
Abstract
Type 2 diabetes mellitus (T2DM) patients exhibit greater susceptibility to vascular calcification (VC), which has a higher risk of death and disability. However, there is no specific drug for VC therapy. NLRP3 inflammasome activation as a hallmark event of medial calcification leads to arterial stiffness, causing vasoconstrictive dysfunction in T2DM. Empagliflozin (EMPA), a sodium-glucose co-transporter 2 inhibitor (SGLT2i), restrains hyperglycemia with definite cardiovascular benefits. Given the anti-inflammatory activity of EMPA, herein we investigated whether EMPA protected against VC in the aorta of T2DM mice by inhibiting NLRP3 inflammasome activation. Since db/db mice receiving a normal diet developed VC at the age of about 20 weeks, we administered EMPA (5, 10, 20 mg·kg−1·d−1, i.g) to 8 week-old db/db mice for 12 weeks. We showed that EMPA intervention dose-dependently ameliorated the calcium deposition, accompanied by reduced expression of RUNX2 and BMP2 proteins in the aortas. We found that EMPA (10 mg·kg−1·d−1 for 6 weeks) also protected against VC in vitamin D3-overloaded mice, suggesting the protective effects independent of metabolism. We showed that EMPA (10 mg·kg−1·d−1) inhibited the abnormal activation of NLRP3 inflammasome in aortic smooth muscle layer of db/db mice. Knockout (KO) of NLRP3 significantly alleviated VC in STZ-induced diabetic mice. The protective effects of EMPA were verified in high glucose (HG)-treated mouse aortic smooth muscle cells (MOVASs). In HG-treated NLRP3 KO MOVASs, EMPA (1 μM) did not cause further improvement. Bioinformatics and Western blot analysis revealed that EMPA significantly increased the expression levels of basic helix-loop-helix family transcription factor e40 (Bhlhe40) in HG-treated MOVASs, which served as a negative transcription factor directly binding to the promotor of Nlrp3. We conclude that EMPA ameliorates VC by inhibiting Bhlhe40-dpendent NLRP3 inflammasome activation. These results might provide potential significance for EMPA in VC therapy of T2DM patients.
Similar content being viewed by others

Introduction
Vascular calcification (VC), the deposition of calcium phosphate in the vessel wall, is associated with aggravation of vascular sclerosis, and coronary artery calcium score is a potentially effective indicator for assessing cardiovascular risk in asymptomatic individuals which serves as a clinical guideline for precise risk stratification and prognostic determination [1,2,3]. VC is a passive regulatory process involving the trans-differentiation of vascular smooth muscle cells (VSMCs) into osteoblast-like cells and the high expression of ossification-related proteins such as runt-related transcription factor 2 (RUNX2) and bone morphogenetic protein 2 (BMP2) [4]. Diabetes mellitus (DM) patients exhibit greater susceptibility to VC, which has a higher risk of death and disability compared to DM without calcification [5]. According to data released by the International Diabetes Federation, DM affected nearly 463 million people aged between 20 and 79 in 2019 and the number is expected to surge up to 702 million by the year 2045. Although the treatment strategies to reduce blood glucose levels are widely used, the development of VC in diabetic patients remained increasing [6]. There is no specific drug for VC therapy in clinical application. Therefore, it is urgent to explore an effective drug therapy for VC and its deep molecular mechanism among hypoglycemia agents.
Empagliflozin (EMPA), a sodium-glucose co-transporter 2 inhibitor (SGLT2i), is currently employed as an oral hypoglycemic drug which inhibits the SGLT2 in the kidneys and increases urinal glucose excretion or glycosuria without insulin dependence [7], and it acts as one of the most common SGLT2i which is about 5 to 10 times more specific for SGLT2 over SGLT1 than dapagliflozin, ertugliflozin, and canagliflozin [8]. A Cardiovascular Outcome Event Trial in DM (EMPA-REG outcome trial) demonstrates EMPA has definite protective properties in cardiovascular disease [9], which some advantages were independent of lowering blood glucose [10]. Notably, EMPA significantly improves arterial stiffness compared to metformin in T1DM patients [11]. It is reported that EMPA can prevent arterial stiffness and lower blood pressure by anti-inflammatory mechanisms in T2DM patients [12]. However, another research reveals that there is no evidence of a favorable change in arterial stiffness with EMPA treatments in a randomized and prospective study of T2DM patients [13]. Thus, the vascular stiffness protective effects of EMPA have remained controversial. Furthermore, the effect of EMPA on VC has never been reported. Here, we investigate whether EMPA has a protective role in VC and consequent vascular stiffness caused by T2DM.
T2DM has been confirmed as a chronic inflammatory disease and this low-grade chronic inflammation can aggravate the development of adverse vascular events [14], particularly acts as a culprit of VC [15]. EMPA reduces inflammation through AMPK activation-mediated energy repletion and contributes to the observed cardiovascular benefits in heart failure (HF) [16]. EMPA inhibits inflammatory and oxidative stress, leading to less pathological cardiomyocyte stiffness [17]. Nucleotide-binding domain-like receptor 3 (NLRP3) inflammasome is the most potent initiator involved in the inflammation pathological process, which includes the nucleotide-binding domain, leucine-rich repeat, pyrin domain-containing protein 3 (NLRP3), the apoptosis-associated speck-like protein containing a CARD (ASC) and caspase-1. The assembly activates caspase-1, leading to the processing of bio-active IL-1β and IL-18 secretion under diabetic conditions [18]. It has been reported that NLRP3 inflammasome is involved in the regulation of cardiovascular diseases [19].
The current study reveals the contribution of NLRP3 inflammasome to VC and identifies that the protective effect of EMPA in VC is associated with the restoration of basic helix-loop-helix family transcription factor e40 (Bhlhe40), which has been classified as a negative transcription regulator of NLRP3 inflammasome by directly binding to the promoter region of the Nlrp3. Taken together, our study provides potential targeting genes and therapeutic glucose-lowering drugs for VC in T2DM.
Materials and methods
Animal studies
5 week-old male widetype (WT) and NLRP3 knockout (KO) mice on a C57BL/6 J background were obtained from GemPharmatech Company (Nanjing, China). 6 week-old male homozygous db/db mice on a C57BL/6 background were purchased from the Model Animal Research Center of Nanjing University (Nanjing, China). All mice were housed in an animal facility at 25 ± 2 °C with 55%–60% humidity and a 12 h light/dark cycle. We processed all animal experiments following the Guidelines for Animal Experiment set by the Bureau of Sciences and Techniques of Jiangsu Province, China [NO. SYXK2007-0025]. Experimental procedures were conducted under protocols approved by the Institutional Animal Care and Use Committee at the Southeast University.
db/db mice VC model
The db/db mice have been used as a model of T2DM and other metabolic disorder including obesity and dyslipidemia [20]. This type of mice can develop VC about 20 week-old with a normal diet (36% corn, 23% triturate wheat, 10% bran, 12% soy bean powder, 3% egg; 12% fish powder, 2% dried yeast, 1% of a mixture of calcium bicarbonate, multi-vitamins and micro-elements) as described previously [21]. In brief, after adaptive feeding for 2 weeks, the interventions with EMPA (5, 10, 20 mg·kg−1·d−1, i.g) were conducted, respectively. Mice of control group were given an equal volume of saline solution containing 0.6% ethanol. The living environment of all mice was maintained at a standard controlled temperature (25 ± 2°C). At the age of 20 weeks, all mice were sacrificed.
STZ-induced diabetic VC(DVC) mice model
5 week-old mice were divided into 4 groups (n = 9). The model mice were fed with a high-fat diet (HFD, 10% saccharose, 10% lard, 10% sugar, 5% egg yolk powder, 0.5% cholesterol, 74.5% basal chow) for 4 weeks. Subsequently, mice fed with HFD were intraperitoneally injected with low-dose streptozotocin (STZ) (40 mg·kg−1, dissolved in cold citrate buffer, pH 4.5) for 5 consecutive days, and the other mice were sham-treated with citrate buffer. One week after the final injection, mice on HFD with low STZ were recognized as diabetic mice when the fasting blood glucose (FBG) level reached 11.1 mmol·L−1. Then, the diabetic mice were fed continually with HFD for 14 weeks before being sacrificed. The living environment of all mice was maintained at a standard controlled temperature (25 ± 2°C).
