
Abstract
All multicellular organisms develop during evolution the highly regulated and interconnected pathways of cell death. This complex network contributes to the pathogenesis of various cardiovascular disorders including ischemia/reperfusion injury, myocardial infarction, heart failure, dysrhythmias and atherosclerosis. Chronic cardiac remodeling response and transition to overt HF have been associated with modestly increased apoptosis, although the actual burden of chronic cell loss attributable to apoptosis is not clear. Central mediators of cardiomyocyte survival and death are the mitochondrial organelles. Based on its morphological characteristics, cell death can be classified into three major types: apoptosis, necrosis and autophagy. Recently, a new pathway of regulated necrosis, necroptosis, has also been reported in the failing heart. The mitochondrial (intrinsic) and the death-receptor-mediated (extrinsic) converge at mitochondria inducing release of mitochondrial apoptogens to initiate the caspase cascade and eventually degradation of the doomed cardiomyocyte. Activation of death receptors can initiate not only extrinsic apoptotic pathway, but also necrosis. On the other hand, autophagy, which is characterized by the massive formation of lysosomal-derived vesicles, containing degenerating cytoplasmic contents, is primarily a survival response to nutrient deprivation, and a selective form of autophagy, mitophagy, is also a protective mechanism that allows to eliminate damaged mitochondria and thereby to attenuate mitochondria-mediated apoptosis and necrosis in the myocardium. Further insight into the molecular mechanisms underlying cell death will increase the efficiency and repertoire of therapeutic interventions available in cardiovascular disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The absolute percentage of apoptotic cardiac myocytes in the failing human heart is low. However, the percentage of cardiac myocyte apoptosis is still 10- to 100-fold higher than that observed in control hearts. These data suggest that in HF low but elevated levels of cardiac myocyte apoptosis result over time in a cumulative loss of cardiac myocytes and therefore HF. Experimental models have substantiated the sufficiency of clinically relevant degrees of apoptosis to induce HF, as well as the necessity of myocyte apoptosis for the development of HF [1].
According to Hein et al. [2], cardiac myocyte necrosis may play a role in HF. Cardiac myocyte-specific transgenic overexpression of the β2-α subunit of the L-type Ca2+ channel resulted in Ca2+ overload, mitochondrial permeability transition pore (MPTP) opening, necrosis and cardiac dysfunction. The phenotype was rescued by deletion of peptidylprolylisomerase F encoding cyclophilin D, but not overexpression of Bcl-2, suggesting that HF in this model is attributable to cardiac myocyte necrosis. Transition to HF occurs by fibrosis and myocyte degeneration partially compensated by hypertrophy involving DNA synthesis and transcription. Cell loss, mainly by autophagy and oncosis, contributes significantly to the progression of LV systolic dysfunction [2].
Autophagy can be considered as a third type of cell death, but in contrast to necrosis, autophagic cell death similar to apoptosis is characterized by the absence of tissue inflammatory response. Also, in contrast to apoptosis, which is characterized by early collapse of cytoskeletal elements but with preservation of organelles until late in the process, autophagic cell death exhibits early degradation of organelles and preservation of cytoskeletal elements until later stages.
Moreover, autophagy-related protein-5 deletion precipitates ventricular enlargement and cardiac dysfunction after hemodynamic overload, implying that autophagy is a compensatory mechanism during HF. Thus, autophagy may be protective in response to some cardiomyopathic stimuli and pathogenic in response to others.
Although various cardiovascular cells, such as cardiomyocytes, endothelial cells, vascular smooth muscle cells, fibroblasts and infiltrating inflammatory cells, are implicated in response to oxidative stress and critical for heart function, in this review, we will focus primarily on cardiomyocytes and discuss recent progress in our understanding of the molecular mechanisms of apoptosis extrinsic pathway, necrosis and autophagy, and these mechanisms combined may be sufficient to account for the progression of HF.
Mechanisms of cell death
Cells die primarily by either apoptosis or necrosis. Apoptosis is a highly regulated mode of cell suicide [3]. Although necrosis has traditionally been regarded as passive, recent data indicate that a substantial proportion of necrotic deaths are actively executed by the cell in a highly regulated manner. This form of necrosis is sometimes referred to as regulated or programmed. Indeed, both apoptosis and necrosis may play critical roles in normal biology including prenatal development and postnatal homeostasis [4].
These pathways, which mediate both apoptosis and necrosis, are linked by multiple biochemical and functional connections. Extrapolating this degree of connectivity, the possibility is raised that these cell death mechanisms comprise a single unified death mechanism.
Apoptosis induced through activation of the extrinsic pathway and necrosis
Since the intrinsic mitochondrial pathway topic is discussed in another review in this spotlight, here it is sufficed to say that this pathway involves both mitochondria and the endoplasmic reticulum.
In the death receptor or extrinsic pathway (Fig. 1), a variety of death ligands bind their cognate receptors to trigger cell death. Some of these ligands are soluble, e.g., tumor necrosis factor [TNF]-α, and some are bound to the surface of other cells, e.g., Fas ligand. The efficiency of these ligands to induce death varies with cell type. Recent observations have shown that the same death ligands may induce apoptosis or necrosis, and the choice is principally mediated by downstream events.

