
Abstract
Hepatocellular carcinoma (HCC) remains challenging due to the lack of efficient therapy. Promoting degradation of certain cancer drivers has become an innovative therapy. The nuclear transcription factor sine oculis homeobox 1 (SIX1) is a key driver for the progression of HCC. Here, we explored the molecular mechanisms of ubiquitination of SIX1 and whether targeting SIX1 degradation might represent a potential strategy for HCC therapy. Through detecting the ubiquitination level of SIX1 in clinical HCC tissues and analyzing TCGA and GEPIA databases, we found that ubiquitin specific peptidase 1 (USP1), a deubiquitinating enzyme, contributed to the lower ubiquitination and high protein level of SIX1 in HCC tissues. In HepG2 and Hep3B cells, activation of EGFR-AKT signaling pathway promoted the expression of USP1 and the stability of its substrates, including SIX1 and ribosomal protein S16 (RPS16). In contrast, suppression of EGFR with gefitinib or knockdown of USP1 restrained EGF-elevated levels of SIX1 and RPS16. We further revealed that SNS-023 (formerly known as BMS-387032) induced degradation of SIX1 and RPS16, whereas this process was reversed by reactivation of EGFR-AKT pathway or overexpression of USP1. Consequently, inactivation of the EGFR-AKT-USP1 axis with SNS-032 led to cell cycle arrest, apoptosis, and suppression of cell proliferation and migration in HCC. Moreover, we showed that sorafenib combined with SNS-032 or gefitinib synergistically inhibited the growth of Hep3B xenografts in vivo. Overall, we identify that both SIX1 and RPS16 are crucial substrates for the EGFR-AKT-USP1 axis-driven growth of HCC, suggesting a potential anti-HCC strategy from a novel perspective.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) is the major type of primary liver cancer with high heterogeneity and mortality worldwide [1]. Due to its extremely heterogeneous characteristics, its early detection and clinical classification are challenging to perform [2,3,4,5]. Thus, most HCC patients present with an advanced stage at the time of diagnosis and have a poor prognosis [3]. Target therapy is one of the most common treatments for advanced HCC. Sorafenib chemotherapy is the first-line therapy for patients with HCC in an advanced stage. Yet, sorafenib has narrow effectiveness considering that less than one-third of patients are sensitive to the therapy. Moreover, HCC patients who initially respond to sorafenib usually develop acquired drug resistance within 6 months [2, 3, 6,7,8]. Therefore, it has become a general goal to continuously deepen the understanding of the accurate molecular mechanism of HCC progression to develop more effective targets.
Sine oculis homeobox 1 (SIX1), a transcription factor to drive cell proliferation, is originally described as a critical regulator of organogenesis during embryonic development [9]. Recently, increasing studies have reported that SIX1 is reactivated or highly expressed in a variety of malignant tumors, including breast cancer [10,11,12], HCC [13,14,15], and ovarian cancer [16]. In HCC, SIX1 regulates the Warburg effect, which further facilitates tumor cell metabolism to match the rapid proliferation of HCC [15]. SIX1 is degraded by the proteasome post the anaphase-promoting complex (APC) mediated ubiquitination [17]. Our previous study revealed that the 75 kDa glucose-regulated protein (GRP75) provides a molecular platform to recruit SIX1 and the ubiquitin-specific peptidase 1 (USP1). USP1 is responsible for removing the ubiquitin linkages on SIX1 and further stabilizes its protein level to drive cyclin D1-dependent cell cycle progression and proliferation of prostate cancer [18]. Herein, we addressed whether the abnormal ubiquitination of SIX1 could be observed in HCC tissues and the way it may regulate the progression of HCC.
The ubiquitin-proteasome system (UPS) can recognize and eliminate the substrates labeled with ubiquitin chains through 26 s proteasome. Deubiquitinating enzymes (DUBs) are critical components of the UPS that reverse the ubiquitination reaction to achieve precise quality control of the substrates [19]. Abnormal activation of certain oncoproteins results from the overexpression of DUBs that is frequently observed in various tumors [20,21,22]. For example, USP1 is significantly increased in HCC and leads to the degradation suppression of ribosomal protein S16 (RPS16), thus further facilitating the growth and metastasis of HCC cells [23]. Yet, it remains unclear whether USP1 may also regulate additional oncoproteins in HCC.
SNS-032 is a potent inhibitor of CDK1/2/7/9; its anti-cancer activity has been found in chronic lymphocytic leukemia [24, 25] and uveal melanoma [26]. Moreover, we previously uncovered that SNS-032 is a suppressor of the USP1-SIX1 axis in prostate cancer [27]. USP1 downregulates the ubiquitination level of SIX1 in HCC tissues. In addition, USP1 simultaneously regulates the stability of both SIX1 and RPS16 to drive HCC progression, which is enhanced by the EGFR-AKT signaling pathway. Inhibition of the EGFR-AKT-USP1 axis with SNS-032 notably induces the degradation of both SIX1 and RPS16, and further suppresses the growth of HCC in cultured cells and xenograft models. In this study, we further investigated the role of the EGFR-AKT-USP1-SIX1/RPS16 axis in driving HCC development, but also proposed a potential anticancer strategy by triggering the degradation of SIX1 and RPS16 with an existing kinase inhibitor.
Materials and methods
HCC samples
Fresh tumor and adjacent normal tissues were collected from patients with HCC. These samples were obtained from the discarded material utilized for routine laboratory tests at the Department of Hepatopancreatic Surgery, First People’s Hospital of Foshan (Foshan, China). This study was performed with the approval of the Medical Ethics Committee of Guangzhou Medical University and the First People’s Hospital of Foshan. The fresh tissues were minced in protease inhibitor cocktails-supplemented tissue RIPA buffer, and prepared into tissue suspension in Precellys® Evolution (Bead Mill Homogenizer, Bertin, France). Tissues were then sonicated and centrifuged at 4 °C and 13,000 r/min for 15 min. The supernatant protein samples were collected for subsequent Co-IP and Western blotting assays. The whole process of protein extraction is operated at 4 °C. The basic information of patients with HCC is listed in Table S1.
