
Abstract
Atherosclerosis, one of the life-threatening cardiovascular diseases (CVDs), has been demonstrated to be a chronic inflammatory disease, and inflammatory and immune processes are involved in the origin and development of the disease. Toll-like receptors (TLRs), a class of pattern recognition receptors that trigger innate immune responses by identifying pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs), regulate numerous acute and chronic inflammatory diseases. Recent studies reveal that TLRs have a vital role in the occurrence and development of atherosclerosis, including the initiation of endothelial dysfunction, interaction of various immune cells, and activation of a number of other inflammatory pathways. We herein summarize some other inflammatory signaling pathways, protein molecules, and cellular responses associated with TLRs, such as NLRP3, Nrf2, PCSK9, autophagy, pyroptosis and necroptosis, which are also involved in the development of AS. Targeting TLRs and their regulated inflammatory events could be a promising new strategy for the treatment of atherosclerotic CVDs. Novel drugs that exert therapeutic effects on AS through TLRs and their related pathways are increasingly being developed. In this article, we comprehensively review the current knowledge of TLR signaling pathways in atherosclerosis and actively seek potential therapeutic strategies using TLRs as a breakthrough point in the prevention and therapy of atherosclerosis.
Similar content being viewed by others

Introduction
Cardiovascular diseases (CVDs), most of which are associated with the onset and progression of atherosclerosis (AS), remain a leading cause of morbidity and mortality around the world [1]. Considering the seriousness of atherosclerotic CVDs, numerous scientists have undertaken extensive studies on the pathophysiology of AS and are working to develop effective treatments. Although the pathogenesis of AS remains to be fully uncovered, it has been demonstrated to be a chronic inflammatory disease, and inflammatory and immune processes are engaged in the occurrence and development of the disease [2,3,4]. Therefore, targeting inflammatory pathways could be a promising new treatment strategy for atherosclerotic CVDs [4]. Toll-like receptors (TLRs), a class of pattern recognition receptors, are linked to inflammation and the innate immune response [5]. Dysregulation of TLRs leads to the development of AS [6] and other cardiovascular and metabolic diseases [7, 8]. Statins are currently the representative drugs in atherosclerotic CVDs [9], but this drug has many limitations. For example, many patients are tolerable or intolerant to statins, and such drugs also have a significant likelihood of causing persistent inflammation, which will result in serious events. In recent years, TLRs have gained special attention as promising therapeutic targets for AS. As the quest for increasingly effective medications is crucial, we discuss the involvement of TLRs in AS and the prospective therapeutic treatment options for these receptors.
Pathogenesis and progression of AS
AS has received widespread attention as a common pathophysiological underpinning for a variety of CVDs like hypertension, coronary artery disease, and acute myocardial infarction [10]. The evolution of AS can be loosely classified into four stages: 1) stimulation of endothelial cells, followed by a sequence of inflammatory reactions; 2) deposition, retention, and modification of endothelial lipoproteins, and formation of foam cells; 3) plaque formation and growth, necrotic core enlargement, fibrosis, thrombosis, and remodeling; and 4) acute clinical events, including myocardial infarction (MI), angina pectoris, sudden coronary death, etc. [11]. AS occurs in areas prone to slow blood flow, or so-called “blood flow disorders” [12]. Early vascular smooth muscle cell (VSMC) aggregation in sensitive locations causes intimal thickening, perhaps as a consequence of platelet-derived growth factor (PDGF) production by activated endothelial cells [13]. When endothelial cells are activated, they secrete adhesion molecules such as E-selectin, P-selectin, vascular cell adhesion molecule-1 (VCAM-1), and the monocyte chemoattractant protein 1 (MCP-1). Among the early inflammatory cells that migrate from the endothelium to the subendothelial region are monocytes and potentially neutrophils. MCP-1 is the most common chemokine that mediates monocyte migration. Pathogen-associated molecular patterns (PAMPs), damage-associated molecular patterns (DAMPs), and a variety of cytokines, including as tumor necrosis factor α (TNF-α), interleukin-1β (IL-1β), and interleukin-6 (IL-6), further stimulate monocytes, which are gradually transformed into macrophages. When preserved and modified lipoproteins are recruited by macrophages and VSMCs and the action continues to be carried out, foam cells, a characteristic of atherosclerotic lesions, are created [14]. In addition, when some inflammatory signaling pathways are activated, fatty streaks will form. In short, when endothelial cells lose their ability to maintain the internal environment’s balance, vasoconstriction, lipid infiltration, leukocyte adhesion, platelet activation, and oxidative stress are a few of the reactions that can result in vessel wall damage and trigger an inflammatory response. The development of fatty streaks is also thought to be the first stage of AS [15]. Fibrotic plaques arise when atherosclerotic plaques change from fatty streaks to intimal expansion, which is indicated by the existence of the acellular, lipid-rich necrotic core. The fibrous covering those forms around the necrotic core provides the plaque with additional stability. A necrotic core and fibrous cap indicate the presence of advanced atherosclerosis [16]. With continued and extensive damage to the vascular endothelium beyond repair, the integrity of the blood vessels is compromised, which leads to rupture and bleeding. And as more and more monocytes adhere simultaneously, plaques are formed. Another characteristic of advanced atherosclerotic lesions is the calcification of atherosclerotic plaques. It starts in inflammatory sections of the plaque and looks like a bone-like structure [17]. The vesicle release from the matrix onto macrophages and the death of synthetic VSMC initiate calcification processes within plaques [18]. As lipid-rich plaques progress, accumulated macrophages, along with other migrating cells and activated endothelial cells, release lots of pro-inflammatory cytokines, matrix metalloproteinases (MMPs), and histone proteases, the massive release of which makes the plaques brittle. Furthermore, interferon gamma (IFN-γ) also prevented the VSMC from producing collagen, weakening the plaques and newly produced fibrous caps [19]. The outcome may have serious consequences, such as thrombosis and downstream tissue infarction. Taken together, inflammatory events participate in the whole process of AS development.
Toll-like receptors (TLRs)
TLRs are pattern recognition receptors (PRRs) found initially in Drosophila. They work by identifying PAMPs and DAMPs to trigger a number of innate immune responses [20]. The majority of cells that express TLRs are mammalian cells, including dendritic cells (DCs), macrophages, neutrophils, monocytes, lymphocytes, fibroblasts, epithelial cells, endothelial cells, neuronal cells, etc. [6]. Mammalian TLRs can be classified as cell membrane receptors as well as intracellular receptors based on their location. TLRs 1, 2, 5, 6, 10, and 11 are surface-expressed and detect predominantly microbial membrane components; only internal vesicles express TLRs 3, 7, 8, 9, 12, and 13, which identify microbial nucleic acids. It is noteworthy that TLR4 localizes to both intracellular vesicles and cell surfaces [21]. In general, all TLRs comprise three main structural domains (Fig. 1): (1) 16–18 leucine-rich repeats, which are hydrophobic leucines concentrated in the extracellular region, and their role is to recognize receptors and can bind to other co-receptors to form the corresponding receptor complexes. (2) The cysteine region, known as the transmembrane region, controls the subcellular location of TLRs. (3) The cytoplasmic area of TLRs and the cytoplasm of the interleukin-1 (IL-1) receptor family are strikingly similar in the toll-IL-1 receptor domain (TIR domain) [22]. The dimerization of TIR structural domain-linked junctions and TLRs can eventually transmit signals to downstream pathways. Previous studies have reported five junctions: myeloid differentiation factor 88 (MyD88), toll receptor-associated interferon activator (TRIF), MyD88 junction-like protein (MAL/TIRAP), TRIF-associated junction molecule (TRAM), and sterile α- and armadillo-motif-containing protein (SARM).

(1) 16–18 leucine-rich repeats, which are hydrophobic leucines concentrated in the extracellular region, and their role is to recognize receptors and can bind to other co-receptors to form the corresponding receptor complexes. (2) The cysteine region, known as the transmembrane region, controls the subcellular location of TLRs. (3) The cytoplasmic area of TLRs and the cytoplasm of the interleukin-1 (IL-1) receptor family are strikingly similar in the toll-IL-1 receptor domain (TIR domain).
PAMPs are constituted of two primary types: cell nucleus components of viral and bacterial products (C-phospho-G DNA, CpGDNA; single-stranded RNA, ssRNA; and double-stranded RNA, ds-RNA) and cell wall components (lipopolysaccharides, peptidoglycans, lipophilic acids, mannose, and lipids) [23]. There is a broad variety of ligands for TLRs (Table 1), and each type has a series of ligands with specificity. On the basis of their cellular location and particular ligands, TLRs that identify PAMPs may be divided into two categories [20]. One of them (TLRs 1, 2, 4, 5, 6, and 11), which seems expressed on this cell surface, identifies lipoproteins and cell membrane proteins as membrane components of microorganisms. The second detects microbial nucleic acids and is found on intracellular vesicles (TLRs 3, 7, 8 and 9) [24, 25].
The signaling pathways mediated by TLRs are intricate, and a few of their mechanisms are well understood (Fig. 2). For the present, TLRs have two main signaling routes: TRIF-dependent pathways (TLRs 3 and 4) and MyD88-dependent pathways (TLRs 1, 2, 4, 5, 6, 7, 8, and 9) [26]. The most common signaling pathways to initiate immune responses are MyD88-dependent. Its mechanism is roughly as follows: MyD88 is recruited to the TLRs’ TIR structural domain following TLR activation, forming a combination that subsequently recruits IRAK. IRAK4, a downstream signaling component, is a kinase that promotes IRAK1 phosphorylation, and its function is to recruit phosphorylated IRAK1 and TRAF6 [27]. The alternative pathway is TRIF-dependent. Multiple proteins, including TRIF, TRAF6, tumor necrosis factor receptor-associated death domain protein (TRARD), receptor-interacting serine/threonine protein kinase 1 (RIP1), etc., comprise the pathway’s signaling complex [28]. It is through this pathway that TLR3 and TLR4 function, with TLR3 connected directly to TRIF and TLR4 connected to TRIF through TRAM. After activation of TANK-binding kinase 1 (TBK1) by TRIF, IRF3 is activated, and after activation, IRF3 is finally transferred into the nucleus. At the moment, IRF3 may play a role in regulating IFN-β synthesis. Similarly, the TRIF-dependent pathway can also induce inflammation by activating the conversion of TRAF6 to the NF-кB pathway [29].

MyD88 interacts with IRAK4, which then transmits signals to NF-κB and MAPK in response to TLR activation, a pathway dependent on MyD88 that activates the expression of inflammatory cytokines. TLRs on endosomes transmit signals via a TRIF-dependent pathway. TRIF, TRAF6 and IRAK1/2/4 act together to activate TAK1; alternatively, TRIF acts together with non-classical TBK1 and IKKε to activate IRF3/7 which in turn induce inflammatory cytokines. Abbreviations: IKKε, I-κB kinase ε; IRAK1/2/4, interleukin 1 Receptor Associated Kinase 1/2/4; IRF3/7 interferon regulatory factor 3/7; MAPK, p38 mitogen-activated protein kinase; MyD88, myeloid differentiation factor 88; NF-κB, nuclear factor kappa B; TAB1/2, TGF-beta Activated Kinase 1 Binding Protein 1/2; TAK1, transforming growth factor activating kinase-1; TBK1, TANK-binding kinase 1; TLRs, toll-like receptors; TRAF6, tumor necrosis factor receptor-associated factor 6; TRIF, toll receptor-associated interferon activator.
Role of TLRs in AS
Numerous recent studies indicate that TLRs play a crucial role in the onset and progression of AS. These include involvement in the onset of endothelial dysfunction, the interaction of various immune cells, and a number of additional inflammatory pathways associated with CVDs (Fig. 3). Targeting TLRs and their regulated inflammatory events could be a promising new AS treatment strategy.

CD80 cluster of differentiation 80, CD86 cluster of differentiation 86, CCL5 RANTES, CD40L CD40 Ligand, CXCL4 C-X-C motif chemokine 4/ Platelet factor 4, ERK1/2 extracellular regulated protein kinases 1/2, ICAM-1 intercellular cell adhesion molecule-1, IL-1 interleukin-1, IL-6 interleukin-6, IL-8 interleukin-8, IL-12 interleukin-12, IP-10 IFN-γ inducible protein-10, MAFB The bZip transcription factor, MCP-1 monocyte chemoattractant protein 1, MMP2 matrix metalloproteinase-2, MMP9 matrix metalloproteinase-9, NF-κB nuclear factor κB, PI3K/AKT Phosphatidylinositide 3-kinases/protein kinase B, ROS reactive oxygen species, TN-C Tenascin-C, TREMs Triggering receptors expressed on myeloid cells, VCAM-1 vascular cell adhesion molecule-1.