Extrinsic apoptotic pathway. The extrinsic or death receptor pathway is triggered by binding of death ligands (e.g., FasL, TNF-α or TRAIL) to their cognate receptors. Engaged death receptors recruit the adaptor protein FADD, which in turn recruits receptor-interacting protein kinase 1 (RIP1), cellular inhibitor of apoptosis proteins (cIAP), Flice inhibitor proteins (FLIP) and procaspase-8 (or procaspase-10), assembling the death-inducing signaling complex (DISC). DISC formation triggers activation of caspase-8 (or caspase-10), which in turn cleaves and activates downstream effector procaspases (e.g., caspase-3, caspase-6 and caspase-7). Caspase-8 can also cleave Bid to generate its truncated form tBid, which translocates to mitochondria to contribute to activation of mitochondrial permeabilization. cIAP attenuate caspase activation
Binding of ligand to receptor induces the formation of either of two multiprotein complexes: the death-inducing signaling complex (DISC) and complex I [5]. The DISC signals apoptosis, whereas complex I can signal either apoptosis, necrosis, or promote cell survival. The DISC has been studied intensively in the context of Fas ligand/Fas signaling and complex I in the setting of TNF/TNF receptor 1 signaling. TNFR1-induced apoptosis involves two sequential signaling complexes [5]. The initial plasma-membrane-bound complex (complex I) consists of TNFR1, the adaptor TRADD, the kinase RIP1, and TRAF2 and rapidly signals activation of NF-κB. In the second step, TRADD and RIP1 associate with FADD and caspase-8 and form a cytoplasmic complex (complex II). When NF-κB is activated by complex I, complex II harbors the caspase-8 inhibitor FLIP(L), and the cell survives. Therefore, TNFR1-mediated-signal transduction includes a checkpoint, resulting in cell death (via complex II) in instances where the initial signal (via complex I, NF-κB) fails to be activated. However, which ligand/receptor combinations use the DISC versus complex I is still unclear.
In DISC formation, the binding of death ligand induces a conformational change in the cytosolic domain of the death receptor (DR), which recruits an adaptor protein (Fas-associated via death domain, TNF receptor-associated death domain) [6]. This adaptor protein, in turn, binds upstream procaspase-8 or procaspase-10 to form the DISC [6, 7]. Procaspases are the zymogen form of caspases, cystenyl proteases that cut after aspartic acid residues [8]. Within the DISC, procaspase-8 and procaspase-10 are activated through a forced proximity mechanism [9, 10]. Once activated, these caspases cleave and then activate downstream procaspase-3 and procaspase-7. Caspase-3 and caspase-7 then cut multiple cellular proteins to bring about apoptotic death through mechanisms that are incompletely understood. In most cells, activation of the extrinsic pathway alone is not enough to kill and requires amplification through the intrinsic pathway. One means by which amplification is achieved is through the cleavage of the B-cell lymphoma-2 (Bcl-2) family protein BH3-interacting domain death agonist (Bid) by caspase-8, after which truncated Bid translocates to the mitochondria and contributes to outer mitochondrial membrane (OMM) apoptotic events that are described below [11].
In the assembly of complex I, the binding of death ligand to receptor recruits TNF receptor-associated death domain, which recruits receptor-interacting protein 1 (RIP1, a serine/threonine kinase), cellular inhibitor of apoptosis proteins (IAP) 1 and 2, and TNF receptor-associated factor 2 and 5 [5]. RIP1 undergoes K63-polyubiquitination by cellular IAP 1 and 2 [12, 13]. This provides a platform for the recruitment of additional kinases that activate nuclear factor-κB, resulting in the transcription of survival proteins. However, after dissociation of DR, endocytosis, deubiquitination of RIP1 and recruitment of a Fas-associated via death domain-RIP3 complex, complex I morphs into complex II [14, 15]. Complex II signals apoptosis when Fas-associated via death domain recruits procaspase-8 leading to its activation by forced proximity [5, 10]. Caspase-8 not only activates downstream caspases to bring about apoptosis, it also cleaves RIP1 and RIP3 abrogating their ability to signal necrosis [16]. If caspase-8 activity is inhibited experimentally or by certain viral or cancer proteins, apoptosis is blocked, obligating the cell to undergo necrosis in this pathway [17, 18]. Necrosis is triggered by the interaction of RIP1 with RIP3 (Fig. 2), a second serine/threonine kinase, resulting in a complex series of crossphosphorylation events. Necrostatin-1, a small molecule inhibitor of the kinase activity of RIP1, ablates necrosis in the DR pathway [19]. Events in this pathway downstream of RIP1 and RIP3 are incompletely understood, but may include phosphorylation by RIP3 of mixed lineage kinase domain-like protein [20]. Phosphoglycerate mutase 5, a mitochondrial phosphatase [21], and certain catabolic enzymes (glutamate dehydrogenase 1, glutamate ammonia ligase and glycogen phosphorylase) potentially elicit necrosis through the generation of reactive oxygen species [22]. Moreover, ROS-mediated DNA damage leads to overactivation of poly(ADP-ribose) polymerase 1, a nuclear enzyme that consumes nicotinamide adenine dinucleotide leading to significant ATP consumption, a key feature of necrosis [19]. Other downstream events that have been implicated in DR necrosis signaling include activation of calpains, phospholipases, lipoxygenases and sphingomyelinases, and permeabilization of lysosomes [23].