Vitamin D3‐induced VC mice model
Vitamin D3 (cholecalciferol, 5.5 × 105 U·kg−1·d−1) was subcutaneously administered to the C57BL/6 J mice for 3 consecutive days at the beginning of the experiment [22,23,24]. Then, vitamin D3-overloaded mice received oral gavages of EMPA (10 mg·kg−1) at once per day for 6 weeks. We gave mice of the control group an equal volume of saline solution containing 0.6% ethanol. At the end of the experiment, we anesthetized the mice with pentobarbital sodium (50 mg·kg−1, i.p).
Primary culture of mouse aortic smooth muscle cells (MOVASs)
Primary MOVASs were isolated and cultured according to the prior protocol [25]. Cells were grown in DMEM basic (1×) (Gibco, Waltham, MA, USA) with 10% fetal bovine serum (Gibco, Waltham, MA, USA), 1% penicillin–streptomycin (Gibco, Waltham, MA, USA) at 37 °C in a 5% CO2 humidified incubator. We replaced the medium twice a week until the cells grew to 80%–90% confluence. MOVASs were identified by immunofluorescence analysis for the specific protein expression of α-SMA (1:200, Affinity, Changzhou, China).
Cell treatments and reagents
In the preliminary experiment, we used mannitol as a high glucose osmotic control and confirmed that it had no effect on relevant stimulators, which was consistent with the normal culture medium. To simulate the diabetes microenvironment, we incubated the cells with high glucose (HG, 30 mM) or AGEs-BSA (AGEs, 100 μg·mL−1) for 14 days, and the cells of the control group were incubated with normal culture medium or BSA (100 μg·mL−1). Empagliflozin (EMPA) was provided by MedChemExpress (Shanghai, China). β-Sodium glycerophosphate (Pi, 10 mM), bovine serum albumin (BSA), and D-glucose were purchased from Sigma-Aldrich (St. Louis, MO, USA). AGEs-BSA preparation: 5 g·L−1 BSA and 50 mmol·L−1 D-glucose were dissolved together in phosphate-buffered saline (PBS), the solution was then sterilised by ultrafiltration and incubated in the dark at 37 °C for 90 days. Next, the incubated AGEs-BSA was dialyzed by filtration membrane that only reserved AGEs-BSA and removed D-glucose and BSA for two days. Lastly, the purified AGEs-BSA was obtained and its concentration was estimated by a BCA protein assay kit.
Von kossa staining
Von Kossa staining was performed to detect the tissue mineralization in animals [26]. The sections of isolated aorta tissues were dewaxed and hydrated firstly. Then, samples were subsequently incubated with 1% silver nitrate solution for 30 min under an intense sunbeam or ultraviolet light. Samples were subsequently washed and treated with 5% sodium thiosulfate. After washing several times, brown to black calcified nodules were observed under the light microscope. Percentage of positively stained area for each aortic section was quantified using ImageJ software (NIH, Littleton, CO, USA).
Quantification of calcium deposition
Homogenized aortic tissues or MOVASs were decalcified with 0.6 mol·L−1 HCl for 24 h. Calcium content was determined by the calcium assay kit according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, Nanjing, China), and the protein concentration was detected by BCA protein assay kit (Beyotime Biotechnology, Jiangsu, China). The calcium content was normalized to its protein content.
Western blot analysis
The protein expressions of the aorta and the MOVASs were analyzed using Western blot as described before [27]. Protein extracts were determined by the BCA protein quantitative kit (KeyGEN BioTECH, Nanjing, China). Following boiling for 5 min at 95 °C in a 5×loading buffer (Saint-Bio, Shanghai, China), the prepared proteins were separated by 6% or 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS PAGE) at 80 V for 30 min and then 120 V for 1 h, transferred onto polyvinylidene fluoride membranes (PDVF membrane, 0.45 μm, Millipore, Burlington, MA, USA.) at 200 mA for 1 h, and then, the membrane was incubated with nonfat milk (5% w·v−1) in Tris-buffered saline with Tween 20 (TBST) for 1 h. After washing three times with TBST, we probed the membranes with primary antibody (1:100–1:5000 dilutions) overnight at 4 °C. After the triple washing with TBST, the membranes were incubated with Goat Anti-Rabbit IgG (1:5000; Affinity, Changzhou, China) for 1 h at room temperature. The immunoreactive bands were detected by chemiluminescent detection systems with LumiGlo and Peroxide (1:1, Univ-Bio, Shanghai, China). The density of the bands was analyzed by ImageJ software (NIH, Littleton, CO, USA).
Measurement of pulse wave velocity (PWV)
Pulse wave velocity (PWV) is a gold diagnosis standard of arterial stiffness [28]. In brief, mice was anesthetized by 2% isoflurane and confirmed the depth of anesthesia by unresponsiveness to toe pinch. Then, mice was placed on a heating board with legs secured to electrocardiogram electrodes (Iworx, Vista, CA, USA). Noninvasive Doppler probes (Indus Instruments, Webster, TX, USA) were used to record the curvilinear distance from the aortic arch to the abdominal (D2 − D1; in mm). The electrocardiogram signal was detected simultaneously to identify the relative time delay (T2 − T1; in ms). PWV was calculated as (D2 − D1)/(T2 − T1) and was presented as m·s−1.
Isometric vascular function
The constriction function of the aorta was examined through a tension detection system (BL-420S, TaiMeng, Chengdu, China). After the mice were anesthetized, aorta was quickly removed and immersed into ice-cold Krebs Henseleit solution (mM) (KH solution, pH 7.4, 119.0 NaCl, 25.0 NaHCO3, 11.1 Glucose, 2.4 CaCl2, 4.7 KCl, 1.2 KH2PO4, 1.2 MgSO4, 0.024 Na2EDTA). The aortas were dissected from free of connective tissue and fat and then cut into three separated aortic rings with a width of approximately 2 mm. All anatomical procedures were done carefully in order to protect the endothelium and smooth muscle layer from unexpected damage. Aortic rings were then suspended in water-jacketed tissue baths and tested for isometric force recorded by a tension detection system. The KH solution was maintained at 37˚C and the mixed gas contained 95% O2 and 5% CO2 was continuously bubbled through the bath. The baseline load placed on the aortic rings was 1.0 g. Changes in isometric tension were recorded using a force-displacement transducer (TaiMeng, Chengdu, China). The aortic rings were washed every 10 min with KH solution until the tension returned to the basal level. After stabilization, the rings were contracted with 60 mM KCl to obtain a maximal response as a reference value (100%) [29]. And then a cumulative dose-response curve to phenylephrine (Phe, 1 × 10−9−1 ×10−4 M) was detected in the aorta.
Biochemical analysis
Blood samples from the mice were collected and centrifuged at 3000 ×g for 10 min at 4 °C. The serum was isolated and stored at −80 °C before use. Fasting blood glucose (FBG), fasting insulin (FIN), triglyceride (TG), IL-1β, IL-18, TNF-ɑ and MCP-1 were assayed following the instructions of the kits provided by the Jiancheng Bioengineering Institute (Nanjing, China) and Proteintech Group (Proteintech™, Wuhan, China). The procedures specified by the manufacturer were followed. The absorbance was detected using Multiskan FC (Thermo Scientific, Waltham, MA, USA). Homeostasis model assessment of insulin resistance (HOMA-IR) was calculated as FBG (mmol·L−1) ×FIN (mU·L−1)/22.5.
MOVASs were seeded in 96-well plates at a concentration of 1 × 104 cells/well and incubated until 50%–60% confluent. After the treatments, cell supernatants were collected, IL-1β and IL-18 production were respectively measured by ELISA kits as described in vivo. Caspase-1 activity was detected according to the manufacturer’s instructions (Solarbio, Beijing, China).
Immunofluorescence analysis
Mice aortas were perfused with PBS and fixed with 4% paraformaldehyde and embedded in paraffin. The paraffin sections of the aortic rings were sliced, dewaxed and washed with PBS three times for 5 min, followed by blocked with 3% H2O2-CH3OH for 30 min, citrate antigen retrieval solution for 18 min and 5% BSA in PBS for 1 h at room temperature. The sections were incubated with anti-Bhlhe40 (1:100, Novus, Centennial, CO, USA), anti-ɑ-SMA (1:200, Affinity, Changzhou, China), anti-NLRP3 (1:100, Abways, Technology, Inc., Shanghai, China), anti-ASC (1:100, Abways Technology Inc., Shanghai, China), respectively, overnight at 4 °C. After washing triple with PBS, the sections were then incubated with Goat Anti-Rabbit IgG (H + L) Alexa Fluor 488 (1:100, Abways Technology Inc., Shanghai, China) or Cy3 Conjugate (1:100, Boster, Wuhan, China) for 2 h at room temperature, respectively. The sections were sealed with neutral balsam (Solarbio, Beijing, China).