Cell culture and reagents
HepG2, Hep3B, HCC LM3, and HEK293T cell lines were all purchased from ATCC (American Type Culture Collection) and validated by the short tandem repeat. The culture method was introduced, as reported previously [23]. Cell culture conditions are listed in Table S2. Information on reagents, including antibodies and chemicals are listed in Table S3 and Table S4.
Western blotting and co-immunoprecipitation (Co-IP)
As classical methods for detecting the expression levels of certain proteins and protein interactions, Western blotting and Co-IP are clearly described in our previous publications [18, 28]. Western blot assay was performed in accordance with the practice guide [29]. Briefly, protein samples with a standardized total protein concentration were transferred to a PVDF membrane after separation by electrophoresis with 12% SDS-PAGE gel. After the PVDF membrane was sealed with 5% skim milk for 1 h, it was allowed to react appropriately with the corresponding primary and secondary antibodies (see Table S3) respectively. Finally, the X-ray film was used to capture the fluorescence generated by a chemical reaction between the ECL reagent and secondary antibodies.
Co-IP assay was performed according to the instructions of Dynabeads™ Co-immunoprecipitation Kit (#14321D) (Carlsbad, CA, USA). A rotary shaker was used to incubate the dynabeads with antibodies (see Table S3) overnight to ensure that the antibodies were fully absorbed in the dynabeads. Next, excessive free antibodies were removed from the dynabeads, followed by incubation with cell lysates for 1 h. After removing the excessive free lysates, the interacting proteins were separated by a 70 °C water bath and high-speed centrifugation.
Transfection of plasmids and siRNAs
The transfection methods have been introduced in our previous reports [30, 31]. The wild type of human USP1 plasmids, the truncated mutant types of USP1 (1-200aa, 201-785aa, 1-400aa, 401-785aa) plasmids (CMV-MCS-Flag-SV40-neomycin), and the human SIX1 plasmids (CMV-MCS-HA-SV40-neomycin) were purchased from GeneChem (Shanghai, China). Transfection was performed as follows: 500 μL RPMI opti-MEM (Gibco, Invitrogen, Paisley, UK), lipofectamine 3000 (Invitrogen) (4 μL of P3000 and 4 μL of lipofectamine 3000) and 2 μg plasmids were gently mixed a 6 cm dish, and then the mixture was allowed to stand for 15 min. Next, the mixture was added to the cells pre-seeded and cultured for 24 h to ensure the final concentration of plasmids was 0.75 μg/mL. Finally, we replaced the medium for the cells after 6–12 h and continued to culture for 48 h.
The si-RNAs used in the study included si-USP1, si-SIX1 and si-RPS16 purchased from RiboBio (Guangzhou, China). We prepared the transfection mixture with 500 μL RPMI opti-MEM, 6 μL lipofectamine RNAiMax (Invitrogen), and 10 μL siRNAs allowing it to stand and react for 15 min. Next, the mixture was added to the cells pre-seeded and cultured for 24 h. The cell medium was replaced after 6–12 h, and the cells were cultured for an additional 48 h. The final concentration of si-RNAs was 50 nM. The sequences of si-RNAs are listed in Table S5.
Immunofluorescence assay
As previously described [32], HCC cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton-X, and blocked with 5% bovine serum albumin (BSA). Then, the cells were incubated with primary antibodies and fluorescent secondary antibodies (see Table S3), respectively. The nucleus was visualized by DAPI (Abcam, #ab104139) staining. Cold PBS solution was used to wash 2 to 3 times in the interval of each operation step. The immunofluorescence images were captured using a confocal microscope (Leica TCS SP8).
Cell proliferation assays
Cell proliferation assays include the determination of cell viability, analysis of colony formation ability and EdU staining assay. For cell viability, cells were seeded in a 96-well plate (2000 cells/well) for 24 h, after which they were treated with the corresponding chemicals indicated in the figures. Next, 20 μL MTS reagent was added to each well for 3 h in the dark. Finally, the absorbance value at 490 nm with a microplate reader (Sunrise, Tecan) was adopted to calculate percent cell viability.
For colony formation assay, the treated cells were digested with trypsin, terminated with a medium containing FBS, centrifuged at 1000 r/min for 5 min, and then resuspended in the medium. The number of cells was calculated with a counting chamber. Next, the resuspended cells were randomly seeded in a 6-well plate and the culture was continued for 2 weeks. Finally, the cells were fixed with 4% paraformaldehyde, washed with PBS, and stained with crystal violet solution.
EdU staining assay was used to detect the DNA replication status of cells, which is also used to determine cell proliferation ability. The increment of positive cells is considered to have a strong proliferation ability. The experiment was carried out in accordance with the instructions of EdU reagents (RiboBio, Guangzhou, China).
Cell migration assay
HCC cells seeded in 6-well plates overnight were exposed to SNS-032 with or without EGF 24 h. Next, the cells were collected and resuspended in 5% serum-containing medium and then reseeded in the upper chamber of the trans-well at the concentration of 1 × 105 cells/well; 10% serum-containing medium was added to the lower chamber of the trans-well. HCC cells migrated downward through the media for 2 days. Cells in the upper chamber that did not pass through the media were gently wiped off with a sterile cotton swab. HCC cells on the lower surface of the media were then fixed with 4 % paraformaldehyde for 15 min, washed with PBS solution and stained with 1 % crystal violet solution for 5 min. All experiments were performed at least in triplicate.
Flow cytometry analysis
Cell cycle distribution and apoptosis were generally analyzed by flow cytometry as reported previously [33, 34]. A Cell Cycle Detection Kit (KeyGEN BioTECH, Nanjing, China) was used for the determination of cell cycle. An Annexin V-FITC/PI Double Stain Apoptosis Detection Kit (KeyGEN BioTECH, Nanjing, China) was used to detect apoptosis as the product instructions.
Animal models
BALB/c nude mice (5–6 weeks old, 18–20 g) were purchased from Vital River Laboratory Animal Technology Co., Ltd (Beijing, China). All the animals were housed in an environment with a temperature of 22 ± 1 °C, relative humidity of 50% ± 1%, and a light/dark cycle of 12/12 h. All animal studies (including the mice euthanasia procedure) were done in compliance with the regulations and guidelines of Guangzhou Medical University institutional animal care and conducted according to the AAALAC and the IACUC guidelines.