Necrotic cell death. Engagement of death receptor TNFR1 triggers the assembly of the membrane-proximal complex I composed of TNFR1, TRADD, RIP1, cIAP1/2 and TRAF2/5. Complex I can mediate through TAK1 recruitment activation of NF-κB pathway leading to upregulation of survival genes. Depending on context, complex I can also undergo endocytosis resulting in dissociation of TNFR1, deubiquitination of RIP1 by CYLD and A20 and recruitment of FADD and procaspase-8 to form complex II. Within complex II, procaspase-8 is activated and can promote apoptosis by activating the caspase cascade. Caspase-8 can also cleave and inactivate RIP1 and RIP3 inhibiting thereby either necrosis or pro-survival NF-κB-mediated pathway. However, if caspase-8 is inhibited or deleted, RIP1 recruits RIP3 to the complex, where they are phosphorylated, and complex II promotes MPTP opening via its subunit CypD activating eventually necrosis
Autophagy
The continuous renewal of cellular components requires the removal of worn-out and damaged macromolecules and defective organelles (e.g., mitochondria) for recycling and replacement with newly synthesized ones (Fig. 3). Several complementary degradation systems are employed in the turnover of cellular components. Proteins are digested by a variety of proteases including calcium-dependent neutral proteases (calpains) and multicatalytic proteinase complexes (proteasomes) [24, 25]. More long-lived proteins, other macromolecules, cell membranes and damaged organelles, such as mitochondria, are turned over by a highly conserved process involving intralysosomal degradation termed autophagy. Multiple specialized proteolytic systems including matrix-associated Lon protease and the membrane-bound AAA proteases are implicated in this pathway [26, 27].

Schematic representation of autophagic cell death. Various sublethal stress stimuli induce autophagy through inhibition of the mTOR complex 1 (mTORC1) or activation of the class III PI3K (Vps34). Upon normal nutrient conditions, mTORC1 is associated with and inhibits the ULK1/2 complex. Starvation inhibits the mTORC1 and induces its dissociation from the ULK1/2 complex. This dissociation is coupled to activation of ULK1 kinase activity leading to the activation of autophagy-mediating machinery via largely unknown mechanism. Beclin 1 (BECN1) along with Vps34, Vps15 and Ambra1 form the BECN1 core complex, which is further activated by the interaction with UVRAG and Bif-1 to initiate vesicle nucleation. Rubicon interacts with the BECN1 core complex negatively regulating autophagy. A complex cascade of the ATG-ubiquitin conjugation events mediates elongation of the pre-autophagosome and attachment to damaged organelles and protein aggregates. Maturation of the pre-autophagosome followed by its closure generates the double-membrane autophagosome. The mature autophagosome fuses with lysosome delivering its contents for degradation by the lysosomal enzymes. p53 regulates autophagy in dual fashion: Basal p53 levels (p53b) inhibit AMPK relieving mTORC1-mediated suppression of autophagy, while stress-activated p53 (p53a) induces autophagy through both AMPK activation and transcriptional upregulation of PTEN and DRAM
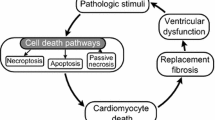
Cell death in heart failure
Apoptosis
The absolute percentage of apoptotic cardiac myocytes in the failing human heart is low (0.08–0.25 % as assessed by TUNEL). However, this percentage of cardiac myocyte apoptosis is 10- to 100-fold higher than that observed in control non-failing hearts (0.001–0.01 %) [28, 29]. These data support the notion that low but increased levels of cardiac myocyte apoptosis result over time in cumulative loss of cardiac myocytes and HF. This possibility was first tested in transgenic mice with a conditionally activated procaspase-8 allele, which showed that rates of cardiac myocyte apoptosis as low as 0.023 % elicited a lethal dilated cardiomyopathy (DCM). Control mice overexpressing an enzymatically dead procaspase-8 remained normal [1]. Interestingly, these levels were up to tenfold lower than those observed in failing human hearts. Conversely, inhibition of cardiac myocyte death in this murine model largely prevents the development of cardiac dilation and contractile dysfunction. These data reveal the sufficiency of clinically relevant levels of apoptosis to induce HF.
The necessity of cardiac myocyte apoptosis for HF has been also tested using pan-caspase inhibition in an experimental model of peripartum cardiomyopathy induced by cardiac-specific overexpression of Gαq, a surrogate for humoral stimuli in HF [30]. Pregnancy precipitated lethal HF in 30 % of Gαq transgenic mice. Pre-treatment with a pan-caspase inhibitor reduced cardiac myocyte apoptosis, preserved heart function and rescued mortality. Moreover, reduction in cardiac myocyte apoptosis by caspase inhibition improved left ventricular function and survival in pregnant Gα(q) mice. These data therefore confirm the necessity of cardiac myocyte apoptosis for HF. This concept was also confirmed in other models; for example, after MI in mice, deletion of Bcl-2/adenovirus E1B 19kD-interacting protein 3, a BH3-like protein, reduced pathological remodeling in the periinfarct zone and the resultant HF in 3 weeks [31]. Immediately following ischemia reperfusion (IR), no significant differences were observed between Bnip3(−/−) and WT mice. However, at 2 days after IR, apoptosis was diminished in Bnip3(−/−) periinfarct and remote myocardium, and at 3 weeks after IR, Bnip3(−/−) mice exhibited preserved LV systolic performance, diminished LV dilation and decreased ventricular sphericalization. Forced cardiac expression of Bnip3 increased cardiomyocyte apoptosis in unstressed mice causing progressive LV dilation and diminished systolic function. On the other hand, conditional Bnip3 overexpression prior to coronary ligation increased apoptosis and infarct size.