Fluorescence images of aortas were photographed by a laser scanning confocal microscope (LSM700, Carl Zeiss, Oberkohen, Germany) and the images were processed by ZEN blue 2.3 software (Carl Zeiss, Oberkohen, Germany). The Pearson’s coefficient which represented the co-localization of Bhlhe40/ɑ-SMA was calculated by Image J software (NIH, Littleton, CO, USA).
Alizarin Red S staining
Alizarin Red S staining was performed to detect the calcification of MOVASs. Cells were fixed in 4% polyoxymethylene for 20 min. After triple washing with PBS, cells were incubated with staining solution for 20 min at room temperature. Then, the cells were observed and photographed under the light microscope. The quantified data were calculated by measuring optical density (OD) at 420 nm with a microplate reader.
Cell transfection
MOVASs were seeded in 6-well plates and the transfection was performed when the cells were 60%–70% confluence. Bhlhe40 siRNA was purchased from Santa Cruz (sc-142950, Dallas, TX, USA). The scrambled siRNA (control siRNA, sc-37007, Dallas, TX, USA) was confirmed as non-silencing double stranded RNA as control in current study. The plasmids of Bhlhe40 (pcDNA3.1-Bhlhe40) were designed through the insertion of mRNA with the mouse Bhlhe40 CDS region into the pcDNA3.1 vector. Transfection of siRNA and plasmids were performed using the LipofactmineTM2000 Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions. pcDNA3.1-Bhlhe40 and pcDNA3.1 vector treated cells were defined as Bhlhe40 overexpression (Bhlhe40 OV) and negative control overexpression (NC OV) group. The transfection efficacy of Bhlhe40 was confirmed by PCR analysis.
Reverse transcription and quantitative real-time PCR (RT-qPCR)
Total RNA was isolated from cultured cells by RNA extraction kit (TianGen, Beijing, China). The concentration of the total RNA was analyzed by Nano100 ultra-micro spectrophotometer (Allsheng, Hangzhou, China). 1 μg RNA was reverse-transcribed into cDNA in 20 μl reaction volumes via the Fast Quant RT kit (TianGen, Beijing, China). SYBR Green reaction mix (TianGen, Beijing, China) in the 20 μl reaction mixtures was used to perform real-time PCR by Real-Time qPCR instrument (Life Tech, QuantStudio3, Carlsbad, CA, USA). The reaction was run at 95 °C for 15 min, followed by 40 cycles of 10 s at 95 °C and 30 s at 60 °C. The mRNA levels were calculated by ΔΔCT method and normalized by β-actin. The PCR primers were: Bhlhe40: forward 5′-GGAGAGCGAGGTTACAGTG-3′ and reverse 5′-AATGCCAGGCACATGACAAG-3′. SGLT2: forward 5′-AACAGCAGTAGCACGCTCTT-3′ and reverse 5′-AAAGACCGCAGACACTGGAG-3′. β-actin: forward 5′-ATCATGTTTGAGACCTTCAACAC-3′ and reverse 5′-TCTGCGCAAGTTAGGTTTTGTC-3′.
RNA-sequencing of MOVASs
The quality sample selection, library preparation and RNA-sequencing were operated by Shanghai Biotechnology Corporation, China. Different genes expression were identified by using edgeR. (filter criteria: P ≤ 0.05 and fold-change ≥2). Then the functional enrichment was presented with the difference in gene expression by Metascape, and several annotation categories were identified. A heatmap was constructed for representative gene expression, using pheatmap in the R package and considering normalized values by z‐scores.
Luciferase reporter assay
The potential binding sites of Cebpb (CCAAT/enhancer-binding protein beta) and the promoter of Nlrp3 gene have been predicted by bioinformatics analysis. Next, we used luciferase reporter assay to further confirm the binding efficacy. The fragment of Nlrp3 promoter including the predicted binding sites of Cebpb was amplified by PCR and inserted into the pGL3 luciferase reporter vector (Promega, Madison, WI, USA). Moreover, QuikChange Multi Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) was used to create a mutant promoter of Nlrp3. MOVASs were seeded into 96-well plates and then transfected with a luciferase reporter containing the wild type Nlrp3 (WT-pGL3-Nlrp3) or the mutant (Mut-pGL3-Nlrp3) promoter and pRL-TK for 48 h. Luciferase activities in cells were quantified using a luciferase reporter assay (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Statistical analysis
Statistical analysis was performed using IBM SPSS Statistics 20 software (SPSS Inc., Chicago, IL, USA). Data were presented as means ± standard deviations (SD). The Student’s t-test was used to determine significant differences between the two groups, and the significant differences within multiple groups were examined using ANOVA for repeated measures, followed by Duncan’s multiple range test. P < 0.05 was considered statistically significant.
Results
EMPA inhibited VC and alleviated vascular dysfunction in db/db mice
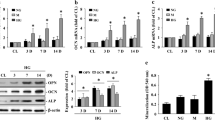
To explore the effects of EMPA on VC in diabetic mice, we treated db/db mice with EMPA in different concentrations (5, 10, 20 mg·kg−1·d−1) for 12 weeks (Fig. 1a). Von Kossa (VK) staining and calcium content analysis (Fig. 1b–d) showed that calcium deposition was apparently enhanced in the aorta of db/db mice, which confirmed the aortic medial calcification can be developed in db/db mice at 20 week-old [21]. However, these effects were significantly inhibited by EMPA treatments. The essence of VC is an active regulatory process, including the trans-differentiation of vascular smooth muscle cells (VSMCs) into osteoblast-like cells [4], and the endogenous ossification markers for VSMCs comprise BMP2 and RUNX2 [30]. Therefore, we further detected these two osteogenic proteins’ expression (Fig. 1e-f), which were dramatically induced in the aorta of db/db mice, whereas were significantly reversed by EMPA treatments. Pulse wave velocity (PWV) is considered as the clinical benchmark for detecting arterial stiffness [15]. Next, to investigate the effect of EMPA on vascular function in db/db mice, the aortic PWV was detected, and the vascular contraction responses to phenylephrine (1 × 10−9−1 ×10−4 M) were tested in the intact aorta of mice. Compared to the control group, the aortic PWV of db/db mice was increased more obviously, and ɑ receptor-dependent contraction responses were significantly impaired in db/db mice. Nevertheless, these effects were apparently reversed by the treatments of EMPA (Fig. 1g–h).

Schematic of the animal study design showing procedure for 6 week-old db/db mice, which were treated with EMPA at the concentration of 5, 10, 20 mg·kg−1·d−1 after 2 weeks adaptive feeding (a). Representative VK staining microscopic images showed the calcium deposition in aortas (b). Quantification of these results was summarized (c). And aortic calcium contents were determined by calcium assay kit (d). Western blot analysis showed protein expression of BMP2 and RUNX2 (e), and the summarized data presented the intensity of these two proteins’ expression (f). PWV was tested in the aorta of mice by Noninvasive Doppler probes (g). Vascular contraction responses to phenylephrine (Phe) (1 × 10−9−1 ×10−4M) showed the role of EMPA on aorta from db/db mice (h). Data are means ± SD from 7–8 experiments. *P < 0.05 vs. Control (Ctrl); #P < 0.05 vs. db/db group.
Furthermore, as shown in sFig. 1a–c, FBG, HOMA-IR and triglyceride in the serum of db/db mice were dramatically increased compared to the control group. The intervention with EMPA throughout 12 weeks resulted in a significant decrease in FBG and HOMA-IR, as well as triglyceride. It suggested EMPA relieved metabolic disorders and insulin resistance in db/db mice. Moreover, we found all these effects of EMPA were in a dose-dependent way. EMPA 10 and 20 mg·kg−1·d−1 can suppress aortic calcification and improve vascular function obviously than 5 mg·kg−1·d−1 treatments. Therefore, 10 mg·kg−1·d−1 EMPA was used to treat mice for the following analysis.