After one week of animal acclimatization, the in vivo experiment was performed. Hep3B cells (3 × 106/100 μL PBS/mouse) were subcutaneously inoculated on the dorsal side of nude mice. When the transplanted tumor grew to an average of 50 mm3, the mice were randomly divided into 3 groups (8 mice/group): mice treated with SNS-032 (15 mg·kg−1·d−1, i.p.), mice treated with ML323 (40 mg·kg−1·d−1, i.p.), and mice treated with the vehicle; the treatment was performed every two days. Moreover, additional mice were divided into 6 more groups (8 mice/group): mice treated with sorafenib (20 mg·kg−1·d−1, p.o.), mice treated with SNS-032 (15 mg·kg−1·d−1, i.p.), mice treated with sorafenib+SNS-032, mice treated with gefitinib (30 mg·kg−1·d−1, p.o.), mice treated with gefitinib+sorafenib, and mice treated with vehicle; the treatment was performed every other day. After 21 days, the mice were also sacrificed by vertebrate dislocation post CO2 inhalation. Finally, the body weight of mice, tumor volume, and tumor weight were recorded. Tumor volume was calculated as length × width2 × 0.5.
Statistical analysis
Data in this study are shown as mean ± S.D. from three independent experiments. The unpaired Student’s t-test or one-way ANOVA was used appropriately. GraphPad Prism 9.0 and SPSS 16.0 software were used for producing charts and statistical analysis. A P value <0.05 was considered statistically significant (#P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001).
Results
The ubiquitination level of SIX1 is decreased and associated with the overexpression of USP1 in HCC tissues
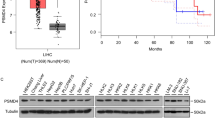
To investigate the abnormal ubiquitination of SIX1 in HCC, we randomly detected the ubiquitination level of SIX1 in HCC tissues and adjacent tissues derived from 6 patients using Co-IP and Western blotting. The results showed that the ubiquitination level of SIX1 was decreased, while the protein level of SIX1 was increased in HCC tissues. Meanwhile, SIX1 interacted with APC (a previously identified E3 ligase of SIX1) and USP1 (a DUB of SIX1 identified in prostate cancer) in HCC tissues (Fig. 1a).

a Co-IP and Western blotting assays were collectively performed to detect ubiquitination levels of SIX1 in HCC tissues and adjacent normal tissues of 6 HCC patients (number #1, #3, #5, #9, #12 and #15). Quantitative data on the ubiquitination levels are shown on the right. Mean ± SD (n = 6). b Western blotting performed to detect differences in the expression levels of SIX1, USP1, and APC in tumor tissues and adjacent normal tissues of 16 HCC patients. c Quantitative data of (b) are shown. Mean ± SD (n = 16). d Correlation analysis of SIX1 with USP1 or APC protein levels based on (c) using Pearson r assay. The data of tumor tissues and adjacent normal tissues are included in the statistics (n = 32). e Comparison of SIX1, USP1 and APC at mRNA expressions in tumor tissues and normal tissues of HCC patients from the TCGA database. f Kaplan–Meier curves from patients with HCC expressing low and high SIX1/APC from the tissue microarray from the TCGA database. #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001.
Next, the immunoblotting assay was performed to detect the expression of SIX1, USP1, and APC in HCC tissues and adjacent tissues derived from 16 patients. The expression of SIX1 and USP1, but not APC, was increased in HCC tissues (Fig. 1b, c). Additionally, the expression of USP1, but not APC, was positively correlated with the protein level of SIX1 in HCC tissues and adjacent tissues (Fig. 1d). Moreover, after analyzing the public TCGA and GEPIA database, it was found that the mRNA level of SIX1 and USP1, but not APC, was increased in HCC tissues (Figs. 1e and S1a, b). In addition, higher expression of SIX1 and USP1 (see previously published data [23]) predicted a poorer survival rate of HCC patients, while the expression of APC failed to predict survival probability in HCC (Fig. 1f).
Through analysis of the UALCAN database, a higher level of SIX1 and RPS16 (a downstream substrate of USP1 in HCC) was associated with a higher-grade HCC (Fig. S1c, d). These findings collectively demonstrate that downregulation of SIX1 ubiquitination, which is associated with USP1, contributes to the high protein level of SIX1 and the development of HCC.
USP1 interacts with SIX1 and RPS16 through its C-terminal
To determine whether SIX1 is a definite substrate of USP1 in HCC, the protein interaction between endogenous USP1 and SIX1 in HCC cell lines was analyzed by Co-IP and immunoblotting assays (Fig. 2a). Briefly, the interaction was observed in both nucleus and cytoplasm (Fig. 2b).

a Co-IP and Western blotting assays were performed to determine the interaction of USP1-SIX1. b An immunofluorescence assay was performed using FLAG and SIX1 antibodies in HepG2 and Hep3B cells transfected with FLAG-USP1 plasmids. Scale bars, 25 μm. c Schematic diagram of FLAG-labeled human USP1 full-length and truncated mutant plasmids. d FLAG-labeled human USP1 full-length and truncated mutant plasmids were co-transfected with the HA-SIX1 plasmids into HEK293T cells for 48 h. Co-IP and Western blotting assays were performed to detect the interaction between FLAG and HA. e Co-IP and Western blotting assays were performed to detect ubiquitination levels of SIX1 in HepG2 cells transfected with USP1 si-RNA-1, −2 or control si-RNA for 48 h, and exposed to MG132 (10 μM) for 6 h before harvest. f Co-IP and Western blotting assays were performed using USP1 antibodies to detect the interaction of USP1-SIX1 and USP1-RPS16 in HepG2 cells transfected with SIX1 si-RNAs, RPS16 si-RNAs, or control si-RNAs for 48 h.