Necrosis
In contrast to apoptosis, necrotic cell death is not energy-requiring and exhibits characteristic features that include swelling of the cell and its organelles, extensive mitochondrial disruption, blebbing and ultimately irreversible disintegration of the plasma membrane. Necrotic disruption of the plasma membrane leads to release of cellular content into the extracellular space (e.g., release of CK from necrotic myocardial cells), which promotes further inflammatory reaction and subsequent damage or death of neighboring cells. Honda et al. [32] have shown that in the absence of phagocytic cells, which remove damaged apoptotic cells, plasma membrane disruption of apoptotic cells can occur leading to secondary necrosis. Furthermore, shared signaling pathway elements between these different modes of cell death include mitochondrial-based events such as mitochondrial permeabilization and dysfunction (i.e., both involve MPTP opening), although the mechanistic basis of mitochondrial injury appears to vary in different settings [33]. ATP level is a critical factor determining which type of cell death will proceed [34–36]. If ATP levels fall profoundly, plasma membrane permeabilization and cell rupture will lead to necrosis. If ATP levels are partially maintained, apoptosis, which requires ATP for its progression, follows MPTP opening. In transgenic mice with enhanced sarcolemmal L-type Ca2+ channel activity, progressive myocyte necrosis has led to pump failure and premature death. Importantly, in this model, necrosis associated with dysregulated Ca2+ handling, β-adrenergic receptor signaling and subsequent HF could be prevented by the targeted loss of cyclophilin D, a regulator of the MPTP [37].
Cardiac myocyte necrosis may also play an important role in HF. Inducible transgenic mice with enhanced sarcolemmal L-type Ca2+ channel (LTCC) activity showed progressive myocyte necrosis that led to pump dysfunction and premature death, effects that were dramatically enhanced by acute stimulation of β-adrenergic receptors. Enhanced Ca2+ influx-induced cellular necrosis and cardiomyopathy were prevented with either LTCC blockers or β-adrenergic receptor antagonists, demonstrating a proximal relationship among β-adrenergic receptor function, Ca2+ handling and HF progression through necrotic cell loss. Indeed, cardiac myocyte-specific transgenic overexpression of the β2-α subunit of the L-type Ca2+ channel resulted in Ca2+ overload, MPTP opening, subsequent necrosis and cardiac dysfunction [37]. This phenotype was rescued by deletion of peptidylprolyl isomerase F encoding cyclophilin D, a mitochondrial matrix peptidylprolyl isomerase known to modulate opening of the MPTP encoded by the Ppif gene, but not overexpression of Bcl-2, suggesting that HF in this model is attributable principally to cardiac myocyte necrosis. Doxorubicin-induced cardiomyopathy was ameliorated by knockout peptidylprolyl isomerase F. In contrast to MI, involvement of necrosis in HF is somewhat unexpected, and its role is probably minor.
Recently, a new pathway of regulated necrosis, necroptosis, has been reported in the failing heart. Since this type of cell death is comprehensively discussed in another review within this thematic issue, here it is suffice to say that this type of cell death shows morphological features of both passive necrosis and autophagic cell death.
Autophagy-associated cell death
While autophagy-associated cell death has been considered the most common form of cellular demise during HF, more robust data concerning the relationship of autophagy and HF have been provided by genetic loss- and gain-of-function studies [38].
Autophagy protein-5 deletion precipitates ventricular enlargement and cardiac dysfunction after hemodynamic overload implying that autophagy is a compensatory mechanism during HF. In keeping with its role in disposing of defective proteins, autophagy plays a protective role in HF initiated by proteotoxicity. Thus, autophagy may be protective in response to some cardiomyopathic stimuli and pathogenic in response to others.
A study of failing human hearts has suggested that autophagy-associated cell death is the most common form of cellular demise in HF [39]. Explanted hearts from 19 patients with idiopathic dilated cardiomyopathy (IDC) and seven control hearts were analyzed. Myocyte apoptosis estimated by caspase-3 activation and TUNEL staining occurred at a rate of 0.002 ± 0.0005 % (P < 0.05 versus control), and oncosis assessed by complement 9 labeling at 0.06 ± 0.001 % (P < 0.05). Cellular degeneration including appearance of ubiquitin containing autophagic vacuoles and nuclear disintegration was present at the ultrastructural level. Nuclear and cytosolic ubiquitin/protein accumulations occurred at 0.08 ± 0.004 % (P < 0.05). The ubiquitin-activating enzyme E1 and the ligase E3 were not different from control. In contrast, ubiquitin mRNA levels were 1.8-fold (P < 0.02) elevated, and the conjugating enzyme E2 was 2.3-fold upregulated (P < 0.005). There was a 2.3-fold downregulation of the deubiquitination enzyme isopeptidase-T and 1.5-fold reduction of the ubiquitin-fusion degradation system-1, which in conjunction with unchanged proteasomal subunit levels and proteasomal activity resulted in massive storage of ubiquitin/protein complexes and in autophagic cell death. However, it should be noted that the markers used to diagnose various forms of cell death were not specific.
Autophagy can be considered as a third type of cell death, but in contrast to necrosis, autophagic cell death, similar to apoptosis, is characterized by the absence of tissue inflammatory response. Also, in contrast to apoptosis, which is characterized by early collapse of cytoskeletal elements with preservation of organelles until late in the process, autophagic cell death exhibits early degradation of organelles and preservation of cytoskeletal elements until later stages. In addition, unlike apoptosis, caspase activation and DNA fragmentation occur very late (if at all) in autophagic cell death [40]. Although the information currently available concerning involved signaling pathways and interaction with mitochondria is rather limited, autophagy appears to be triggered by mitochondrial ROS. The role(s) of defective mitochondria in autophagosome biogenesis and in selective cell removal in aging are being actively explored [41].
Steps involved in autophagy include: (1) formation of a double-membrane-enclosed vacuole (autophagosome) within the cell; (2) sequestering of the material to be degraded into an autophagosome; (3) fusion of the autophagosome with a lysosome or a late endosome and (4) the enzymatic degradation of the sequestered materials by a large spectrum of lysosomal enzymes (i.e., acid hydrolase or lysosomal acid lipase). Variations on this theme include the lysosomal incorporation of cytoplasmic components without a preliminary sequestration through invaginations of the membrane (microautophagy as compared to macroautophagy) and a process termed chaperone-mediated autophagy in which particular cytosolic proteins are selectively transported to lysosomes by molecular chaperones, including several heat-shock proteins (HSP) [42, 43]. Multiple signaling pathways including target of rapamycin (TOR) or mammalian target of rapamycin (mTOR), phosphatidylinositol 3-kinase-I (PI3K-I)/PKB, various GTPases, calcium and protein synthesis contribute to autophagy. Evolutionary conserved autophagy-related proteins (Atg proteins), involved in autophagosome formation and function, have been identified in yeast, worms, fruit flies and mammals [44].