To verify if these protection roles of EMPA on VC are dependent on hypoglycemic and anti-metabolic effects, vitamin D3 induced VC mice were used to confirm the effects of EMPA without diabetic condition (sFig. 2a). Interestingly, VK staining presented that EMPA also obviously decreased calcium deposition in aorta of vitamin D3 overloaded mice (sFig. 2b, c). Western blot analysis showed EMPA inhibited the expression of osteogenic proteins in vitamin D3 induced calcified aorta (sFig. 2d, e). The calcium contents and PWV of aorta were significantly ameliorated by EMPA as well (sFig. 2f, g). These results were consistent with the trend of previous diabetic VC model, which demonstrated the beneficial effects of EMPA on VC were independent on regulating the blood glucose and metabolic disorders.
EMPA inhibited NLRP3 inflammasome activation in the arterial smooth muscle layer of db/db mice
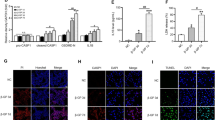
T2DM was considered as a low-grade chronic inflammatory syndrome which aggravated the development of adverse vascular events such as VC [14, 15]. Moreover, vitamin D3 induced VC still accompanied inflammation [31]. Therefore, we investigated whether this protective effect of EMPA on VC was related to inflammation in diabetic mice. The expression of IL-1β, IL-18, TNF-ɑ and MCP-1 were detected in serum with commercially available ELISA kit. As illustrated in Fig. 2b-e, all these inflammatory cytokines were obviously increased in db/db mice. However, these changes were significantly reversed by EMPA treatments, which showed no remarkable effects under control conditions. It confirmed that EMPA displayed anti-inflammatory potential with the cardiovascular disease benefit [32]. Notably, we found both IL-1β and IL-18 can be inhibited by EMPA simultaneously, which were considered as the critical downstream indicators of NLRP3 inflammasome activation that serves as a principal pathogenesis to trigger the inflammatory response [33]. NLRP3 inflammasome comprises NLRP3, ASC and recruited pro-caspase-1. Upon recruitment to an inflammasome complex, caspase-1 is activated and processed into mature caspase-1 subunit (p20), which is secreted into the culture supernatants [34]. We consequently analyzed the protein expression of NLRP3, ASC, p20/pro-caspase-1 and IL-1β/pro-IL-1β in aorta which were dramatically induced in db/db mice, whereas these impairments were apparently decreased by EMPA intervention, which showed no obvious effects under control conditions (Fig. 2f, g). The co-localization of NLRP3 and ASC is a hallmark of NLRP3 inflammasome activation and was assessed by immunofluorescence analysis (Fig. 2h). Yellow punctuates or patches which merged by red ones (NLRP3) and green ones (ASC) in cytoplasmic regions under the confocal microscope represented the formation of NLRP3 inflammasome in db/db mice. Compared with these diabetic mice, barely yellow dots were detected in aortas when treated with EMPA, showing less NLRP3 inflammasome was assembled. Thus, we speculated EMPA inhibited the assembly of NLRP3 inflammasome, which ultimately blocked caspase-1 and IL-1β maturation. ɑ-SMA is a well-recognized marker of smooth muscle cells [35]. Using confocal microscopy, the co-localizations of NLRP3 vs. ɑ-SMA and ASC vs. ɑ-SMA were apparently increased, as shown in yellow color in the aorta of db/db mice (Fig. 2i, j). It suggested the location of T2DM-induced NLRP3 inflammasome activation occurred in the aortic smooth muscle layer of mice.

Diabetic VC model was constructed in 6 week-old db/db mice, which were treated with or without EMPA at the concentration of 10 mg·kg−1·d−1 (a). The activity of IL-1β (b), IL-18 (c), TNF-ɑ (d) and MCP-1 (e) levels in the serum by ELISA. The protein expression of NLRP3, ASC, p20/pro-caspase-1 and IL-1β/pro-IL-1β with qualification data (f, g), Representative confocal microscopic images showed the colocalization of NLRP3 and ASC (h), NLRP3 and ɑ-SMA (i), ASC and ɑ-SMA (j) in aorta of mice. Data are means ± SD from 7–8 experiments. *P < 0.05 vs. Ctrl; #P < 0.05 vs. db/db mice group.
Next, we used NLRP3 KO mice to investigate the effects of NLRP3 inflammasome on VC in vivo (sFig. 3a). Mice were treated by STZ to induce diabetic VC (DVC) as described before [36]. The levels of IL-1β and IL-18 were increased significantly in the serum of DVC mice, but were rescued by NLRP3 KO (sFig. 3b, c). As shown in sFig. 3d–f, DVC group had a higher protein expression of BMP2 and RUNX2 in the aortas of WT mice. However, NLRP3 KO significantly decreased both of these proteins compared to DVC mice, which showed no remarkable effects under control mice. VK staining presented NLRP3 KO decreased calcium deposition obviously in the aorta of DVC mice (sFig. 3g, h). Moreover, the calcium contents and PWV of the aorta in DVC mice were significantly ameliorated by NLRP3 KO as well (sFig. 3i, j). These results demonstrated that NLRP3 deletion dramatically reduced arterial calcification in diabetic mice.
EMPA attenuated HG-induced calcification in MOVASs by inhibiting NLRP3 inflammasome
WT and NLRP3 KO MOVASs were used to confirm the protective effects of EMPA on VC by regulating NLRP3 inflammasome activation in vitro. Western blotting was performed to detect the protein expression of RUNX2 and BMP2 in MOVASs (Fig. 3a). The summarized data in Fig. 3b, c showed that HG obviously increased both of the RUNX2 and BMP2 proteins’ levels compared to the control group, but were inhibited by EMPA in WT cells. Interestingly, these proteins’ expression of NLRP3 KO cells were obviously decreased in the HG group, which was consistent with the former detection in vivo. However, compared to the HG + NLRP3 KO group, EMPA had no significant effect on the protein expression of RUNX2 and BMP2 in NLRP3 KO cells. Next, we used Alizarin Red S staining to further confirm the effects of EMPA on HG-induced calcium deposition between WT and NLRP3 KO cells. The cells were additionally added with Pi due to the characteristic calcium deposition in vitro. The positively stained area showed a reddish color. As shown in Fig. 3d, e, compared to the control group, the calcium deposition of WT cells were apparently increased by HG+Pi, which was substantially recovered by pretreatment with EMPA, or by NLRP3 KO. However, no significant changes were observed in NLRP3 KO cells between HG+Pi and HG+Pi+EMPA group. Figure 3f, g showed IL-1β and IL-18 in culture supernatants were both increased obviously under HG conditions, which were inhibited by EMPA in WT cells. Nevertheless, there were no remarkable changes between each group in NLRP3 KO cells.

According to the result of preliminary experiment, WT and NLRP3 KO MOVASs were treated with 1 μM EMPA under high glucose (HG, 30 mM). Representative Western blot gel documents and summarized data showed the role of EMPA on the protein expression of RUNX2 and BMP2 (a–c). Alizarin Red S staining images showed calcium deposition in cells with β-sodium glycerophosphate (Pi, 10 mM) (d). Quantification of these results were summarized (e). ELISA kit analysis showed the expression of IL-1β and IL-18 in the culture supernatants of MOVASs (f, g). Data are means ± SD from 3–4 experiments. *P < 0.05 vs. Ctrl; #P < 0.05 vs. HG or HG + Pi treated WT group.
AGEs are identified as an important factor in the progression of diabetes and diabetes-induced complications which represents one of the products in the end state of diabetes [37]. To confirm the exhaustive role of EMPA on osteogenic differentiation and calcium deposition in the persistent state of T2DM in vitro, cells were treated by AGEs. Not surprisingly, Western blot analysis (sFig. 4a–c) and Alizarin Red S staining (sFig. 4d, e) as well as ELISA kit analysis (sFig. 4f, g) showed EMPA had inhibitory effects on the AEGs-induced calcification and NLRP3 inflammasome activation in WT MOVASs. No obvious differences were found between each group in NLRP3 KO cells. These results were consistent with the HG-induced model as before.