To further determine the binding region of USP1 and SIX1, 4 USP1 truncated mutants labeled with the FLAG-tag at their C-terminals (including FLAG-USP1-MT1, FLAG-USP1-MT2, FLAG-USP1-MT3, and FLAG-USP1-MT4) were constructed, and then co-transfected with the HA-SIX1 plasmids into HEK293T cells. Co-IP analysis showed that SIX1 interacts with FLAG-USP1-MT2 and FLAG-USP1-MT4, but not FLAG-USP1-MT1 or FLAG-USP1-MT3, which suggests that SIX1 binds to the C-terminal of USP1 (Fig. 2c, d). These results are consistent with our previous findings, which suggest that USP1 binds to RPS16 through its C-terminal [23].
Co-IP and immunoblotting assays were then used to determine the role of USP1 in SIX1 ubiquitination in HCC cells. The knockdown of USP1 aggravated K48-ubiquitination and pan-ubiquitination of SIX1 in HepG2 cells (Fig. 2e), suggesting that USP1 as a DUB mediates the deubiquitination and stabilization of SIX1 in HCC.
Next, we examined whether RPS16 and SIX1 may competitively bind to USP1 and whether USP1 is sufficient to support the protein stability of both SIX1 and RPS16. To address this question, we performed a Co-IP experiment to determine the protein interaction of USP1-SIX1 in HCC cells treated with si-RPS16, and the protein interaction of USP1-RPS16 in HCC cells treated with si-SIX1. The results showed that knockdown of RPS16 did not alter the interaction of USP1-SIX1 and the expression of SIX1; conversely, knockdown of SIX1 did not affect the interaction of USP1-RPS16 and the expression of RPS16 (Fig. 2f), indicating that cellular USP1 is sufficient to simultaneously maintain the protein stability of SIX1 and RPS16 in HCC.
EGFR signaling pathway promotes the expression of USP1 and its substrates
The EGFR signaling pathway is critical for developing HCC and is an important target for cancer therapy [35, 36]. Therefore, we examined whether its pathway may control the USP1-SIX1/RPS16 axis. Immunoblotting and immunofluorescence assays showed that the activation of EGFR triggered by EGF notably increases the expression of USP1, SIX1, and RPS16 (Fig. 3a–c). Additionally, inactivation of the signaling pathway with gefitinib (an EGFR inhibitor) or MK-2206 (an AKT inhibitor) inhibits the EGF-induced upregulation of USP1, SIX1, and RPS16 (Fig. 3d). Also, the knockdown of USP1 suppresses the upregulation of SIX1, and RPS16 induced by the activation of EGFR signaling pathway (Fig. 3e). Moreover, Co-IP and immunoblotting assays showed that EGF increases the bindings of USP1-SIX1/RPS16 and decreases the pan-/K48 linked- ubiquitination levels of SIX1 and RPS16, whereas this process is blocked by the treatment of gefitinib (Fig. 3f), suggesting that the activation of EGFR suppresses the ubiquitination and degradation of SIX1 and RPS16. Together, these findings indicate that the EGFR signaling pathway promotes the USP1-SIX1/RPS16 axis to drive the development of HCC.

a Western blotting of p-EGFR, EGFR, p-AKT, AKT, USP1, SIX1, and RPS16 in HepG2 and Hep3B cells exposed to EGF for 24 h. b Western blotting of p-EGFR, EGFR, p-AKT, AKT, USP1, SIX1, and RPS16 in HepG2 cells exposed to EGF for 6, 12, and 24 h. c An immunofluorescence assay was performed using USP1, SIX1 and RPS16 antibodies in HepG2 cells exposed to EGF for 24 h. Scale bars, 10 μm. Quantitative data are shown below. Mean ± SD (n = 3). d Western blotting of p-EGFR, EGFR, p-AKT, AKT, USP1, SIX1 and RPS16 in HepG2 and Hep3B cells treated with gefitinib or MK-2206 or DMSO for 9 h, and then exposed to EGF for 24 h. e Western blotting of USP1, SIX1 and RPS16 in HepG2 cells transfected with USP1 si-RNAs or control si-RNAs for 24 h, and then exposed to EGF for 12 h. f Co-IP and Western blotting assays were performed to detect the ubiquitination levels of SIX1 and RPS16 in HepG2 cells treated with gefitinib or DMSO for 9 h and then exposed to EGF for 12 h. Cells were exposed to MG132 (10 μM) for 6 h before harvest. ##P < 0.01, ###P < 0.001.
SNS-032 induces degradation of SIX1 and RPS16 by the inhibition of the EGFR-AKT-USP1 axis
We previously identified SNS-032 as a new inhibitor of USP1 in prostate cancer cells. In this study, we further investigated whether SNS-032 may alter the expression and stability of SIX1 and RPS16 in HCC cells and discovered that SNS-032 downregulates the protein levels of USP1, SIX1 and RPS16 in a concentration- and time-dependent manner (Fig. 4a). Next, we speculated that SNS-032 might destroy protein stability of the substrates of USP1. Cycloheximide (CHX)-tracking assay was performed to determine the speed of SIX1 and RPS16 degradation in HCC cells treated with SNS-032. SNS-032 accelerated the degradation rate of both SIX1 and RPS16 (Fig. 4b, c). Additionally, the SNS-032-induced reduction of SIX1 and RPS16 was significantly reversed by the proteasome inhibitor bortezomib (BTZ) (Fig. S2a, b). Furthermore, Co-IP analysis showed that SNS-032 triggers the accumulation of ubiquitination of SIX1 and RPS16 in HCC cells (Fig. 4d). Overexpression of FLAG-USP1 reverses the SNS-032-induced downregulation of SIX1 and RPS16 (Fig. 4e). Moreover, SNS-032 inhibits EGFR signaling pathway-mediated upregulation of SIX1 and RPS16 (Fig. 4f). Together, these results indicate that SNS-032 promotes the ubiquitin-mediated degradation of SIX1 and RPS16 by depressing the EGFR-AKT-USP1 axis.