Two Atg proteins (Atg12 and Atg8) have been found to be ubiquitin-like proteins indicating that ubiquitination may be involved in autophagic degradation, in addition to its well-documented role in targeting proteins for degradation by the proteasome [45].
Autophagy of defective mitochondria, peroxisomes and possibly other organelles may be selective rather than random. The term “mitophagy” has been introduced to describe selective autophagic removal of damaged mitochondria [46]. It will be of interest to find out whether this selective mechanism is affected in HF and whether it plays a role in selective degradation of mitochondria containing high levels of mtDNA mutations preventing thereby their accumulation [46].
Quantification of autophagic activity using “autophagy reporter” was carried out by Zhu et al. [47] in mice and found that cardiomyocyte autophagy can be induced by short-term nutrient deprivation in vivo. Pressure overload induced by aortic banding was followed by HF associated with markedly increased cardiac autophagy. Load-induced autophagic activity peaked at 48 h and remained significantly elevated for at least 3 weeks. Interestingly, autophagic activity has not been spatially homogeneous, but rather has been seen at particularly high levels in specific areas of the heart such as the basal septum. Heterozygous disruption of the gene coding Beclin 1, a protein required for early autophagosome formation, decreased cardiomyocyte autophagy and diminished pathological remodeling induced by severe pressure stress. Conversely, Beclin 1 overexpression heightened autophagic activity and accentuated pathological remodeling. These findings suggest that autophagy is implicated in the pathogenesis of load-induced HF, and autophagy-mediated factors may be important targets for therapeutic intervention.
In a hamster model of cardiomyopathy, myocardial autophagy-related proteins, such as ubiquitin, cathepsin D and Rab7, have been upregulated and autophagic degeneration and death have been found to be important contributors to the loss of cardiomyocyte function [48]. Treatment with granulocyte colony-stimulating factor (G-CSF) has significantly improved survival, cardiac function and remodeling in these animals, and such beneficial effects have been accompanied by a reduction in autophagy, an increase in cardiomyocyte size and a reduction in myocardial fibrosis.
The interaction between deficient mitochondria and lysosomes has received attention as a potentially critical element of aging, since both organelles appear to suffer from significant age-related alterations in post-mitotic cells. Interestingly, in multiple aging cell-types, an increasing number of senescent mitochondria undergo both enlargement and gradual structural alterations including loss of cristae and swelling associated with decreased capacity to produce energy. In addition, the aging heart markedly increases the amount of oxidative damaged cytosolic proteins and results in a surfeit of increasingly indigestible (due to cross-linking) macromolecules, which comprise the lysosomal substrates including the aging-related deposition of the undegradable pigment lipofuscin, most of which is refractory to removal by exocytosis [49]. Moreover, the activities of both the proteasomal system and the lysosomal proteases have been shown to be markedly decreased in post-mitotic human fibroblasts cultured under hyperoxic conditions to facilitate age-related oxidative senescence [50]. Lysosomes from 22-month-old rats show lower rates of chaperone-mediated autophagy, and both substrate binding (due to lower levels of specific receptors) to the lysosomal membrane and transport into lysosomes decline with age [43]. Therefore, given that lysosomes responsible for mitochondrial removal experience a decrease in their activity, it is not surprising that the rate of total mitochondrial protein turnover declines with age [51]. Several mechanisms may potentially contribute to the age-related accumulation of damaged mitochondria following initial oxidative injury, including the clonal expansion of defective mitochondria, a reduction in the number of mitochondria targeted for mitophagy, suppressed autophagy because of the increased load of undegradable and irreversibly oxidized substrate, and decreased efficiency of specific proteases [52].
Abnormal autophagic degradation of damaged macromolecules and organelles, termed biological “garbage,” is also considered as an important contributor to death of post-mitotic cells, including cardiomyocytes. A dramatic accumulation of small mitochondria has been found in cardiomyocytes treated with 3-methyladenine (3-MA) to inhibit autophagocytosis, whereas the number of large organelles is increased only slightly suggesting that small mitochondria are autophagocytosed more efficiently [53]. However, the mechanism of the selective recruitment of damaged mitochondria to autophagosomes remains to be determined.
A further consequence of aging-induced accumulation of oxidized substrates is lysosomal rupture. Due to autophagy and degradation of iron-containing proteins, such as iron-containing metalloproteins, including cytochromes and ferritin, lysosomes contain a pool of redox-active iron, which makes these organelles particularly susceptible to oxidative damage; chelation of the intralysosomal pool of redox-active iron prevents these effects. OS above a certain limit causes lysosomal rupture due to intralysosomal iron-catalyzed peroxidative reactions [54, 55]. Moderate release of hydrolytic lysosomal enzymes to the cytosol can initiate mitochondrial apoptotic pathway, whereas a more pronounced release of lysosomal enzymes can result in necrotic cell death [56]. This interrelated mitochondrial and lysosomal damage can eventually lead to functional failure and death of cardiomyocytes. Hence, while under some conditions (e.g., ischemia), autophagy may provide cardioprotection and stress resistance to chronic ischemic insult [57], and autophagy can also lead to detrimental loss of cardiomyocytes. Autophagic cell death has been well documented in myocardial cells from hypertrophied, failing and hibernating myocardium, although its role in the cardiomyopathy of the aging heart remains to be determined and quantitated [58].
