
Abstract
Heart disease is a worldwide health menace. Both intractable primary and secondary cardiomyopathies contribute to malignant cardiac dysfunction and mortality. One of the key cellular processes associated with cardiomyopathy is cardiomyocyte death. Cardiomyocytes are terminally differentiated cells with very limited regenerative capacity. Various insults can lead to irreversible damage of cardiomyocytes, contributing to progression of cardiac dysfunction. Accumulating evidence indicates that majority of cardiomyocyte death is executed by regulating molecular pathways, including apoptosis, ferroptosis, autophagy, pyroptosis, and necroptosis. Importantly, these forms of regulated cell death (RCD) are cardinal features in the pathogenesis of various cardiomyopathies, including dilated cardiomyopathy, diabetic cardiomyopathy, sepsis-induced cardiomyopathy, and drug-induced cardiomyopathy. The relevance between abnormity of RCD with adverse outcome of cardiomyopathy has been unequivocally evident. Therefore, there is an urgent need to uncover the molecular and cellular mechanisms for RCD in order to better understand the pathogenesis of cardiomyopathies. In this review, we summarize the latest progress from studies on RCD pathways in cardiomyocytes in context of the pathogenesis of cardiomyopathies, with particular emphasis on apoptosis, necroptosis, ferroptosis, autophagy, and pyroptosis. We also elaborate the crosstalk among various forms of RCD in pathologically stressed myocardium and the prospects of therapeutic applications targeted to various cell death pathways.
Similar content being viewed by others

Introduction
Cardiomyopathies encompass broad spectrum of cardiac diseases, which are multifactorial in the underlying causes and highly heterogeneous in their symptomatic manifestations [1]. In general, cardiomyopathies can be divided into two categories, primary and secondary based on the origin of the disease. When the disease progresses into an overt heart failure, it is an intractable health burden to worldwide population and a leading cause of mortality [2]. Primary cardiomyopathies whose pathology is mainly confined to the heart can be caused by genetic factors, such as familiar Hypertrophic Cardiomyopathy (HCM), and Arrhythmogenic Right Ventricular Dysplasia Cardiomyopathy (ARVD/C). They can also be caused by systemic insults or injuries in acquired cardiomyopathies, such as peripartum cardiomyopathy. However, the genetic and acquired forms are not mutual exclusive, as in some cases of dilated and restrictive cardiomyopathies [3]. Heart muscle diseases caused by extra-cardiovascular pathologic factors are often categorized as secondary cardiomyopathies. Examples are diabetic cardiomyopathy (DMCM), sepsis-induced cardiomyopathy (SIC), and doxorubicin-induced cardiomyopathy (DOX). Currently, the main therapeutic strategy for cardiomyopathy focuses on symptomatic relief through pharmacological reagents and surgery [4, 5]. Even with much progress in genetic editing and gene-based therapies, genetic intervention as an effective and clinically available treatment for cardiomyopathies remains unfeasible currently [6,7,8]. There is an urgent need for new and therapeutic targets for the diseases.
Cell death is a ubiquitous and conserved feature in both physiological and pathological states [9, 10]. Controlled cell death orchestrated by precise molecular processes contributes to normal development and organism homeostasis. However, in pathological conditions, overwhelming physical, chemical, or mechanical stimuli can trigger adverse cell death that can lead to abnormal organ function [11,12,13,14,15]. In 1972, Kerr creatively used the term “apoptosis” to describe a programmed form of cell death that did not elicit immune activation [16]. Later, multiple forms of cell death have been uncovered, including apoptosis, necroptosis, autophagy, pyroptosis, ferroptosis as well as copper-induced cell death [17]. At first, cell death was classified according to three diverse morphologies [18]. In 2018, the cell death classification system was updated by the Committee on Cell Death Nomenclature (NCCD) to its current structure [19]. Based on functional differences, cell death can be divided into accidental cell death (ACD) and RCD. ACD is an uncontrolled process that is triggered by fatal stimuli that exceed the cell’s ability to adapt and survive. In contrast, RCD involves cascades of signaling events carried out by specific effector molecules, including various modes of apoptosis such as autophagy-dependent cell death, necroptosis, autophagy, pyroptosis, ferroptosis, and more [13] (Table 1).
It is clear now various forms of cell death have essential roles in numerous human diseases including cancer, nervous system disease, autoimmune diseases, cardiovascular disorders, and so on [20, 21]. In terms of cardiovascular disease, myocardial infarction, and myocardial ischemia-reperfusion injury have been widely studied, and inhibiting excessive apoptosis and ferroptosis are being pursued as potential therapeutic targets. Intriguingly, accumulating evidence has also indicated that multiple forms of cell death are significantly implicated in various cardiomyopathies, such as HCM, DMCM, and DOX [14]. Herein, we review the latest research progress on the significant roles of different forms of cell death in cardiomyopathy. By elaborating on the cellular mechanisms of RCD in the disease pathogenesis, we will discuss the possibility of cell-death targeted therapy for cardiomyopathies.
Apoptosis and cardiomyopathies
Apoptosis pathway
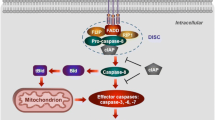
Apoptosis is the primary and most well-characterized form of RCD. Two established signaling pathways can launch the course of apoptosis: the extrinsic pathway (also known as the death receptor-mediated pathway) and the intrinsic pathway (also called the mitochondria-mediated pathway) [22] (Fig. 1a). The triggering event of the extrinsic pathway is the binding of death ligands to their canonical death receptors, and subsequent formation of death-inducing signaling complex, including complex I or complex II, which then leads to a cascade of reactions to activate procaspases [23]. The main death receptors are cell surface death receptor Fas and Tumor Necrosis Factor (TNF) receptor superfamily including TNFR1, TRAILR1, and TRAILR2 [19]. When TNFα binds to TNFR1, TNF receptor-associated death domain (TRADD) is recruited to form Complex I which generally includes TNF receptor-associated factor 2, receptor-interacting protein kinase 1 (RIPK1), and cellular inhibitor of apoptosis protein and the linear ubiquitin chain assembly complex (LUBAC). When RIPK1 is ubiquitinated by cIAPs, nuclear factor (NF)-κB signaling is subsequently activated which protects cells from apoptotic death. Alternatively, in the absence of cIAPs, activated RIPK1 associates with FADD and procaspase 8 (CASP8) to form Complex II. Subsequently, procaspase 8 is activated through autocatalytic cleavage, leading to the activation of the death effector caspase 3 and caspase 7 [24]. Similarly, when FasL and TRAIL are respectively engaged with their death receptors Fas and TRAILR1, FAS-associated death domain protein (FADD) and TRADD are recruited to assemble the complex containing procaspase 8 and cellular FLICE-like inhibitory protein (c-FLIP). As long as the pro-apoptotic caspase 8 surpasses the inhibition from anti-apoptotic c-FLIP, the downstream proteolytic cascade will be activated, resulting in apoptosis [25].

a Apoptosis pathway. The extrinsic pathway is initiated by the binding of death ligands to their canonical death receptors and subsequent formation of DISC, which then activates caspase 8 and the death effector caspases 3 and 7, leading to apoptosis. In the intrinsic pathway, numerous stimuli lead to permeabilization of the mitochondrial outer membrane (MOMP) and further leakage of proapoptotic proteins such as Cytochrome c. Afterward, released Cytochrome c binds Apaf-1 to form apoptosome which then activates caspase 9 and subsequently triggers the activation of the downstream effector caspase 3 and caspase 7, leading to apoptosis. BAK/BAX which can be activated by tBID promotes to the MOMP while BCL-2 inhibits its function. b Apoptosis in cardiomyopathy. In DMCM, accumulated AGEs promote apoptosis while Melatonin, Lin28a, miR675, or deficiency of Mst1 inhibit apoptosis. In DIC, CDK2-dependent FOXO1 phosphorylation, as well as, JNK and ERK phosphorylation promote apoptosis while activation of FNDC5 Wnt/PCP-JNK pathway or SESN2 inhibits apoptosis. In DCM, activation of TP53 pathway and BAG3 mutation promote apoptosis.
In contrast to receptor mediated extrinsic apoptotic pathway, the intrinsic apoptotic pathway can be trigged by numerous stimuli such as oxidative stress, DNA damage, calcium overload, and endoplasmic reticulum stress. It is characterized by mitochondrial outer membrane permeabilization (MOMP) [26], and dissipation of mitochondrial transmembrane potential (Δψm), leading to mitochondrial swelling and leakage of proapoptotic proteins such as cytochrome c and second mitochondria-derived activator of caspase (SMAC) into the cytosol. The released cytochrome c interacts with apoptosis protease activating factor-1 (APAF-1) to form a quaternary apoptosome complex which then recruits as well as activates caspase 9 and subsequently triggers the activation of the downstream effector caspase 3 and caspase 7 [23]. The protein family members of B cell lymphoma 2 (BCL-2) have pro-apoptosis and anti-apoptosis activities by regulating MOMP. BCL-2 family members include three subsets: the pro-apoptotic regulators (including BAK, BAX, and BOK), pro-apoptotic BH3-only proteins (including BID, BIM, BAD, BMF, NOXA, and PUMA) and the anti-apoptotic proteins (including BCL-2, BCL-W, BCL-XL, A1, and MCL1) [27]. Under physiological state, anti-apoptotic proteins interact with pro-apoptotic proteins so apoptosis is inhibited. Following stress stimulation, BH3-only proteins are induced by transcriptional or post-transcriptional mechanisms. Their inductions liberate BAX and BAK from inhibition, and lead to MOMP and apoptosis [28]. Of note, the interplay between extrinsic and intrinsic apoptotic pathways is also significant. In the extrinsic apoptotic pathway, activated caspase 8 can also trigger the cleavage of BID, which generates a truncated form of BID (tBID) to induce MOMP-mediated intrinsic apoptosis [27].
Apoptosis in cardiomyopathies
The existence of apoptosis in dilated cardiomyopathy has been demonstrated in clinical studies for more than 20 years [29, 30] (Fig. 1b). Dilated cardiomyopathy (DCM) is one of the most prevalent cardiomyopathies globally [31], characterized by dilation of ventricles and compromised systolic dysfunction. DCM harbors heterogenous etiologies, categorized into primary and secondary causes. The latter consists of immune response, exposure to alcohol, drugs, and toxins, as well as metabolic and endocrine disturbances [32,33,34]. DCM can be a result of inherited mutations affecting contraction-related genes or abnormal RNA splicing of titin due to the deletion of the corresponding RNA binding protein, RBM24 [35]. Currently, a large number of mutations related to DCM have been discovered, affecting genes including BAG3, LMNA, MYH6, and MYH7 [32]. Both biochemical and ultrastructural analyses from idiopathic dilated cardiomyopathy and ischemic cardiomyopathy hearts demonstrate the evidence of mitochondrial cytochrome c release and caspase-3 activation in diseased hearts [36]. As with the observation from Narula et al. [37], mice carrying DCM-related LMNA mutation develop DCM phenotype associated with TP53 activation, apoptosis, fibrosis, and contractile dysfunction [38]. A similar phenomenon is reported in titin-deficient and Orai3 deletion mice [39, 40]. Transgenic mice with cardiac-specific overexpression of an FKBP–caspase-8 fusion protein was normal at birth, but the administration of the bivalent dimer FK1012H2 led to activation of both caspase-8 and caspase-3, resulting in massive cardiomyocyte apoptosis and premature death, demonstrating myocyte apoptosis is sufficient to induce DCM. Low-grade apoptosis was also observed in the absence of dimer administration and lethal DCM developed at a later stage further supporting the casual role of apoptosis to DCM [41]. In another mouse model of heart-specific knockout of gp130, when applied to acute pressure overload via trans-aortic constriction (TAC) surgery, the mice developed significant cardiac apoptosis (≈34%), and most (>90%) died of DCM within a few weeks [42]. These early studies in mouse models suggest that apoptosis does indeed play an important role in cardiomyopathy. As a member of BAG anti-apoptotic family, BAG3 coupled with anti-apoptotic Bcl-2 to inhibit apoptosis [43]. Toro found that the mutation of BAG3 correlates to a more severe DCM phenotype, suggesting BAG3 may contribute to the initiation and progression of DCM [44]. Further studies in mice show the mutation of BAG3 increases apoptosis [45] while overexpression of BAG3 reduces cardiomyocyte apoptosis [46]. In addition, MSC-derived exosomes were found to alleviate DCM via reducing cardiomyocyte apoptosis and decreasing the inflammatory signaling [47]. Beyond DCM, apoptosis is also observed in other categories of cardiomyopathy such as diabetic cardiomyopathy, acromegalic cardiomyopathy, HCM, and ARVD [48,49,50,51].