a Western blotting of USP1, SIX1 and RPS16 in HepG2 and Hep3B cells exposed to SNS-032 for 24 h or 6, 12, 24 h. b Western blotting of SIX1 level in HepG2 cells treated with SNS-032 (1 μM) or DMSO for 3 h, and then exposed to cycloheximide (CHX) for 0 to 9 h. Quantitative data are shown on the right. Mean ± SD (n = 3). c Western blotting of RPS16 level in HepG2 cells treated with SNS-032 (1 μM) or DMSO for 12 h, and then exposed to cycloheximide (CHX) for 0 to 36 h. Quantitative data are shown on the right. Mean ± SD (n = 3). d Co-IP and Western blotting assays were performed to detect the ubiquitination levels of SIX1 and RPS16 in HepG2 cells treated with SNS-032. Cells were exposed to MG132 (10 μM) for 6 h before harvest. To detect SIX1 ubiquitination, cells were treated with SNS-032 for 6 h. To detect RPS16 ubiquitination, cells were treated with SNS-032 for 18 h. e Western blotting of FLAG, USP1, SIX1 and RPS16 in HepG2 cells transfected with FLAG-USP1 plasmids or Mock (control plasmids) for 24 h, and then exposed to SNS-032 for 24 h. f Western blotting of p-EGFR, EGFR, p-AKT, AKT, USP1, SIX1 and RPS16 in HepG2 and Hep3B cells exposed to SNS-032 with or without EGF for 24 h. ##P < 0.01, ###P < 0.001, ####P < 0.0001.
Activation of EGFR reverses the SNS-032-mediated growth inhibition of HCC cells
To estimate the anti-cancer property of SNS-032 in HCC, the effect of SNS-032 on cell viability, colony formation, and proliferation ability in HCC cells was analyzed. SNS-032 notably restrained the cell viability, colony formation, and cell proliferation in HCC cells (Fig. 5a–c). Next, we examined whether cell cycle arrest might be involved in the SNS-032-induced growth inhibition. To verify this hypothesis, flow cytometry and Western blotting were used to detect cell cycle distribution and cell cycle-related molecules, respectively. SNS-032 increased the distribution at G0/G1 phase (Fig. S3a). The immunoblotting analysis further showed that SNS-032 increased the protein level of cell cycle inhibitor p21, and decreased the protein level of cell cycle promoter Cyclin D1 (Fig. S3b, c). Together, our data indicate that SNS-032 impedes the proliferation of HCC cells partially via cell cycle arrest.

a Cell viability assay of HepG2, Hep3B, and HCCLM3 cells exposed to SNS-032 for 24, 48, and 72 h. Mean ± SD (n = 3). b Colony formation assays were performed in HepG2 and Hep3B cells exposed to SNS-032 for 24 h, followed by re-seeded and cultured in the 6-well plates for 2 weeks. Quantitative data are shown on the right. Mean ± SD (n = 3). c EdU staining assays were performed in HepG2 and Hep3B cells treated with SNS-032 for 24 h. Quantitative data are shown. Mean ± SD (n = 3). d Cell viability assay of HepG2 and Hep3B cells exposed to SNS-032 with or without EGF for 24 h. Mean ± SD (n = 3). e Migration assays were performed in HepG2 and Hep3B cells exposed to SNS-032 with or without EGF for 24 h, followed by re-seeded and cultured in the trans-wells for 2 days. Scale bars, 50 μm. Quantitative data are shown on the right. Mean ± SD (n = 3). f Western blotting of PARP and CC3 (cleaved caspase 3) in HepG2 and Hep3B cells exposed to SNS-032 with or without EGF for 24 h. #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001.
During experiments, we observed that SNS-032 could trigger cell death in HCC under an optical microscope. To quantitatively determine the SNS-032-induced cell death, we used flow cytometry to count apoptotic cells. Apoptosis was significantly induced by SNS-032 (Fig. S4a, b). We further detected the apoptosis-related proteins through immunoblotting, and found that SNS-032 causes PARP cleavage, Caspase 3 activation, and the decrease of mitochondria-associated apoptosis inhibitors, including Bcl-xl and Bcl-2 (Fig. S4c), which indicates that SNS-032 can trigger apoptosis in HCC cells. To further explore whether the SNS-032-triggered apoptosis may depend on caspase activation and whether the alteration of USP1, SIX1, and RPS16 may result from apoptosis, the caspase inhibitor Z-VAD-FMK was used to inactivate the caspase cascade in HCC cells treated with SNS-032. SNS-032-induced apoptosis and apoptotic indicators, but not the expression of USP1, SIX1, or RPS16, were reversed by Z-VAD-FMK (Fig. S4d, e). Our data suggest that SNS-032 induces a canonical caspase-dependent apoptosis; however, the apoptosis does not affect SNS-032-mediated reduction of USP1, SIX1, and RPS16 in HCC cells.
We next wondered whether activation of the EGFR signaling pathway might reverse the SNS-032-induced inhibition of HCC cells. Our cell viability, transwell migration, and Western blotting assays showed that EGF might rescue the SNS-032-mediated growth inhibition, migration suppression, and apoptosis induction of HCC cells (Fig. 5d–f). These findings demonstrate that SNS-032 induces inhibition of HCC cells by suppressing EGFR signaling pathways.
SNS-032 suppresses the proliferation of HCC in vivo
To determine the in vivo anti-cancer activity of SNS-032 in HCC, we further established the xenograft models by inoculating Hep3B cells subcutaneously onto the nude mice. Mice carrying transplanted tumors were randomly divided into three groups, and treated with SNS-032, ML323 (a previously reported inhibitor of USP1), or vehicle every other day. SNS-032 and ML323 significantly suppressed the growth of Hep3B xenografts, yet, the anti-cancer effect of SNS-032 was more obvious than that of ML323 (Fig. 6a–c). Besides, there was no difference in body weight among the three groups (P > 0.05, Fig. 6d). H&E staining assay showed no obvious impairment in the liver and kidney tissues (Fig. S5), indicating that the USP1 inhibitors induce low toxicity. Moreover, immunohistochemical analysis of the transplanted tumor tissues showed that the expression levels of USP1, SIX1, RPS16, and Ki67 were downregulated, while the expression level of cleaved caspase3 was upregulated in the treatment group (Fig. 6e, f), which is highly consistent with our in vitro experiments.