Furthermore, to eliminate whether the EMPA protective effect on VC was associated with SGLT2 inhibition, we detected the expression of SGLT2 in the aorta and MOVASs. Compared to the kidney group, the SGLT2 mRNA expression was very weak (sFig. 5a).And Western blot analysis indicated there were no obvious bands expression of SGLT2 in MOVASs compared with mouse aortic endothelial cells (MAOECs) or kidney group (sFig. 5b, c). In brief, SGLT2 was not expressed in the arterial smooth muscle layer of the aorta. These data demonstrated EMPA attenuated HG-induced calcifications in MOVASs was independent of SGLT2.
EMPA inhibited NLRP3 inflammasome by reversing Bhlhe40 diminution in HG-induced MOVASs
To better explore the regulatory mechanism underlying the VC protection effect of EMPA, RNA sequencing analysis was performed in MOVASs. The differential gene data were loaded into Metascape for gene ontology (GO) enrichment analysis and selected gene expression was represented in a heatmap, bioinformatics testing and analysis have revealed Bhlhe40 as a transcription factor was up-regulated by EMPA which had common base binding sites with the promoter of Nlrp3 (Fig. 4a–c). Next, the protein levels of Bhlhe40 were investigated by Western blot. As shown in Fig. 4d, compared to the control group, the protein expression of Bhlhe40 was apparently decreased by HG, which was substantially recovered by EMPA. Next, Chromatin Immunoprecipitation (ChIP) was used to determine the direct binding between Bhlhe40 and the promoter of Nlrp3. We found anti-Bhlhe40 antibody (but not normal IgG) successfully precipitated the Nlrp3 promoter which the corresponding band was identified below 250 bp (Fig. 4e). We then transfected Bhlhe40 plasmid (Bhlhe40 OV) into MOVASs, and the transfection efficacy of Bhlhe40 was three times higher compared with negative control overexpression group (NC OV) (Fig. 4f). Moreover, Luciferase reporter analysis confirmed that transfection of Bhlhe40 significantly inhibited the relative luciferase activity of wild type Nlrp3 gene, but did not reduce the luciferase activity of mutant Nlrp3 reporter gene (Fig. 4g). To elucidate the role of Bhlhe40 on HG-induced NLRP3 inflammasome activation in MOVASs, the mRNA level and protein expression of NLRP3 in MOVASs, as well as caspase-1 activity, IL-1β and IL-18 production in culture supernatants were analyzed respectively (Fig. 4h–l). As expected, Bhlhe40 gene overexpression inhibited the formation and activation of NLRP3 inflammasome in MOVASs under HG treatments. Combined with Fig. 3, it suggested that Bhlhe40 can directly bind to the promoter of Nlrp3 and inhibit HG-induced NLRP3 inflammasome activation, which participated in the protective role of EMPA on MOVASs calcification.

The selected gene expression was represented in a heatmap (a). Gene Ontology (GO) enrichment analysis for mRNA profile in MOVASs treated with EMPA (b, c). Representative Western blot gel documents and summarized data showed the role of EMPA on the protein expression of Bhlhe40 (d). Chromatin Immunoprecipitation (ChIP) was used to determine the direct binding between Bhlhe40 and the promoter of Nlrp3 (e). MOVASs were transfected with the NC OV and Bhlhe40 OV, the mRNA expression of Bhlhe40 was detected by RT-qPCR (f). Luciferase reporter analysis detected the transfection efficacy of luciferase reporter carrying WT-pGL3-Nlrp3 or Mut-pGL3-Nlrp3 and their co-transfection with the Bhlhe40-OV or not. Data were presented as firefly luciferase values, which were normalized for Renilla luciferase (g). RT-qPCR and Western blot analysis showed the role of Bhlhe40 on the mRNA level and protein expression of NLRP3 (h, i), ELISA kit analysis showed the expression of caspase-1, IL-1β and IL-18 in the culture supernatants of MOVASs (j–l). Data are means ± SD from 3-4 experiments. *P < 0.05 vs. Vehicle (Vehl)/Negative control (NC) or Negative control overexpression vector (NC OV); #P < 0.05 vs. HG or WT-pGL3-Nlrp3+Bhlhe40 OV treated group.
EMPA attenuated HG-induced osteogenic differentiation and calcium deposition by increasing Bhlhe40 in MOVASs
To confirm the role of Bhlhe40 on EMPA protective effect of osteogenic differentiation and calcium deposition caused by HG in MOVASs, we further transfected Bhlhe40 siRNA into MOVASs, and the transfection efficacy was 75% (Fig. 5a). After Bhlhe40 siRNA transfection, HG incubation obviously increased the protein expressions of RUNX2 and BMP2. However, compared to the HG group, no significant changes were observed in EMPA treatments when lacking Bhlhe40 under HG (Fig. 5b, c). Next, we used calcium assay kit to detect the calcium content of the cell supernatants through a fluorescent micro-plate reader. As shown in Fig. 5d, we observed calcium contents were increased obviously by HG, but cannot be reversed by EMPA under the Bhlhe40 silence condition. Alizarin Red S staining images and summarized data were shown in Fig. 5e, f, compared to the scramble group, the calcium deposition was apparently increased in HG+Pi, which cannot be recovered by EMPA after Bhlhe40 siRNA transfection. However, when Bhlhe40 OV was transfected into MOVASs, the calcium deposition was significantly decreased compared with HG incubation (sFig. 6a, b). These results above suggested that EMPA attenuated HG-induced osteogenic differentiation and calcium deposition by increasing Bhlhe40 in MOVASs.

Bhlhe40 siRNA was transfected into MOVASs. The mRNA expression of Bhlhe40 in MOVASs was detected by RT-qPCR (a). MOVASs were incubated in HG or HG + Pi after the Bhlhe40 siRNA transfection with or without EMPA (1 μM). Representative Western blot gel documents and summarized data showed the role of EMPA on the protein expression of RUNX2 and BMP2 when lacking Bhlhe40 (b, c), Calcium contents in the cell supernatants were determined by calcium assay kit (d). Alizarin Red S staining images showed the calcium deposition in cells (e). Quantification of these results were summarized (f). Data are means ± SD from 3–4 experiments. *P < 0.05 vs. Scram.
We also examined the expressions of Bhlhe40/NLRP3 axis in primary human aortic smooth muscle cells (HAoSMCs). According to the previous study on the dosage of EMPA in HAoSMCs [38]. Cells were treated by HG (30 mM) with or without EMPA (1 μM) for 14 days. EMPA reversed HG induced calcium deposition and Bhlhe40 protein decrease in HAoSMCs (sFig. 7a–d). Further, we transfected Bhlhe40 siRNA into cells, and Bhlhe40 mRNA levels were inhibited by 70% (sFig. 7e). After Bhlhe40 siRNA transfection, HG incubation obviously increased the protein expression of NLRP3 in HAoSMCs. However, EMPA had no significant effect on HG induced NLRP3 protein expression (sFig. 7f). These results further clarified that VC protective role of EMPA was dependent on Bhlhe40/NLRP3 axis under HG.
EMPA reversed Bhlhe40 diminution in the arterial smooth muscle layer of db/db mice
To further clarify whether EMPA can reverse Bhlhe40 expression in the arterial smooth muscle layer of diabetic mice. RT-qPCR was used to determine the role of EMPA on the mRNA level of Bhlhe40 in the aortas of db/db mice (Fig. 6b). Western blotting was performed to further detect the protein expression of Bhlhe40 in the aortas of mice (Fig. 6c), the qualification data was summarized (Fig. 6d). These showed that Bhlhe40 mRNA and protein levels were obviously decreased in the aortas of db/db mice. However, these changes were markedly reversed by EMPA intervention exclusive of the normal mice. Moreover, we using immunofluorescence staining to detect the distribution and the co-localization of Bhlhe40 and ɑ-SMA in the aortas of mice, the confocal microscopy images with the qualification data (Fig. 6e, f) showed the co-localization of Bhlhe40 and ɑ-SMA were apparently decreased, which were recovered by EMPA intervention as shown in yellow color in the arterial smooth muscle layer of the db/db mice. Taken together, EMPA can reverse Bhlhe40 decrease in the aortas caused by diabetes, no obvious changes were observed in normal mice.

Diabetic VC model was constructed in 6 week-old db/db mice, which were treated with or without EMPA at the concentration of 10 mg·kg−1·d−1 (a). The mRNA expression of Bhlhe40 was detected by RT-qPCR (b). Representative Western blot gel documents and summarized data showed the role of EMPA on the protein expression of Bhlhe40 in the aortas of mice (c, d). Representative confocal microscopic images showed the colocalization of Bhlhe40 and ɑ-SMA (e), quantification of these results were summarized (f). Data are means ± SD from 7–8 experiments. *P < 0.05 vs. Vehicle (Vehl); #P < 0.05 vs. db/db mice group.