DMCM is a well-documented secondary cardiomyopathy induced by diabetes mellitus (DM) [52, 53]. The pathological features include adverse cardiac remodeling such as hypertrophy and fibrosis. Early diastolic or systolic dysfunction can finally turn into overt heart failure. DMCM is the most serious diabetic complication and a significant risk factor for patient fatality. The etiologies of DMCM are multifactorial, involving hyperglycemia, inflammation, oxidative stress, mitochondrial dysfunction, and endoplasmic reticulum stress as well as cell death [54]. In DMCM, high glucose and disordered metabolism lead to advance glycation end products (AGEs) accumulation, which stimulates reactive oxygen species (ROS), impairs mitochondrial function and finally triggers both extrinsic and intrinsic apoptotic pathways [55]. Elevated apoptosis has been detected both in the hearts of diabetic patients and in animal models of diabetes [48, 56]. Kuethe et al. found that there were more apoptotic cardiomyocytes in diabetic hearts according to DM patients’ autopsy reports [57]. Zhang et al. explored the role of mammalian sterile 20-like kinase 1 (MST1) in DMCM. They found the deficiency of MST1 enhanced the interaction between anti-apoptotic protein Bcl-2 and with Bax to inhibit apoptosis [58]. They further demonstrated that melatonin alleviated DMCM via inhibiting MST1 phosphorylation, and the same mechanism was observed for Lin28a-mediated protection against DMCM [59, 60]. P53 is a well-characterized molecule central to apoptosis [61]. Administration of a p53-specific inhibitor pifithrin-α (PFT-α) in streptozotocin-induced diabetes model showed palliative effects on remodeling and apoptosis. What is more, inactivated p53 increases the stability of hypoxia-induced factor-1α which is necessary for angiogenesis [62]. A microarray carried out by Li et al. noted that lncRNA H19 whose exon1 also encoded miR675 was downregulated in diabetes models. miR675 restrained apoptosis by directly targeting voltage-dependent anion channel 1 (VDAC1), in important mediator of intrinsic apoptosis [63].
Intriguingly, activated caspase cascade under DOX treatment was also observed by Tadokoro et al. and Kalyanaraman et al. [64]. Since its discovery in the 1960s, DOX has become a mainstay in the treatment of malignant tumors, but its use has been limited by its main side effect, cardiotoxicity [65]. The administration of DOX gives rise to excessive accumulation of ROS, DNA damage, and mitochondrial dysfunction, which appear to be the major culprit for initiating apoptosis [66]. DOX treatment downregulates the level of SESN2 as well as the interaction with Parkin and p62, leading to mitochondria dysfunction and cardiomyocyte apoptosis [67]. Fibronectin type III domain-containing 5 (FNDC5) was reported to activate AKT/mTOR signaling and inhibited DOX-induced cardiomyocyte apoptosis [68]. The treatment of DOX led to CDK2-dependent phosphorylation of the transcription factor forkhead box O1 (FOXO1) at Ser-249, which also lead to induction of apoptosis [69]. Mitogen-activated protein kinase are important molecules in intracellular signaling [70]. Activation of p38 cascade induces apoptosis in cardiomyocytes via BAX, Bcl-2, and p53 [70]. Exposure of DOX to H9c2 rat myocytes increased the level of JNK and ERK phosphorylation, upregulated nuclear factor-κB (NF-κB), associated with enhanced cardiomyocyte apoptosis [68, 71].
In short, apoptotic cell death is a well-documented phenomenon in broad range of cardiomyopathy hearts and a significant contributor to adverse outcomes of the disease. One of the major challenges in the field is the complexity of the signaling network linking the highly conserved apoptotic execution machinery with different pathological conditions. Establishing the key molecules with specific impact for different etiology of cardiomyopathies will require more comprehensive and unbiased analysis in future.
Necroptosis and cardiomyopathies
Necroptosis pathway
The term “necroptosis” was first proposed by Degterev in 2005 to describe a form of programmed necrosis which differs from but also interplays with apoptosis [72]. Currently, necroptosis is characterized as a form of RCD when necrotic membrane rupture, release of cellular content and exposure to Damage-associated Molecular Patterns (DAMPs) are triggered in a caspase independent manner. Necroptosis can be triggered by ligand-receptor signaling, including TNF-α (Fig. 2a). Similar to its role in the extrinsic apoptosis, TNFR1 is activated by TNF-α and then TRADD, RIPK1, as well as other regulatory proteins are recruited to form complex I. RIPK1 is a key molecule for determine cell viability. Under normal physiological state when RIPK1 is predominately ubiquitinated by LUBAC and cIAPs, the canonical NF-κB pathway is activated by transforming growth factor-β-activated kinase 1 (TAK1) and phosphorylated IKK, and cells are protected from death [73]. In contrast, when RIPK1 is deubiquitinated by linear ubiquitin chain assembly complex (CYLD) and spermatogenesis-associated 2 (SPATA2), NF-κB signaling is restricted, and then Complex II consisting of RIPK1, TRADD, caspase 8 and FADD will be assembled to execute apoptosis or necroptosis. In the case of apoptosis, caspase 8 is activated and it cleaves both RIPK1 and RIPK3, leading to conventional apoptosis pathway [74]. Alternatively, when caspase 8 is inhibited, phosphorylated RIPK1 dimerizes and interacts with RIPK3 via RIP homotypic interaction motifs (RHIMs), and subsequently recruits and phosphorylates its substrate Mixed Lineage Kinase-Domain Like (MLKL) protein to form necrosome. Activated MLKL oligomerizes and inserts into plasma membrane to execute necroptosis [75]. In addition to MLKL, Ca2+/calmodulin-dependent protein kinase (CaMKII) is also reported as another substrate for RIP3. Phosphorylated CaMKII impacts mitochondrial permeability transition pore (mPTP) to regulate necroptosis as well as apoptosis [76, 77].

a Necroptosis pathway. TNF-α activates TNFR1 and then recruits regulatory proteins such as TRADD and RIPK1 to form complex I as well as subsequent Complex II. When caspase 8 is inhibited, phosphorylated RIPK1 and phosphorylated RIPK3 recruit as well as phosphorylate its substrate MLKL. Activated MLKL oligomerizes and inserts into plasma membrane to execute necroptosis, finally causing plasma membrane rupture and release of DAMPs and PAMPs. b Necroptosis in cardiomyopathy. In DMCM, abnormal splicing of CaMKII and SIRT3 as well as H2S deficiency promotes necroptosis. In DIC, TAK1 downregulation promotes necroptosis. In DCM, increased MLKL phosphorylation promotes necroptosis. In SIC, activation of PPAR-γ inhibits necroptosis.
Necroptosis in cardiomyopathies
By comparing the samples collected from myocardial infarction or DCM-induced end-stage heart failure with controls, Szobi et al. found that necroptosis-associated genes including RIPKs and phosphorylated MLKL were significantly upregulated [78]. Similarly, increased MLKL phosphorylation was observed in another report from clinical specimens and heart tissues of animal models of DCM [79] (Fig. 2b). In addition, decreased caspase-8 expression as well as the unchanged caspase-3 were observed in the same study [78], which implies that necroptosis rather than apoptosis was the main form of death involved in the progression of heart failure. TEAD1 is an important regulator in embryonic cardiac development. In animal studies, deficiency of TEAD1 led to lethality due to acute-onset DCM, while administration of necrostatin-1 (Nec-1) significantly alleviated cardiac dysfunction [80], supporting again the potential role of necroptosis in the pathogenesis of DCM.
In DMCM, overwhelming superoxide production is able to trigger the initiation of necroptosis. Pro-necroptosis molecules such as RIPKs are upregulated in H9c2 treated with high glucose (HG) as well as in mice injected with streptozotocin [81, 82]. Moreover, HG insult also leads to the abnormal splicing of CaMKII which further promotes necroptosis [83]. By treatment of inhibition for RIP1 autophosphorylation using Nec-1 or CaMKII activity using inhibitor 1 of protein phosphatase 1 (I1PP1) treatment can restrict necroptosis and protect against HG‑induced injury [81, 83]. Sirtuin 3 (SIRT3) and hydrogen sulfide (H2S) are potent antioxidants, however, their levels are reduced in streptozotocin-induced diabetic mouse hearts. SIRT3 and H2S deficiency increases ROS accumulation, worsens mitochondrial damage, and aggravates necroptosis [82, 84]. Oppositely, H2S supplementation remarkably alleviated cardiac injury [84].
Severe streptococcus pneumoniae infection could lead to lethal cardiac injury [85, 86]. Beno et al. uncovered that pneumolysin executed cardiomyocyte death by the process of necroptosis. Besides, inhibition of necroptosis via targeting key necroptosis-associated genes using MLKL and RIPK3 KO in mice showed prominent cardiac protection. As with directly blocking necroptosis by necrostatin-5 [87], activation of RIPK3 and MLKL was also identified in rats subjected to sepsis induction via cecal ligation and punctuation (CLP), and application of peroxisome proliferator-activated receptor- (PPAR-) γ agonist (rosiglitazone) significantly alleviated sepsis-induced cardiac dysfunction via activating NF-κB while downregulating RIPKs and MLKL [88].
DOX-induced ROS accumulation downregulated the expression of TAK1 and thus initiated apoptosis as well as necroptosis in H9c2. Administration of anti-oxidant phosphocreatine decreased the expression of cleaved-Caspase 3 and RIP3, and restrained DOX toxicity in cardiomyocytes [89]. However, another report showed that although RIPK3 knockout mice had lower mortality caused by DOX than wild-type littermates, no significant changes in DIC were observed [90]. It suggests that necroptosis may be more important in other organs such as the liver than the heart in the pathogenesis of DOX-induced damage. Reliable evidence for necroptosis in DIC is limited.
While some encouraging progress has been made in necroptosis research for cardiomyopathy, much remains to be learnt about its specific contribution at different stages of cardiomyopathy development. As more new players of necroptosis pathway being uncovered from non-cardiac system, more research will be needed to demonstrate their potential role in the onset and progression of cardiomyopathy.
Ferroptosis and cardiomyopathies
Ferroptosis pathway
In 2012, Dixon first reported an iron-dependent form of non-apoptotic cell death, formally proposing the term “Ferroptosis” [91]. In general, ferroptosis is a form of RCD that is characterized by iron-dependent lipid peroxidation and decompensated anti-oxidant system (Fig. 3a).

a Ferroptosis pathway. Both Iron-dependent lipid peroxidation and decompensated anti-oxidation system can launch the course of ferroptosis. Iron in circulation can be taken up in the form of Fe3+ and converted to Fe2+ inside of endosome. GPX4 is the major antioxidant defense and GSH is an indispensable cofactor for its activity. Cysteine utilization, after its conversion from cystine, is a major limiting factor for the GSH biosynthesis while cystine uptake mainly relies on System Xc- which imports extracellular cystine by exchanging intracellular glutamate. AA-PE is the main substrates of peroxidation, its synthesis needs the catalyzation by ACSL4 and LPCAT3. b Ferroptosis in cardiomyopathy. In DMCM, Nrf2 inactivation, IncRNA-ZFAS1 and AGEs accumulation promote ferroptosis. In DIC, PRMT4 and HMGB1 promote ferroptosis, while Acot1 inhibits ferroptosis. In SIC, Nrf2 activation and administrations of dexmedetomidine or Fer-1 inhibit ferroptosis.
Iron plays an essential role in almost all organisms. It participates in oxygen transport, energy metabolism as well as biosynthesis [92]. Iron in the systemic circulation is in the form of ferric ion (Fe3+) carried by transferrin protein, it can be taken up by cells through iron transporters, such as transferrin receptor 1 (TfR1). Once inside of endosome through receptor-mediated endocytosis, the Fe3+ is converted to ferrous ion (Fe2+) via ferri-reductase activity of STEAP3. By divalent metal transporter 1 (DMT1/SLC11A2), Fe2+ is then exported out of endosome into cytosol [93,94,95,96]. Under normal condition, iron is stored by iron-storage protein ferritin, consisting of FTL and FTH1 subunits [97]. Disruption of normal iron homeostasis can trigger overwhelming ROS, leading to lipid peroxidation, a hallmark of ferroptosis.
Lipids, predominantly phospholipids (PLs), form the basic structure of cellular membranes. Polyunsaturated fatty acids (PUFAs) especially phosphatidylethanolamines (PEs) containing arachidonic acid (AA) are the main substrates of peroxidation [98, 99]. The incorporation of PUFAs into cellular membrane requires both Acyl-CoA synthase long-chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3). ACSL4 acylates free AA to form AA-CoA, LPCAT3 catalyzes the generation of arachidonic acid-phosphatidylethanolamines (AA-PE). They are highly sensitive to peroxidation [100]. Finally, lipoxygenases (Lox), a dioxygenase that is activated by iron, drives the oxidation of PUFAs. Eventually, elevated lipid peroxidation leads to the destruction of lipid bilayer during ferroptosis [101, 102].