Endothelial cells
Vascular endothelial cells are monolayers of cells that serve as the circulatory system’s initial line of defense against CVDs. Endothelial cells, which serve as the link between both the vessel wall and blood flow, contribute to the regulation of vascular tension by producing and releasing vasoactive substances such as nitric oxide (NO), prostacyclin-2 (PGI2), and endothelin (ET)-1, as well as by regulating platelet function, the inflammatory response, and vasodilation [30]. Different TLRs are differentially expressed in the endothelium; for example, TLR2 and TLR4 are prevalent in the vasculature, but TLR7 and TLR9 are less expressed, while TLR1, TLR3, TLR5, TLR6, TLR8, and TLR10 are selectively expressed according to environmental changes [25]. By triggering the release of IL-1β from monocyte cells, promoting CD14 expression, and boosting macrophage survival, TLR-activated endothelium cells indirectly control local inflammation [31]. Porphyromonas gingivalis is an anaerobic Gram-negative bacterium that causes periodontal disease [32]. A recent study found some significant associations between Pseudomonas gingivalis and CVDs, as all CAD patients had arterial colonization by P. gingivalis. Additionally, Pseudomonas gingivalis oral infection speeds up the development of atherosclerotic infection [33]. It is widely recognized that vascular endothelial cells’ normal morphology and function are necessary to preserve vascular health and that their dysfunction is crucial for the onset and development of CVDs. Vascular endothelial cells can become attached to and invaded by Pseudomonas gingivalis [30]. It triggers endothelial cells to produce a number of pro-inflammatory factors, chemokines, adhesion molecules, and nitric oxide, all of which have significant effects on vascular function [34]. Pseudomonas gingivalis primarily affects atherogenesis by activating the TLR-NF-κB signaling pathway, inhibiting endothelial cell growth, inducing endothelial cell mesenchymal transition, and inducing apoptosis [35]. The most prevalent TLRs for Pseudomonas gingivalis lipopolysaccharide and bacterial hair responses in the host are TLR2 and TLR4 [36]. Cytokine production during inflammation (i.e., CRP, IL-6, TNF-α, and MCP-1) was found to be significantly increased in rabbit serum and TLR2 and TLR4 expression in local aortic tissue after intravenous administration of Pseudomonas gingivalis [37]. More importantly, Xie et al. [35] demonstrated that endothelial homeostasis disrupted by Pseudomonas gingivalis was repaired after the use of antagonists or inhibitors blocking the TLR-NF-κB pathway. Pseudomonas gingivalis accelerates atherogenesis by inhibiting endothelial cell growth, inducing endothelial cell mesenchymal transition (EndoMT), and facilitating inflammation and apoptosis, all of which appear to be associated with the activation of TLR-NF-κB signaling pathway. In inflammatory or autoimmune diseases, TLR3 disrupts vascular endothelial homeostasis after inappropriate stimulation [35]. In addition, TLR3 disrupts the function of cord blood-derived endothelial cell progenitors; as a result, angiogenesis in vitro is hindered, cell proliferation is inhibited, and pro-inflammatory cytokines are produced [38]. Furthermore, P-selectin and vascular cell adhesion molecule-1 (VCAM-1) are upregulated by TLR3 in injured endothelial cells, which facilitates leukocyte (mainly lymphocytes and monocytes) adherence in the blood and eventual infiltration into endothelial cells [39]. Consequently, it is conceivable that TLR3 will be utilized more in future research and will also serve as an effective therapeutic target for endothelial dysfunction and associated AS. Taken together, these findings indicate a critical role of TLRs in inflammation and dysfunction of endothelial cells as well as the development of AS.
Platelets
Platelets in innate immunity attract inflammatory cells to vascular lesions, causing endothelial dysfunction and AS [40]. Some platelet functions are achieved through the secretion of cytokines and others through the expression of surface proteins/receptors [41]. Platelets possess the potential to synthesize proteins, even though they are anucleated cells. The role of platelets under physiological stimulation is dependent on the platelet transcriptome, as they are equipped with all the organelles required for the translation of mRNA from macrophage sources into proteins [42]. Rondina et al. [43] analyzed the mRNA expression of Toll receptor superfamily members in platelets. They found that the transmembrane protein Toll receptor plays a role in coordinating platelet function when activated, in part by increasing the splicing and translation of platelet pre-mRNA proteins. TLRs mediate the thrombotic inflammatory response and may serve as a link between platelet function and the innate immune system [44]. So far, nine TLR family members have been identified as being expressed on platelets, with each TLR member recognizing its own DAMP and PAMP and initiating its own unique biological response in succession. Platelet TLR2 and TLR4 bind gram-positive and gram-negative bacteria, respectively, and perform a crucial function [45]. TLR2 forms heterodimers with either TLR2 or TLR6, whereas TLR4 is normally generated as a homodimer. Platelets also often express TLR3, TLR7, and TLR9 in vivo in order to recognize viral infections [46]. A pro-inflammatory response is established as a result of platelet TLR activation and responds to stimulation upon injection of the appropriate agonist [47].
Essential thrombocythemia (ET) is a type of chronic myeloproliferative neoplasm (MPN) that causes megakaryocyte proliferation and significant thrombocytosis. We know that P-selectin and CD40L, alpha-derived adhesion molecules, mediate platelet contacts with leukocytes and endothelial cells, respectively [48]. In addition to expressing the alpha particle-derived adhesion molecules described above, activated platelets also release a series of molecules that store alpha particles, such as the inflammatory chemokine RANTES (CCL5), which has potent chemotactic effects on a variety of cells, including leukocyte and monocyte recruitment to the vessel wall, a key step in the formation of atherosclerosis [49]. Von Willebrand factor (VWF), a molecule that plays a new role in vascular inflammation and is one of the inflammatory molecules housed in alpha particles, has both a hemostatic and an inflammatory effect [50]. TLR stimulation activates many signaling pathways in platelets, including extracellularly regulated protein kinases 1/2 (ERK1/2), phosphatidylinositide 3-kinases/protein kinase B (PI3K/AKT), and NF-κB, which all regulate TLR-triggered platelet responses [51, 52]. TLR2 and TLR4 were expressed normally on the surface of the platelets, but their downstream effector molecules, ERK1/2, had higher phosphorylation levels both at rest and upon incubation with Pam3CSK4, indicating that this may help explain the heightened TLR2 response. As a result of TLR stimulation, the increased response may increase platelet activation in ET as well as platelet/leukocyte/endothelial cell interactions and the generation of inflammatory mediators, aggravating the thrombotic inflammatory state. Thus, controlling or reversing platelet activation through regulation of TLR signaling may be an attractive therapeutic target for AS.
Monocytes
After infection, huge numbers of monocytes are attracted to the relevant tissues from the bone marrow [53]. Monocytes are capable of rapidly eliminating pathogens, in addition to differentiating into monocyte-derived macrophages (mo-Mac) and monocyte-derived dendritic cells (mo-DC) [54, 55]. Mo-mac acts largely to replenish tissue-resident macrophages that have perished due to an infection [56, 57], also able to maintain enhanced responsiveness to subsequent inflammation [58, 59]. The role of Mo-DC is to present antigens to T cells in tissues to enhance their effector function, which plays a very important role in complementing classical DC in inducing adaptation [60]. Monocyte differentiation is not random but responds accordingly to an assessment of the surrounding environment. Coillard et al. [61] found that recognition of pathogens by the mTORC1 pathway, TLRs encourage mo-Mac differentiation, which elevates the expression of MAFB (the bZip transcription factor). In contrast, activation of nucleotide-binding oligomerization domain (NOD) receptors promoted mo-DC differentiation through the autocrine action of TNF-α. Divergent bacilli containing both TLR and NOD ligands differentiate monocytes toward mo-DC, implying that NOD activation in monocytes during Divergent bacilli recognition blocks TLR signaling. This result is in keeping with other studies showing that mouse bone marrow macrophages and splenic myeloid lineage cells have NOD receptor-inhibitory effects on TLR-induced NF-κB activation [62]. Collectively, these studies contribute to an in-depth understanding of the mechanisms of monocyte differentiation and the role played by TLR in this process.
The mononuclear macrophage system (MPS) is a component of the inflammatory response and is composed of tissue-resident macrophages and their related cells [63]. In mice, the conventional monocyte subtype, Ly6Chi monocytes, is a crucial component of this system. Originating from progenitor cells in the bone marrow (BM), they circulate in the peripheral blood (PB) and can differentiate into at least 3 different MPS effector phagocyte profiles to adapt to changing conditions: macrophages, dendritic cells, and patrolling monocytes [64, 65]. A cell contact-dependent signaling pathway called notch signaling may help control the innate immune system’s cell destiny [66]. TLR7 is a pathogen receptor that is found on myeloid cells. Long-term stimulation of TLR7 causes Ly6Chi monocytes to change into specialized macrophages and anemia to develop [67]. Gamrekelashivili et al. [63] found that TLR and Notch signaling play independent and synergistic roles in promoting the development of Ly6Chi monocytes into Ly6Clow monocytes. Loss of Notch2 function during TLR stimulation promotes gene expression in macrophages and monocytes and the development of Ly6Chi monocytes. Overall, Notch signaling has the potential to regulate inflammation in the vasculature by regulating the differentiation of cells in the innate immune system.
Among all TLRs-induced gene clusters, stimulation of monocytes and MDM by TLR4 and TLR7/8 demonstrated regulation of both spectrum-specific and TLR-specific transcriptional responses. Song et al. [68] revealed that interferon-stimulated genes (ISGs) in monocytes and MDM activated by TLR4 or TLR7/8 ligands revealed an unanticipated dichotomy. Thus, stimulation of TLR7 and TLR8, but not TLR4, induces ISG production in monocytes. The IRF family, which is crucial for type I interferon production and its downstream actions, provides the most prominent and abundant motif in the open chromatin area of MDM, in contrast to MDM, where TLR4 activation induces the chromatin opening of the ISG motif [69,70,71]. Moreover, it has been demonstrated that tumor necrosis factor receptor (TNFR) signaling in mouse macrophages induces interferon-β (IFN-β) in an IRF1-dependent way, which synergizes with other tumor necrosis factor-inducing ligands such as TLRs to sustain the expression of inflammatory genes [72]. Together, these findings offer new insights into the study of inflammation formation and indicate that inhibition of the TLRs/TNFR-IRF-IFN-β axis may become a treatment for inflammatory diseases such as AS.
Macrophages
Macrophages appear in the early stages of atherogenesis, where endothelial cell chemokine-receptor interactions and cell adhesion molecules, including expression of ICAM-1 and VCAM-1, aggregate monocytes into the arterial wall [73]. Monocytes migrate into the arterial ducts, where they develop into macrophages and display a variety of phenotypes, mostly in response to cues from the local microenvironment. Macrophages have two phenotypes: the M1-like macrophages are of the inflammatory phenotype, which is mainly characterized by the production of large amounts of pro-inflammatory cytokines, reactive oxygen species (ROS), and reactive nitrogen species, whereas the M2-type is of the catabolic phenotype, which is characterized by the inhibition of inflammation and the enhancement of tissue repair [74]. Due to increased activity of the cholesterol transport pathway in cells, macrophages with pro-inflammatory injury take up a large amount of low-density lipoprotein (LDL)-derived cholesterol [75]. Macrophage scavenger receptors promote the storage of ingested oxidized cholesterol in the cell, which gradually evolves into the typical “foam” appearance in macrophages [76].
TLR3 promotes the instability of atherosclerotic plaques by modulating the activities of MMP2 and MMP9 in macrophages [77]. In addition, immunological cells play a crucial role in the progression of AS. TLR5 is the ubiquitously expressed extracellular receptor for bacterial flagellin that is found in almost all tissues [78]. Exogenous TLR ligands are abundant in human atherosclerotic lesions. And TLR5 triggers a disease characterized by sterile inflammation, so it is predictable that both endogenous and exogenous ligands can cause TLR5-dependent inflammation. TLR5 is also found on several immune cells, with macrophages and T cells being more typical [79]. MyD88 recruits and activates several intracellular kinases during TLR5 receptor activation, which eventually results in nuclear localization of NF-κB and inflammatory reactions [80]. It has been demonstrated that loss of hematopoietic TLR5 inhibits AS development in LDL receptor knockout mice [79], whose plaques include fewer macrophages and smaller necrotic sites. In line with the finding that deficiency of TLRs generally attenuates inflammatory signaling to reduce the burden of AS, in response to flagellin stimulation, macrophages from TLR5-deficient mice exhibited no change in the production of monocyte chemoattractant protein-1 (MCP-1) and interleukin 6 (IL-6) compared to macrophages from wild-type animals.
Animal studies in immunodeficient mice show that TLR7 and MyD88-deficient macrophages do not react to stimulation with R-848 and do not produce the associated cytokines [81]. This suggests that TLR7 plays a role in AS formation; however, a surprising aspect is that TLR7 may have a role in preventing the formation of AS by blocking the stimulation of inflammatory macrophages and the generation of cytokines, according to a previous study [82]. Recent studies have demonstrated that TLR9 is also implicated in the pathophysiology of the development of several illnesses, including CVDs. TLR9 functions in the innate immune system by recognizing exogenous DNA fragments containing unmethylated CpG DNA [83]. Activation of TLR9 enhances the proinflammatory activity of macrophages via phosphorylating the p38MAPK signaling pathway [84]. In addition, the attachment of TLR9 agonists to macrophages induced the generation of a number of inflammatory markers, including TNF-α, ICAM-1, and VCAM-1, which was not observed in TLR9-deleted macrophages. These results indicate that inhibiting TLR7 and TLR9 may inhibit the production of certain inflammatory factors by macrophages, thereby exerting an anti-AS effect. However, the contradictory perspectives on TR7 suggest that additional research on TLR7 is required.