a BALB/c nude mice with Hep3B xenografts were treated with SNS-032 (15 mg·kg−1·d−1) (i.p.) or ML323 (40 mg·kg−1·d−1) (i.p.) or vehicle every other day for a total of 21 days. Xenografts grown in the nude mice are shown. b Tumor size, (c) tumor weight, and (d) body weight of nude mice. Mean ± SD (n = 8). e IHC assays of USP1, SIX1, RPS16, Ki67, and CC3 (cleaved caspase 3) were performed in the tissues from xenografts. Representative images per group are shown at 200×. Scale bars, 50 μm. f Quantitative data of (e). Mean ± SD (n = 3). #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001.
SNS-032 enhances the sensitivity of HCC cells to targeted therapy
USP1-mediated stabilization of oncoproteins has a critical role in promoting the progression of HCC. Targeting USP1 with SNS-032 alone suppresses HCC in cultured cells and xenografts. Next, we wondered whether SNS-032 could sensitize HCC cells to the clinically used anti-cancer drugs, thereby providing a combination strategy for HCC therapy. Sorafenib is the first-line targeted drug for the clinical HCC treatment. However, the effectiveness of sorafenib in improving the survival rate is limited. Therefore, we were prompted to determine the combined effect of SNS-032 or gefitinib and sorafenib in HCC. Cell viability assay showed that the combination of SNS-032 or gefitinib and sorafenib more obviously suppressed the growth of HepG2 and Hep3B cells, compared to either SNS-032, gefitinib or sorafenib alone treatment group (Fig. S6a, b). The colony formation assay was consistent with the viability results (Fig. 7a), indicating that combination treatment with SNS-032 and sorafenib exerts a synergistic effect in suppressing the proliferation of HCC cells. Next, we investigated whether the anticancer effect of sorafenib could be enhanced by the inactivation of EGFR with SNS-032 or gefitinib in vivo. Our animal experiments showed that sorafenib plus SNS-032 or gefitinib synergistically inhibited the growth of Hep3B xenografts (Fig. 7b–d). Meanwhile, these treatments led to common toxic side effects as the body weight of mice was not significantly reduced (Fig. 7e). Our findings demonstrate that the inactivation of the EGFR signaling pathway increases the sensitivity of HCC to sorafenib treatment.

a Colony formation assays were performed in HepG2 and Hep3B cells exposed to SNS-032 with or without sorafenib for 24 h, followed by re-seeded and cultured in the 6-well plates for 2 weeks. Quantitative data are shown on the right. Mean ± SD (n = 3). b BALB/c nude mice with Hep3B xenograft were randomly divided into 6 groups: mice were treated with vehicle, sorafenib (20 mg·kg−1·d−1) (p.o.), SNS-032 (15 mg·kg−1·d−1) (i.p.), gefitinib (30 mg·kg−1·d−1) (p.o.), sorafenib + SNS-032, or sorafenib + gefitinib every other day respectively, for a total 21 days. Xenografts grown in nude mice are shown. c Tumor size, (d) tumor weight, and (e) body weight of nude mice. Mean ± SD (n = 8). f A hypothetical model by which SNS-032 targets the EGFR/USP1 axis-mediated stabilization of SIX1 and RPS16 to suppress HCC. ###P < 0.001, ####P < 0.0001.
Discussion
First-line systemic treatment options for unresectable HCC have increased in the last two decades, and several drugs, such as batezolizumab plus bevacizumab, sorafenib, and lenvatinib, are currently recommended for selected patients. Yet, the clinical benefits of these drugs are still modest for HCC patients. Meanwhile, acquired drug resistance of these drugs or toxic side effects can hardly be avoided during the treatment [2, 7, 8]. Therefore, a deeper our understanding of the HCC occurrence and development is urgently needed to establish better anti-cancer strategies.
DUBs are essential players in cancer development that control deubiquitination and stabilize oncoproteins. We previously reported that the USP1-RPS16 axis promotes the growth and migration of HCC cells by inducing the expression of Twist1 and Snail [23]. It has been gradually accepted that multiple substrates of DUB drive the development of cancer. In another study, we found that SIX1 is a new substrate of USP1 in prostate cancer [18]. However, it is unclear whether USP1 still regulates the deubiquitination of SIX1 in HCC and how USP1 simultaneously regulates two substrates. In this study, co-IP analysis on clinical HCC samples showed that the ubiquitination level of SIX1 was decreased in HCC tissues; this largely depends on USP1-mediated deubiquitination, but not APC-mediated ubiquitination. Mechanically, we showed that USP1 regulates the protein ubiquitination and stability of both SIX1 and RPS16. The C-terminal of USP1 is required for the binding of SIX1 and RPS16. Further investigations showed that cellular USP1 is sufficient to maintain the protein stability of SIX1 and RPS16, which may be due to their different half-life. These data renew our understanding of how USP1 drives the progression of HCC. It will be of interest to further clarify how the USP1-SIX1/RPS16 axis is mastered during certain conditions.
This study also highlights the role of the EGFR-AKT-USP1-SIX1/RPS16 axis in driving the progression of HCC. Activation of the EGFR signaling pathway inhibits the degradation of SIX1 and RPS16 via upregulating USP1. We further identified a small molecular compound that can effectively suppress this signaling pathway to inhibit the development of HCC. Indeed, we have previously found that SNS-032, a well-characterized CDKs inhibitor, can potently suppress the USP1-SIX1 axis in prostate cancer cells [27]. Herein, we explored whether SNS-032 could inhibit the EGFR-mediated activation of the USP1-SIX1/RPS16 axis and exert anti-cancer activity in HCC. Recent studies have been mainly focused on investigating the anti-cancer effects of SNS-032 in hematologic malignancies, including leukemia [24, 37, 38] and melanoma [26, 39]. Mechanically, SNS-032 may suppress RNA synthesis during gene transcription by inactivating RNA polymerase II and CDK7/9 [24]. In this study, we revealed that SNS-032 decreases the expression of SIX1 and RPS16 via suppressing the EGFR-AKT-USP1 axis-mediated stabilization of these oncoproteins based on the following key evidence: (1) our cycloheximide-chasing experiments confirmed that SNS-032 notably shortens the half-life of SIX1 and RPS16; (2) the SNS-032-induced downregulation of SIX1 and RPS16 can be reversed by the proteasome inhibitor and overexpression of USP1; (3) SNS-032 notably induces polyubiquitination of SIX1 and RPS16; (4) EGF-induced stabilization or upregulation of SIX1 and RPS16 can be reversed by gefitinib, MK2206, SNS-032, and siUSP1. Thus, these findings may broaden our perspective on the roles of SNS-032 and the EGFR signaling pathway in HCC. Subsequent studies should determine whether the EGFR-AKT-USP1-SIX1/RPS16 axis confers cancer development in other models.