The best characterized antioxidant defense is glutathione peroxidase 4 (GPX4). GPX4 is a glutathione (GSH)-dependent enzyme that converts lipid hydroperoxides to non-toxic lipid alcohols, thus blocking the onset of ferroptosis [91]. GSH is an indispensable cofactor for GPX4 activity. GSH is synthesized from three amino acids–cysteine, glycine, and glutamate. Cysteine utilization, after its conversion from cystine, is a major limiting factor for the GSH biosynthesis process [103]. Cystine uptake by mammalian cells mainly relies on a plasma membrane cystine/glutamate reverse transporter, System Xc-. System Xc-, consists of two subunits, a specific light chain subunit encoded by solute carrier family 7 member 11 (SLC7A11, also called xCT) and a heavy chain subunit encoded by SLC3A2 (also called 4F2) [104]. Numerous factors regulate SLC7A11 expression, such as TP53, NRF2, and BECN1, which then influence GPX4 activity. Direct (e.g., via RSL3) or indirect (e.g., System Xc-) inhibition of GPX4 will compromise cellular anti-oxidant system and trigger ferroptosis. Another intracellular guardian for ferroptosis is ferroptosis suppressor protein 1 (FSP1). Using NAD(P)H as substrate, FSP1 catalyzes the regeneration of coenzyme Q (CoQ10), which serves as a reversible reducing agent to protect cells from lipid peroxidation [105, 106]. Finally, emerging evidence indicates that ferroptosis may also interact with autophagy as in the form of ferroptosis nuclear receptor coactivator 4 (NCOA4)-dependent ferritinophagy and lipophagy [107, 108].
Ferroptosis in cardiomyopathies
Considering that excess accumulation of iron is a direct pathological feature of iron overload cardiomyopathy [109], ferroptosis should be a potential candidate mechanism. Conditional FTH knockout mice manifest increased oxidative stress and decreased GSH levels while overexpressing Slc7a11 is able to restore the anti-oxidant system and alleviate cardiac injury [110]. Sickle cell disease upregulates heme oxygenase 1 (Hmox1) and unresolved free iron overload eventually inducing cardiac cardiomyopathy via ferroptosis [111]. Cardiac L-type voltage-dependent Ca2+ channels are key transporters of iron into cardiomyocytes under iron-overloaded conditions [112], there is some clinic evidence for benefit from the usage of calcium channel blockers [113].
DOX impairs mitochondrial function and elevates the level of ROS which is tightly associated with ferroptosis. In a mouse model of DIC, Fang et al. indicated that treatment with DOX intensified the accumulation of iron as well as lipid peroxidation of the membranes. Besides, compared with other cell death inhibitors such as 3-MA, emricasan, and Nec-1, ferroptosis inhibitor ferrostatin-1 (Fer-1) prominently improved the animal survival rate (Fig. 3b). Further RNA-sequencing found that activated Nrf2/Hmox1 pathway was the major mechanism of DOX-induced cardiac ferroptosis. Up-regulated Hmox1 catalyzes heme degradation and thus promotes the release of free iron, leading to excessive iron accumulation [90]. This finding is in line with the report from Menon [111]. However, although Nrf2 leads to the upregulation of Hmox1, it functions in a more protective role in ferroptosis. Suppression of Nrf2 decreases the expression of GPX4 and thus promotes ferroptosis [114]. On the other hand, transcriptome profiling performed by Liu et al. identifies Acyl-CoA thioesterase 1 which functions opposite to ACSL4 is most significantly downregulated in DOX-treated heart and may serve as a possible mechanism in ferroptosis induction [115]. In addition, Fang also noted that Fe2+ chelator Dexrazoxane (DXZ) reduces cardiac nonheme iron thus suppressing DOX-induced lipid peroxidation, which is in good agreement with Tadokoro’s report [64]. They showed that DOX treatment markedly aggravates Fe2+ overload and downregulation of GPX4, especially in mitochondria. Administration of DXZ or overexpression GPX4 conferred significant protection against DOX-induced ferroptosis. Furthermore, DXZ could also affect high mobility group box 1 (HMGB1) to restrain ferroptosis both in vitro and in vivo [116].
Clear evidence for ferroptosis in primary cardiomyopathy such as DCM is limited. L-type Ca2+ channels (LTCC) and T-type Ca2+ channels engage in the uptake of cardiac iron. In iron-overloaded myocytes, the transferrin-dependent iron uptake pathway is inhibited and an alternative mechanism involving transferrin-independent pathway (non-transferrin-bound iron, NTBI) is activated [117]. In HCM, LTCC was increased and CaMKII was activated, inducing elevated intracellular Ca2+ and diastolic intracellular calcium ([Ca2+]) concentrations, initiating the arrhythmogenic delayed after-depolarizations which may contribute to sudden cardiac death [118]. However, whether ferroptosis is directly implicated in the pathogenesis of the disease remains to be better demonstrated.
In terms of DMCM, excessive AGEs exposure leads to downregulation of SLC7A11 and lower GSH levels, triggering cardiomyocyte ferroptosis [119]. A case-control study concludes that patients with T2DM have a lower level of GSH), which may compromise GPX4 activity and sensitize cells to peroxidation [120]. Intriguingly, mouse models of DMCM also manifest higher levels of lipid peroxidation and ferroptosis marker PTGS2, which may be a result of impaired anti-oxidant system due to downregulated SLC7A11 and GPX4 [119, 121], overexpression of GPX4 can indeed ameliorate diabetic heart injury [122].
Extensive research has led to the recognition that DAMPs/PAMPs, storm of inflammatory cytokines, impaired calcium handling as well as mitochondrial disruption are the potential mechanisms underlying sepsis induced cardiac dysfunction [123, 124]. Ferroptosis was activated when mice were injected with lipopolysaccharide (LPS) or subjected to CLP based on increasing levels of MDA and PTGS2 as well as elevated iron deposition [125, 126]. Studies from Li et al. and Zheng et al. both showed that administration of Fer-1 improved cardiac function, alleviated lipid peroxidation and reduced inflammation in sepsis-induced cardiomyopathy [127]. Similar protection was reported from restoring impaired anti-oxidant system including overexpression of GPX4 and Nrf2 [126, 128]. Of note, autophagy may interact with ferroptosis under sepsis induction. LPS stimulation upregulates NCOA4, which degrades ferritin by mediating ferritinophagy, thus releasing excess Fe2+ in H9c2. Despite of these interesting findings, further research is necessary to reveal the underlying mechanism and pathophysiological role of ferroptosis in different forms of cardiomyopathies.
Autophagy death and cardiomyopathies
Autophagic death pathway
In 1963, Christian De Duve first coined the term “Autophagy” based on his discovery of lysosomal-dependent degradation in cells. It is until 1993, when Tsukada first uncovered part of the regulatory mechanisms mediated by autophagy-related genes (ATGs) [129]. These pioneer work lays the foundation of current research on autophagy. Autophagy can be activated in response to a variety of stresses including protein misfolding, organelle damage, nutrient energy deficiency, and pathogen invasion. While the primordial role of autophagy is to deliver cargo into lysosomes for degradation and recycling in order to maintain cell homeostasis, unbridled or excessive autophagy is proved to associate with or mediate cell death [130], according to the Nomenclature Committee of Cell Death statement, autophagy-dependent cell death is defined as a type of RCD that has a strict requirement of autophagic machinery or components thereof [13]. According to different types of delivery mechanisms, autophagy can be classified into three subtypes: (1) macro-autophagy (hereafter referred to as autophagy) is the canonical form involving the formation of double-membrane vesicles, called autophagosomes, for wrapping and delivering cargo into lysosomes; (2) micro-autophagy, in which lysosomes directly engulf cargo via membrane invagination; (3) chaperone-mediated autophagy where proteins with a KefrQ-like motif are directly transported into lysosomes via lysosome-associated membrane protein 2A (lamP-2A) with the help of a chaperone protein HSP70 [131].
The canonical autophagy is initiated by the formation of new membranes at specific sites, also called phagophores when the mammalian target of rapamycin complex 1 (mTORC1) activity is suppressed and the mammalian uncoordinated-51-like protein kinase 1 (ULK1) complex is activated (Fig. 4a). ULK1/2 in complex with ATG101, ATG13, and FIP200 activates the downstream class III phosphoinositide 3 kinase (PI3K) which includes a catalytic core encoded by vacuolar protein sorting 34 (VPS34) and regulatory subunits including VPS15, ATG14, and BECN1. The class III PI3K complex activities were positively regulated by UVRAG and AMBRA1 but were negatively regulated by Bcl-2 and RUBCN during phagophore nucleation [132]. Two distinct ubiquitin-like conjugation systems participate in the elongation of the phagophore: one involves ATG5-ATG12-ATG16L. Through the catalyzation of ATG7 and ATG10, ATG12 covalently binds to ATG5 and then interacts with ATG16L. Phosphatidylinositol-3-phosphate produced from class III PI3K recruits downstream WD-repeat PtdIns(3)P effector proteins (WIPI) at phagophore nucleation site to interact with ATG16L1, leading to phagophore elongation [133]. Another autophagophore elongation system involves the light chain 3-phosphatidylethanolamine. The formation of lipidated form of LC3-II is sequentially catalyzed by ATG4, ATG7, and ATG3 to conjugate LC3 with PE. The autophagosomes then fuse with lysosomes with the help of SNARE protein syntaxin 17 and tethering factors, to form autophagolysosomes where cargoes are degraded [134, 135].

a Autophagy pathway. The canonical autophagy is initiated when the mTORC1 activity is suppressed and ULK1 complex is activated, which subsequently activate Class III PI3K Complex as well as lead to the formation of phagophores. Two distinct ubiquitin-like conjugation systems participate in the elongation of the phagophores: one involves ATG5-ATG12-ATG16L and another involves the LC3-PE. Afterwards, autophagosomes fuse with lysosomes to form autophagolysosomes where cargoes are degraded. b Autophagy in cardiomyopathy. In DMCM, AMPK, DCRF and Heme oxygenase-1 promote autophagy. In DIC, TFEB and prior starvation promote autophagy while GATA4 inhibits autophagy. In DCM, mutation of LMNA or PLEKHM2 inhibits autophagy. In HCM, TSC1 promotes autophagy while Mybpc3-targeted knock-in or loss of Vps34 inhibits autophagy. In SIC, Beclin-1 and miR-22 knock-out promote autophagy while ALDH2 inhibits autophagy.
Autophagic death in cardiomyopathies
Autophagy is compromised in the heart under various pathological states [136]. Although abnormal autophagy is clearly observed in clinical specimens such as HCM and DCM, emerging results from experimental models yielded opposite conclusions about its role in pathogenesis versus cardio-protection. HCM is a common primary cardiomyopathy that affects nearly 0.2% of the population [137]. Ultrastructural analysis of HCM patient tissue uncovered the presence of typical autophagic vacuoles. miR-451 was found to be the most down-regulated miRNAs in HCM heart. Tuberous sclerosis complex 1 (TSC1) is a direct target of miR-451 and a known positive regulator of autophagy. miR-451 is shown to regulate cardiac autophagy and hypertrophy via modulating TSC1 expression [138] (Fig. 4b). HCM’s main phenotypic expression is left ventricular hypertrophy and impaired compliance while its clinical symptoms are highly variable [139]. Mutations in sarcomeric genes for example, cardiac β-myosin heavy chain (MYH7) and myosin-binding protein C (MYBPC), are major pathogenic mechanism [140, 141]. Besides, non-sarcomeric gene mutations such as ACTN2 and ALPK3 are also reported for HCM [142, 143]. Singh et al. found that autophagy was activated in the HCM patients with MYBPC3 mutation and in Mybpc-targeted knock-in mouse hearts, which may be a therapeutic target to rescue HCM induced by MYBPC3 mutations [144]. In contrast, Vps34 was reported to be downregulated in human HCM as demonstrated by Hirotaka Kimura et al. and cVps34–/– mice manifested an HCM-like phenotype as well as sudden death. cVps34 inactivation led to aggregation of K63-linked polyubiquitinated proteins and damage to autophagy flux [145]. Similarly, restoring autophagy by blocking mTORC1 activity improved cardiac function in lamin A/C gene (LMNA) mutation-induced cardiomyopathy [146]. Muhammad et al. elaborated that PLEKHM2 mutation inhibited autophagy flux by perturbing lysosomal function in recessive DCM [147]. Intriguingly, Tsunenori Saito et al. reported that autophagic vacuoles were present in cardiomyocytes of DCM patients. However, more autophagic vacuoles seemed to be correlated with less myocardial degeneration and better clinical outcome [148]. Therefore, both activation and inhibition of autophagy flux have been implicated in HCM and DCM, raising questions about the importance of tightly regulated autophagy activity in cardiac physiology.