Discussion
The above study for the first time demonstrated that EMPA protected VC and improved vascular dysfunction in diabetic mice through inhibition of ossification-related proteins RUNX2 and BMP2 in the vascular smooth muscle layer, which was independent of glycemic control. Notably, the restoration of Bhlhe40 by EMPA contributed to the inhibitory effects on the NLRP3 inflammasome, which was associated with the protective role of VC under T2DM. Our data thus unveiled that the important mechanism of VC protection with EMPA was related to the inhibition of NLRP3 inflammasome, which depended directly on Bhlhe40 upregulation and consequently suppressed MOVASs trans-differentiation into osteoblast-like cells in diabetes.
VC is a common complication of T2DM patients which has been considered as a predominant factor in arterial stiffness hence reinforcing one another [39], and coronary arterial calcification is an independent predictor for all-cause mortality independent of diabetic status [40]. However, there are currently no effective drugs for the specific management of VC in clinical application, and most T2DM patients remain exposed to VC with high morbidity and mortality. We thus explore the VC protection drugs from the perspective of some antidiabetic agents. Empagliflozin (EMPA), a SGLT2i, is an effective oral hypoglycemic drug which increases urinal glucose excretion or glycosuria without insulin dependence [7]. It has been shown established cardiovascular benefits in vulnerable patients with T2DM and chronic kidney disease in clinical trials [41]. In addition, SGLT2i therapies might become the future essential management of people with or without T2DM, patients with renal disease and those with heart failure (HF) [42]. However, the VC protective effects of EMPA in T2DM have remained unclear.
The current study demonstrated that EMPA inhibited the T2DM-induced VC formation as shown by the inhibition of calcium deposition in the aorta as well as RUNX2 and BMP2 protein expression. Here, our data from PWV indicated that EMPA significantly improved arterial stiffness in the aorta of db/db mice. We found that the contraction of aortic rings responding to phenylephrine was impaired in db/db mice, whereas the impairment could be prevented by EMPA medication. It confirmed that EMPA had an anti-arterial stiffness role in T2DM patients in some clinical studies [12, 43]. The intervention with EMPA also significantly decreased the HOMA-IR and triglyceride in db/db mice, which indicated EMPA can relieve metabolic disorders and insulin resistance in diabetic mice. Moreover, our data revealed that EMPA can inhibit vitamin D3 induced VC. Therefore, it demonstrated the VC protective role of EMPA was independent of hypoglycemic and anti-metabolic effects. These may shed a light on expanding the therapeutic scope of EMPA in clinical cardiovascular diseases.
Inflammation is one of the major causes of VC, several mediators of inflammation such as oxidation, carbonyl stress, C-reactive protein, and cytokines may directly stimulate VC, inflammation as a natural inducer of VC by inhibition of fetuin-A [44]. Here, EMPA decreased inflammatory cytokines activation in the serum of db/db mice. As demonstrated by clinical trials, EMPA has been reported to mitigate inflammation, platelet activity and oxidative stress in T2DM patients, which is associated with clinical cardiovascular benefits [45]. Importantly, our present study indicated EMPA reversed both IL-1β and IL-18 activation in db/db mice, and the two inflammatory cytokines served as the typical downstream indicators of the NLRP3 inflammasome activation [46,47,48]. Despite both the caspase-1 and caspase-11 activations cause the release of IL-1β and IL-18, only caspase-1 directly cleaves IL-1β and IL-18 [49]. We also found that EMPA inhibited the formation and activation of NLRP3 inflammasome as shown in the inhibition of NLRP3, ASC, p20/pro-caspase-1 and IL-1β/pro-IL-1β protein expression in the aorta and the colocalization of NLRP3 with ASC in the arterial smooth muscle layer of db/db mice. It was consistent with some reports that NLRP3 inflammasome can be inhibited by EMPA in cultured human aortic smooth muscle cells or human macrophages [38, 50]. However, whether the inhibitory effect of EMPA on NLRP3 inflammasome is associated with VC has never been reported. In the present study, diabetic VC (DVC) model was induced by a high-fat diet (HFD) with low-dose STZ treatments in mice. During this process, the NLRP3 KO mice showed an obvious decrease in RUNX2 and BMP2 protein expression and calcium deposition, as well as arterial stiffness in the aorta compared with the DVC group of WT mice. Some studies reveal NLRP3 blockade in mice might protect against HFD-induced diabetes and insulin resistance [51]. Here, we found NLRP3 deficiency displayed inhibition features of VC. Combining with the data in vitro, our study demonstrated EMPA ameliorated VC through inhibiting NLRP3 inflammasome. It was also in accordance with that NLRP3 inflammasome had a critical role on aortic valve calcification in human and mice study [52]. The locations of SGLT2 were currently identified to be unexpressed in the arterial smooth muscle layer of mice, which was consistent with some studies about the localization of SGLT2 in different tissues of mice and human [53, 54]. Combining the reports and our data explained the protective effect of EMPA on VC was related to the inhibition of NLRP3 inflammasome. The insufficiency is that we should use diverse gene KO such as ASC, Caspase-1 and IL-1β to further clarify the role of NLRP3 inflammasome in the EMPA protection of VC.
The NLRP3 inflammasome can be triggered by many different stimuli such as ionic flux, mitochondrial dysfunction, the production of reactive oxygen species, and lysosomal damage. It is a critical component of the innate immune system [46]. However, the molecular mechanism under EMPA responds to the NLRP3 inflammasome activation for the subsequent VC protection in T2DM is not fully understood. Basic helix-loop-helix family transcription factor e40 (Bhlhe40), also named as Dec1, Stra13 or Sharp2, is a transcription factor which participates in several pathological processes such as inflammation, apoptosis and hypertrophic remodeling [55,56,57]. Bhlhe40 has been acted as inhibitor for the proliferation and oxidative stress of VSMCs and protects intimal hyperplasia [58]. A cross-sectional study revealed high serum Bhlhe40 levels are associated with subclinical atherosclerosis in patients with T2DM [59]. Here, the bioinformatics analysis unveiled the Bhlhe40 as the most obvious predicted transcription factor was associated with the treatment of EMPA under HG induced MOVASs calcification. Notably, it acted as a transcription repressor which had common base binding sites with the promoter of Nlrp3 gene, and the direct binding between Bhlhe40 and Nlrp3 promoter was confirmed by ChIP. The experimental study further indicated EMPA recovered HG-induced Bhlhe40 inhibition and consequently suppressed NLRP3 inflammasome activation. Interestingly, Bhlhe40 deficiency attenuated the therapeutic effect of EMPA in MOVASs calcification. However, Bhlhe40 overexpression displayed inhibition features of calcium deposition in MOVASs. These indicated the protective role of EMPA in MOVASs calcification would be associated with the inhibition of Bhlhe40-dependent NLRP3 inflammasome activation. In accordance with the results in vitro, we found EMPA increased Bhlhe40 expression in the aorta of diabetic mice. ɑ-SMA is considered as a well-recognized marker of smooth muscle cells [35]. In our present study, the co-localizations of Bhlhe40/ɑ-SMA were apparently decreased but reversed by EMPA in the aortas of the db/db mice. Taken together, it indicated that VC protective role of EMPA was dependent on the Bhlhe40 reversion, which can directly bind to Nlrp3 and suppressed NLRP3 inflammasome activation in T2DM.
Bhlhe40 has been considered as a key regulator of immunity during infection, autoimmunity, and inflammatory conditions [60]. Previous study clarified that TNF-α decreased Bhlhe40 expression and consequently stimulated mouse vascular smooth muscle cell (MVSMC) proliferation and oxidative stress [58]. Moreover, a clinical observation revealed that EMPA can significantly decrease the pro-inflammatory cytokines including TNF-α, C-reactive protein and IL-6 with oxidative stress which were regarded as coronary risk factors in diabetes [61]. Therefore, we detected the TNF-α mRNA and intracellular O2.- production by RT-qPCR and the fluorescent spectrometry with a fluorescent probe DHE in the preliminary experiment. Combined with the results, it is speculated that TNF-α and oxidative stress are responsible for the regulation of Bhlhe40 by EMPA. These could give us insights to verify the exact mechanism of EMPA regulating Bhlhe40 expression in the future study.