It is well recognized that endotoxin activation of macrophages alters cellular metabolism, which in turn alters the action of effectors with features like inflammation, bactericidal activity, and motility [85]. Activated macrophages exhibit a strong aerobic glycolytic effect, also known as Warburg metabolism, and in addition, ATP generation via OXPHOS is replaced by the creation of biosynthetic precursors and in the mitochondrial metabolic pathway of macrophages [86]. In contrast to macrophage-activated Warburg metabolism, TLR4 signaling rapidly increases glucose uptake, promoting glycolysis and tricarboxylic acid (TCA) cycle capacity, which enables citric acid production. And through increased expression of ATP-citrate lyase (ACLY) activity, endotoxin signaling directly stimulates glucose-dependent acetyl coenzyme A (CoA) incorporation into histones, hence enhancing the activation of certain gene sets related to a robust pro-inflammatory response [87]. We know that the transition of macrophages from a pro-inflammatory to a reparative state is essential to eliminate inflammation and restore endostasis. Two steps comprise the transformation of reparative macrophages, the first of which involves TLR and NF-κB activation and culminates in the generation of inflammatory chemicals. In step two, macrophages undergo metabolic conversion, resulting in aerobic glycolysis and lactate generation [88,89,90]. The cells use the accumulated lactate to induce histone glycation, which is an effective way to promote the transformation of reparative macrophages [90]. BCAP is the adaptor that links TLR signaling to optimum aerobic glycolysis in macrophages, and the lactate produced is required for histone lactosylation, which stimulates the expression of repair genes and, eventually, the transition of macrophages into repair type cells [91]. Collectively, metabolism and its associated metabolic pathways are a current research hot topic, and it appears to be of great importance to investigate the influence of possible inflammation triggered by metabolic processes on the development of AS using TLRs as a research subject.
Dendritic cells (DCs)
AS is thought to arise mostly from inflammatory cells, including T cells, monocytes/macrophages, and DCs, among which DCs are undoubtedly the most powerful antigen-presenting cells because of their ability to efficiently extract, process, and present antigens in vivo [92]. Although CD4+ T cells and DCs congregate in clusters in atherosclerotic plaques, there are few DCs in the subendothelial layer of normal artery intima. These activated DCs produce many atherogenic cytokines, including interleukin-6 and 12 (IL-6 and IL-12) and express co-stimulatory molecules such as CD80 and CD86, which are considered to be mature DCs and are capable of triggering Th1 and Th17 immune responses, both of which have been associated with accelerated AS formation [93]. IL-37 is an inflammatory factor belonging to the IL-1 cytokine family [94]. Exogenous IL-37 inhibits DC maturation and induces T regulatory (Treg) cell responses, thereby reducing atherosclerotic lesions in mice [95]. Liu et al. [96] demonstrated that when plaque-infiltrated DCs developed, TLR4 expression was considerably greater in apolipoprotein E (ApoE) deficiency (ApoE-/-) mice, but significantly lower in IL-37tg mice. Interestingly, TLR4 expression is also inhibited in the absence of IL-1R8. Since the importance of NF-κB for TLR4-induced DC is also becoming increasingly clear, it can be concluded that IL-37 can inhibit DCs maturation via the IL-1R8-TLR4-NF-κB pathway. Collectively, these findings expand our knowledge of how TLRs affect DC function and atherosclerotic lesion development.
Foam cells
Foam cells are essential for the formation of atherosclerotic lesions, and macrophages serve as their primary source cells [97]. Foam cells cause the formation of fatty streaks, which are a prominent feature of atherosclerosis. The increased influx of modified oxidized LDL and the buildup of cholesterol in the intima are the major causes of foam cell formation [98]. Foam cell generation is advantageous in the early stages of atherosclerosis, but late lesions have foam cells that die and discharge DAMPs into the extracellular space as part of their contents [99, 100]. Tenascin-C (TN-C) is a glycoprotein that is related to four multimeric extracellular matrix (ECM) proteins in the Tenascin family [101]. TN-C is an endogenous ligand of TLR4, which is recognized as a crucial signaling molecule in chronic inflammatory disorders. Since TN-C is created close to where a plaque ruptures, it plays a part in neovascularization, in addition to altering the phenotype of VSMCs and their capacity to generate pro-inflammatory cytokines/ MMPs [102]. Notably, TN-C production by oxLDL-treated macrophages increases foam cell development through TLR4 and the scavenger receptor CD36 [103]. Activating transcription factor 3 (ATF3) seems to be a component of the family of transcription factors known as ATF/cyclic AMP response element-binding (ATF/CREB). ATF3 can adversely affect the transcription and synthesis of pro-inflammatory cytokines; it is a negative regulator of TLR4, for instance [104]. Luo et al. [103] discovered that ATF3 suppresses TN-C-induced THP-1 macrophage foam cell production via inhibiting TLR4. Therefore, ATF3 is anticipated to be a significant molecular target for treating AS.
VSMCs have macrophage characteristics and are a major source of foam cells and inflammatory responses in the component plaques [105]. Using TLR4, oxLDL stimulated pro-inflammatory signaling pathways and increased the expression and generation of inflammatory cytokines in VSMC. Notably, oxLDL enhanced TLR4 binding to Src kinase, stimulated macrophage lipid absorption, and stimulated foam cell production [106]. In oxLDL-induced atherosclerosis, the TLR4-Src pathway may represent a co-regulatory mechanism for lipid buildup and foam cell development. This is because the possible cytoskeletal rearrangement mediated by the activation of Src is a typical cellular change in endocytosis [106]. Additionally, activation of c-Jun N-terminal kinase by the Src signaling pathway boosts c-Jun’s trans response to LPS, thereby upregulating the expression of inflammatory markers [107]. ROS is a common mediator that enhances intracellular oxidative stress, promotes inflammatory responses, and even includes cellular phenotypic transitions [108]. TLR4 regulates the dynamic balance of ROS to mediate oxLDL-induced ROS recruitment, which may be another possible mechanism of TLR4 in the formation of VSMC-derived foam cells [109]. These findings provide a conceptual understanding of the molecular pathway of TLR4 in foam cell formation, which can be targeted in future research to develop effective therapeutic medications for AS.
Triggering receptors expressed on myeloid cells (TREMs) are members of the immunoglobulin family [110]. As a result of increased endotoxin or other microbial products, TREM-1 expression by neutrophils and monocytes/macrophages is increased. Additionally, TREM-1 has been related to the development of foam cells. TREMs stimulate the release of pro-inflammatory cytokines and chemokines, including IL-8, CCL-2, and CCL-7, and rapid neutrophil degranulation, after binding to the DAP12 junction protein [111]. TREMs promote the production of pro-inflammatory cytokines (TNF-α and IL-1β) while TLR2 or TLR4 ligands inhibit the release of the anti-inflammatory cytokine IL-10 [111]. Joffre et al. [112] demonstrated that, as an essential upstream pro-atherogenic receptor, its activation orchestrates the pro-inflammatory response of monocytes/macrophages and foam cell production by activating CD36 and TLR4 in a synergistic manner. These findings support the idea that TLR-mediated inflammation hastens the formation of foam cells and accelerate AS. Based on above, the coordination of inflammatory signals among TREMs, TLRs and other receptor signal transduction pathways has significant implications for CVD research, and if a drug targeting both TREM-1 and TLR4 could be developed, it would be a promising treatment for AS.
Inflammatory signaling pathways associated with TLRs
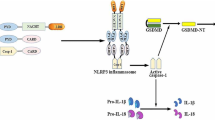
NLR family pyrin domain containing 3 (NLRP3) inflammasome
Inflammasomes are multiprotein complexes that assemble in the cytosol of the innate immune system upon exposure to particular PAMPs or DAMPs that trigger inflammatory responses. NLRP3, the best characterized inflammasome complex, plays a crucial role in the development of AS [113]. Inflammatory cells in the central nervous system, such as mononuclear macrophages, dendritic cells, and microglia, are the primary producers of the NLRP3 inflammasome[114]. Normally, there are two stages to the activation of the NLRP3 inflammasome [113]. The first is signal 1, leading to the initiation signal for transcription of IL-1β and NLRP3 precursors. The initiation signal is usually induced by activated PRPs, such as TLRs, which further activate transcriptional NF-κB. In cellular experiments, LPS is a specific agonist of TLR4 and a typical signal 1 [115]. The second involves further stimulation of immune cells by antigen, causing NLRP3 proteins oligomerization and binding to intracellular apoptosis-associated speck-like proteins containing a CARD (ASC) and procaspase-1 to assemble into inflammasomes. During this process, numerous active types of inflammatory cytokines, including IL-1β and IL-18, are generated and released. Many factors trigger or promote NLRP3 inflammatory vesicle activation, including ROS, lysosomal rupture, potassium efflux, calcium inward flow, and mitochondrial damage [116]. In the ApoE-/- mouse model, the oxidative stress response transcription factor Nrf2 was discovered to be required for the CC-induced activation of NLRP3 inflammatory vesicles. After Nrf2 knockdown, the activation of NLRP3 inflammatory vesicles was significantly suppressed, and AS was somewhat alleviated [117]. In addition, LDL, especially modified LDL including acLDL and oxLDL, greatly contributes to the role played by NLRP3 inflammatory vesicles in AS formation.
Accumulating evidence indicates that pyroptosis and necroptosis play a critical role in atherogenesis and plaque instability [118, 119]. The activation of the inflammasome and the subsequent assembly of NLRP3 inflammasome complexes not only trigger inflammatory cascades, but also stimulate efferocytosis, mitophagy, pyroptosis and necroptosis during the exacerbation of AS [118,119,120]. TLRs-mediated activation of the NLRP3 inflammasome and other NLR members can induce a non-inflammatory autophagic machinery in less dangerous situations or trigger pyroptosis under overwhelming stress conditions. In addition, TLRs induce necroptosis by interacting with TRIF on the necrosome (Fig. 4). Independent of RIPK1, RIPK3 can be directly activated by TRIF or interferon signaling to induce necroptosis via ZBP1 activation. Moreover, necroptosis can induce a RIPK-independent inflammatory response in the absence of infection by activating DAMPs during the development of AS. Further studies are required to determine whether TLR signaling-induced inflammasome activation is also the driver of other cell responses during the initiation and progression of AS, such as pinocytosis, NETosis, ferroptosis, and copper-dependent death.

ASC apoptosis-associated speck-like protein, DAMPs danger-associated molecular patterns, FADD Fas-associated protein with death domain, IKKα IκB kinase-α, IKKβ IκB kinase-β, IL-1β interleukin 1-beta, LPS lipopolysaccharide, MLKL mixed lineage kinase domain-like, NLRP3 NLR family pyrin domain containing 3, PAPMs pathogen-associated molecular patterns, ZBP1 Z-DNA Binding Protein 1.
Nuclear factor E2 related factor 2 (Nrf2)
The body’s resistance to oxidative stress is mostly regulated by the Nrf2-ARE signaling pathway, and it has a role in reducing inflammation [121]. The expression of Nrf2-driven antioxidant molecules may be induced in response to stimulation by different TLR agonists, a process that links the body’s immune response to its antioxidant system [122]. For example, resiquimod, an agonist of TLR7, upregulates NADPH oxidase (NOX-4), which in turn induces Nrf2-mediated expression of Cu/ZnSOD, a process that contributes to the alleviation of ovalbumin-induced sepsis [123]. In a mouse model, activation of Nrf2/HO-1 (heme oxygenase 1) ameliorated endotoxemia/sepsis-induced hypothermia and vascular leakage [124]. Moreover, the Nrf2-ARE (antioxidant response element) signaling pathway is among the principal regulators of endogenous and external stress-induced cytoprotective responses caused by ROS, RNS, and electrophiles [125]. According to in vivo and in vitro investigations, TLR4 (lipopolysaccharide), TLR2 (peptidoglycan), and TLR3 (Poly.C) inducers are able to activate the Nrf2-ARE pathway and its downstream antioxidant enzymes (HO-1, GST) [126]. When Nrf2 was activated, endotoxin-induced HO-1 expression was noticed in THP-1 monocyte cells. Accumulating data indicates that additional downstream targets (SOD) of Nrf2 are also involved in the control of TLR-induced inflammation. Roughly speaking, coupling of TLR signaling to the Nrf2 pathway is achieved through the autophagy-lysosome pathway p62-mediated degradation of Keap1. The activation of autophagy, which is mediated by p62, contributes to the regulation of the inflammatory response. In keratin-forming cells, activation of the TLR pathway, for instance, induces ROS generation, p62 expression, and autophagy via NADPH oxidase 2 and 4 [127]. In conclusion, the TLR/p62/autophagy/Nrf2 axis represents a crucial connection between the immune response and the antioxidant system. This interaction is made possibly by the protein p62. Nrf2 inducers counteract the inflammatory response triggered by the TLR pathway [128]. In an ex vivo mouse model, LPS-induced activation of TLR4 in pancreatic cells caused increased lymphocyte infiltration as well as inflammation, which resulted in ROS production, inducer of the SAPK/JNK death pathway, and activation of NF-κB, activator protein 1, and c-Jun/Fos transcription factors, which ultimately led to inflammation [129]. Studying the regulatory role of TLR-Nrf2 on inflammation can help understand the pathogenesis of inflammatory disease such as AS, which is essential for the development of new drugs and preventive measures.