Autophagy is a relatively new research target for DIC in recent years. Whether DOX activates or inhibits, cardiomyocyte autophagy remains also controversial. Multiple studies reported a decrease [149, 150] or an increase [151, 152] of autophagy activity in DIC. Based on report by Li et al., DOX treatment attenuates cardiomyocyte autophagic flux and promotes the accumulation of undegraded autolysosomes both in vivo and in vitro. It depends not on the activity of mTORC1 but on the V-ATPase-Driven Lysosome Acidification. Conversely, lowering autophagy initiation in Beclin-1+/– mice resulted in reduced ROS accumulation and better protection against DOX-induced cardiotoxicity [153]. Kobayashi S et al. investigated the ability of transcription factor GATA4 in cultured rat ventricular myocytes treated with DOX and found that it eased DOX damage by significantly upregulating Bcl-2 as well as inhibiting ATGs [152]. The cardiac benefit from Caloric Restriction (CR) partly lies in the limitation of detrimental autophagy and preservation of ATP content in DOX-induced cardiomyocyte death [154]. On the contrary, however, other reports consider autophagy as a beneficial process in DOX-induced cardiotoxicity. Bartlett et al. noted that transcription factor EB (TFEB) was repressed after DOX insult. Notably, it is correlative to autophagy inhibition and more severe cardiac injury both in vivo and in vitro [155]. More troubling to the field, the autophagic response seems to be highly species-specific. While an increase in autophagy was observed in rat hearts in vivo and in vitro, opposite results of a decrease was observed in mice under DOX treatment [150].
In DMCM, the homeostasis of autophagy is frequently compromised. The autophagy status in cardiomyocytes varies in different types of diabetic mellitus. It is enhanced in heart from type 1 DM but suppressed in type 2 DM [156]. Emerging evidence showed that several transcriptional factors and molecules are involved in the changes of autophagy in DMCM. Activation of AMPK facilitates autophagy by dissociating the interaction between Beclin1 and Bcl-2 and protect against cardiac cell apoptosis in H9c2 treated with high glucose [157]. Feng et al. reported that a lncRNA termed DCM-related factor (DCRF) was upregulated in streptozotocin-induced diabetes model. DCRF serves as a ceRNA to sponge miR-551b-5p, and it promotes autophagy to alleviate high glucose insult by increasing the expression of PCDH17 [158]. Furthermore, enhancement of autophagy by overexpression of Heme oxygenase-1 (HO-1) or activating AMPK signaling could protect against DMCM [159]. Consistently, adiponectin deficiency increases the level of p62 which inhibits autophagy and aggravates cardiac dysfunction associated with obesity and diabetes [160]. Sciarretta et al. also reported that the HFD mouse model exhibited inactivation of Rheb which was salutary because it downregulated Atg7 and blocked autophagy [161]. However, diabetic cardiac dysfunction was shown to be attenuated in Atg16-deficient and beclin 1-deficient mice. Oppositely, overexpression of beclin 1 facilitated autophagic flux but then aggravated oxidative stress, fibrosis, and cardiac dysfunction [162]. There is much crosstalk among distinct RCD pathways. In DMCM, the activity of AMPK was reduced and the m-TOR signaling was activated, which led to a decrease in cardiomyocyte autophagy. In turn, the reduced autophagy caused more accumulation of damaged mitochondria and polyubiquitinated proteins, which resulted in a further increase of ROS production and cytochrome c release, all leading to an exacerbated apoptosis [163].
Autophagy is also activated and is protective in the sepsis model [164, 165]. Mitochondrial aldehyde dehydrogenase (ALDH2) functions as a safeguard in heart by blocking LPS-induced CAMKKβ-AMPK-mTOR-signaling axis and suppressing uncontrolled autophagy [166]. Lately, more detailed research was performed by Sun et al., which made an interesting observation of dose-dependent bidirectional modulation of autophagic flux by LPS in the heart. At low dose, LPS moderately promoted autophagy while a high dose remarkably inhibited it in cardiomyocytes. In addition, overexpression of Beclin-1 promoted autophagy and mitophagy, preserved mitochondria, improved cardiac function, and alleviated inflammation and fibrosis [167]. Finally, miR-22 inactivation showed cardio-protection against sepsis by enhancing autophagy and reducing apoptosis through sirt1 activation [168].
Despite of a significant volume of research being devoted to autophagic cell death in cardiomyopathies, the overall picture remains controversial and unclear. Considering the highly dynamic nature of autophagy flux in both programmed cell death and normal cellular homeostasis, it is not surprising that it is implicated as both cardioprotective and detrimental in heart. The challenge for future research is to dissect the molecular basis that differentiate the good vs. bad outcomes of autophagy activity in stressed myocardium, including the activation profile, the flux level and the cargo specificity of autophagy under specific cardiomyopathies.
Pyroptosis and cardiomyopathies
Pyroptosis pathway
The term “pyroptosis” was firstly proposed by D’Souza in 2001 to describe a highly inflammatory form of RCD [169]. In the canonical form, pyroptosis is mediated by inflammasome-activated caspase [170] and gasdermin protein family gasdermin D (GSDMD) as a final effector [171], resulting in pore formation in the plasma membrane and leakage of inflammatory mediators (Fig. 5a). The key process is the assembly of the so called inflammasome complex consisting of sensors (e.g., nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) family, absent in melanoma 2 (AIM2)-like receptor (ALR) family), adaptors such as apoptosis-associated speck-like protein containing CARD (ASC) and effector pro-caspase-1, to cleave GSDMD. Inflammasome sensors are recruited to pattern recognition receptors (PRRs) upon exposure to PAMPs and DAMPs [172]. Following that the inflammasome sensor recruits adapter ASC by homotypic interactions, which functions as a link between the sensor and the effector pro-caspase 1. Subsequently, activated caspase 1 mediates GSDMD cleavage and IL-1β/IL-18 maturation [173]. The truncated N-terminal fragment of GSDMD (N-GSDMD) is the final executioner of pyroptosis by forming pores in plasma membrane and releasing intracellular contents [174].

a Pyroptosis pathway. In the canonical form, various insults lead to the assembly of the inflammasome complex consisting of sensors, adaptors, and effector, which then activates caspase 1. Activated caspase 1 mediates the pro IL-1β/IL-18 maturation and GSDMD cleavage. The truncated N-GSDMD is the final executioner of pyroptosis by forming pores in plasma membrane and releasing intracellular contents. In noncanonical inflammasome pathway, LPS directly actives pro-caspase 4/5/11, which further cleaves GSDMD to generate N-GSDMD. b Pyroptosis in cardiomyopathy. In DMCM, miR-30d, KCNQ1OT1 and CACR promote pyroptosis. In DIC, Dox directly binds to GSDMD or upregulated NOX1 and NOX4 promote pyroptosis. In DCM, activated NLRP3 inflammasome promotes pyroptosis. In SIC, STING and SOX9 promote pyroptosis while irisin or JQ1 inhibit pyroptosis.
The noncanonical inflammasome pathway is also called the caspase‐1–independent pathway. LPS derived from Gram-negative bacteria directly actives pro-caspase 4/5/11 without the help of inflammasome or caspase‐1. Activated caspase 4/5/11 can then cleave GSDMD to generate N-GSDMD. Meanwhile, N-GSDMD indirectly promotes NLRP3 inflammasome assembly through efflux of K+ that further aggravates pyroptosis [175, 176]. Further complicating the process, a report by Sarhan lab has revealed that, protein yopj derived from Yersinia binds to TAK1 and activates caspase 8 which can also cleave GSDMD to induce pore formation [177]. In addition, chemotherapeutic-activated caspase 3 can also cleave GSDME (similar to GSDMD) at the conserved site Asp270, to generate N-GSDME as another path leading to pyroptosis [178]. These findings indicate that there may exist more independent signaling pathways in the induction of pyroptosis.
Pyroptosis in cardiomyopathies
NLRP3 inflammasomes are activated and pyroptosis are present in heart tissues of non-ischemic DCM patients [179] (Fig. 5b). Similarly, through targeted analysis of gene expression from public datasets, Wang et al. identified pyroptosis related genes are altered in DCM associated with microenvironment of immune modulation [180]. Other evidence suggests that the onset and progression of DMCM are accompanied by pyroptosis. In DMCM, AGEs accumulation is inflammatory and leads to augmented expression of key players of pyroptosis such as NLRP3 and caspase 1. Jeyabal et al. noted that caspase-1 expression was increased both in human diabetic hearts and in human ventricular cardiomyocytes treated with high glucose [181]. Luo et al. established a type 2 diabetic rat model via streptozotocin injection and a high-fat diet, then found that pyroptosis-related key molecules such as NLRP3, ASC, and caspase-1 were significantly activated. They further demonstrated that inhibition of NLRP3 prominently attenuated cardiac dysfunction in a rat model and blocked pyroptosis in H9c2 cells treated with high glucose [182]. Another independent study also showed that inhibition of NLRP3 expression ameliorated experimental diabetic cardiomyopathy [183]. Sodium-glucose cotransporter 1 (SGLT1) inhibitor sotagliflozin reduced the overall risk of cardiovascular death [184]. Its cardioprotective effect was investigated by Sun et al., who showed that SGLT1 knockdown attenuated the induction of NLRP3, ASC, and caspase-1 and subsequently inhibited the release of inflammatory factors in a form of DMCM heart [185]. Meanwhile, Li et al. reported that upregulated miR-30d in DMCM heart was involved in the induction of caspase-1 dependent cardiomyocyte pyroptosis by targeted inhibition of Forkhead box O3 [186]. Similarly several other studies have also implicated different non-coding RNAs with a role in DMCM, including s lncRNA MIAT [187], lncRNA Kcnq1ot1 [188], caspase-1-associated circRNA (CACR) [189] and miR-223-3p [190].
There has been limited evidence for pyroptosis in DIC. GSDMD, is a key molecule in pyroptosis. Ye et al. found GSDMD knockout mice had attenuated DIC [191]. Furthermore, they found that DOX interacted with GSDMD and led to GSDMD-N-mediated pyroptosis. DOX administration upregulates the expressions of NOX1 and NOX4. Upregulated NOX1 and NOX4 activate Drp1 and promote mitochondrial fission, which leads to excessive ROS accumulation in mitochondria and finally activates NLRP3 inflammasome and caspase 1 dependent pyroptosis [179]. The DOX treatment increased the expression of pyroptotic markers (caspase-1, IL1-beta, and IL-18), and the resulting pyroptosis and cardiac remodeling can be ameliorated by embryonic stem cell-derived exosomes (ES-Exos) or ES-cells (ESCs) [192].
GSDMD-NT expression is increased in the sepsis model according to Dai et al. Correspondingly, Gsdmd-knockout (Gsdmd-/-) mice showed improved survival rate and ameliorated cardiac dysfunction in response to septic injury. Further transcriptome analysis revealed that deficiency of GSDMD significantly reduced the expression of NLRP3 and caspase-1 [193]. In the meantime, elevated expression of NLRP3 and Caspase 1 in the sepsis model was also reported by Xiong et al. In CLP mouse, transcription factor sex-determining region Y (SRY)-box 9 (SOX9) is upregulated because of deubiquitination from ubiquitin-specific peptidase 7 (USP7), upregulated SOX9 activates NLRP3 by reducing miR-96-5p expression [194]. What’s more, LPS directly affected the nuclear location of STING and IRF3, which then activated STING. Activated STING then activates NLRP3, leading to cardiac dysfunction as well as pyroptosis [195]. Administration of irisin alleviated sepsis-induced cardiac pyroptosis [196]. In LPS-induced cardiac dysfunction, JQ1 pre-treatment improved cardiac function, upregulated SIRT1, and inhibited the release of NLRP3, caspase-1, and GSDMD, JQ1 improved LPS-induced cardiac insufficiency by suppressing SIRT1-dependent activation of NLRP3 inflammasomes [197]. Li et al. examined the role of stimulator of interferon genes (STING) and interferons (IFN) regulatory factor 3 (IRF3) in LPS-treated mice. They found that LPS affected the nuclear location of STING and IRF3, which then activated STING. Activated STING then activates NLRP3, leading to cardiac dysfunction, apoptosis as well as pyroptosis [195]. Overall, pyroptosis appears to be induced in most forms of cardiomyopathies and blocking pyroptosis through direct or indirect approaches by targeting pyroptotic machinery or upstream regulators seems to yield protective effects. It would be important. Considering the close link between pyroptosis and inflammation, more research would be needed to investigate the potential role of pyroptotic pathway in inflammatory heart diseases, including myocarditis and cardiomyopathy associated with viral infection.