The vascular smooth muscle layer calcification known as medial calcification is linked to artery stiffness, systolic hypertension, and increased PWV, leading to increased vascular dysfunction and heart failure [62]. Our current results were first time demonstrated that Bhlhe40 could be regarded as a “beneficial target gene” to inhibit NLRP3 inflammasome activation in the complication of diabetic VC. EMPA specifically up-regulated the expression of Bhlhe40 and blocked the signal transduction of NLRP3 inflammasome activation. The “initiation” and “activation” of NLRP3 inflammasome stimulated by diminished local Bhlhe40 in the diabetic vessels were effectively reversed by EMPA, and therefore EMPA’s protective role on VC was expected, which in favor of relieving the arterial stiffness and constriction dysfunction.
A limitation of this study is that we did not confirm whether the efficacy of EMPA on Bhlhe40-dependent NLRP3 inflammasome activity could be related to the improvement in VC of clinical patients. Another limitation is that we should use vessel-specific NLRP3 KO mice to precisely figure out the mechanism of VC protective effects with EMPA. Although our present study mainly focuses on NLRP3 inflammasome induced-VC in T2DM, there is evidence that the inflammatory cytokine IL-29 has an in vivo role in vitamin D3 induced-VC model, suggests that at least one inflammatory cytokine may be involved in this non-diabetic VC model [63]. However, whether EMPA protects the VC process through the anti-inflammatory effects in the vitamin D3 model needs further investigation. It has been confirmed that vitamin D3 with therapeutic doses inhibited NLRP3 inflammasome and consequently protected the inflammatory related diseases in basic and clinical trials [64, 65]. By contrary, a previous study demonstrated that the expression of TNF-α and IL-1β were increased in vitamin D3 induced calcific model [66]. These may provide us an enlightenment to verify the activation of NLRP3 inflammasome in vitamin D3 overloaded mice in the next research. Nevertheless, the present study provided distinct evidence that EMPA suppresses VC and protects arterial stiffness by recovering the expression of Bhlhe40 which acts as a negative regulator of NLRP3 inflammasome in mice with T2DM at high risk of cardiovascular disease.
In conclusion, our data suggested that NLRP3 inflammasome mediated VC in diabetic conditions. EMPA ameliorated diabetic VC by inhibiting NLRP3 inflammasome activation, which depended directly on the promotion of Bhlhe40. These findings identified Bhlhe40 may be a new therapeutic target and enhanced our understanding of the mechanisms underlying the anti-VC effects of EMPA on vascular pathology. These pharmacological properties might bring potential significance for EMPA in VC protection in diabetic patients.
References
Lehker A, Mukherjee D. Coronary calcium risk score and cardiovascular risk. Curr Vasc Pharmacol. 2021;19:280–4.
Shanahan CM, Crouthamel MH, Kapustin A, Giachelli CM. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697–711.
Nicoll R, Henein MY. The predictive value of arterial and valvular calcification for mortality and cardiovascular events. Int J Cardiol Heart Vessel. 2014;3:1–5.
Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. 2015;25:267–74.
Everhart JE, Pettitt DJ, Knowler WC, Rose FA, Bennett PH. Medial arterial calcification and its association with mortality and complications of diabetes. Diabetologia. 1988;31:16–23.
Yao H, Sun Z, Zang G, Zhang L, Hou L, Shao C, et al. Epidemiological research advances in vascular calcification in diabetes. J Diabetes Res. 2021;2021:4461311.
Lu Q, Yang L, Xiao JJ, Liu Q, Ni L, Hu JW, et al. Empagliflozin attenuates the renal tubular ferroptosis in diabetic kidney disease through AMPK/NRF2 pathway. Free Radic Biol Med. 2023;195:89–102.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–57.
Fitchett D, Inzucchi SE, Cannon CP, McGuire DK, Scirica BM, Johansen OE, et al. Empagliflozin reduced mortality and hospitalization for heart failure across the spectrum of cardiovascular risk in the EMPA-REG OUTCOME trial. Circulation. 2019;139:1384–95.
Chen S, Wang Q, Christodoulou A, Mylonas N, Bakker D, Nederlof R, et al. Sodium glucose cotransporter-2 inhibitor empagliflozin reduces infarct size independently of sodium glucose cotransporter-2. Circulation. 2023;147:276–9.
Lunder M, Janić M, Japelj M, Juretič A, Janež A, Šabovič M. Empagliflozin on top of metformin treatment improves arterial function in patients with type 1 diabetes mellitus. Cardiovasc Diabetol. 2018;17:153.
Bosch A, Ott C, Jung S, Striepe K, Karg MV, Kannenkeril D, et al. How does empagliflozin improve arterial stiffness in patients with type 2 diabetes mellitus? Sub analysis of a clinical trial. Cardiovasc Diabetol. 2019;18:44.
Bechlioulis A, Markozannes G, Chionidi I, Liberopoulos E, Naka KK, Ntzani EE, et al. The effect of SGLT2 inhibitors, GLP1 agonists, and their sequential combination on cardiometabolic parameters: a randomized, prospective, intervention study. J Diabetes Compl. 2023;37:108436.
Sharif S, Van der Graaf Y, Cramer MJ, Kapelle LJ, de Borst GJ, Visseren FLJ, et al. Low-grade inflammation as a risk factor for cardiovascular events and all-cause mortality in patients with type 2 diabetes. Cardiovasc Diabetol. 2021;20:220.
Bessueille L, Magne D. Inflammation: a culprit for vascular calcification in atherosclerosis and diabetes. Cell Mol Life Sci. 2015;72:2475–89.
Koyani CN, Plastira I, Sourij H, Hallström S, Schmidt A, Rainer PP, et al. Empagliflozin protects heart from inflammation and energy depletion via AMPK activation. Pharmacol Res. 2020;158:104870.
Kolijn D, Pabel S, Tian Y, Lódi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Gα oxidation. Cardiovasc Res. 2021;117:495–507.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Silvis MJM, Demkes EJ, Fiolet ATL, Dekker M, Bosch L, van Hout GPJ, et al. Immunomodulation of the NLRP3 inflammasome in atherosclerosis, coronary artery disease, and acute myocardial infarction. J Cardiovasc Transl Res. 2021;14:23–34.
Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, Chan L. The db/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of Western diet feeding. Metabolism. 2000;49:22–31.
Ren HL, Cai R, Xue R, Zhang Y, Xu Q, Zhang X, et al. Growth hormone-releasing hormone agonist attenuates vascular calcification in diabetic db/db mice. Front Cardiovasc Med. 2023;10:1102525.
Bhat OM, Yuan X, Cain C, Salloum FN, Li PL. Medial calcification in the arterial wall of smooth muscle cell-specific Smpd1 transgenic mice: a ceramide-mediated vasculopathy. J Cell Mol Med. 2020;24:539–53.
Ha CM, Park S, Choi YK, Jeong JY, Oh CJ, Bae KH, et al. Activation of Nrf2 by dimethyl fumarate improves vascular calcification. Vasc Pharmacol. 2014;63:29–36.
Zeng P, Yang J, Liu L, Yang X, Yao Z, Ma C, et al. ERK1/2 inhibition reduces vascular calcification by activating miR-126-3p-DKK1/LRP6 pathway. Theranostics. 2021;11:1129–46.
Hubert MO, Rodriguez-Vita J, Wiedmann L, Fischer A. Isolation of murine primary aortic smooth muscle cells. Bio Protoc. 2021;11:e3907.
Schneider MR. Von Kossa and his staining technique. Histochem Cell Biol. 2021;156:523–6.
Zhang WX, Tai GJ, Li XX, Xu M. Inhibition of neointima hyperplasia by the combined therapy of linagliptin and metformin via AMPK/Nox4 signaling in diabetic rats. Free Radic Biol Med. 2019;143:153–63.
Di Lascio N, Kusmic C, Stea F, Faita F. Ultrasound-based pulse wave velocity evaluation in mice. J Vis Exp. 2017;120:54362.
Lee SH, Kwon SC, Ok SH, Ahn SH, Bae SI, Hwang Y, et al. Linolenic acid enhances contraction induced by phenylephrine in isolated rat aorta. Eur J Pharmacol. 2021;890:173662.