Next, we investigated the anticancer effects of SNS-032 in HCC. Our in vitro proliferation assays showed that SNS-32 suppressed cell viability, DNA replication, and colony formation in HCC cells. Activation of EGFR reverses SNS-032-induced growth inhibition, migration suppression, and apoptosis induction. Furthermore, SNS-032 showed a superior inhibitory effect on HCC xenografts growth, compared to the USP1 inhibitor ML323. Furthermore, this study provided a novel combination strategy to treat HCC as we found that inactivation of the EGFR signaling pathway with SNS-032/gefitinib and sorafenib synergistically suppressed the proliferation of HCC, which is consistent with a previous study suggesting that EGFR activation contributes to the resistance of HCC cells to sorafenib [40]. Thus, it may be critical for HCC patients to inhibit the EGFR-AKT-USP1-SIX1/RPS16 axis during the sorafenib treatment.
Conclusion
Our findings confirm that the EGFR-AKT-USP1 axis can simultaneously mediate the stability of two critical substrates (SIX1 and RPS16) to boost the progression of HCC. We also identified that SNS-032 could be used as a potent USP1 inhibitor to induce the degradation of SIX1 and RPS16 with a favorable anti-cancer activity, which may further provide a novel perspective in developing anti-cancer strategies for HCC therapy (Fig. 7f).
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet. 2018;391:1301–14.
Fu J, Wang H. Precision diagnosis and treatment of liver cancer in China. Cancer Lett. 2018;412:283–8.
Sia D, Villanueva A, Friedman SL, Llovet JM. Liver cancer cell of origin, molecular class, and effects on patient prognosis. Gastroenterology. 2017;152:745–61.
Schulze K, Nault JC, Villanueva A. Genetic profiling of hepatocellular carcinoma using next-generation sequencing. J Hepatol. 2016;65:1031–42.
Tang W, Chen Z, Zhang W, Cheng Y, Zhang B, Wu F, et al. The mechanisms of sorafenib resistance in hepatocellular carcinoma: theoretical basis and therapeutic aspects. Signal Transduct Target Ther. 2020;5:87.
Huang A, Yang XR, Chung WY, Dennison AR, Zhou J. Targeted therapy for hepatocellular carcinoma. Signal Transduct Target Ther. 2020;5:146.
Vogel A, Saborowski A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treat Rev. 2020;82:101946.
Kumar JP. The sine oculis homeobox (SIX) family of transcription factors as regulators of development and disease. Cell Mol Life Sci. 2009;66:565–83.
Coletta RD, Christensen KL, Micalizzi DS, Jedlicka P, Varella-Garcia M, Ford HL. Six1 overexpression in mammary cells induces genomic instability and is sufficient for malignant transformation. Cancer Res. 2008;68:2204–13.
Iwanaga R, Wang CA, Micalizzi DS, Harrell JC, Jedlicka P, Sartorius CA, et al. Expression of Six1 in luminal breast cancers predicts poor prognosis and promotes increases in tumor initiating cells by activation of extracellular signal-regulated kinase and transforming growth factor-beta signaling pathways. Breast Cancer Res. 2012;14:R100.
Kong D, Zhou H, Neelakantan D, Hughes CJ, Hsu JY, Srinivasan RR, et al. VEGF-C mediates tumor growth and metastasis through promoting EMT-epithelial breast cancer cell crosstalk. Oncogene. 2021;40:964–79.
Ng KT, Man K, Sun CK, Lee TK, Poon RT, Lo CM, et al. Clinicopathological significance of homeoprotein Six1 in hepatocellular carcinoma. Br J Cancer. 2006;95:1050–5.
Kong J, Zhou X, Han L, Quan C, Cui X, Lin Z. [Clinicopathological significance of ezrin and SIX1 protein expression in alpha fetoprotein-negative hepatocellular carcinoma]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi. 2016;32:236–9.
Li L, Liang Y, Kang L, Liu Y, Gao S, Chen S, et al. Transcriptional regulation of the Warburg effect in cancer by SIX1. Cancer Cell. 2018;33:368–85.
Behbakht K, Qamar L, Aldridge CS, Coletta RD, Davidson SA, Thorburn A, et al. Six1 overexpression in ovarian carcinoma causes resistance to TRAIL-mediated apoptosis and is associated with poor survival. Cancer Res. 2007;67:3036–42.
Christensen KL, Brennan JD, Aldridge CS, Ford HL. Cell cycle regulation of the human Six1 homeoprotein is mediated by APC(Cdh1). Oncogene. 2007;26:3406–14.
Liao Y, Liu Y, Shao Z, Xia X, Deng Y, Cai J, et al. A new role of GRP75-USP1-SIX1 protein complex in driving prostate cancer progression and castration resistance. Oncogene. 2021;40:4291–306.
Yau RG, Doerner K, Castellanos ER, Haakonsen DL, Werner A, Wang N, et al. Assembly and function of Heterotypic Ubiquitin chains in cell-cycle and protein quality control. Cell. 2017;171:918–33.
Xia X, Liao Y, Huang C, Liu Y, He J, Shao Z, et al. Deubiquitination and stabilization of estrogen receptor alpha by ubiquitin-specific protease 7 promotes breast tumorigenesis. Cancer Lett. 2019;465:118–28.
Liao Y, Xia X, Liu N, Cai J, Guo Z, Li Y, et al. Growth arrest and apoptosis induction in androgen receptor-positive human breast cancer cells by inhibition of USP14-mediated androgen receptor deubiquitination. Oncogene. 2018;37:1896–910.
Dai X, Lu L, Deng S, Meng J, Wan C, Huang J, et al. USP7 targeting modulates anti-tumor immune response by reprogramming tumor-associated macrophages in lung cancer. Theranostics. 2020;10:9332–47.
