei HY. Sepsis-induced apoptosis of the thymocytes in mice. J Immunol. 1994;152:5014–21.
Hick RW, Gruver AL, Ventevogel MS, Haynes BF, Sempowski GD. Leptin selectively augments thymopoiesis in leptin deficiency and lipopolysaccharide-induced thymic atrophy. J Immunol 2006;177:169–76.
Mocarski ES, Bonyhadif M, Salimif S, McCune JM, Kaneshima H. Human cytomegalovirus in a SCID-hu mouse: thymic epithelial cells are prominent targets of viral replication. Med Sci 1993;90:104–8.
Koning C, de, Admiraal R, Nierkens S, Boelens JJ. Human herpesvirus 6 viremia affects T-cell reconstitution after allogeneic hematopoietic stem cell transplantation. Blood Adv 2018;2:428–32.
Svaldi M, Lanthaler AJ, Dugas M, Lohse P, Pescosta N, Straka C, et al. T-cell receptor excision circles: a novel prognostic parameter for the outcome of transplantation in multiple myeloma patients. Br J Haematol 2003;122:795–801.
Koch U, Fiorini E, Benedito R, Besseyrias V, Schuster-Gossler K, Pierres M, et al. Delta-like 4 is the essential, nonredundant ligand for Notch1 during thymic T cell lineage commitment. J Exp Med 2008;205:2515–23.
Hozumi K, Mailhos C, Negishi N, Hirano K, Yahata T, Ando K, et al. Delta-like 4 is indispensable in thymic environment specific for T cell development. J Exp Med 2008;205:2507–13.
Taghon T, Waegemans E, Van de Walle I. Notch signaling during human T cell development. Curr Top Microbiol Immunol. 2012;360:75–97.
Hosokawa H, Rothenberg EV. Cytokines, Transcription Factors, and the Initiation of T-cell development. Cold Spring Harb Perspect Biol. 2018;1:1–20.
Six EM, Benjelloun F, Garrigue A, Bonhomme D, Morillon E, Rouiller J, et al. Cytokines and culture medium have a major impact on human in vitro T-cell differentiation. Blood Cells Mol Dis 2011;47:72–8.
Schmitt TM, Zúñiga-Pflücker JC. Thymus-derived signals regulate early T-cell development. Crit Rev Immunol 2005;25:141–59.
Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med 2010;16:232–6.
Reimann C, Six E, Dal-Cortivo L, Schiavo A, Appourchaux K, Lagresle-Peyrou C, et al. Human T-lymphoid progenitors generated in a feeder-cell-free delta-like-4 culture system promote T-cell reconstitution in NOD/SCID/γc-/-mice. Stem Cells. 2012;30:1771–80.
Six EM, Bonhomme D, Monteiro M, Beldjord K, Jurkowska M, Cordier-Garcia C, et al. A human postnatal lymphoid progenitor capable of circulating and seeding the thymus. J Exp Med 2007;204:3085–93.
Simons L, Ma K, de Chappedelaine C, Elkaim E, Olivré J, Susini S, et al. Generation of adult human T-cell progenitors for immunotherapeutic applications. J Allergy Clin Immunol 2018;141:1491–1494.e4.
Hacein-Bey-Abina S, Pai S-Y, Gaspar HB, Armant M, Berry CC, Blanche S, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med 2014;371:1407–17.
Bernadin O, Amirache F, Girard-Gagnepain A, Moirangthem RD, Lévy C, Ma K et al. Baboon envelope LVs efficiently transduced human adult, fetal, and progenitor T cells and corrected SCID-X1 T-cell deficiency. Blood Adv. 2019;3:461–75.
Acknowledgements
We thank Pauline Huguenin for help in bibliography.
Funding
The study was funded by the French National Institute of Health and Medical Research (INSERM), a European Research Council grant (ERC Regenerative Therapy, 269037), a European Union FP7 grant (CELL-PID, 261387), a European Union H2020 grant (SCIDNet, 666908), Imagine Institute, a Clinical Research Hospital Program (PHRC) (Ministry of Health and Social Affairs), an INCA-Plan Cancer grant (2009–2013) and a public grant overseen by the French National Research Agency (ANR) as part of the program “Investissements d’Avenir” (reference: ANR-10-IAHU-01). LS was funded by Imagine Institute. KM was funded by a China Scholarship Council (CSC). EE was funded by an INSERM-Plan Cancer fellowship. CR was funded by postdoctoral scholarships from the Deutsche Forschungsgemeinschaft (DFG). TT was funded by the Fund for Scientific Research Flanders (FWO Vlaanderen research projects G0B2913N). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Publication of this supplement was sponsored by Gilead Sciences Europe Ltd, Cell Source, Inc., The Chorafas Institute for Scientific Exchange of the Weizmann Institute of Science, Kiadis Pharma, Miltenyi Biotec, Celgene, Centro Servizi Congressuali, Almog Diagnostic.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
MC and IA-S own equity in Smart Immune and hold two patents in this area, about the in vitro process of production of T-cell progenitors. KM is co-inventor of patent WO 2018/146297 A1, Methods for generating T-cells progenitors. The remaining authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
André, I., Simons, L., Ma, K. et al. Ex vivo generated human T-lymphoid progenitors as a tool to accelerate immune reconstitution after partially HLA compatible hematopoietic stem cell transplantation or after gene therapy. Bone Marrow Transplant 54 (Suppl 2), 749–755 (2019). https://doi.org/10.1038/s41409-019-0599-9
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41409-019-0599-9
- Springer Nature Limited
This article is cited by
-
Terminal deoxynucleotidyl transferase and CD84 identify human multi-potent lymphoid progenitors
Nature Communications (2024)