Li M, Wu P, Shao J, Ke Z, Li D, Wu J. Losartan inhibits vascular calcification by suppressing the BMP2 and Runx2 expression in rats in vivo. Cardiovasc Toxicol. 2016;16:172–81.
Cai X, Tintut Y, Demer LL. A potential new link between inflammation and vascular calcification. J Am Heart Assoc. 2023;12:e028358.
Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021;20:150.
Wen C, Yang X, Yan Z, Zhao M, Yue X, Cheng X, et al. Nalp3 inflammasome is activated and required for vascular smooth muscle cell calcification. Int J Cardiol. 2013;168:2242–7.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801.
Chen Z, Li R, Pei LG, Wei ZH, Xie J, Wu H, et al. High-mobility group box-1 promotes vascular calcification in diabetic mice via endoplasmic reticulum stress. J Cell Mol Med. 2021;25:3724–34.
Khalid M, Petroianu G, Adem A. Advanced glycation end products and diabetes mellitus: mechanisms and perspectives. Biomolecules. 2022;12:542.
Sukhanov S, Higashi Y, Yoshida T, Mummidi S, Aroor AR, Jeffrey Russell J, et al. The SGLT2 inhibitor empagliflozin attenuates interleukin-17A-induced human aortic smooth muscle cell proliferation and migration by targeting TRAF3IP2/ROS/NLRP3/Caspase-1-dependent IL-1β and IL-18 secretion. Cell Signal. 2021;77:109825.
Van den Bergh G, Opdebeeck B, D’Haese PC, Verhulst A. The vicious cycle of arterial stiffness and arterial media calcification. Trends Mol Med. 2019;25:1133–46.
Raggi P, Shaw LJ, Berman DS, Callister TQ. Prognostic value of coronary artery calcium screening in subjects with and without diabetes. J Am Coll Cardiol. 2004;43:1663–9.
Wanner C, Lachin JM, Inzucchi SE, Fitchett D, Mattheus M, George J, et al. Empagliflozin and clinical outcomes in patients with type 2 diabetes mellitus, established cardiovascular disease, and chronic kidney disease. Circulation. 2018;137:119–29.
Khunti K. SGLT2 inhibitors in people with and without T2DM. Nat Rev Endocrinol. 2021;17:75–6.
Chilton R, Tikkanen I, Cannon CP, Crowe S, Woerle HJ, Broedl UC, et al. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes Metab. 2015;17:1180–93.
Moe SM, Chen NX. Inflammation and vascular calcification. Blood Purif. 2005;23:64–71.
Gohari S, Reshadmanesh T, Khodabandehloo H, Karbalaee-Hasani A, Ahangar H, Arsang-Jang S, et al. The effect of EMPAgliflozin on markers of inflammation in patients with concomitant type 2 diabetes mellitus and Coronary Artery Disease: the EMPA-CARD randomized controlled trial. Diabetol Metab Syndr. 2022;14:170.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20:3328.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–22.
Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009;10:241–7.
Man SM, Kanneganti TD. Regulation of inflammasome activation. Immunol Rev. 2015;265:6–21.
Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. 2020;11:2127.
Rheinheimer J, de Souza BM, Cardoso NS, Bauer AC, Crispim D. Current role of the NLRP3 inflammasome on obesity and insulin resistance: a systematic review. Metabolism. 2017;74:1–9.
The E, de Graaf DM, Zhai Y, Yao Q, Ao L, Fullerton DA, et al. Interleukin 38 alleviates aortic valve calcification by inhibition of NLRP3. Proc Natl Acad Sci USA. 2022;119:e2202577119.
El-Daly M, Pulakazhi Venu VK, Saifeddine M, Mihara K, Kang S, Fedak PWM, et al. Hyperglycaemic impairment of PAR2-mediated vasodilation: prevention by inhibition of aortic endothelial sodium-glucose-co-transporter-2 and minimizing oxidative stress. Vasc Pharmacol. 2018;109:56–71.
Vrhovac I, Balen Eror D, Klessen D, Burger C, Breljak D, Kraus O, et al. Localizations of Na+-D-glucose cotransporters SGLT1 and SGLT2 in human kidney and of SGLT1 in human small intestine, liver, lung, and heart. Pflug Arch. 2015;467:1881–98.
Huynh JP, Lin CC, Kimmey JM, Jarjour NN, Schwarzkopf EA, Bradstreet TR, et al. Bhlhe40 is an essential repressor of IL-10 during Mycobacterium tuberculosis infection. J Exp Med. 2018;215:1823–38.
Sun L, Gao G, Wang X, Zhang X, Li Y. Protective effect of basic helix-loop-helix family member e40 on cerebral ischemia/reperfusion injury: Inhibition of apoptosis via repressing the transcription of pleckstrin homology-like domain family A, member 1. Adv Clin Exp Med. 2023;32:655–66.
Ren KW, Yu XH, Gu YH, Xie X, Wang Y, Wang SH, et al. Cardiac-specific knockdown of Bhlhe40 attenuates angiotensin II (Ang II)-induced atrial fibrillation in mice. Front Cardiovasc Med. 2022;9:957903.
Feng DD, Zheng B, Yu J, Zhang ML, Ma Y, Hao X, et al. 17β-Estradiol inhibits proliferation and oxidative stress in vascular smooth muscle cells by upregulating BHLHE40 expression. Front Cardiovasc Med. 2021;8:768662.
Xu H, Xiang QY, Li S, Liu YS. High serum Bhlhe40 levels are associated with subclinical atherosclerosis in patients with type 2 diabetes mellitus: a cross-sectional study. Diab Vasc Dis Res. 2023;20:14791641231169246.
Cook ME, Jarjour NN, Lin CC, Edelson BT. Transcription factor Bhlhe40 in immunity and autoimmunity. Trends Immunol. 2020;41:1023–36.
Singh RB, Fatima G, Kumar P, Fedacko J, Mojto V, Isaza A, et al. Effects of empagliflozin on proinflammatory cytokines and other coronary risk factors in patients with type 2 diabetes mellitus: a single-arm real-world observation. Int J Clin Pharmacol Ther. 2021;59:17–25.
Chow B, Rabkin SW. The relationship between arterial stiffness and heart failure with preserved ejection fraction: a systemic meta-analysis. Heart Fail Rev. 2015;20:291–303.
Hao N, Zhou Z, Zhang F, Li Y, Hu R, Zou J, et al. Interleukin-29 accelerates vascular calcification via JAK2/STAT3/BMP2 signaling. J Am Heart Assoc. 2023;12:e027222.
Dong X, He Y, Ye F, Zhao Y, Cheng J, Xiao J, et al. Vitamin D3 ameliorates nitrogen mustard-induced cutaneous inflammation by inactivating the NLRP3 inflammasome through the SIRT3-SOD2-mtROS signaling pathway. Clin Transl Med. 2021;11:e312.
Chen M, He Y, Hu X, Dong X, Yan Z, Zhao Q, et al. Vitamin D3 attenuates SARS-CoV-2 nucleocapsid protein-caused hyperinflammation by inactivating the NLRP3 inflammasome through the VDR-BRCC3 signaling pathway in vitro and in vivo. MedComm (2020). 2023;4:e318.
Alesutan I, Luong TTD, Schelski N, Masyout J, Hille S, Schneider MP, et al. Circulating uromodulin inhibits vascular calcification by interfering with pro-inflammatory cytokine signalling. Cardiovasc Res. 2021;117:930–41.
Acknowledgements
This work was supported by National Natural Science Foundation of China (No. 81970381), the Fundamental Research Funds for the Central Universities, the Postgraduate Research & Practice Innovation Program of Jiangsu Province (No.5024002205).
Author information
Authors and Affiliations
Contributions
XXL investigated, designed and performed the experiments; ZDC, XJS and YQY participated in part of the experiments and formal analysis; HJ investigated and modified the format; NFL conceived and supervised the study; XXL analyzed the data and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Li, Xx., Chen, Zd., Sun, Xj. et al. Empagliflozin ameliorates vascular calcification in diabetic mice through inhibiting Bhlhe40-dependent NLRP3 inflammasome activation. Acta Pharmacol Sin 45, 751–764 (2024). https://doi.org/10.1038/s41401-023-01217-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-023-01217-0
- Springer Nature Singapore Pte Ltd.