Crosstalk network among different RCDs
Despite that each of the RCD pathways has been well elucidated with unique molecular characteristics, different forms of RCD do share common pathways and molecular features, and interact extensively in an intricate crosstalk network in cells.
Extensive interaction between apoptosis and necroptosis exists involving common key regulatory molecules such as TNFR1 and caspase 8. What is more, CaMKII which is reported as another substrate for RIP3 in addition to MLKL can be phosphorylated and then impacts on mPTP to regulate necroptosis as well as apoptosis [76, 77]. In addition, the energetic state of cells (in a form of ratio between ATP and ADP) seems to influence the final mode of cell death as well. High ATP level leads to apoptosis rather than necrosis [198]. Similarly, energy sensor AMPK is molecular master to regulate apoptosis and autophagy. According to Song’ s study, multidrug-induced cell death in colon cancer cells is associated with mitochondrial dysfunction and AMPK activation. At early time point of AMPK activation, autophagy is induced while persistent AMPK activation leads to Beclin-1 phosphorylation and subsequent cleavage by caspase-8, which finally induces apoptosis [199]. The key mediator of necroptosis RIPK1 is also involved in the modulation of autophagic signaling [200], the same study indicates that autophagy also influences necroptosis, and indeed impaired autophagic flux promotes the activation of RIPK1 and MLKL [201]. Another study also indicates that moderate autophagy activation recruits necrosome components to modulate apoptosis vs. necroptosis [202], suggesting that the effects among these forms of RCD are multi-directional.
Autophagy interplays with ferroptosis mainly in the form of NCOA4-dependent ferritinophagy and lipophagy [107, 108]. Ferritinophagy which is mediated by ferritin autophagic cargo-receptor NCOA4 leads to the excessive release of iron from ferritin and eventual onset of Fenton reaction. Lipophagy which is mediated by lipid droplets cargo receptor RAB7A results in the lipid degradation and lipid peroxidation [203]. Consistently, administration of lysosome inhibitors to restrict autophagy showed better ferroptosis resistance and lower ROS accumulation [204]. In the meantime, other research has indicated that lysosomal activity may be impaired by ferroptosis as well [205]. Although it has clearly been proved that caspase activation does not take part in ferroptosis, treatment with erastin significantly aggravates apoptotic agent-induced apoptosis by BAX-associated pathway. This study provides potential evidence of crosstalk between apoptosis and ferroptosis [206].
Finally, emerging evidence indicates that pyroptosis may also interact with apoptosis. Chemotherapeutic-activated caspase 3 can cleave GSDME (similar to GSDMD) at the conserved site Asp270, to generate N-GSDME as another path leading to pyroptosis [178]. Another study indicates that GSDME rather than GSDMD mediates pyroptosis downstream of the ROS/JNK/Bax apoptotic pathway as well as caspase-3/9 activation [207]. These findings suggest that caspase 3/GSDME signaling might function as a switch between pyroptosis and apoptosis.
Although interdependent interactions among different RCD pathways are clearly demonstrated in cells, most of these cross-talks are characterized in non-cardiac settings. Considering the extensive heterogeneity in etiologies and disease manifestations among different forms of cardiomyopathies, the major challenge is to establish how each RCD and their potential interactions can contribute to the onset as well as the progression of the disease. This is a key question needs to be addressed in future studies.
Therapeutic potential targeted to intervene regulated cell death
Currently, the main therapeutic approach for cardiomyopathies focuses on standard therapy including neural hormonal blockade such as Beta blockers and angiotensin-converting enzyme inhibitors (ACEI), and, statins. However, the remedy is far from satisfaction and cardiomyopathy remains a critical unmet clinical need. Considering the significant contribution of RCD to the onset and progression of cardiomyopathies, and various cell death signaling pathways have been identified, it seems to highly rationale that synthetic or natural products directly or indirectly impacting the key regulatory molecules in cell death pathway may serve as a potential effective therapy for the disease (Fig. 6). Several encouraging examples have been demonstrated in preclinical studies. RIP1 autophosphorylation inhibitor Nec-1 blocks the process of necroptosis and shows a potent cardiac protection against DCM and DIC. Similar efficacy has been demonstrated for Fer-1 as an established inhibitor for ferroptosis [208]. BAX activation inhibitor 1 (BAI1) is another example that alleviates cardiomyocyte necrosis as well as apoptosis in the setting of DIC [209]. Fe2+ chelator DXZ reduces cardiac nonheme iron thus suppresses DOX-induced lipid peroxidation [90]. Administration of DXZ restores the expression of GPX4 and FTH1 which is downregulated in DIC [116]. Finally, Rapamycin inactivates mTOR and restores autophagy in SIC to remit cardiac dysfunction [210]. However, regulated cell death also has physiological role in homeostasis, particularly in wound healing and anti-infection. More studies are needed to assess the long-term impact of these treatments on other organs and systemic health, as well as the ability to cope with potential external injuries and infections.

a Standard therapy including classic drugs is far from satisfaction. b ncRNAs have emerged as potential therapeutic targets in broad range of cardiomyopathy hearts: miR-21-3p antisense inhibitor alleviates inappropriate autophagy in SIC. In DCM, cardiac protection was achieved by inhibition of the ncRNA ZFAS1, miR-30d, or overexpression of circHIPK3, which respectively attenuates ferroptosis, pyroptosis and apoptosis. c Directly impact the key regulatory molecules in cell death pathway may serve as a potential effective therapy for the disease: Nec-1 blocks the process of necroptosis against DCM and DIC; BAI1 alleviates cardiomyocyte necrosis as well as apoptosis while Fer-1 and DXZ suppress ferroptosis in the setting of DIC; Rapamycin restores autophagy in SIC to remit cardiac dysfunction. d Indirectly intervening RCD by aiming at upstream signaling may also prove to be effective: Natural products such as Flavonoids, Spermine, and Luteolin eliminate excess ROS accumulation; Sirt3 and MitoTEMPO protect mitochondrial homeostasis.
Indirectly intervening RCD by aiming at upstream signaling may also prove to be effective. Natural products such as Flavonoids, Spermine, and Luteolin reduce cardiomyocyte loss by eliminating excess ROS accumulation [211,212,213]. Mitochondrial disruption plays an irreplaceable role in various cardiomyopathies. Visnagin restores mitochondrial function and lightens DIC [214, 215]. Sirtuin-3 (Sirt3) and MitoTEMPO protect mitochondrial homeostasis by respectively promoting Parkin-mediated mitophagy and scavenging lipid peroxidation [90, 216]. Similar cardiac protection is also exhibited in reactivation of the p53 pathway and AMPK pathway through natural products, such as sulforaphane, melatonin, and resveratrol [119, 217, 218].
Recently, non‐coding RNAs (ncRNAs) have emerged as potential therapeutic targets because of their underlying functions in human diseases [219]. ncRNAs including microRNA (miRNA), long ncRNA (ln‐cRNA), and circular RNA (circRNA) have been widely studied in the context of different forms of cardiomyopathies. Human miR-29a could be one of the biomarkers in HCM [220]. Upregulated miR-21-3p aggravates SIC while its antisense inhibitor protects cardiomyocytes by inhibiting inappropriate autophagy [221]. Inhibition of the lncRNA ZFAS1 attenuates ferroptosis, repression of miR-30d manifests alleviation of pyroptosis while overexpression of circHIPK3 suppresses apoptosis in DCM [121, 186, 222]. ncRNA targeted therapy may open a new dimension to modulate RCD program against pathological progression of cardiomyopathies.
Conclusion and perspective
Globally, heart disease remains the leading cause of death. Both primary and secondary cardiomyopathy greatly increase the risk of malignant cardiac dysfunction and worsen patients’ prognosis. Current literature clearly indicate that regulated cell death plays a significant role in cardiomyopathy such as DCM, DMCM, SIC, DIC, etc. In summary, apoptosis is the most well-documented form of RCD in the pathogenesis of cardiomyopathies associated with inherited mutations, metabolic disorder and drug toxicity [38,39,40]. Necroptosis, however, differs from but also interplays with apoptosis in the setting of DCM [79], and DMCM associated with ROS accumulation, antioxidants deficiency or abnormal splicing of CaMKII [81,82,83]. Furthermore, PPAR-γ is inhibited in SIC and TAK1 is downregulated in DIC, which also lead to cardiomyocyte necroptosis and subsequent cardiac dysfunction [88, 89]. Both iron-dependent lipid peroxidation and decompensated anti-oxidation system can launch the course of ferroptosis. Inhibition of Nrf2 signaling and downregulation of GPX4 are cardinal features in the pathogenesis of DIC, DMCM and SIC [114, 119, 128]. Moderate autophagy initiated by various stresses functions to degrade as well as recycle cargo to maintain cell homeostasis while uncontrolled autophagy can also be detrimental. TSC1 promotes cardiomyocyte autophagy while key regulatory molecule Vps34 inactivation has autophagy inhibition effect in HCM [138, 145]. Autophagy flux is impaired in DCM patients with inherited mutations of LMNA or PLEKHM2 [146, 147]. Activation of AMPK can restore autophagy in DIC and DMCM [154, 157]. Transcription factor GATA4 inhibits autophagy while TFEB has opposite effect in DIC [152, 155]. In SIC, overexpression of Beclin-1 promotes autophagy [167]. In the canonical inflammasome pathway, pyroptosis is mediated by the assembly of the inflammasome complex and subsequent caspase 1 activation as well as GSDMD cleavage. In DMCM and SIC, pyroptosis-related key molecules are significantly activated [182, 193]. Pyroptosis is also present in DIC because doxorubicin can activate GSDMD via direct interaction [191]. As multiple forms of RCD are observed and identified as a common feature of cardiomyopathies in both clinical specimens as well as animal models, therapies targeted to directly or indirectly intervening programmed cell death by synthetic, natural products or targeted ncRNAs hold great potential to have a real impact in future clinical management of the diseases.
Despite of these significant findings, much work remains to uncover the full spectrum of the molecular network and physiological effects on RCD in the pathogenesis of cardiomyopathies. Evidences show that different forms of RCD seem to have an intricated crosstalk and some common features and even may coexist in the same diseased heart and contribute independently or synergistically to the pathogenesis of cardiomyopathy. In addition, the role of RCD in the disease may be very dynamic depending on the different stages of cardiomyopathy development. Thus, it is vital to identify what kind of RCD predominates in the specific pathogenic processes of cardiomyopathies. Finally, there is still a long way to go to effectively intervene highly conserved regulatory molecules in cell death pathway and to translate the basic scientific research into clinical practice. Despite of these challenges, there is plenty reason to be optimistic that the rich knowledge in RCD will yield better therapies for cardiomyopathies in the future.
References
McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121:722–30.
Lannou S, Mansencal N, Couchoud C, Lassalle M, Dubourg O, Stengel B, et al. The Public Health Burden of Cardiomyopathies: insights from a Nationwide Inpatient Study. J Clin Med. 2020;9:920–33.
Yamada T, Nomura S. Recent findings related to cardiomyopathy and genetics. Int J Mol Sci. 2021;22:15222–34.
Elliott PM, Anastasakis A, Borger MA, Borggrefe M, Cecchi F, Charron P, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014;35:2733–79.
Sorajja P, Pedersen WA, Bae R, Lesser JR, Jay D, Lin D, et al. First experience with percutaneous mitral valve plication as primary therapy for symptomatic obstructive hypertrophic cardiomyopathy. J Am Coll Cardiol. 2016;67:2811–8.
Ma H, Marti-Gutierrez N, Park SW, Wu J, Lee Y, Suzuki K, et al. Correction of a pathogenic gene mutation in human embryos. Nature. 2017;548:413–9.
Ho CY, Charron P, Richard P, Girolami F, Van Spaendonck-Zwarts KY, Pinto Y. Genetic advances in sarcomeric cardiomyopathies: state of the art. Cardiovasc Res. 2015;105:397–408.
Repetti GG, Toepfer CN, Seidman JG, Seidman CE. Novel therapies for prevention and early treatment of cardiomyopathies. Circ Res. 2019;124:1536–50.
Christgen S, Tweedell RE, Kanneganti TD. Programming inflammatory cell death for therapy. Pharmacol Ther. 2022;232:108010.
Tummers B, Green DR. The evolution of regulated cell death pathways in animals and their evasion by pathogens. Physiol Rev. 2022;102:411–54.
Loftus LV, Amend SR, Pienta KJ. Interplay between cell death and cell proliferation reveals new strategies for cancer therapy. Int J Mol Sci. 2022;23:4723–48.