Proprotein convertase subtilisin/kexin type 9 (PCSK9)
The proprotein convertase family contains nine members, and PCSK9 is the ninth serine protease member. This leads to a rise in LDL cholesterol (LDL-C) levels, which is caused by the degradation of the LDL cholesterol receptor (LDLR) [130]. According to immunohistochemistry and immunoblotting, PCSK9 is mainly expressed in VSMCs within atherosclerotic plaques [131]. Pro-inflammatory factors TNF-α and LPS can both increase PCSK9 expression in vascular endothelial cells and VSMCs. Evidence supports that PCSK9 overexpression increases the size of atherosclerotic plaques [132]. Ox-LDL is a process within an effective stimulus that causes AS, and together with other pro-inflammatory mediators such as LPS, it can cause the expression of PCSK9 in macrophages, endothelial cells, VSMCs, and dendritic cells, all of which are closely associated with the development of AS [133]. Notably, TLRs, NLRP3 inflammatory vesicles, and NF-κB are activated concurrently with the synthesis and release of PCSK9. PCSK9 probably promotes the production of pro-inflammatory cytokines by binding to the C-terminal structural domain of TLR4, hence increasing TLR4 expression and activating the TLR4/NF-κB signaling pathway [134]. NLRP3 activation is followed by the release of IL-1β and IL-18, which are essential for PCSK9 transcription. Similarly, TLR4 induces PCSK9 transcription in the aorta via the TRIF and MyD88 pathways [135]. Recently, Ding et al. discovered a beneficial feedback connection among VSMC-derived PCSK9 and mitochondrial DNA damage, which induces apoptosis as a hallmark of plaque formation and rupture and is mediated by mitochondrial ROS (mtROS) [136]. PCSK9 on the artery wall, in addition to decreasing LDL, encourages monocyte migration toward atherosclerotic plaque [137]. It is assumed that PCSK9 activation is responsible for increased LDL uptake by macrophages and reverse cholesterol transport [138]; recombinant PCSK9 also stimulates the expression of different scavenger receptors (SRs), including lectin-like oxidized lipoprotein-1 (LOX-1), SR-A, and CD36 [139] which are key mediators of oxidized LDL absorption by macrophages and contribute to the development of AS.
Potential therapeutic targets associated with TLR signaling pathways
Drugs for AS associated with TLR signaling pathway
Since TLRs play a crucial role in the progression of atherosclerosis, targeting TLRs for treatment has unlimited therapeutic potential. Experimental animals have shown that treatment or prevention of AS can be achieved by decreasing TLR signaling or disrupting PAMPs [140]. Drugs that exert therapeutic effects on AS through TLRs and their related pathways have also been gradually developed (Fig. 5). For example, the traditional cardiovascular therapeutic drugs, statins [141], and TLR antagonists/inhibitors, have been developed to exert therapeutic strategies primarily via inhibiting TLR ligand-receptor interaction and interfering with intracellular signaling. Other drugs targeting TRL signaling pathway-related molecules, such as PCSK9 antagonists [142], are able to bind to PCSK9 through active vaccination and inhibit its interaction with LDLR to improve systemic and vascular inflammation and the advancement of AS. Furthermore, inhibitors of IRAK1/4 represent a potentially useful therapeutic strategy for the treatment of AS (Table 2).

GNP gold nanoparticles, HDL-like NP high-density lipoprotein nanoparticles, IRAKs interleukin-1 receptor kinases, IRF3 interferon regulatory factor 3, IRF7 interferon regulatory factor 7, LPS lipopolysaccharide; miRNA, MicroRNA, NF-κB nuclear factor kappa B, PCSK9 Proprotein convertase subtilisin/kexin type 9, SMIs Small molecule inhibitors, TIRAP toll-interleukin 1 receptor domain-containing adapter protein, TRAF3 tumor necrosis factor receptor-associated factor 3, TRAF6 tumor necrosis factor receptor-associated factor 6, TRAM TRIF-associated junction molecule, TRIF toll receptor-associated interferon activator.
TLR antagonists/inhibitors
Numerous inflammatory diseases are linked to the overactivation of TLRs, and blocking this pathway can be used as an effective treatment. In general, there are two main approaches [143]: (1) disrupting intracellular signaling and preventing signal transduction; (2) preventing TLR ligands from attaching to their receptors.
Small molecule inhibitors (SMIs)
Small molecule inhibitors (SMIs) are a family of naturally generated or manufactured pharmacologic compounds that suppress TLR signaling by acting on particular intracellular adaptor proteins or compartments in the TLR signaling pathways [144]. SMI of TLR7/8/9 can be applied to diseases related to uncontrollable acute or chronic inflammation. As an illustration, the anti-malarial drug CQ has the ability to block acidification and inflammation in vivo and also significantly improves cerebral ischemic symptoms in rats with transient total cerebral ischemia, suggesting that SMI of these TLRs in vivo may be beneficial for patients with CVDs [145]. Importantly, a recent study reported that the small molecule SMU-CX24, which is derived from O. elliptica, the carbazole alkaloid 9-methoxy-elliciptine NSC69187 (CX1), is a novel potent inhibitor of TLR3. SMU-CX24 inhibits TLR3 with a specificity 10 times greater than that of CX1. This may be because the changed side chain of SMU-CX24 improves the compound’s alkalinity, making it simpler to bind to TLR3 in the weakly acidic in vivo environment [146]. Chen et al. [147] conducted relevant tests on an AS model given a Western diet and showed that SMU-CX24 dramatically lowered the expression of TLR3 in mice. In addition, the expression levels of associated inflammatory factors were considerably reduced in atherosclerotic lesions. Given the preceding information, small molecule compounds that target TLRs will be a potential class of medications for the treatment of AS.
MicroRNAs (miRNAs)
MiRNAs are endogenous, non-coding RNAs of around 22 nucleotides, and because of their post-transcriptional regulatory function, they can often fine-tune gene expression [148]. MiRNAs have been shown to regulate TLR signaling pathways. For instance, TLR4 is a direct target of both miR-217 [149] and miR-20a [150], which are capable of reducing apoptosis and inflammation in atherosclerotic endothelial cells, respectively. In addition, MiR-21 enhances the synthesis of the anti-inflammatory cytokine IL-10 by inhibiting the expression of the programmed cell death 4 (PDCD4) gene, hence inhibiting the inflammatory response [151]. Furthermore, MS2 VLP-based delivery of MiR-146a has been reported to suppress TRAF6 and IRAK1 downstream of TLR4 signaling [152], indicating that induction of miR-146a using an MS2 VLP-based delivery system may result in the development of novel therapies for AS. Considering that MiRNAs play an important role in endothelial inflammation and atherogenesis, it appears that the development of TLR inhibitors by targeting miRNAs for the prevention of AS formation is promising.
Nanodevices
With the development of technology, nanotechnology is increasingly coming into the public eye. When nanotechnology is combined with drugs, it can produce many surprising effects. Similarly, nanodevices are a novel class of potent TLR inhibitors [153]. A small database of physiologically stable peptide-gold nanoparticle (GNP) hybrids has been established by screening. The researchers identified a potent inhibitor of TLRs, P12, which inhibited not only the activation of two pathways of TLR4 signaling, namely MyD88-dependent NF-κB and TRIF-dependent IRF3, but also TLR2-, TLR3-, and TLR5-related pathways. The potent anti-inflammatory effects of P12 are manifested in the correction of LPS-induced gene expression, a reduction in the production of subsequent pro-inflammatory factors (IL-12p40, MCP-1, and IFN-γ), and an increased production of the anti-inflammatory cytokine IL-1RA [154]. In addition, by isolating LPS as a TLR4 antagonist, high-density lipoprotein (HDL-like) nanoparticles (HDL-like NP) were created [155]. HDL is well-known to bind and neutralize LPS naturally, and thus HDLNP has the ability to scavenge LPS molecules to exert anti-inflammatory actions. Moreover, the reason for these physiological processes is the ability of P12 to regulate pH in the body and to prevent acidification in the body [156]. The “nano-drug” is flexible in that the inhibitory activity can be adjusted by selecting alternative amino acids for the GNPs. However, there is concern that gold nanoparticles are not biodegradable [157], which raises the question of the long-term safety of such drugs. How to use materials that can degrade will be the focus of research.
Antibodies
Antibodies are highly specific, so they are widely used as biological tools for the precise detection of certain molecules. When antibodies are designed as TLR antagonists, they can prevent ligands from binding to specific TLRs. T2.5 is a neutralizing anti-TLR2 antibody that increased the survival of mice attacked with Pam3CSK4 or Bacillus subtilis in a mouse model experiment. In addition, T2.5 inhibits macrophage recruitment and inflammation in the heart, preventing angiotensin II-induced cardiac fibrosis [158]. These results demonstrated that T2.5 has a beneficial therapeutic impact on cardiovascular disorders. OPN-305 is another potent antibody against TLR2, as demonstrated by its action on CD14+and CD45+ cells (monocytes) to completely block TLR2 receptors [159]. In addition to being the first completely humanized TLR2-specific IgG4 monoclonal antibody, OPN-305 inhibits the generation of TLR2-mediated pro-inflammatory factors in animal models of ischemia-reperfusion (IR) damage [160]. In addition, NI-0101 is a monoclonal antibody against TLR4. Being the first TLR4 monoclonal antibody to reach clinical development, it inhibits TLR4 dimerization to have a therapeutic effect [161]. Importantly, NI-0101 inhibits the generation of pro-inflammatory factors by synovia-induced monocytes in rheumatoid arthritis (RA) patients [162]. In conclusion, antibodies targeting TLRs have a high level of specificity, but there are constraints, such as high production costs and immunogenic hazards, that prevent their widespread use. We expect future TLRs to overcome these shortcomings and play a greater role in the prevention and treatment of cardiovascular illnesses.
Statins
Statins are the most commonly used medications for treating hyperlipidemia caused by CVDs. It influences the mevalonate pathway by competitively suppressing 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme in cholesterol synthesis [163]. Overall, statins achieve this by optimizing the lipid profile, inhibiting LDL oxidation, providing anti-inflammatory function, inhibiting immune cell migration, and suppressing VSMC proliferation. Statins are the most often prescribed medications for treating hyperlipidemia. A recent study revealed that the combination of atorvastatin and mevastatin reduced NF-κB activity and inhibited TLR4 expression in LPS-stimulated mice [164]. Notably, they also had a protective effect against atherosclerotic lesions by reducing tissue factor (TF) expression to limit the growth of new lesions and stabilize existing plaques. Several studies have shown that statins also have an effect on IL-1β and NLRP3 inflammatory vesicles [165]. Statin treatment reduces NLRP3, cathepsin-B, and downstream mediators such as IL-1β, which play an important role in the formation of AS [166]. Moreover, simvastatin regulates and inhibits TLR4 expression through the control of lipids. Wang et al. [167] found that atorvastatin downregulated TLR mRNA expression via two distinct signaling networks: one that stabilized IκB-α to achieve direct inhibition of NF-κB activation; the other that indirectly inhibited NF-κB activity by involving inactivation of ERK phosphorylation. It has been demonstrated that high-dose simvastatin reduces membrane expression of TLR4 and TLR2 on the surface of monocytes, consequently lowering generation of the vasoprotective cytokines IL-6 and IL-1β [168]. Overall, these findings suggest that statins protect against atherosclerotic CVDs, at least partly, by inhibiting TLR signaling pathways, beyond their hypolipidemic effects.
PCSK9 antagonists
Through active immunization, PCSK9 antagonists could attach to PCSK9 and block its interaction with LDLR. Alirocumab is an antagonist of PCSK9, and its usage in mouse tests decreased endothelial expression of ICAM-1 and Ly6Chi monocytes. Other indicators of vascular inflammation, such as T-cell abundance in the aortic root, the quantity of macrophage necrosis, and cholesterol breakdown in the arterial plaque, also decreased [169]. In addition, patients with familial hypercholesterolemia (FH) showed reduced CCR2 expression following PCSK9 inhibition, which is a consequence of the reduced transendothelial migration capacity of monocytes [170]. In a mouse study, the PCSK9 AT04A vaccine induced immunosuppression of circulating PCSK9, a marked decline in plasma cholesterol levels, but rather decreased densities of macrophage colony-stimulating factor 1 (M-CSF-1) and vascular endothelial growth factor A (VEGF-A), which led to downregulation of ICAM-1, hence reducing the recruitment and adhesion of monocytes to vascular endothelial cells [171]. All of these findings indicate that PCSK9 inhibitors can reduce systemic and vascular inflammation along with AS development.
Interleukin-1 receptor associated kinase (IRAK) inhibitors
IRAK, also known as IRAK1, IRAK2, IRAK3, and IRAK4, is a protein kinase that belongs to the serine/threonine kinase family [172]. MyD88 attracts IRAK4 to form the myddosome, which in turn phosphorylates IRAK1 when TLRs are activated [173]. Therefore, IRAK participates in the TLR signaling pathways, which are essential for the innate immune response to infections [174]. Accumulated evidence has shown that the IL1-R/TLR signaling pathway is involved in the development of AS. Canakinumab, the anti-IL-1β antibody, reduces mortality in myocardial infarction (MI) patients with elevated CRP levels [175]. We recently investigated the effects of IRAK1/4i, an IRAK1 and IRAK4 dual inhibitor, on endothelial function and AS development in ApoE-/- mice [176]. We found that IRAK1/4i decreased LPS-induced endothelial inflammation in human endothelial cells via inhibition of NF-κB transcriptional activity. Of note, IRAK1/4i is more effective than monotherapy using an IRAK1 inhibitor (IRAK1i) or an IRAK4 inhibitor (IRAK4i). In vivo, IRAK1/4i possesses anti-inflammatory properties and the capacity to reduce and stabilize atherosclerotic plaques and lower LDL-C levels via boosting the expression of the LDLR protein. These findings imply that pharmacological inhibitors of IRAK1/4 are a viable therapeutic strategy for the treatment of inflammation and dyslipidemia in atherosclerotic CVDs.