Loss of membrane potential, fission of mitochondria and ubiquitination are important steps that direct specific autophagic degradation of mitochondria [59], a mechanism which is paramount for normal cardiac function during a life span [60].
Transgenic mice haplo-insufficient for the autophagy gene Beclin 1 have reduced autophagic activity and improved cardiac function in HF induced by pressure overload. Consistently, mice carrying a cardiac-specific aMHC-Beclin1 transgene, to increase autophagic capacity, are more sensitive to pressure overload [47]. This finding suggests that in the context of pressure overload, autophagy may be maladaptive [59]. On the other hand, in the failing heart, upregulation of autophagy may be an adaptive response [40]. Complete loss of autophagy, as in the mice with a cardiomyocyte-specific disruption of Atg5, results in increased sensitivity to pressure overload [41].
Conceivably, mitophagy might be a significant target in the treatment of HF and other cardiovascular diseases. However, as it has been suggested by Iglewski et al. [59], it is critical for the initiation of efficient therapeutic treatment to determine at which stage mitophagy is adaptive and when it is maladaptive. Indeed, excessive mitophagy may deplete the mitochondrial population, and if it falls below the required level to maintain cardiomyocyte integrity will lead to deterioration in cardiac function and eventually to the death of individual cardiomyocytes. On the other hand, failure to remove damaged mitochondria could increase cellular damage from excessive ROS generated by defective mitochondria. Obviously, further research on how to control these processes in the management of HF appears to be a significant challenge to be overcome.
Discussion
In comparison with the massive, short-lived burst of cell death happening during myocardial infarction (MI), the absolute magnitude of cell death (by TUNEL) in failing human hearts is quite low (0.08–0.25 % of cardiac myocytes), but is of orders of magnitude higher than in control hearts (0.001–0.01 %) [29]. Moreover, cardiac myocytes continue to die over the course of advanced HF, suggesting that low levels of cell death could lead to significant cumulative cardiac myocyte loss. Multiple studies have demonstrated that cardiac myocyte apoptosis is a critical component in the pathogenesis of HF. Transgenic experiments demonstrate that very low levels of cardiac myocyte apoptosis are sufficient to cause a lethal cardiomyopathy [1]. Moe et al. [61] have previously reported that early and continuous activation of myocardial apoptosis and pro-apoptotic factors may be an important mechanism that contributes to the progression of HF in canine pacing-induced cardiomyopathy. Conversely, genetic inhibition of cardiac myocyte apoptosis ameliorates HF induced by a variety of stimuli.
The frequency of necrosis in HF models has not yet been studied intensively. Because abnormalities in Ca2 handling are a component of HF and also a trigger of MPTP opening, transgenic mice have been created with inducible, cardiac-specific overexpression of the β2-α subunit of the L-type Ca2 channel [37].These mice exhibit Ca2 overload, spontaneous myocardial necrosis and HF. Interestingly, this phenotype is rescued by the deletion of cyclophilin D (but not Bcl-2 overexpression), implicating necrosis (but not apoptosis) as a causal component in pathogenesis. This conclusion was also tested in a more clinically relevant model of HF, doxorubicin-induced cardiomyopathy. The absence of cyclophilin D confers significant protection against HF in this model. Taken together, these data raise the possibility of a role for cardiac myocyte necrosis in HF.
Besides ROS-mediated damage and cell death leading to cardiac remodeling and eventually to HF, other processes in mitochondria may affect the development and progression of this devastating disease. In cardiomyocytes, attenuation of autophagy, which appears to be cardioprotective, can lead to apoptosis during IR injury, contributing thereby to the development of HF. Mitophagy, induced in response to ischemic stress, mediates the selective autophagic elimination of dysfunctional mitochondria [62, 63]. Albeit an essential role for mitochondrial dynamics in autophagy has been suggested, the mechanistic link of these processes to the pathogenesis of cardiac disease remains to be established [64, 65].
That the genetic machinery of autophagy may be essential for cell death in certain settings was recently confirmed by Liu and Levine [66]. They identified a novel form of autophagy gene-dependent cell death, termed autosis, which is mediated by the Na+K+-ATPase pump and has specific morphological features. As occurs with treatment with autophagy-inducing peptides, starvation, or in vivo during certain types of ischemia, high levels of cellular autophagy can trigger autosis. As the authors indicated, these findings provide new insights into the mechanisms and new strategies to prevent cell death during extreme stress conditions.
All things considered, the complexity of the signaling pathways and regulatory mechanisms impinging on cell death will require an integrative approach to bring together diverse information obtained from functional and genome-scale data (genomics, transcriptomics, proteomics, interactomics) analyses across a number of experimental and clinical contexts [67].
Conclusions
-
All multicellular organisms develop during evolution the highly regulated and interconnected pathways of cell death.
-
While cell death has initially been considered as an accidental process, it is now well recognized that many aspects of cell death are tightly regulated.
-
Based on its morphological characteristics, cell death can be classified into three types: apoptosis, necrosis and autophagy.
-
In comparison with the massive, short-lived burst of cell death that occurs during MI, the absolute magnitude of cell death in the failing human heart is quite low, but is of orders of magnitude higher than in control hearts
-
Apoptosis and necrosis are mediated by distinct, while overlapping central pathways
-
Experimental models have substantiated the sufficiency of clinically relevant degrees of apoptosis to induce HF, as well as the necessity of myocyte apoptosis for the development of HF.
-
Activation of death receptors can initiate not only extrinsic apoptotic pathway, but also necrosis.