Garg JP, Vucic D. Targeting cell death pathways for therapeutic intervention in kidney diseases. Semin Nephrol. 2016;36:153–61.
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541.
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99:1765–817.
Fuchs Y, Steller H. Programmed cell death in animal development and disease. Cell. 2011;147:742–58.
Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972;26:239–57.
Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M, et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science. 2022;375:1254–61.
Schweichel JU, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–66.
Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. 2019;29:347–64.
Jensen M, Engert A, Weissinger F, Knauf W, Kimby E, Poynton C, et al. Phase I study of a novel pro-apoptotic drug R-etodolac in patients with B-cell chronic lymphocytic leukemia. Invest New Drugs. 2008;26:139–49.
Fricker M, Tolkovsky AM, Borutaite V, Coleman M, Brown GC. Neuronal cell death. Physiol Rev. 2018;98:813–80.
Moujalled D, Strasser A, Liddell JR. Molecular mechanisms of cell death in neurological diseases. Cell Death Differ. 2021;28:2029–44.
Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–41.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012;19:107–20.
Chang DW, Xing Z, Pan Y, Algeciras-Schimnich A, Barnhart BC, Yaish-Ohad S, et al. c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21:3704–14.
Kalkavan H, Green DR. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018;25:46–55.
Bock FJ, Tait SWG. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol. 2020;21:85–100.
Singh R, Letai A, Sarosiek K. Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins. Nat Rev Mol Cell Biol. 2019;20:175–93.
Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182–9.
Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med. 1997;336:1131–41.
Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–47.
Liu L, Sun K, Zhang X, Tang Y, Xu D. Advances in the role and mechanism of BAG3 in dilated cardiomyopathy. Heart Fail Rev. 2021;26:183–94.
Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752–62.
Bozkurt B, Colvin M, Cook J, Cooper LT, Deswal A, Fonarow GC, et al. Current diagnostic and treatment strategies for specific dilated cardiomyopathies: a scientific statement From the American Heart Association. Circulation. 2016;134:e579–e646.
Liu J, Kong X, Zhang M, Yang X, Xu X. RNA binding protein 24 deletion disrupts global alternative splicing and causes dilated cardiomyopathy. Protein Cell. 2019;10:405–16.
Narula J, Pandey P, Arbustini E, Haider N, Narula N, Kolodgie FD, et al. Apoptosis in heart failure: release of cytochrome c from mitochondria and activation of caspase-3 in human cardiomyopathy. Proc Natl Acad Sci USA 1999;96:8144–9.
Narula J, Kolodgie FD, Virmani R. Apoptosis and cardiomyopathy. Curr Opin Cardiol. 2000;15:183–8.
Chen SN, Lombardi R, Karmouch J, Tsai JY, Czernuszewicz G, Taylor MRG, et al. DNA damage response/TP53 pathway is activated and contributes to the pathogenesis of dilated cardiomyopathy associated with LMNA (Lamin A/C) mutations. Circ Res. 2019;124:856–73.
Gramlich M, Michely B, Krohne C, Heuser A, Erdmann B, Klaassen S, et al. Stress-induced dilated cardiomyopathy in a knock-in mouse model mimicking human titin-based disease. J Mol Cell Cardiol. 2009;47:352–8.
Gammons J, Trebak M, Mancarella S. Cardiac-specific deletion of Orai3 leads to severe dilated cardiomyopathy and heart failure in mice. J Am Heart Assoc. 2021;10:e019486.
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497–504.
Hirota H, Chen J, Betz UA, Rajewsky K, Gu Y, Ross J Jr., et al. Loss of a gp130 cardiac muscle cell survival pathway is a critical event in the onset of heart failure during biomechanical stress. Cell. 1999;97:189–98.
Behl C. Breaking BAG: the Co-chaperone BAG3 in health and disease. Trends Pharmacol Sci. 2016;37:672–88.
Norton N, Li D, Rieder MJ, Siegfried JD, Rampersaud E, Zuchner S, et al. Genome-wide studies of copy number variation and exome sequencing identify rare variants in BAG3 as a cause of dilated cardiomyopathy. Am J Hum Genet. 2011;88:273–82.
Myers VD, Tomar D, Madesh M, Wang J, Song J, Zhang XQ, et al. Haplo-insufficiency of Bcl2-associated athanogene 3 in mice results in progressive left ventricular dysfunction, beta-adrenergic insensitivity, and increased apoptosis. J Cell Physiol. 2018;233:6319–26.
Zhang J, He Z, Xiao W, Na Q, Wu T, Su K, et al. Overexpression of BAG3 attenuates hypoxia-induced cardiomyocyte apoptosis by inducing autophagy. Cell Physiol Biochem. 2016;39:491–500.
Sun X, Shan A, Wei Z, Xu B. Intravenous mesenchymal stem cell-derived exosomes ameliorate myocardial inflammation in the dilated cardiomyopathy. Biochem Biophys Res Commun. 2018;503:2611–8.
Cai L, Li W, Wang G, Guo L, Jiang Y, Kang YJ. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome c-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–48.
Frustaci A, Chimenti C, Setoguchi M, Guerra S, Corsello S, Crea F, et al. Cell death in acromegalic cardiomyopathy. Circulation. 1999;99:1426–34.
Ino T, Nishimoto K, Okubo M, Akimoto K, Yabuta K, Kawai S, et al. Apoptosis as a possible cause of wall thinning in end-stage hypertrophic cardiomyopathy. Am J Cardiol. 1997;79:1137–41.
Mallat Z, Tedgui A, Fontaliran F, Frank R, Durigon M, Fontaine G. Evidence of apoptosis in arrhythmogenic right ventricular dysplasia. N Engl J Med. 1996;335:1190–6.
Dillmann WH. Diabetic cardiomyopathy: what is it and can it Be fixed? Circ Res. 2019;124:1160–2.
Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602.
Marwick TH, Ritchie R, Shaw JE, Kaye D. Implications of underlying mechanisms for the recognition and management of diabetic cardiomyopathy. J Am Coll Cardiol. 2018;71:339–51.
Bugger H, Abel ED. Molecular mechanisms of diabetic cardiomyopathy. Diabetologia. 2014;57:660–71.
Li Z, Zhang T, Dai H, Liu G, Wang H, Sun Y, et al. Involvement of endoplasmic reticulum stress in myocardial apoptosis of streptozocin-induced diabetic rats. J Clin Biochem Nutr. 2007;41:58–67.
Kuethe F, Sigusch HH, Bornstein SR, Hilbig K, Kamvissi V, Figulla HR. Apoptosis in patients with dilated cardiomyopathy and diabetes: a feature of diabetic cardiomyopathy? Horm Metab Res. 2007;39:672–6.
Zhang M, Zhang L, Hu J, Lin J, Wang T, Duan Y, et al. MST1 coordinately regulates autophagy and apoptosis in diabetic cardiomyopathy in mice. Diabetologia. 2016;59:2435–47.
Zhang M, Lin J, Wang S, Cheng Z, Hu J, Wang T, et al. Melatonin protects against diabetic cardiomyopathy through Mst1/Sirt3 signaling. J Pineal Res. 2017;63:e12418.
You P, Cheng Z, He X, Deng J, Diao J, Chen H, et al. Lin28a protects against diabetic cardiomyopathy through Mst1 inhibition. J Cell Physiol. 2020;235:4455–65.
Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell. 2006;126:107–20.
Gu J, Wang S, Guo H, Tan Y, Liang Y, Feng A, et al. Inhibition of p53 prevents diabetic cardiomyopathy by preventing early-stage apoptosis and cell senescence, reduced glycolysis, and impaired angiogenesis. Cell Death Dis. 2018;9:82.
Li X, Wang H, Yao B, Xu W, Chen J, Zhou X. lncRNA H19/miR-675 axis regulates cardiomyocyte apoptosis by targeting VDAC1 in diabetic cardiomyopathy. Sci Rep. 2016;6:36340–9.
Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. 2020;5:e132747.
Chatterjee K, Zhang J, Honbo N, Karliner JS. Doxorubicin cardiomyopathy. Cardiology. 2010;115:155–62.
Ichikawa Y, Ghanefar M, Bayeva M, Wu R, Khechaduri A, Naga Prasad SV, et al. Cardiotoxicity of doxorubicin is mediated through mitochondrial iron accumulation. J Clin Invest. 2014;124:617–30.
Wang P, Wang L, Lu J, Hu Y, Wang Q, Li Z, et al. SESN2 protects against doxorubicin-induced cardiomyopathy via rescuing mitophagy and improving mitochondrial function. J Mol Cell Cardiol. 2019;133:125–37.
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu SC, et al. FNDC5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating AKT. Cell Death Differ. 2020;27:540–55.
Xia P, Chen J, Liu Y, Fletcher M, Jensen BC, Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box O1. J Biol Chem. 2020;295:4265–76.
Kitakata H, Endo J, Ikura H, Moriyama H, Shirakawa K, Katsumata Y, et al. Therapeutic targets for DOX-induced cardiomyopathy: role of apoptosis vs. ferroptosis. Int J Mol Sci. 2022;23:1414–30.
Hu YH, Liu J, Lu J, Wang PX, Chen JX, Guo Y, et al. sFRP1 protects H9c2 cardiac myoblasts from doxorubicin-induced apoptosis by inhibiting the Wnt/PCP-JNK pathway. Acta Pharmacol Sin. 2020;41:1150–7.
Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–9.
Park SM, Yoon JB, Lee TH. Receptor interacting protein is ubiquitinated by cellular inhibitor of apoptosis proteins (c-IAP1 and c-IAP2) in vitro. FEBS Lett. 2004;566:151–6.
Lin Y, Devin A, Rodriguez Y, Liu ZG. Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev. 1999;13:2514–26.
Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148:213–27.
Nomura M, Ueno A, Saga K, Fukuzawa M, Kaneda Y. Accumulation of cytosolic calcium induces necroptotic cell death in human neuroblastoma. Cancer Res. 2014;74:1056–66.
Zhang T, Zhang Y, Cui M, Jin L, Wang Y, Lv F, et al. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat Med. 2016;22:175–82.
Szobi A, Goncalvesova E, Varga ZV, Leszek P, Kusmierczyk M, Hulman M, et al. Analysis of necroptotic proteins in failing human hearts. J Transl Med. 2017;15:86–92.
Fujita Y, Yano T, Kanamori H, Nagahara D, Muranaka A, Kouzu H, et al. Enhanced nuclear localization of phosphorylated MLKL predicts adverse events in patients with dilated cardiomyopathy. ESC Heart Fail. 2022;9:3435–51.
Liu J, Wen T, Dong K, He X, Zhou H, Shen J, et al. TEAD1 protects against necroptosis in postmitotic cardiomyocytes through regulation of nuclear DNA-encoded mitochondrial genes. Cell Death Differ. 2021;28:2045–59.
Fang T, Cao R, Wang W, Ye H, Shen L, Li Z, et al. Alterations in necroptosis during ALDH2mediated protection against high glucose-induced H9c2 cardiac cell injury. Mol Med Rep. 2018;18:2807–15.
Song S, Ding Y, Dai GL, Zhang Y, Xu MT, Shen JR, et al. Sirtuin 3 deficiency exacerbates diabetic cardiomyopathy via necroptosis enhancement and NLRP3 activation. Acta Pharmacol Sin. 2021;42:230–41.
Sun L, Chen Y, Luo H, Xu M, Meng G, Zhang W. Ca(2+)/calmodulin-dependent protein kinase II regulation by inhibitor 1 of protein phosphatase 1 alleviates necroptosis in high glucose-induced cardiomyocytes injury. Biochem Pharmacol. 2019;163:194–205.
Gong W, Zhang S, Chen Y, Shen J, Zheng Y, Liu X, et al. Protective role of hydrogen sulfide against diabetic cardiomyopathy via alleviating necroptosis. Free Radic Biol Med. 2022;181:29–42.
Reyes LF, Restrepo MI, Hinojosa CA, Soni NJ, Anzueto A, Babu BL, et al. Severe pneumococcal pneumonia causes acute cardiac toxicity and subsequent cardiac remodeling. Am J Respir Crit Care Med. 2017;196:609–20.
Shenoy AT, Beno SM, Brissac T, Bell JW, Novak L, Orihuela CJ. Severity and properties of cardiac damage caused by Streptococcus pneumoniae are strain dependent. PLoS One. 2018;13:e0204032.
Beno SM, Riegler AN, Gilley RP, Brissac T, Wang Y, Kruckow KL, et al. Inhibition of necroptosis to prevent long-term cardiac damage during pneumococcal pneumonia and invasive disease. J Infect Dis. 2020;222:1882–93.