Natural compounds
Currently, the incidence and mortality of CVDs show an unpromising trend and involve complex etiology and pathophysiology, and the therapeutic effect of single-target compounds is relatively limited. Traditional Chinese medicine has been handed down for thousands of years, with a long history and a deep cultural heritage. Due to its nature of “multiple ingredients-multiple targets,” many chronic diseases can be cured by some herbal medicines, we are also looking to herbal medicines for help (Table 3) [177].
Tanshinone IIA (tan-IIA)
Danshen (Salvia miltiorrhiza) is a Chinese herbal cure composed of the dried rhizome and root of Danshen [178]. The mechanisms of action of compounds isolated from Salvia miltiorrhiza, especially tan-IIA, in cardiovascular protection have been widely reported. Anti-inflammatory, antioxidant, anti-thrombotic, anti-proliferation of VSMCs, suppression of vascular endothelial cell and leukocyte adhesion molecule expression, and amelioration of myocardial ischemia are some of the effects of this compound [179]. Modern pharmacological research has revealed that salvia has a variety of therapeutic benefits, such as dilatation of coronary arteries, reduction in myocardial oxygen consumption, avoidance of myocardial ischemia and myocardial infarction, and enhancement of microcirculation [180, 181]. Tan-IIA contributes considerably to the activation, development, and maintenance of proper immune cell function in both innate and acquired immunological responses. Meng et al. [182] revealed that tan-IIA can inhibit endotoxin-induced inflammatory responses by partially inhibiting the TLR4/TAK1/NF-κB signaling pathway in VSMCs. Tan-IIA significantly inhibited the transcript levels of several matrix metalloproteinases and pro-inflammatory factors in RA-FLS stimulated by TNF-α, resulting in an anti-inflammatory effect [183]. Additionally, tan-IIA can modulate the synthesis of microRNA and the production of a variety of cytokines by blocking the TLR4-MyD88-NF-κB signaling pathway, leading to an anti-inflammatory effect on endotoxin-stimulated RAW264.7 cells [184]. In summary, tan-IIA has powerful anti-inflammatory effects, at least in part by targeting TLR4, thereby becoming an outstanding representative of traditional Chinese medicine with protective effects against cardiovascular and cerebrovascular diseases.
Dan-Lou prescription
Traditional Chinese medicine makes use of a concoction called Gualou-Xiebai-Banxia. This concoction is where the Dan-Lou prescription comes from. Now it has a history of nearly 2000 years. Gallic acid, salvianic acid, paeoniflorin, daidzin, puerarin, salvianolic acid B, tan-IIA and cryptotanshinone, are the eight components with the greatest concentration in the ethanol extract of Dan-Lou (EEDL) [185]. It is primarily used in clinical practice to treat angina pectoris, acute myocardial infarction, and other CVDs [185]. EEDL exerts its anti-inflammatory actions by reducing the production of iNOS/NO, COX-2/prostaglandin (PG), E2, and cytokines. The highest levels of puerarin in EEDL reduce AS through leptin and low-density lipoprotein. Macrophages abnormally digest oxLDL, leading to fat accumulation as well as promoting the growth of foam cells [3]. EEDL antagonizes the binding as well as internalization of oxidized LDL and reduces the oxidized LDL-induced buildup of lipids in RAW 264.7 cells. Notably, TLR4, SRs including SRA1, SRB1, and lectin-like oxLDL receptor (LOX)-1 are pattern recognition molecules for oxLDL. The reduction of ox-LDL uptake by EEDL was closely associated with the downregulation of SRB1 and TLR4 [186]. EEDL also exerted an anti-apoptotic effect, which was mainly achieved by inhibiting Bax expression and upregulating Bcl-2 expression. Gao et al. [187] discovered that EEDL antagonizes the binding as well as internalization of oxLDL and reduces the oxLDL-induced buildup of lipids in RAW 264.7 cells, and the Dan-Lou prescription successfully reduced the production of macrophage foam cells via numerous signaling pathways, including TLR4/NF-κB and PPARγ. Collectively, TLR4, PPARγ, NF-κB, MCP-1, COX-2, TNF-α, Bcl-2, and Bax are some of the molecules that are intended to serve as Dan-Lou prescription targets and that influence cellular processes such as apoptosis, inflammatory response, energy metabolism, lipid metabolism, and oxidative stress.
Rhamnetin
Rhamnetin, which is found in Rhamnus petiolaris, is a member of the flavonoid family and is extracted from the plant [188]. Rhamnetin has potential anti-inflammatory activity and cytoprotective effects. In addition, rhamnolipid reduces oxidative stress and inflammatory mediators to protect neurons from damage [189]. It is known that plasma ApoE is mainly of hepatic origin and is an essential ligand for the clearance of celiac, LDL, and very-low-density-lipoprotein (VLDL) residual proteins, and that defects in the ApoE gene are connected to elevated levels of residual lipoprotein in both humans and mice [190]. These effects are intimately connected to AS prevention and therapy. Rhamnetin, like the aforementioned herbs, inhibited the production of foam cells and the expression of CD36, TLR4, and P-IκBα/IκBα proteins in macrophages activated by oxLDL [191]. Rhamnetin reportedly protects liver function and lipid profile in rats fed a high cholesterol diet by reducing oxidative stress and superoxide anion production [192]. In addition, rhamnetin ameliorated the increased levels of NF-κBp65, TLR4 and MMP13 in the aortic tissue homogenates of ApoE-/- mice. That is, rhamnetin treatment inhibited inflammatory and proatherosclerotic pathways in ApoE-/- mice by modulating the TLR-4/IκBα/NF-κBp65 pathway [188].
Conclusions and future prospects
TLRs are essential for both innate and acquired immunity, serving as the host’s initial line of defense against microbial infections. Although TLRs play a significant role in recognizing pathogens and enhancing the body’s defenses, excessive activation destabilizes the immune system and ultimately leads to the onset of inflammatory diseases. There is no doubt that TLRs are involved in the formation of AS from the very beginning of endothelial cell activation and inflammation generation, to endothelial damage, and finally to the onset of plaque instability (Fig. 3).
The limitation and challenge of targeting TLRs as a therapy strategy for AS: (1) Although most of the reported TLRs are contributing to the formation of AS, there are some inconsistent views. For example, some investigators found that in ApoE-/- mice given a high-fat diet (HFD), genetic deletion of TLR9 promoted the development of atherosclerotic lesions and increased the formation of macrophages, dendritic cells, and CD4+ T cells in plaques, indicating that TLR9 has an anti-atherosclerotic effect [193]. This appears to contradict the majority of reports that TLR9 activation can accelerate the expression of pro-inflammatory molecules, thereby promoting the production of AS. In addition, the role of TLR7 in atherosclerotic plaque formation is somewhat controversial. TLR7-activated platelets, for instance, can raise the risk of thrombosis through neutrophil aggregation, but their release of granulocyte-macrophage colony-stimulating factor protects against thrombosis [8]. Conflicting views such as these deserve more in-depth exploration to clarify what role TLRs actually play in the formation of AS. In this way, we can precisely and effectively develop novel and effective drugs targeting TLRs that can treat or prevent AS. (2) As traditional cardiovascular therapies, statins are by far the most widely used and cost-effective option, but they have numerous limitations, including the fact that many patients are tolerable or intolerant to statins and a high risk of residual inflammation, which can result in serious events. Therefore, it is crucial to discover alternative, more effective medications. TLR agonists and inhibitors are currently undergoing clinical testing for the treatment of inflammatory disorders, including asthma, cancer, and autoimmune diseases, but research on these compounds for the treatment of CVDs is still in its infancy [194]. The safety, efficacy, and stability of therapeutic pharmaceuticals must be given greater weight in the future drug development process. (3) As some medications have the potential to compromise local immune defense function, it is undeniable that a holistic approach is also crucial to consider. In addition, the fact that pathogens can activate more than one TLR suggests that the development of antagonists/inhibitors that target multiple TLR signaling pathways may be more effective. (4) Due to the extensive use of novel materials, nanotechnology is also being applied to therapeutic medications. Nanodevices are a new class of effective TLR inhibitors; they feature precise targeting, but the fact that gold nanoparticles are not biodegradable raises concerns about the long-term safety of such medications. This will prompt us to concentrate our future research on achieving a safe and dependable biodegradation procedure. (5) As mentioned in Endothelial cells, Pseudomonas gingivalis accelerates the formation of AS. This suggests that, from the perspective of the relationship between pathogenic microorganisms and TLRs, the use of vaccines or immunomodulatory therapies for the treatment of AS may be a proven approach. However, there are some limitations to this approach, for example, the direct role of many pathogens in the development of AS is not clear. More research is needed to prove the hypothesis that pathogenic microorganisms cause AS by interacting with specific TLRs. (6) Likewise, Chinese herbal medicine, a cultural treasure of China, has multiple therapeutic effects; for example, many herbal medicines can play a role in preventing and treating AS by fighting TLRs. New applications of old medicines are also a form of innovation and development, so it is particularly necessary to increase research on herbal medicines. (7) Finally, in addition to TLR itself, other inflammatory signaling pathways associated with TLRs deserve special consideration. For instance, overexpression of PCSK9, which can induce the production of pro-inflammatory cytokines by activating TLR4/NF-κB pathway, increases the size of atherosclerotic plaques, whereas PCSK9 antagonists substantially impeded the development of AS in mouse experiments; and the TLR-driven NLRP3 inflammasome, a specific and important form of inflammation and innate immunity, is also a key trigger of pyroptosis and necroptosis, all of which are involved in the formation and development of AS. If corresponding therapeutic agents can be developed for the aforementioned substances and pathways, more novel therapeutic strategies and approaches will be available for the treatment of AS.
In summary, as the emphasis on inflammation continues to grow and the research on TLRs in atherosclerotic CVDs continues to intensify, many new ideas and insights continue to come to light. TLRs have great potential as pharmacological targets for AS, and we anticipate that targeting TLRs will serve as a proper and successful therapy strategy for AS.
References
Tsao CW, Aday AW, Almarzooq ZI, Alonso A, Beaton AZ, Bittencourt MS, et al. Heart disease and stroke statistics-2022 update: a report from the American Heart Association. Circulation. 2022;145:e153–e639.
Roy P, Orecchioni M, Ley K. How the immune system shapes atherosclerosis: roles of innate and adaptive immunity. Nat Rev Immunol. 2022;22:251–65.
Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74.
Soehnlein O, Libby P. Targeting inflammation in atherosclerosis-from experimental insights to the clinic. Nat Rev Drug Discov. 2021;20:589–610.
Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32:305–15.
Wang Y, Song E, Bai B, Vanhoutte PM. Toll-like receptors mediating vascular malfunction: Lessons from receptor subtypes. Pharmacol Ther. 2016;158:91–100.
Bayan N, Yazdanpanah N, Rezaei N. Role of toll-like receptor 4 in diabetic retinopathy. Pharmacol Res. 2022;175:105960.
Shafeghat M, Kazemian S, Aminorroaya A, Aryan Z, Rezaei N. Toll-like receptor 7 regulates cardiovascular diseases. Int Immunopharmacol. 2022;113:109390.
Almeida SO, Budoff M. Effect of statins on atherosclerotic plaque. Trends Cardiovasc Med. 2019;29:451–5.
Kobiyama K, Ley K. Atherosclerosis. Circ Res. 2018;123:1118–20.
Kales SN, Soteriades ES, Christophi CA, Christiani DC. Emergency duties and deaths from heart disease among firefighters in the United States. N Engl J Med. 2007;356:1207–15.
Koskinas KC, Feldman CL, Chatzizisis YS, Coskun AU, Jonas M, Maynard C, et al. Natural history of experimental coronary atherosclerosis and vascular remodeling in relation to endothelial shear stress: a serial, in vivo intravascular ultrasound study. Circulation. 2010;121:2092–101.
Allahverdian S, Pannu PS, Francis GA. Contribution of monocyte-derived macrophages and smooth muscle cells to arterial foam cell formation. Cardiovasc Res. 2012;95:165–72.
Hopkins PN. Molecular biology of atherosclerosis. Physiol Rev. 2013;93:1317–542.
Sun HJ, Wu ZY, Nie XW, Bian JS. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front Pharmacol. 2019;10:1568.
Mori H, Torii S, Kutyna M, Sakamoto A, Finn AV, Virmani R. Coronary artery calcification and its progression: what does it really mean? JACC Cardiovasc Imaging. 2018;11:127–42.
Shi X, Gao J, Lv Q, Cai H, Wang F, Ye R, et al. Calcification in atherosclerotic plaque vulnerability: friend or foe? Front Physiol. 2020;11:56.
Kapustin AN, Chatrou MLL, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. 2015;116:1312–23.
Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res. 2009;50:S352–S7.
Vijay K. Toll-like receptors in immunity and inflammatory diseases: past, present, and future. Int Immunopharmacol. 2018;59:391–412.
Fitzgerald KA, Kagan JC. Toll-like receptors and the control of immunity. Cell. 2020;180:1044–66.