-
In the death receptor or extrinsic pathway, a variety of death ligands bind their cognate receptors to trigger cell death.
-
Binding of ligand to receptor induces the formation of either of two multiprotein complexes: the death-inducing signaling complex (DISC) and complex I. The DISC signals apoptosis, whereas complex I can signal either apoptosis, necrosis, or promote cell survival.
-
Cardiac myocyte necrosis may also play a role in HF. Cardiac myocyte-specific transgenic overexpression of the β2-α subunit of the L-type Ca2+ channel resulted in Ca2+ overload, MPTP opening, necrosis and cardiac dysfunction
-
Necrosis is triggered by the interaction of RIP1 with RIP3, a second serine/threonine kinase, resulting in a complex series of crossphosphorylation events.
-
Autophagy can be considered as a third type of cell death, but in contrast to necrosis, autophagic cell death similar to apoptosis is characterized by the absence of tissue inflammatory response.
-
Autophagy, which is characterized by the massive formation of lysosomal-derived vesicles, containing degenerating cytoplasmic contents, is primarily a survival response to nutrient deprivation.
-
A study of failing human hearts suggested that autophagy-associated cell death is the most common form of cellular demise in HF.
-
A selective form of autophagy, mitophagy, is a protective mechanism that allows to eliminate damaged mitochondria and thereby to attenuate mitochondria-mediated apoptosis and necrosis in the myocardium.
-
Mitophagy might be a significant target in the treatment of HF and other cardiovascular diseases. However, it has been suggested that it is critical for the initiation of efficient treatment to determine at which stage mitophagy is adaptive and when it is maladaptive.
-
Since autophagy is implicated in the pathogenesis of load-induced HF, autophagy-mediated factors may be important targets for therapeutic intervention.
-
Heterozygous disruption of the gene coding Beclin 1, a protein required for early autophagosome formation, decreased cardiomyocyte autophagy and diminished pathological remodeling induced by severe pressure stress.
-
Autosis, a novel form of autophagy gene-dependent cell death, has particular morphological features and is mediated by the Na+K+-ATPase pump.
References
Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S et al (2003) A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Investig 111:1497–1504
Hein S, Arnon E, Kostin S, Schonburg M, Elsasser A et al (2003) Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation 107:984–991
Danial NN, Korsmeyer SJ (2004) Cell death: critical control points. Cell 116:205–219
Fuchs Y, Steller H (2011) Programmed cell death in animal development and disease. Cell 147:742–758
Micheau O, Tschopp J (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114:181–190
Peter ME, Krammer PH (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 10:26–35
Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M et al (1995) Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. The EMBO journal 14:5579–5588
Pop C, Salvesen GS (2009) Human caspases: activation, specificity, and regulation. J Biol Chem 284:21777–21781
Bao Q, Shi Y (2007) Apoptosome: a platform for the activation of initiator caspases. Cell Death Differ 14:56–65
Boatright KM, Renatus M, Scott FL, Sperandio S, Shin H et al (2003) A unified model for apical caspase activation. Mol Cell 11:529–541
Li H, Zhu H, Xu CJ, Yuan J (1998) Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94:491–501
Bertrand MJ, Milutinovic S, Dickson KM, Ho WC, Boudreault A et al (2008) cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell 30:689–700
Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ (2006) Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell 22:245–257
Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A et al (2008) Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 135:1311–1323
Wang L, Du F, Wang X (2008) TNF-alpha induces two distinct caspase-8 activation pathways. Cell 133:693–703
Lin Y, Devin A, Rodriguez Y, Liu ZG (1999) Cleavage of the death domain kinase RIP by caspase-8 prompts TNF-induced apoptosis. Genes Dev 13:2514–2526
Holler N, Zaru R, Micheau O, Thome M, Attinger A et al (2000) Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol 1:489–495
Vercammen D, Beyaert R, Denecker G, Goossens V, Van Loo G et al (1998) Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med 187:1477–1485
Konstantinidis K, Whelan RS, Kitsis RN (2012) Mechanisms of cell death in heart disease. Arterioscler Thromb Vasc Biol 32:1552–1562
Sun L, Wang H, Wang Z, He S, Chen S et al (2012) Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 148:213–227
Wilkins JM, McConnell C, Tipton PA, Hannink M (2014) A conserved motif mediates both multimer formation and allosteric activation of phosphoglycerate mutase 5. J Biol Chem 289:25137–25148
Perrone GG, Tan SX, Dawes IW (2008) Reactive oxygen species and yeast apoptosis. Biochim Biophys Acta 1783:1354–1368
Vandenabeele P, Melino G (2012) The flick of a switch: which death program to choose? Cell Death Differ 19:1093–1095
Yang Y, Fang S, Jensen JP, Weissman AM, Ashwell JD (2000) Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science 288:874–877
Suzuki Y, Nakabayashi Y, Takahashi R (2001) Ubiquitin-protein ligase activity of X-linked inhibitor of apoptosis protein promotes proteasomal degradation of caspase-3 and enhances its anti-apoptotic effect in Fas-induced cell death. Proc Natl Acad Sci USA 98:8662–8667
Ngo JK, Pomatto LC, Davies KJ (2013) Upregulation of the mitochondrial Lon Protease allows adaptation to acute oxidative stress but dysregulation is associated with chronic stress, disease, and aging. Redox Biol 1:258–264
Tatsuta T, Langer T (2009) AAA proteases in mitochondria: diverse functions of membrane-bound proteolytic machines. Res Microbiol 160:711–717