Peng S, Xu J, Ruan W, Li S, Xiao F. PPAR-gamma activation prevents septic cardiac dysfunction via inhibition of apoptosis and necroptosis. Oxid Med Cell Longev. 2017;2017:8326749.
Wang C, Hu L, Guo S, Yao Q, Liu X, Zhang B, et al. Phosphocreatine attenuates doxorubicin-induced cardiotoxicity by inhibiting oxidative stress and activating TAK1 to promote myocardial survival in vivo and in vitro. Toxicology. 2021;460:152881.
Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci USA 2019;116:2672–80.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Roemhild K, von Maltzahn F, Weiskirchen R, Knuchel R, von Stillfried S, Lammers T. Iron metabolism: pathophysiology and pharmacology. Trends Pharmacol Sci. 2021;42:640–56.
Morales M, Xue X. Targeting iron metabolism in cancer therapy. Theranostics. 2021;11:8412–29.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–81.
Ohgami RS, Campagna DR, McDonald A, Fleming MD. The Steap proteins are metalloreductases. Blood. 2006;108:1388–94.
Zhang N, Yu X, Xie J, Xu H. New Insights Into The Role Of Ferritin In Iron Homeostasis And Neurodegenerative Diseases. Mol Neurobiol. 2021;58:2812–23.
Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90.
Doll S, Conrad M. Iron and ferroptosis: a still ill-defined liaison. IUBMB Life. 2017;69:423–34.
Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10:1604–9.
Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA 2016;113:E4966–75.
Kuhn H, Humeniuk L, Kozlov N, Roigas S, Adel S, Heydeck D. The evolutionary hypothesis of reaction specificity of mammalian ALOX15 orthologs. Prog Lipid Res. 2018;72:55–74.
Fujii J, Homma T, Kobayashi S. Ferroptosis caused by cysteine insufficiency and oxidative insult. Free Radic Res. 2020;54:969–80.
Tuo QZ, Zhang ST, Lei P. Mechanisms of neuronal cell death in ischemic stroke and their therapeutic implications. Med Res Rev. 2022;42:259–305.
Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92.
Morre DJ, Morre DM. Non-mitochondrial coenzyme Q. Biofactors. 2011;37:355–60.
Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509:105–9.
Du J, Wang T, Li Y, Zhou Y, Wang X, Yu X, et al. DHA inhibits proliferation and induces ferroptosis of leukemia cells through autophagy dependent degradation of ferritin. Free Radic Biol Med. 2019;131:356–69.
Rhee JW, Yi H, Thomas D, Lam CK, Belbachir N, Tian L, et al. Modeling secondary iron overload cardiomyopathy with human induced pluripotent stem cell-derived cardiomyocytes. Cell Rep. 2020;32:107886.
Fang X, Cai Z, Wang H, Han D, Cheng Q, Zhang P, et al. Loss of cardiac ferritin H facilitates cardiomyopathy via Slc7a11-mediated ferroptosis. Circ Res. 2020;127:486–501.
Menon AV, Liu J, Tsai HP, Zeng L, Yang S, Asnani A, et al. Excess heme upregulates heme oxygenase 1 and promotes cardiac ferroptosis in mice with sickle cell disease. Blood. 2022;139:936–41.
Oudit GY, Sun H, Trivieri MG, Koch SE, Dawood F, Ackerley C, et al. L-type Ca2+ channels provide a major pathway for iron entry into cardiomyocytes in iron-overload cardiomyopathy. Nat Med. 2003;9:1187–94.
Khaled A, Salem HA, Ezzat DA, Seif HM, Rabee H. A randomized controlled trial evaluating the effects of amlodipine on myocardial iron deposition in pediatric patients with thalassemia major. Drug Des Devel Ther. 2019;13:2427–36.
Wang Y, Yan S, Liu X, Deng F, Wang P, Yang L, et al. PRMT4 promotes ferroptosis to aggravate doxorubicin-induced cardiomyopathy via inhibition of the Nrf2/GPX4 pathway. Cell Death Differ. 2022;29:1982–95.
Liu Y, Zeng L, Yang Y, Chen C, Wang D, Wang H. Acyl-CoA thioesterase 1 prevents cardiomyocytes from doxorubicin-induced ferroptosis via shaping the lipid composition. Cell Death Dis. 2020;11:756–69.
Zhang H, Wang Z, Liu Z, Du K, Lu X. Protective effects of dexazoxane on rat ferroptosis in doxorubicin-induced cardiomyopathy through regulating HMGB1. Front Cardiovasc Med. 2021;8:685434.
Knutson MD. Non-transferrin-bound iron transporters. Free Radic Biol Med. 2019;133:101–11.
Li D, Pi W, Sun Z, Liu X, Jiang J. Ferroptosis and its role in cardiomyopathy. Biomed Pharmacother. 2022;153:113279.
Wang X, Chen X, Zhou W, Men H, Bao T, Sun Y, et al. Ferroptosis is essential for diabetic cardiomyopathy and is prevented by sulforaphane via AMPK/NRF2 pathways. Acta Pharm Sin B. 2022;12:708–22.
Lutchmansingh FK, Hsu JW, Bennett FI, Badaloo AV, McFarlane-Anderson N, Gordon-Strachan GM, et al. Glutathione metabolism in type 2 diabetes and its relationship with microvascular complications and glycemia. PLoS One. 2018;13:e0198626.
Ni T, Huang X, Pan S, Lu Z. Inhibition of the long non-coding RNA ZFAS1 attenuates ferroptosis by sponging miR-150-5p and activates CCND2 against diabetic cardiomyopathy. J Cell Mol Med. 2021;25:9995–10007.
Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, Nichols CE, Shepherd DL, et al. Reversal of mitochondrial proteomic loss in Type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. Am J Physiol Regul Integr Comp Physiol. 2013;304:R553–65.
Hollenberg SM, Singer M. Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol. 2021;18:424–34.
Martin L, Derwall M, Al Zoubi S, Zechendorf E, Reuter DA, Thiemermann C, et al. The septic heart: current understanding of molecular mechanisms and clinical implications. Chest. 2019;155:427–37.
Li N, Wang W, Zhou H, Wu Q, Duan M, Liu C, et al. Ferritinophagy-mediated ferroptosis is involved in sepsis-induced cardiac injury. Free Radic Biol Med. 2020;160:303–18.
Wang C, Yuan W, Hu A, Lin J, Xia Z, Yang CF, et al. Dexmedetomidine alleviated sepsis-induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22:175–84.
Xiao Z, Kong B, Fang J, Qin T, Dai C, Shuai W, et al. Ferrostatin-1 alleviates lipopolysaccharide-induced cardiac dysfunction. Bioengineered. 2021;12:9367–76.
Jiang CS, Zhuang CL, Zhu K, Zhang J, Muehlmann LA, Figueiro Longo JP, et al. Identification of a novel small-molecule Keap1-Nrf2 PPI inhibitor with cytoprotective effects on LPS-induced cardiomyopathy. J Enzym Inhib Med Chem. 2018;33:833–41.
Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F, et al. Molecular definitions of autophagy and related processes. EMBO J. 2017;36:1811–36.
Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018;19:349–64.
Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221:3–12.
Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31.
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–91.
Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. 2017;17:528–42.
Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer. 2018;124:3307–18.
Sciarretta S, Yee D, Nagarajan N, Bianchi F, Saito T, Valenti V, et al. Trehalose-induced activation of autophagy improves cardiac remodeling after myocardial infarction. J Am Coll Cardiol. 2018;71:1999–2010.
Maron BJ, Rowin EJ, Casey SA, Link MS, Lesser JR, Chan RH, et al. Hypertrophic cardiomyopathy in adulthood associated with low cardiovascular mortality with contemporary management strategies. J Am Coll Cardiol. 2015;65:1915–28.
Song L, Su M, Wang S, Zou Y, Wang X, Wang Y, et al. MiR-451 is decreased in hypertrophic cardiomyopathy and regulates autophagy by targeting TSC1. J Cell Mol Med. 2014;18:2266–74.
Bos JM, Towbin JA, Ackerman MJ. Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol. 2009;54:201–11.
Spudich JA, Aksel T, Bartholomew SR, Nag S, Kawana M, Yu EC, et al. Effects of hypertrophic and dilated cardiomyopathy mutations on power output by human beta-cardiac myosin. J Exp Biol. 2016;219:161–7.
Spudich JA. Three perspectives on the molecular basis of hypercontractility caused by hypertrophic cardiomyopathy mutations. Pflug Arch. 2019;471:701–17.
Ziane R, Huang H, Moghadaszadeh B, Beggs AH, Levesque G, Chahine M. Cell membrane expression of cardiac sodium channel Na(v)1.5 is modulated by alpha-actinin-2 interaction. Biochemistry. 2010;49:166–78.
Almomani R, Verhagen JM, Herkert JC, Brosens E, van Spaendonck-Zwarts KY, Asimaki A, et al. Biallelic truncating mutations in ALPK3 cause severe pediatric cardiomyopathy. J Am Coll Cardiol. 2016;67:515–25.
Singh SR, Zech ATL, Geertz B, Reischmann-Dusener S, Osinska H, Prondzynski M, et al. Activation of autophagy ameliorates cardiomyopathy in Mybpc3-targeted knockin mice. Circ Heart Fail. 2017;10:e004140.
Kimura H, Eguchi S, Sasaki J, Kuba K, Nakanishi H, Takasuga S, et al. Vps34 regulates myofibril proteostasis to prevent hypertrophic cardiomyopathy. JCI Insight. 2017;2:e89462.
Choi JC, Muchir A, Wu W, Iwata S, Homma S, Morrow JP, et al. Temsirolimus activates autophagy and ameliorates cardiomyopathy caused by lamin A/C gene mutation. Sci Transl Med. 2012;4:144ra02.
Muhammad E, Levitas A, Singh SR, Braiman A, Ofir R, Etzion S, et al. PLEKHM2 mutation leads to abnormal localization of lysosomes, impaired autophagy flux and associates with recessive dilated cardiomyopathy and left ventricular noncompaction. Hum Mol Genet. 2015;24:7227–40.
Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, et al. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579–87.
Ding Y, Sun X, Huang W, Hoage T, Redfield M, Kushwaha S, et al. Haploinsufficiency of target of rapamycin attenuates cardiomyopathies in adult zebrafish. Circ Res. 2011;109:658–69.
Kawaguchi T, Takemura G, Kanamori H, Takeyama T, Watanabe T, Morishita K, et al. Prior starvation mitigates acute doxorubicin cardiotoxicity through restoration of autophagy in affected cardiomyocytes. Cardiovasc Res. 2012;96:456–65.
Lu L, Wu W, Yan J, Li X, Yu H, Yu X. Adriamycin-induced autophagic cardiomyocyte death plays a pathogenic role in a rat model of heart failure. Int J Cardiol. 2009;134:82–90.
Kobayashi S, Volden P, Timm D, Mao K, Xu X, Liang Q. Transcription factor GATA4 inhibits doxorubicin-induced autophagy and cardiomyocyte death. J Biol Chem. 2010;285:793–804.
Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. 2016;133:1668–87.
Chen K, Xu X, Kobayashi S, Timm D, Jepperson T, Liang Q. Caloric restriction mimetic 2-deoxyglucose antagonizes doxorubicin-induced cardiomyocyte death by multiple mechanisms. J Biol Chem. 2011;286:21993–2006.
Bartlett JJ, Trivedi PC, Yeung P, Kienesberger PC, Pulinilkunnil T. Doxorubicin impairs cardiomyocyte viability by suppressing transcription factor EB expression and disrupting autophagy. Biochem J. 2016;473:3769–89.
Kanamori H, Takemura G, Goto K, Tsujimoto A, Mikami A, Ogino A, et al. Autophagic adaptations in diabetic cardiomyopathy differ between type 1 and type 2 diabetes. Autophagy. 2015;11:1146–60.
He C, Zhu H, Li H, Zou MH, Xie Z. Dissociation of Bcl-2-Beclin1 complex by activated AMPK enhances cardiac autophagy and protects against cardiomyocyte apoptosis in diabetes. Diabetes. 2013;62:1270–81.
Feng Y, Xu W, Zhang W, Wang W, Liu T, Zhou X. LncRNA DCRF regulates cardiomyocyte autophagy by targeting miR-551b-5p in diabetic cardiomyopathy. Theranostics. 2019;9:4558–66.
Zhao Y, Zhang L, Qiao Y, Zhou X, Wu G, Wang L, et al. Heme oxygenase-1 prevents cardiac dysfunction in streptozotocin-diabetic mice by reducing inflammation, oxidative stress, apoptosis and enhancing autophagy. PLoS One. 2013;8:e75927.
Guo R, Zhang Y, Turdi S, Ren J. Adiponectin knockout accentuates high fat diet-induced obesity and cardiac dysfunction: role of autophagy. Biochim Biophys Acta. 2013;1832:1136–48.