Asami J, Shimizu T. Structural and functional understanding of the toll-like receptors. Protein Sci. 2021;30:761–72.
Jaén RI, Val-Blasco A, Prieto P, Gil-Fernández M, Smani T, López-Sendón JL, et al. Innate immune receptors, key actors in cardiovascular diseases. JACC Basic Transl Sci. 2020;5:735–49.
Wang YC, Lin S, Yang QW. Toll-like receptors in cerebral ischemic inflammatory injury. J Neuroinflammation. 2011;8:134.
Salvador B, Arranz A, Francisco S, Córdoba L, Punzón C, Llamas MÁ, et al. Modulation of endothelial function by Toll like receptors. Pharmacol Res. 2016;108:46–56.
Alonso-Pérez A, Franco-Trepat E, Guillán-Fresco M, Jorge-Mora A, López V, Pino J, et al. Role of toll-like receptor 4 on osteoblast metabolism and function. Front Physiol. 2018;9:504.
Vollmer S, Strickson S, Zhang T, Gray N, Lee KL, Rao VR, et al. The mechanism of activation of IRAK1 and IRAK4 by interleukin-1 and Toll-like receptor agonists. Biochem J. 2017;474:2027–38.
Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3.
Leulier F, Lemaitre B. Toll-like receptors-taking an evolutionary approach. Nat Rev Genet. 2008;9:165–78.
Cahill PA, Redmond EM. Vascular endothelium-Gatekeeper of vessel health. Atherosclerosis. 2016;248:97–109.
Ciesielska A, Matyjek M, Kwiatkowska K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci. 2021;78:1233–61.
Xu W, Zhou W, Wang H, Liang S. Roles of Porphyromonas gingivalis and its virulence factors in periodontitis. Adv Protein Chem Struct Biol. 2020;120:45–84.
Mougeot JLC, Stevens CB, Paster BJ, Brennan MT, Lockhart PB, Mougeot FKB. Porphyromonas gingivalis is the most abundant species detected in coronary and femoral arteries. J Oral Microbiol. 2017;9:1281562.
Joshi C, Bapat R, Anderson W, Dawson D, Hijazi K, Cherukara G. Detection of periodontal microorganisms in coronary atheromatous plaque specimens of myocardial infarction patients: A systematic review and meta-analysis. Trends Cardiovasc Med. 2021;31:69–82.
Xie M, Tang Q, Yu S, Sun J, Mei F, Zhao J, et al. Porphyromonas gingivalis disrupts vascular endothelial homeostasis in a TLR-NF-κB axis dependent manner. Int J Oral Sci. 2020;12:28.
Inaba H, Yoshida S, Nomura R, Kato Y, Asai F, Nakano K, et al. Porphyromonas gulae lipopolysaccharide elicits inflammatory responses through Toll-like receptor 2 and 4 in human gingivalis epithelial cells. Cell Microbiol. 2020;22:e13254.
Lin G, Chen S, Lei L, You X, Huang M, Luo L, et al. Effects of intravenous injection of porphyromonas gingivalis on rabbit inflammatory immune response and atherosclerosis. Mediators Inflamm. 2015;2015:364391.
Grelier A, Cras A, Balitrand N, Delmau C, Lecourt S, Lepelletier Y, et al. Toll-like receptor 3 regulates cord blood-derived endothelial cell function in vitro and in vivo. Angiogenesis. 2013;16:821–36.
Gimbrone MA, García-Cardeña G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. 2016;118:620–36.
Yeung J, Li W, Holinstat M. Platelet signaling and disease: targeted therapy for thrombosis and other related diseases. Pharmacol Rev. 2018;70:526–48.
Schrottmaier WC, Mussbacher M, Salzmann M, Assinger A. Platelet-leukocyte interplay during vascular disease. Atherosclerosis. 2020;307:109–20.
Gremmel T, Frelinger AL 3rd, Michelson AD. Platelet physiology. Semin Thromb Hemost. 2016;42:191–204.
Rondina MT, Schwertz H, Harris ES, Kraemer BF, Campbell RA, Mackman N, et al. The septic milieu triggers expression of spliced tissue factor mRNA in human platelets. J Thromb Haemost. 2011;9:748–58.
Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867–79.
D’ Atri LP, Schattner M. Platelet toll-like receptors in thromboinflammation. Front Biosci (Landmark Ed). 2017;22:1867–83.
Holinstat M. Normal platelet function. Cancer Metastasis Rev. 2017;36:195–8.
Niklaus M, Klingler P, Weber K, Koessler A, Boeck M, Kobsar A, et al. The involvement of toll-like receptors 2 and 4 in human platelet signalling pathways. Cell Signal. 2020;76:109817.
Koupenova M, Clancy L, Corkrey HA, Freedman JE. Circulating platelets as mediators of immunity, inflammation, and thrombosis. Circ Res. 2018;122:337–51.
Bakogiannis C, Sachse M, Stamatelopoulos K, Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. 2019;122:154157.
Jakob A, Schachinger E, Klau S, Lehner A, Ulrich S, Stiller B, et al. Von Willebrand factor parameters as potential biomarkers for disease activity and coronary artery lesion in patients with Kawasaki disease. Eur J Pediatr. 2020;179:377–84.
Rivadeneyra L, Carestia A, Etulain J, Pozner RG, Fondevila C, Negrotto S, et al. Regulation of platelet responses triggered by Toll-like receptor 2 and 4 ligands is another non-genomic role of nuclear factor-kappaB. Thromb Res. 2014;133:235–43.
Damien P, Cognasse F, Payrastre B, Spinelli SL, Blumberg N, Arthaud CA, et al. NF-κB links TLR2 and PAR1 to soluble immunomodulator factor secretion in human platelets. Front Immunol. 2017;8:85.
Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. 2011;11:762–74.
Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–62.
Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. 2018;49:595–613.
Blériot C, Dupuis T, Jouvion G, Eberl G, Disson O, Lecuit M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity. 2015;42:145–58.
Machiels B, Dourcy M, Xiao X, Javaux J, Mesnil C, Sabatel C, et al. Author Correction: A gammaherpesvirus provides protection against allergic asthma by inducing the replacement of resident alveolar macrophages with regulatory monocytes. Nat Immunol. 2018;19:1035.
Aegerter H, Kulikauskaite J, Crotta S, Patel H, Kelly G, Hessel EM, et al. Influenza-induced monocyte-derived alveolar macrophages confer prolonged antibacterial protection. Nat Immunol. 2020;21:145–57.
Guilliams M, Svedberg FR. Does tissue imprinting restrict macrophage plasticity? Nat Immunol. 2021;22:118–27.
Honda T, Egen JG, Lämmermann T, Kastenmüller W, Torabi-Parizi P, Germain RN. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity. 2014;40:235–47.
Coillard A, Guyonnet L, De Juan A, Cros A, Segura E. TLR or NOD receptor signaling skews monocyte fate decision via distinct mechanisms driven by mTOR and miR-155. Proc Natl Acad Sci USA. 2021;118:1–12.
Watanabe T, Kitani A, Murray PJ, Strober W. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nat Immunol. 2004;5:800–8.
Gamrekelashvili J, Kapanadze T, Sablotny S, Ratiu C, Dastagir K, Lochner M, et al. Notch and TLR signaling coordinate monocyte cell fate and inflammation. Elife. 2020;9:e57007.
Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. 2019;20:902–14.
Bonnardel J, Guilliams M. Developmental control of macrophage function. Curr Opin Immunol. 2018;50:64–74.
Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13:427–37.
Akilesh HM, Buechler MB, Duggan JM, Hahn WO, Matta B, Sun X, et al. Chronic TLR7 and TLR9 signaling drives anemia via differentiation of specialized hemophagocytes. Science. 2019;363:eaao5213.
Song R, Gao Y, Dozmorov I, Malladi V, Saha I, McDaniel MM, et al. IRF1 governs the differential interferon-stimulated gene responses in human monocytes and macrophages by regulating chromatin accessibility. Cell Rep. 2021;34:108891.
Mogensen TH. IRF and STAT transcription factors - from basic biology to roles in infection, protective immunity, and primary immunodeficiencies. Front Immunol. 2018;9:3047.
Negishi H, Taniguchi T, Yanai H. The interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb Perspect Biol. 2018;10:a028423.
Platanitis E, Decker T. Regulatory networks involving STATs, IRFs, and NFκB in inflammation. Front Immunol. 2018;9:2542.
Rajagopalan S, Lee EC, DuPrie ML, Long EO. TNFR-associated factor 6 and TGF-β-activated kinase 1 control signals for a senescence response by an endosomal NK cell receptor. J Immunol. 2014;192:714–21.
Sager HB, Dutta P, Dahlman JE, Hulsmans M, Courties G, Sun Y, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. 2016;8:342ra80.
Jinnouchi H, Guo L, Sakamoto A, Torii S, Sato Y, Cornelissen A, et al. Diversity of macrophage phenotypes and responses in atherosclerosis. Cell Mol Life Sci. 2020;77:1919–32.
Yan J, Horng T. Lipid metabolism in regulation of macrophage functions. Trends Cell Biol. 2020;30:979–89.
Liu X, Guo JW, Lin XC, Tuo YH, Peng WL, He SY, et al. Macrophage NFATc3 prevents foam cell formation and atherosclerosis: evidence and mechanisms. Eur Heart J. 2021;42:4847–61.
Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–16.
Yang X, Yin Y, Yan X, Yu Z, Liu Y, Cao J. Flagellin attenuates experimental sepsis in a macrophage-dependent manner. Crit Care. 2019;23:106.
Ellenbroek GHJM, van Puijvelde GHM, Anas AA, Bot M, Asbach M, Schoneveld A, et al. Leukocyte TLR5 deficiency inhibits atherosclerosis by reduced macrophage recruitment and defective T-cell responsiveness. Sci Rep. 2017;7:42688.
Hussain S, Johnson CG, Sciurba J, Meng X, Stober VP, Liu C, et al. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. Elife. 2020;9:e50458.
Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200.
Salagianni M, Galani IE, Lundberg AM, Davos CH, Varela A, Gavriil A, et al. Toll-like receptor 7 protects from atherosclerosis by constraining “inflammatory” macrophage activation. Circulation. 2012;126:952–62.
Ishida H, Ohto U, Shibata T, Miyake K, Shimizu T. Structural basis for species-specific activation of mouse Toll-like receptor 9. FEBS Lett. 2018;592:2636–46.
Fukuda D, Nishimoto S, Aini K, Tanaka A, Nishiguchi T, Kim-Kaneyama JR, et al. Toll-like receptor 9 plays a pivotal role in angiotensin II-induced atherosclerosis. J Am Heart Assoc. 2019;8:e010860.
Behmoaras J. The versatile biochemistry of iron in macrophage effector functions. FEBS J. 2021;288:6972–89.
Nonnenmacher Y, Hiller K. Biochemistry of proinflammatory macrophage activation. Cell Mol Life Sci. 2018;75:2093–109.
Lauterbach MA, Hanke JE, Serefidou M, Mangan MSJ, Kolbe CC, Hess T, et al. Toll-like receptor signaling rewires macrophage metabolism and promotes histone acetylation via ATP-citrate lyase. Immunity. 2019;51:997–1011.e7.
Sharif O, Brunner JS, Vogel A, Schabbauer G. Macrophage rewiring by nutrient associated PI3K dependent pathways. Front Immunol. 2019;10:2002.
Krawczyk CM, Holowka T, Sun J, Blagih J, Amiel E, DeBerardinis RJ, et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115:4742–9.
Wang Y, Li N, Zhang X, Horng T. Mitochondrial metabolism regulates macrophage biology. J Biol Chem. 2021;297:100904.
Irizarry-Caro RA, McDaniel MM, Overcast GR, Jain VG, Troutman TD, Pasare C. TLR signaling adapter BCAP regulates inflammatory to reparatory macrophage transition by promoting histone lactylation. Proc Natl Acad Sci USA. 2020;117:30628–38.
Artis D, Spits H. The biology of innate lymphoid cells. Nature. 2015;517:293–301.
Cybulsky MI, Cheong C, Robbins CS. Macrophages and dendritic cells: partners in atherogenesis. Circ Res. 2016;118:637–52.
Gallenga C, Pandolfi F, Caraffa A, Kritas S, Ronconi G, Toniato E, et al. Interleukin-1 family cytokinesand mast cells: activation and inhibition. J Biol Regul Homeost Agents. 2019;33:1–6.
Ji Q, Meng K, Yu K, Huang S, Huang Y, Min X, et al. Exogenous interleukin 37 ameliorates atherosclerosis via inducing the Treg response in ApoE-deficient mice. Sci Rep. 2017;7:3310.
Liu T, Liu J, Lin Y, Que B, Chang C, Zhang J, et al. IL-37 inhibits the maturation of dendritic cells through the IL-1R8-TLR4-NF-κB pathway. Biochim Biophys Acta Mol Cell Biol Lipids. 2019;1864:1338–49.
Liebner S, Dijkhuizen RM, Reiss Y, Plate KH, Agalliu D, Constantin G. Functional morphology of the blood-brain barrier in health and disease. Acta Neuropathol. 2018;135:311–36.
Hutchins PM, Heinecke JW. Cholesterol efflux capacity, macrophage reverse cholesterol transport and cardioprotective HDL. Curr Opin Lipido. 2015;26:388–93.
Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–67.