Liao Y, Shao Z, Liu Y, Xia X, Deng Y, Yu C, et al. USP1-dependent RPS16 protein stability drives growth and metastasis of human hepatocellular carcinoma cells. J Exp Clin Cancer Res. 2021;40:201.
Chen R, Wierda WG, Chubb S, Hawtin RE, Fox JA, Keating MJ, et al. Mechanism of action of SNS-032, a novel cyclin-dependent kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2009;113:4637–45.
Chen R, Chubb S, Cheng T, Hawtin RE, Gandhi V, Plunkett W. Responses in mantle cell lymphoma cells to SNS-032 depend on the biological context of each cell line. Cancer Res. 2010;70:6587–97.
Zhang J, Liu S, Ye Q, Pan J. Transcriptional inhibition by CDK7/9 inhibitor SNS-032 abrogates oncogene addiction and reduces liver metastasis in uveal melanoma. Mol Cancer. 2019;18:140.
Liao Y, Sun W, Shao Z, Liu Y, Zhong X, Deng Y, et al. A SIX1 degradation inducer blocks excessive proliferation of prostate cancer. Int J Biol Sci. 2022;18:2439–51.
Liu Y, Yu C, Shao Z, Xia X, Hu T, Kong W, et al. Selective degradation of AR-V7 to overcome castration resistance of prostate cancer. Cell Death Dis. 2021;12:857.
Tie L, Xiao H, Wu DL, Yang Y, Wang P. A brief guide to good practices in pharmacological experiments: Western blotting. Acta Pharmacol Sin. 2021;42:1015–7.
Liao Y, Liu N, Hua X, Cai J, Xia X, Wang X, et al. Proteasome-associated deubiquitinase ubiquitin-specific protease 14 regulates prostate cancer proliferation by deubiquitinating and stabilizing androgen receptor. Cell Death Dis. 2017;8:e2585.
Liao Y, Liu N, Xia X, Guo Z, Li Y, Jiang L, et al. USP10 modulates the SKP2/Bcr-Abl axis via stabilizing SKP2 in chronic myeloid leukemia. Cell Discov. 2019;5:24.
Liao Y, Liu Y, Xia X, Shao Z, Huang C, He J, et al. Targeting GRP78-dependent AR-V7 protein degradation overcomes castration-resistance in prostate cancer therapy. Theranostics. 2020;10:3366–81.
Liao Y, Guo Z, Xia X, Liu Y, Huang C, Jiang L, et al. Inhibition of EGFR signaling with Spautin-1 represents a novel therapeutics for prostate cancer. J Exp Clin Cancer Res. 2019;38:157.
Tan Y, Zhong X, Wen X, Yao L, Shao Z, Sun W, et al. Bilirubin restrains the anticancer effect of Vemurafenib on BRAF-mutant melanoma cells through ERK-MNK1 signaling. Front Oncol. 2021;11:698888.
Jin H, Shi Y, Lv Y, Yuan S, Ramirez CFA, Lieftink C, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. 2021;595:730–4.
Ji X, Chen X, Zhang B, Xie M, Zhang T, Luo X, et al. T-box transcription factor 19 promotes hepatocellular carcinoma metastasis through upregulating EGFR and RAC1. Oncogene. 2022;41:2225–38.
Meng H, Jin Y, Liu H, You L, Yang C, Yang X, et al. SNS-032 inhibits mTORC1/mTORC2 activity in acute myeloid leukemia cells and has synergistic activity with perifosine against Akt. J Hematol Oncol. 2013;6:18.
Tong WG, Chen R, Plunkett W, Siegel D, Sinha R, Harvey RD, et al. Phase I and pharmacologic study of SNS-032, a potent and selective Cdk2, 7, and 9 inhibitor, in patients with advanced chronic lymphocytic leukemia and multiple myeloma. J Clin Oncol. 2010;28:3015–22.
Guhan SM, Shaughnessy M, Rajadurai A, Taylor M, Kumar R, Ji Z, et al. The molecular context of vulnerability for CDK9 suppression in triple wild-type melanoma. J Invest Dermatol. 2021;141:2018–27.
Ezzoukhry Z, Louandre C, Trécherel E, Godin C, Chauffert B, Dupont S, et al. EGFR activation is a potential determinant of primary resistance of hepatocellular carcinoma cells to sorafenib. Int J Cancer. 2012;131:2961–9.
Acknowledgements
This work was supported by National Natural Science Foundation of China (82002481, 82072810), the open research funds from the Sixth Affiliated Hospital of Guangzhou Medical University, Qingyuan People’s Hospital (202011-304, 202011-204), the Science and Technology Program of Guangzhou (202102020931), Natural Science Foundation Research Team of Guangdong Province (2018B030312001), Cultivation Program of National Natural Science Foundation for Distinguished Young Scholars of Guangzhou Medical University (JP2022002), Discipline Construction Funds of Guangzhou Medical University (JCXKJS2021C04, JCXKJS2021D03, and JCXKJS2021D06), Special Fund of Foshan Summit Plan (2020G010) and Guangzhou Key Medical Discipline Construction Project Fund.
Author information
Authors and Affiliations
Contributions
YNL, HBH, and GXC raised the conception and designed the experiments. YL, WYK, CFY, ZLS, QCL, YFD, GXC, XFZ, WSS, SGW, RW, and XC performed the experiments. YNL, HBH, and GXC wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
The original online version of this article was revised: The statistical symbols are wrong in primary figure. So Fig 1, 3, 4, 5, 6, 7 and Supplementary materials need replacement with the new one.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, Y., Kong, Wy., Yu, Cf. et al. SNS-023 sensitizes hepatocellular carcinoma to sorafenib by inducing degradation of cancer drivers SIX1 and RPS16. Acta Pharmacol Sin 44, 853–864 (2023). https://doi.org/10.1038/s41401-022-01003-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01003-4
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Biological impact and therapeutic implication of tumor-associated macrophages in hepatocellular carcinoma
Cell Death & Disease (2024)
-
Retinal determination gene networks: from biological functions to therapeutic strategies
Biomarker Research (2023)
-
Ubiquitin-specific peptidase 1: assessing its role in cancer therapy
Clinical and Experimental Medicine (2023)