Sciarretta S, Zhai P, Shao D, Maejima Y, Robbins J, Volpe M, et al. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–46.
Xu X, Kobayashi S, Chen K, Timm D, Volden P, Huang Y, et al. Diminished autophagy limits cardiac injury in mouse models of type 1 diabetes. J Biol Chem. 2013;288:18077–92.
Yao Q, Ke ZQ, Guo S, Yang XS, Zhang FX, Liu XF, et al. Curcumin protects against diabetic cardiomyopathy by promoting autophagy and alleviating apoptosis. J Mol Cell Cardiol. 2018;124:26–34.
Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, et al. LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009;296:H470–9.
Hsieh CH, Pai PY, Hsueh HW, Yuan SS, Hsieh YC. Complete induction of autophagy is essential for cardioprotection in sepsis. Ann Surg. 2011;253:1190–200.
Pang J, Peng H, Wang S, Xu X, Xu F, Wang Q, et al. Mitochondrial ALDH2 protects against lipopolysaccharide-induced myocardial contractile dysfunction by suppression of ER stress and autophagy. Biochim Biophys Acta Mol Basis Dis. 2019;1865:1627–41.
Sun Y, Yao X, Zhang QJ, Zhu M, Liu ZP, Ci B, et al. Beclin-1-dependent autophagy protects the heart during sepsis. Circulation. 2018;138:2247–62.
Wang R, Xu Y, Zhang W, Fang Y, Yang T, Zeng D, et al. Inhibiting miR-22 alleviates cardiac dysfunction by regulating Sirt1 in septic cardiomyopathy. Front Cell Dev Biol. 2021;9:650666.
D’Souza CA, Heitman J. Dismantling the Cryptococcus coat. Trends Microbiol. 2001;9:112–3.
Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10:417–26.
Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5.
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Kovacs SB, Miao EA. Gasdermins: effectors of pyroptosis. Trends Cell Biol. 2017;27:673–84.
Bulek K, Zhao J, Liao Y, Rana N, Corridoni D, Antanaviciute A, et al. Epithelial-derived gasdermin D mediates nonlytic IL-1beta release during experimental colitis. J Clin Invest. 2020;130:4218–34.
Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–21.
Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277:61–75.
Sarhan J, Liu BC, Muendlein HI, Li P, Nilson R, Tang AY, et al. Caspase-8 induces cleavage of gasdermin D to elicit pyroptosis during Yersinia infection. Proc Natl Acad Sci USA 2018;115:E10888–E97.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103.
Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. NLRP3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. 2020;34:101523.
Wang K, Lv Z, Fang C, Xu C, Yu Z, Gao L, et al. Pyroptosis patterns are involved in immune microenvironment regulation of dilated cardiomyopathy. Dis Markers. 2022;2022:4627845.
Jeyabal P, Thandavarayan RA, Joladarashi D, Suresh Babu S, Krishnamurthy S, Bhimaraj A, et al. MicroRNA-9 inhibits hyperglycemia-induced pyroptosis in human ventricular cardiomyocytes by targeting ELAVL1. Biochem Biophys Res Commun. 2016;471:423–9.
Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS One. 2014;9:e104771.
Gao G, Fu L, Xu Y, Tao L, Guo T, Fang G, et al. Cyclovirobuxine D ameliorates experimental diabetic cardiomyopathy by inhibiting cardiomyocyte pyroptosis via NLRP3 in vivo and in vitro. Front Pharmacol. 2022;13:906548.
Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384:117–28.
Sun Z, Chai Q, Zhang Z, Lu D, Meng Z, Wu W. Inhibition of SGLT1 protects against glycemic variability-induced cardiac damage and pyroptosis of cardiomyocytes in diabetic mice. Life Sci. 2021;271:119116.
Li X, Du N, Zhang Q, Li J, Chen X, Liu X, et al. MicroRNA-30d regulates cardiomyocyte pyroptosis by directly targeting foxo3a in diabetic cardiomyopathy. Cell Death Dis. 2014;5:e1479.
Xiao W, Zheng D, Chen X, Yu B, Deng K, Ma J, et al. Long non-coding RNA MIAT is involved in the regulation of pyroptosis in diabetic cardiomyopathy via targeting miR-214-3p. iScience. 2021;24:103518.
Yang F, Qin Y, Wang Y, Li A, Lv J, Sun X, et al. LncRNA KCNQ1OT1 mediates pyroptosis in diabetic cardiomyopathy. Cell Physiol Biochem. 2018;50:1230–44.
Yang F, Li A, Qin Y, Che H, Wang Y, Lv J, et al. A novel circular RNA mediates pyroptosis of diabetic cardiomyopathy by functioning as a competing endogenous RNA. Mol Ther Nucleic Acids. 2019;17:636–43.
Zhao S, Tan Y, Qin J, Xu H, Liu L, Wan H, et al. MicroRNA-223-3p promotes pyroptosis of cardiomyocyte and release of inflammasome factors via downregulating the expression level of SPI1 (PU.1). Toxicology. 2022;476:153252.
Ye B, Shi X, Xu J, Dai S, Xu J, Fan X, et al. Gasdermin D mediates doxorubicin-induced cardiomyocyte pyroptosis and cardiotoxicity via directly binding to doxorubicin and changes in mitochondrial damage. Transl Res. 2022;248:36–50.
Singla DK, Johnson TA, Tavakoli Dargani Z. Exosome treatment enhances anti-inflammatory M2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cells. 2019;8:1224–44.
Dai S, Ye B, Zhong L, Chen Y, Hong G, Zhao G, et al. GSDMD mediates LPS-induced septic myocardial dysfunction by regulating ROS-dependent NLRP3 inflammasome activation. Front Cell Dev Biol. 2021;9:779432.
Gong X, Li Y, He Y, Zhou F. USP7-SOX9-miR-96-5p-NLRP3 network regulates myocardial injury and cardiomyocyte pyroptosis in sepsis. Hum Gene Ther. 2022;33:1073–90.
Li N, Zhou H, Wu H, Wu Q, Duan M, Deng W, et al. STING-IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215.
Xiong X, Lu L, Wang Z, Ma J, Shao Y, Liu Y, et al. Irisin attenuates sepsis-induced cardiac dysfunction by attenuating inflammation-induced pyroptosis through a mitochondrial ubiquitin ligase-dependent mechanism. Biomed Pharmacother. 2022;152:113199.
Li W, Shen X, Feng S, Liu Y, Zhao H, Zhou G, et al. BRD4 inhibition by JQ1 protects against LPS-induced cardiac dysfunction by inhibiting activation of NLRP3 inflammasomes. Mol Biol Rep. 2022;49:8197–207.
Leist M, Single B, Castoldi AF, Kuhnle S, Nicotera P. Intracellular adenosine triphosphate (ATP) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. 1997;185:1481–6.
Song X, Kim SY, Zhang L, Tang D, Bartlett DL, Kwon YT, et al. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014;5:e1504.
Lu JV, Walsh CM. Programmed necrosis and autophagy in immune function. Immunol Rev. 2012;249:205–17.
Xu C, Wu J, Wu Y, Ren Z, Yao Y, Chen G, et al. TNF-alpha-dependent neuronal necroptosis regulated in Alzheimer’s disease by coordination of RIPK1-p62 complex with autophagic UVRAG. Theranostics. 2021;11:9452–69.
Goodall ML, Fitzwalter BE, Zahedi S, Wu M, Rodriguez D, Mulcahy-Levy JM, et al. The autophagy machinery controls cell death switching between apoptosis and necroptosis. Dev Cell. 2016;37:337–49.
Schroeder B, Schulze RJ, Weller SG, Sletten AC, Casey CA, McNiven MA. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61:1896–907.
Torii S, Shintoku R, Kubota C, Yaegashi M, Torii R, Sasaki M, et al. An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem J. 2016;473:769–77.
Ollinger K, Brunk UT. Cellular injury induced by oxidative stress is mediated through lysosomal damage. Free Radic Biol Med. 1995;19:565–74.
Lee YS, Kalimuthu K, Park YS, Luo X, Choudry MHA, Bartlett DL, et al. BAX-dependent mitochondrial pathway mediates the crosstalk between ferroptosis and apoptosis. Apoptosis. 2020;25:625–31.
Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J, et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193.
Takahashi N, Duprez L, Grootjans S, Cauwels A, Nerinckx W, DuHadaway JB, et al. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437.
Amgalan D, Garner TP, Pekson R, Jia XF, Yanamandala M, Paulino V, et al. A small-molecule allosteric inhibitor of BAX protects against doxorubicin-induced cardiomyopathy. Nat Cancer. 2020;1:315–28.
Han W, Wang H, Su L, Long Y, Cui N, Liu D. Inhibition of the mTOR pathway exerts cardioprotective effects partly through autophagy in CLP rats. Mediators Inflamm. 2018;2018:4798209.
Wang Y, Chen J, Li S, Zhang X, Guo Z, Hu J, et al. Exogenous spermine attenuates rat diabetic cardiomyopathy via suppressing ROS-p53 mediated downregulation of calcium-sensitive receptor. Redox Biol. 2020;32:101514.
Li L, Luo W, Qian Y, Zhu W, Qian J, Li J, et al. Luteolin protects against diabetic cardiomyopathy by inhibiting NF-kappaB-mediated inflammation and activating the Nrf2-mediated antioxidant responses. Phytomedicine. 2019;59:152774.
Jubaidi FF, Zainalabidin S, Taib IS, Hamid ZA, Budin SB. The potential role of flavonoids in ameliorating diabetic cardiomyopathy via alleviation of cardiac oxidative stress, inflammation and apoptosis. Int J Mol Sci. 2021;22:5094–115.
Liu Y, Asnani A, Zou L, Bentley VL, Yu M, Wang Y, et al. Visnagin protects against doxorubicin-induced cardiomyopathy through modulation of mitochondrial malate dehydrogenase. Sci Transl Med. 2014;6:266ra170.
Asnani A, Zheng B, Liu Y, Wang Y, Chen HH, Vohra A, et al. Highly potent visnagin derivatives inhibit Cyp1 and prevent doxorubicin cardiotoxicity. JCI Insight. 2018;3:e96753.
Yu W, Gao B, Li N, Wang J, Qiu C, Zhang G, et al. Sirt3 deficiency exacerbates diabetic cardiac dysfunction: Role of Foxo3A-Parkin-mediated mitophagy. Biochim Biophys Acta Mol Basis Dis. 2017;1863:1973–83.
Liu D, Ma Z, Di S, Yang Y, Yang J, Xu L, et al. AMPK/PGC1alpha activation by melatonin attenuates acute doxorubicin cardiotoxicity via alleviating mitochondrial oxidative damage and apoptosis. Free Radic Biol Med. 2018;129:59–72.
Chen TS, Chuang SY, Shen CY, Ho TJ, Chang RL, Yeh YL, et al. Antioxidant Sirt1/Akt axis expression in resveratrol pretreated adipose-derived stem cells increases regenerative capability in a rat model with cardiomyopathy induced by diabetes mellitus. J Cell Physiol. 2021;236:4290–302.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–74.
Angelopoulos A, Oikonomou E, Vogiatzi G, Antonopoulos A, Tsalamandris S, Georgakopoulos C, et al. MicroRNAs as biomarkers in hypertrophic cardiomyopathy: current state of the art. Curr Med Chem. 2021;28:7400–12.
Wang H, Bei Y, Shen S, Huang P, Shi J, Zhang J, et al. miR-21-3p controls sepsis-associated cardiac dysfunction via regulating SORBS2. J Mol Cell Cardiol. 2016;94:43–53.
Jiang J, Gao G, Pan Q, Liu J, Tian Y, Zhang X. Circular RNA circHIPK3 is downregulated in diabetic cardiomyopathy and overexpression of circHIPK3 suppresses PTEN to protect cardiomyocytes from high glucose-induced cell apoptosis. Bioengineered. 2022;13:6272–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sheng, Sy., Li, Jm., Hu, Xy. et al. Regulated cell death pathways in cardiomyopathy. Acta Pharmacol Sin 44, 1521–1535 (2023). https://doi.org/10.1038/s41401-023-01068-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-023-01068-9
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Diabetic Cardiomyopathy and Cell Death: Focus on Metal-Mediated Cell Death
Cardiovascular Toxicology (2024)
-
The E2F family: a ray of dawn in cardiomyopathy
Molecular and Cellular Biochemistry (2024)
-
Therapeutic Approaches Targeting Ferroptosis in Cardiomyopathy
Cardiovascular Drugs and Therapy (2023)
-
What is the impact of ferroptosis on diabetic cardiomyopathy: a systematic review
Heart Failure Reviews (2023)