Martinet W, Coornaert I, Puylaert P, De Meyer GRY. Macrophage death as a pharmacological target in atherosclerosis. Front Pharmacol. 2019;10:306.
Santer D, Nagel F, Gonçalves IF, Kaun C, Wojta J, Fagyas M, et al. Tenascin-C aggravates ventricular dilatation and angiotensin-converting enzyme activity after myocardial infarction in mice. ESC Heart Fail. 2020;7:2113–22.
Podesser BK, Kreibich M, Dzilic E, Santer D, Förster L, Trojanek S, et al. Tenascin-C promotes chronic pressure overload-induced cardiac dysfunction, hypertrophy and myocardial fibrosis. J Hypertens. 2018;36:847–56.
Luo H, Wang J, Qiao C, Zhang X, Zhang W, Ma N. ATF3 inhibits tenascin-C-induced foam cell formation in LPS-stimulated THP-1 macrophages by suppressing TLR-4. J Atheroscler Thromb. 2015;22:1214–23.
Liu Y, Hu Y, Xiong J, Zeng X. Overexpression of activating transcription factor 3 alleviates cardiac microvascular ischemia/reperfusion injury in rats. Front Pharmacol. 2021;12:598959.
Bennett MR, Sinha S, Owens GK. Vascular smooth muscle cells in atherosclerosis. Circ Res. 2016;118:692–702.
Yang K, Wang X, Liu Z, Lu L, Mao J, Meng H, et al. Oxidized low-density lipoprotein promotes macrophage lipid accumulation via the toll-like receptor 4-Src pathway. Circ J. 2015;79:2509–16.
Kasahara K, Nakayama Y, Sato I, Ikeda K, Hoshino M, Endo T, et al. Role of Src-family kinases in formation and trafficking of macropinosomes. J Cell Physiol. 2007;211:220–32.
Moldogazieva NT, Mokhosoev IM, Mel’nikova TI, Porozov YB, Terentiev AA. Oxidative stress and advanced lipoxidation and glycation end products (ALEs and AGEs) in aging and age-related diseases. Oxid Med Cell Longev. 2019;2019:3085756.
Chen Z, Xue Q, Cao L, Wang Y, Chen Y, Zhang X, et al. Toll-like receptor 4 mediated oxidized low-density lipoprotein-induced foam cell formation in vascular smooth muscle cells via Src and Sirt1/3 pathway. Mediators Inflamm. 2021;2021:6639252.
Vandestienne M, Zhang Y, Santos-Zas I, Al-Rifai R, Joffre J, Giraud A, et al. TREM-1 orchestrates angiotensin II-induced monocyte trafficking and promotes experimental abdominal aortic aneurysm. J Clin Invest. 2021;131:e142468.
Nguyen TTT, Yoon HK, Kim YT, Choi YH, Lee WK, Jin M. Tryptophanyl-tRNA synthetase 1 signals activate TREM-1 via TLR2 and TLR4. Biomolecules. 2020;10:1283.
Joffre J, Potteaux S, Zeboudj L, Loyer X, Boufenzer A, Laurans L, et al. Genetic and pharmacological inhibition of TREM-1 limits the development of experimental atherosclerosis. J Am Coll Cardiol. 2016;68:2776–93.
Zahid A, Li B, Kombe AJK, Jin T, Tao J. Pharmacological inhibitors of the NLRP3 inflammasome. Front Immunol. 2019;10:2538.
Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol. 2021;18:1141–60.
Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Zhao C, Zhao W. NLRP3 inflammasome-a key player in antiviral responses. Front Immunol. 2020;11:211.
Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, et al. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. 2011;41:2040–51.
He X, Fan X, Bai B, Lu N, Zhang S, Zhang L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol Res. 2021;165:105447.
Zhang X, Ren Z, Xu W, Jiang Z. Necroptosis in atherosclerosis. Clin Chim Acta. 2022;534:22–8.
Jin Y, Liu Y, Xu L, Xu J, Xiong Y, Peng Y, et al. Novel role for caspase 1 inhibitor VX765 in suppressing NLRP3 inflammasome assembly and atherosclerosis via promoting mitophagy and efferocytosis. Cell Death Dis. 2022;13:512.
Kobayashi EH, Suzuki T, Funayama R, Nagashima T, Hayashi M, Sekine H, et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat Commun. 2016;7:11624.
Nadeem A, Siddiqui N, Al-Harbi NO, Al-Harbi MM, Ahmad SF. TLR-7 agonist attenuates airway reactivity and inflammation through Nrf2-mediated antioxidant protection in a murine model of allergic asthma. Int J Biochem Cell Biol. 2016;73:53–62.
Wang Y, Zhang S, Li H, Wang H, Zhang T, Hutchinson MR, et al. Small-molecule modulators of Toll-like receptors. Acc Chem Res. 2020;53:1046–55.
Lee JW, Bae CJ, Choi YJ, Kim SI, Kwon YS, Lee HJ, et al. 3,4,5-Trihydroxycinnamic acid inhibits lipopolysaccharide (LPS)-induced inflammation by Nrf2 activation in vitro and improves survival of mice in LPS-induced endotoxemia model in vivo. Mol Cell Biochem. 2014;390:143–53.
Joo MS, Kim WD, Lee KY, Kim JH, Koo JH, Kim SG. AMPK facilitates nuclear accumulation of Nrf2 by phosphorylating at serine 550. Mol Cell Biol. 2016;36:1931–42.
Yin S, Cao W. Toll-like receptor signaling induces Nrf2 pathway activation through p62-triggered Keap1 degradation. Mol Cell Biol. 2015;35:2673–83.
Sun Q, Fan J, Billiar TR, Scott MJ. Inflammasome and autophagy regulation - a two-way street. Mol Med. 2017;23:188–95.
Lei L, Chai Y, Lin H, Chen C, Zhao M, Xiong W, et al. Dihydroquercetin activates AMPK/Nrf2/HO-1 signaling in macrophages and attenuates inflammation in LPS-induced endotoxemic mice. Front Pharmacol. 2020;11:662.
Choudhury S, Ghosh S, Gupta P, Mukherjee S, Chattopadhyay S. Inflammation-induced ROS generation causes pancreatic cell death through modulation of Nrf2/NF-κB and SAPK/JNK pathway. Free Radic Res. 2015;49:1371–83.
Sabatine MS. PCSK9 inhibitors: clinical evidence and implementation. Nat Rev Cardiol. 2019;16:155–65.
Giunzioni I, Tavori H, Covarrubias R, Major AS, Ding L, Zhang Y, et al. Local effects of human PCSK9 on the atherosclerotic lesion. J Pathol. 2016;238:52–62.
Roche-Molina M, Sanz-Rosa D, Cruz FM, García-Prieto J, López S, Abia R, et al. Induction of sustained hypercholesterolemia by single adeno-associated virus-mediated gene transfer of mutant hPCSK9. Arterioscler Thromb Vasc Biol. 2015;35:50–9.
Ding Z, Liu S, Wang X, Theus S, Deng X, Fan Y, et al. PCSK9 regulates expression of scavenger receptors and ox-LDL uptake in macrophages. Cardiovasc Res. 2018;114:1145–53.
Tang ZH, Peng J, Ren Z, Yang J, Li TT, Li TH, et al. New role of PCSK9 in atherosclerotic inflammation promotion involving the TLR4/NF-κB pathway. Atherosclerosis. 2017;262:113–22.
Liu S, Deng X, Zhang P, Wang X, Fan Y, Zhou S, et al. Blood flow patterns regulate PCSK9 secretion via MyD88-mediated pro-inflammatory cytokines. Cardiovasc Res. 2020;116:1721–32.
Ding Z, Liu S, Wang X, Deng X, Fan Y, Sun C, et al. Hemodynamic shear stress via ROS modulates PCSK9 expression in human vascular endothelial and smooth muscle cells and along the mouse aorta. Antioxid Redox Signal. 2015;22:760–71.
Tavori H, Giunzioni I, Predazzi IM, Plubell D, Shivinsky A, Miles J, et al. Human PCSK9 promotes hepatic lipogenesis and atherosclerosis development via apoE- and LDLR-mediated mechanisms. Cardiovasc Res. 2016;110:268–78.
Shen L, Peng HC, Nees SN, Zhao SP, Xu DY. Proprotein convertase subtilisin/kexin type 9 potentially influences cholesterol uptake in macrophages and reverse cholesterol transport. FEBS Lett. 2013;587:1271–4.
Adorni MP, Cipollari E, Favari E, Zanotti I, Zimetti F, Corsini A, et al. Inhibitory effect of PCSK9 on Abca1 protein expression and cholesterol efflux in macrophages. Atherosclerosis. 2017;256:1–6.
Bahrami A, Parsamanesh N, Atkin SL, Banach M, Sahebkar A. Effect of statins on Toll-like receptors: a new insight to pleiotropic effects. Pharmacol Res. 2018;135:230–8.
Koushki K, Shahbaz SK, Mashayekhi K, Sadeghi M, Zayeri ZD, Taba MY, et al. Anti-inflammatory action of statins in cardiovascular disease: the role of inflammasome and Toll-like receptor pathways. Clin Rev Allergy Immunol. 2021;60:175–99.
Kuzmich N, Andresyuk E, Porozov Y, Tarasov V, Samsonov M, Preferanskaya N, et al. PCSK9 as a target for development of a new generation of hypolipidemic drugs. Molecules. 2022;27:434.
Gao W, Xiong Y, Li Q, Yang H. Inhibition of Toll-like receptor signaling as a promising therapy for inflammatory diseases: a journey from molecular to nano therapeutics. Front Physiol. 2017;8:508.
Yu Y, Liu J, Liu C, Liu R, Liu L, Yu Z, et al. Post-translational modifications of cGAS-STING: a critical switch for immune regulation. Cells. 2022;11:3043.
Cui G, Ye X, Zuo T, Zhao H, Zhao Q, Chen W, et al. Chloroquine pretreatment inhibits toll-like receptor 3 signaling after stroke. Neurosci Lett. 2013;548:101–4.
Liu S, Jiang Q, Zhao X, Zhao R, Wang Y, Wang Y, et al. Publisher correction: a DNA nanodevice-based vaccine for cancer immunotherapy. Nat Mater. 2021;20:431–3.
Cen X, Wang B, Liang Y, Chen Y, Xiao Y, Du S, et al. Small molecule SMU-CX24 targeting toll-like receptor 3 counteracts inflammation: A novel approach to atherosclerosis therapy. Acta Pharm Sin B. 2022;12:3667–81.
Naqvi AR, Sarwat M. MicroRNAs and immunity. Semin Cell Developmental. Biol. 2022;124:1–2.
Zhang B, Zhang YF, Li R, Zhao L, Qin SG, Pan LF, et al. MiR-217 inhibits apoptosis of atherosclerotic endothelial cells via the TLR4/PI3K/Akt/NF-κB pathway. Eur Rev Med Pharmacol Sci. 2020;24:12867–77.
Chen M, Li W, Zhang Y, Yang J. MicroRNA-20a protects human aortic endothelial cells from Ox-LDL-induced inflammation through targeting TLR4 and TXNIP signaling. Biomed Pharmacother. 2018;103:191–7.
Yan H, Huang W, Rao J, Yuan J. miR-21 regulates ischemic neuronal injury via the p53/Bcl-2/Bax signaling pathway. Aging. 2021;13:22242–55.
Pan Y, Jia T, Zhang Y, Zhang K, Zhang R, Li J, et al. MS2 VLP-based delivery of microRNA-146a inhibits autoantibody production in lupus-prone mice. Int J Nanomed. 2012;7:5957–67.
Wang L, Zhang H, Sun L, Gao W, Xiong Y, Ma A, et al. Manipulation of macrophage polarization by peptide-coated gold nanoparticles and its protective effects on acute lung injury. J Nanobiotechnol. 2020;18:38.
Yang H, Fung SY, Xu S, Sutherland DP, Kollmann TR, Liu M, et al. Amino acid-dependent attenuation of Toll-like receptor signaling by peptide-gold nanoparticle hybrids. ACS Nano. 2015;9:6774–84.
Foit L, Thaxton CS. Synthetic high-density lipoprotein-like nanoparticles potently inhibit cell signaling and production of inflammatory mediators induced by lipopolysaccharide binding Toll-like receptor 4. Biomaterials. 2016;100:67–75.
Yang H, Kozicky L, Saferali A, Fung SY, Afacan N, Cai B, et al. Endosomal pH modulation by peptide-gold nanoparticle hybrids enables potent anti-inflammatory activity in phagocytic immune cells. Biomaterials. 2016;111:90–102.
Shinchi H, Yamaguchi T, Moroishi T, Yuki M, Wakao M, Cottam HB, et al. Gold nanoparticles coimmobilized with small molecule Toll-like receptor 7 ligand and α-mannose as adjuvants. Bioconjugate Chem. 2019;30:2811–21.
Wang L, Li YL, Zhang CC, Cui W, Wang X, Xia Y, et al. Inhibition of Toll-like receptor 2 reduces cardiac fibrosis by attenuating macrophage-mediated inflammation. Cardiovasc Res. 2014;101:383–92.
Reilly M, Miller RM, Thomson MH, Patris V, Ryle P, McLoughlin L, et al. Randomized, double-blind, placebo-controlled, dose-escalating phase I, healthy subjects study of intravenous OPN-305, a humanized anti-TLR2 antibody. Clin Pharmacol Ther. 2013;94:593–600.