Guerra S, Leri A, Wang X, Finato N, Di Loreto C et al (1999) Myocyte death in the failing human heart is gender dependent. Circ Res 85:856–866
Saraste A, Pulkki K, Kallajoki M, Heikkila P, Laine P et al (1999) Cardiomyocyte apoptosis and progression of heart failure to transplantation. Eur J Clin Invest 29:380–386
Hayakawa Y, Chandra M, Miao W, Shirani J, Brown JH et al (2003) Inhibition of cardiac myocyte apoptosis improves cardiac function and abolishes mortality in the peripartum cardiomyopathy of Galpha(q) transgenic mice. Circulation 108:3036–3041
Diwan A, Krenz M, Syed FM, Wansapura J, Ren X et al (2007) Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Investig 117:2825–2833
Honda O, Kuroda M, Joja I, Asaumi J, Takeda Y et al (2000) Assessment of secondary necrosis of Jurkat cells using a new microscopic system and double staining method with annexin V and propidium iodide. Int J Oncol 16:283–288
Malhi H, Gores GJ, Lemasters JJ (2006) Apoptosis and necrosis in the liver: a tale of two deaths? Hepatology 43:S31–S44
Kim JS, He L, Lemasters JJ (2003) Mitochondrial permeability transition: a common pathway to necrosis and apoptosis. Biochemical and biophysical research communications 304:463–470
Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP et al (1998) The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366:177–196
Zamzami N, Hirsch T, Dallaporta B, Petit PX, Kroemer G (1997) Mitochondrial implication in accidental and programmed cell death: apoptosis and necrosis. J Bioenerg Biomembr 29:185–193
Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X et al (2007) Ca2 + - and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Investig 117:2431–2444
Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M et al (2012) The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nature communications 3:1078
Kostin S, Pool L, Elsasser A, Hein S, Drexler HC et al (2003) Myocytes die by multiple mechanisms in failing human hearts. Circ Res 92:715–724
Nishida K, Otsu K (2008) Cell death in heart failure. Circ J 72(Suppl A):A17–A21
Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S et al (2007) The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med 13:619–624
Cuervo AM, Dice JF (2000) Age-related decline in chaperone-mediated autophagy. J Biol Chem 275:31505–31513
Cuervo AM (2004) Autophagy: many paths to the same end. Mol Cell Biochem 263:55–72
Levine B, Klionsky DJ (2004) Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6:463–477
Ohsumi Y, Mizushima N (2004) Two ubiquitin-like conjugation systems essential for autophagy. Semin Cell Dev Biol 15:231–236
Lemasters JJ (2005) Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation research 8:3–5
Zhu H, Tannous P, Johnstone JL, Kong Y, Shelton JM et al (2007) Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Investig 117:1782–1793
Takemura G, Miyata S, Kawase Y, Okada H, Maruyama R et al (2006) Autophagic degeneration and death of cardiomyocytes in heart failure. Autophagy 2:212–214
Terman A, Brunk UT (1998) On the degradability and exocytosis of ceroid/lipofuscin in cultured rat cardiac myocytes. Mech Ageing Dev 100:145–156
Grune T, Merker K, Jung T, Sitte N, Davies KJ (2005) Protein oxidation and degradation during postmitotic senescence. Free Radic Biol Med 39:1208–1215
Rooyackers OE, Adey DB, Ades PA, Nair KS (1996) Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci USA 93:15364–15369
Brunk UT, Terman A (2002) The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. European journal of biochemistry/FEBS 269:1996–2002
Terman A, Brunk UT (2005) Autophagy in cardiac myocyte homeostasis, aging, and pathology. Cardiovasc Res 68:355–365
Kurz T, Eaton JW, Brunk UT (2011) The role of lysosomes in iron metabolism and recycling. The international journal of biochemistry & cell biology 43:1686–1697
Brunk UT, Neuzil J, Eaton JW (2001) Lysosomal involvement in apoptosis. Redox report: communications in free radical research 6:91–97
Terman A, Gustafsson B, Brunk UT (2006) The lysosomal-mitochondrial axis theory of postmitotic aging and cell death. Chem Biol Interact 163:29–37
Yan L, Sadoshima J, Vatner DE, Vatner SF (2006) Autophagy: a novel protective mechanism in chronic ischemia. Cell Cycle 5:1175–1177
Kunapuli S, Rosanio S, Schwarz ER (2006) “How do cardiomyocytes die?” apoptosis and autophagic cell death in cardiac myocytes. J Cardiac Fail 12:381–391
Iglewski M, Hill JA, Lavandero S, Rothermel BA (2010) Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hypertens Rep 12:418–425
Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T et al (2010) Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6:600–606
Moe GW, Naik G, Konig A, Lu X, Feng Q (2002) Early and persistent activation of myocardial apoptosis, bax and caspases: insights into mechanisms of progression of heart failure. Pathophysiology 8:183–192
Hamacher-Brady A, Brady NR, Gottlieb RA, Gustafsson AB (2006) Autophagy as a protective response to Bnip3-mediated apoptotic signaling in the heart. Autophagy 2:307–309
Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H et al (2007) Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res 100:914–922
Gottlieb RA, Gustafsson AB (2011) Mitochondrial turnover in the heart. Biochim Biophys Acta 1813:1295–1301
Kuzmicic J, Del Campo A, Lopez-Crisosto C, Morales PE, Pennanen C et al (2011) Mitochondrial dynamics: a potential new therapeutic target for heart failure. Rev Esp Cardiol 64:916–923
Liu Y, Levine B (2015) Autosis and autophagic cell death: the dark side of autophagy. Cell Death Differ 22:367–376
Ng AC (2010) Integrative systems biology and networks in autophagy. Seminars in immunopathology 32:355–361
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Moe, G.W., Marín-García, J. Role of cell death in the progression of heart failure. Heart Fail Rev 21, 157–167 (2016). https://doi.org/10.1007/s10741-016-9532-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-016-9532-0