Arslan F, Houtgraaf JH, Keogh B, Kazemi K, de Jong R, McCormack WJ, et al. Treatment with OPN-305, a humanized anti-Toll-Like receptor-2 antibody, reduces myocardial ischemia/reperfusion injury in pigs. Circ Cardiovasc Interv. 2012;5:279–87.
Monnet E, Lapeyre G, Poelgeest EV, Jacqmin P, Graaf K, Reijers J, et al. Evidence of NI-0101 pharmacological activity, an anti-TLR4 antibody, in a randomized phase I dose escalation study in healthy volunteers receiving LPS. Clin Pharmacol Ther. 2017;101:200–8.
Hatterer E, Shang L, Simonet P, Herren S, Daubeuf B, Teixeira S, et al. A specific anti-citrullinated protein antibody profile identifies a group of rheumatoid arthritis patients with a toll-like receptor 4-mediated disease. Arthritis Res Ther. 2016;18:224.
Hirota T, Fujita Y, Ieiri I. An updated review of pharmacokinetic drug interactions and pharmacogenetics of statins. Expert Opin Drug Metab Toxicol. 2020;16:809–22.
Guo X, Wang L, Xia X, Wang P, Li X. Effects of atorvastatin and/or probucol on recovery of atherosclerosis in high-fat-diet-fed apolipoprotein E-deficient mice. Biomed Pharmacother. 2019;109:1445–53.
Moutzouri E, Tellis CC, Rousouli K, Liberopoulos EN, Milionis HJ, Elisaf MS, et al. Effect of simvastatin or its combination with ezetimibe on Toll-like receptor expression and lipopolysaccharide - induced cytokine production in monocytes of hypercholesterolemic patients. Atherosclerosis. 2012;225:381–7.
Altaf A, Qu P, Zhao Y, Wang H, Lou D, Niu N. NLRP3 inflammasome in peripheral blood monocytes of acute coronary syndrome patients and its relationship with statins. Coron Artery Dis. 2015;26:409–21.
Wang Y, Zhang MX, Meng X, Liu FQ, Yu GS, Zhang C, et al. Atorvastatin suppresses LPS-induced rapid upregulation of Toll-like receptor 4 and its signaling pathway in endothelial cells. Am J Physiol Heart Circ Physiol. 2011;300:H1743–52.
Kapelouzou A, Giaglis S, Peroulis M, Katsimpoulas M, Moustardas P, Aravanis CV, et al. Overexpression of Toll-like receptors 2, 3, 4, and 8 is correlated to the vascular atherosclerotic process in the hyperlipidemic rabbit model: the effect of statin treatment. J Vasc Res. 2017;54:156–69.
Hoogeveen RM, Opstal TSJ, Kaiser Y, Stiekema LCA, Kroon J, Knol RJJ, et al. PCSK9 antibody alirocumab attenuates arterial wall inflammation without changes in circulating inflammatory markers. JACC Cardiovasc Imaging. 2019;12:2571–3.
Bernelot Moens SJ, Neele AE, Kroon J, van der Valk FM, Van den Bossche J, Hoeksema MA, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. 2017;38:1584–93.
Landlinger C, Pouwer MG, Juno C, van der Hoorn JWA, Pieterman EJ, Jukema JW, et al. The AT04A vaccine against proprotein convertase subtilisin/kexin type 9 reduces total cholesterol, vascular inflammation, and atherosclerosis in APOE*3Leiden.CETP mice. Eur Heart J. 2017;38:2499–507.
Flannery S, Bowie AG. The interleukin-1 receptor-associated kinases: critical regulators of innate immune signalling. Biochem Pharmacol. 2010;80:1981–91.
Akar-Ghibril N. Defects of the innate immune system and related immune deficiencies. Clin Rev Allergy Immunol. 2022;63:36–54.
Alfaidi M, Acosta CH, Wang D, Traylor JG, Orr AW. Selective role of Nck1 in atherogenic inflammation and plaque formation. J Clin Invest. 2020;130:4331–47.
Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–31.
Wu X, Xu M, Liu Z, Zhang Z, Liu Y, Luo S, et al. Pharmacological inhibition of IRAK1 and IRAK4 prevents endothelial inflammation and atherosclerosis in ApoE-/- mice. Pharmacol Res. 2022;175:106043.
Ye T, Li Y, Xiong D, Gong S, Zhang L, Li B, et al. Combination of Danshen and ligustrazine has dual anti-inflammatory effect on macrophages and endothelial cells. J Ethnopharmacol. 2021;266:113425.
Li Z-M, Xu S-W, Liu P-Q. Salvia miltiorrhizaBurge (Danshen): a golden herbal medicine in cardiovascular therapeutics. Acta Pharmacol Sin. 2018;39:802–24.
Luo J, Song W, Yang G, Xu H, Chen K. Compound Danshen (Salvia miltiorrhiza) dripping pill for coronary heart disease: an overview of systematic reviews. Am J Chin Med. 2015;43:25–43.
Hao DC, Ge GB, Xiao PG. Anticancer drug targets of Salvia phytometabolites: chemistry, biology and omics. Curr Drug Targets. 2018;19:1–20.
Irmak F, Kurt Yazar S, Şirvan SS, Serin M, Özağarı A. Karasoy, et al. Beneficial effects of Salvia miltiorrhiza in the healing of burn wounds: an experimental study in rats. J Plast Surg Hand Surg. 2018;52:229–33.
Meng Z, Si CY, Teng S, Yu XH, Li HY. Tanshinone IIA inhibits lipopolysaccharide-induced inflammatory responses through the TLR4/TAK1/NF-κB signaling pathway in vascular smooth muscle cells. Int J Mol Med. 2019;43:1847–58.
Du H, Wang Y, Zeng Y, Huang X, Liu D, Ye L, et al. Tanshinone IIA suppresses proliferation and inflammatory cytokine production of synovial fibroblasts from rheumatoid arthritis patients induced by TNF-α and attenuates the inflammatory response in AIA mice. Front Pharmacol. 2020;11:568.
Fan G, Jiang X, Wu X, Fordjour PA, Miao L, Zhang H, et al. Anti-inflammatory activity of tanshinone IIA in LPS-stimulated RAW264.7 macrophages via miRNAs and TLR4-NF-κB pathway. Inflammation. 2016;39:375–84.
Gao LN, Zhou X, Zhang Y, Cui YL, Yu CQ, Gao S. The anti-inflammatory activities of ethanol extract from Dan-Lou prescription in vivo and in vitro. BMC Complement Alter Med. 2015;15:317.
Ichimura T, Asseldonk EJ, Humphreys BD, Gunaratnam L, Duffield JS, Bonventre JV. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J Clin Invest. 2008;118:1657–68.
Gao LN, Zhou X, Lu YR, Li K, Gao S, Yu CQ, et al. Dan-Lou prescription inhibits foam cell formation induced by ox-LDL via the TLR4/NF-κB and PPARγ signaling pathways. Front Physiol. 2018;9:590.
Medeiros DL, Lima ETG, Silva JC, Medeiros MA, Pinheiro EBF. Rhamnetin: a review of its pharmacology and toxicity. J Pharm Pharmacol. 2022;74:793–9.
Zhang W, Li B, Guo Y, Bai Y, Wang T, Fu K, et al. Rhamnetin attenuates cognitive deficit and inhibits hippocampal inflammatory response and oxidative stress in rats with traumatic brain injury. Cent Eur J Immunol. 2015;40:35–41.
Kritharides L, Nordestgaard BG, Tybjærg-Hansen A, Kamstrup PR, Afzal S. Effect of APOE ε genotype on lipoprotein(a) and the associated risk of myocardial infarction and aortic valve stenosis. J Clin Endocrinol Metab. 2017;102:3390–9.
Wang M, Wu Y, Li W. Rhamnetin ameliorates macrophage-mediated inflammation and pro-atherosclerosis pathways in apolipoprotein E-deficient mice. J Physiol Pharmacol 2021;72. https://doi.org/10.26402/jpp.2021.2.10.
Aquila G, Morelli MB, Vieceli Dalla Sega F, Fortini F, Nigro P, Caliceti C, et al. Heart rate reduction with ivabradine in the early phase of atherosclerosis is protective in the endothelium of ApoE-deficient mice. J Physiol Pharmacol. 2018;69:35–52.
Krogmann AO, Lüsebrink E, Steinmetz M, Asdonk T, Lahrmann C, Lütjohann D, et al. Proinflammatory stimulation of Toll-like receptor 9 with high dose CpG ODN 1826 impairs endothelial regeneration and promotes atherosclerosis in mice. PLoS One. 2016;11:e0146326.
Zhuang C, Chen R, Zheng Z, Lu J, Hong C. Toll-like receptor 3 in cardiovascular diseases. Heart, Lung. Circulation. 2022;31:e93–e109.
Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF-κB signaling in PMA-stimulated THP-1 monocytes. Biomed Pharmacother. 2016;82:167–72.
Koike A, Tsujinaka K, Fujimori K. Statins attenuate antiviral IFN-β and ISG expression via inhibition of IRF3 and JAK/STAT signaling in poly(I:C)-treated hyperlipidemic mice and macrophages. FEBS J. 2021;288:4249–66.
Chen JY, Zhu GY, Su XH, Wang R, Liu J, Liao K, et al. 7-deacetylgedunin suppresses inflammatory responses through activation of Keap1/Nrf2/HO-1 signaling. Oncotarget. 2017;8:55051–63.
Cong ZX, Wang HD, Zhou Y, Wang JW, Pan H, Zhang DD, et al. Temozolomide and irradiation combined treatment-induced Nrf2 activation increases chemoradiation sensitivity in human glioblastoma cells. J Neuro-Oncol. 2014;116:41–8.
Mohan S, Gupta D. Crosstalk of toll-like receptors signaling and Nrf2 pathway for regulation of inflammation. Biomed Pharmacother. 2018;108:1866–78.
Gómez-Guzmán M, Jiménez R, Romero M, Sánchez M, Zarzuelo MJ, Gómez-Morales M, et al. Chronic hydroxychloroquine improves endothelial dysfunction and protects kidney in a mouse model of systemiclupus erythematosus. Hypertension. 2014;64:330–7.
El-Salamouni NS, Gowayed MA, Younis SE, Abdel-Bary A, Kamel MA, Labib GS. Pentoxifylline/Valsartan co-delivery in liposomal gel alters the inflammatory HMGB-1/TLR pathway and promotes faster healing in burn wounds: A promising repurposed approach. Int J Pharm. 2022;625:122129.
Atwa AM, Abd El-Ghafar OAM, Hassanein EHM, Mahdi SE, Sayed GA, Alruhaimi RS, et al. Candesartan attenuates cisplatin-induced lung injury by modulating oxidative stress, inflammation, and TLR-4/NF-κB, JAK1/STAT3, and Nrf2/HO-1 signaling. Pharmaceuticals (Basel, Switzerland). 2022;15:1222. https://doi.org/10.3390/ph15101222.
Guo R, Li L, Su J, Li S, Duncan SE, Liu Z, et al. Pharmacological activity and mechanism of tanshinone IIA in related diseases. Drug Des Devel Ther. 2020;14:4735–48.
Song X, Tan L, Wang M, Ren C, Guo C, Yang B, et al. Myricetin: A review of the most recent research. Biomed Pharmacother. 2021;134:111017.
Liu ZW, Wang JK, Qiu C, Guan GC, Liu XH, Li SJ, et al. Matrine pretreatment improves cardiac function in rats with diabetic cardiomyopathy via suppressing ROS/TLR-4 signaling pathway. Acta Pharmacol Sin. 2015;36:323–33.
Liu K, Li M, Ren X, You QS, Wang F, Wang S, et al. Huang Qi Tong Bi Decoction attenuates myocardial ischemia-reperfusion injury via HMGB1/TLR/NF-κB pathway. Mediators Inflamm. 2019;2019:8387636.
Kim WS, Kim K, Byun EB, Song HY, Han JM, Park WY, et al. RM, a novel resveratrol derivative, attenuates inflammatory responses induced by lipopolysaccharide via selectively increasing the Tollip protein in macrophages: A partial mechanism with therapeutic potential in an inflammatory setting. Int Immunopharmacol. 2020;78:106072.
Chen L, Yu J. Modulation of Toll-like receptor signaling in innate immunity by natural products. Int Immunopharmacol. 2016;37:65–70.
Zhao H, Zhang M, Zhou F, Cao W, Bi L, Xie Y, et al. Cinnamaldehyde ameliorates LPS-induced cardiac dysfunction via TLR4-NOX4 pathway: The regulation of autophagy and ROS production. J Mol Cell Cardiol. 2016;101:11–24.
Acknowledgements
This work is supported by National Natural Science Foundation of China (grant numbers 82270500, 81870324, 82203304, U21A20419), Science and Technology Project of Huadu District in Guangzhou (21-HDWS-007) and Local Innovative and Research Teams Project of Guangdong Pearl River Talents Program (grant numbers 2017BT01Y093, 2017BT01Y036).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Jin, M., Fang, J., Wang, Jj. et al. Regulation of toll-like receptor (TLR) signaling pathways in atherosclerosis: from mechanisms to targeted therapeutics. Acta Pharmacol Sin 44, 2358–2375 (2023). https://doi.org/10.1038/s41401-023-01123-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-023-01123-5
- Springer Nature Singapore Pte Ltd.