
Abstract
Imatinib represents the standard therapy for gastrointestinal stromal tumor (GIST) patients with metastatic/unresectable disease. Despite the excellent results achieved with its introduction, the majority of patients quite invariably experience disease progression. The aim of this study was to understand the contribution of germline DNA polymorphisms in discriminating between imatinib clinical response [evaluated as progression free survival (PFS)] and toxicity. In particular, a discovery cohort (34 GIST with a KIT exon 11 primary mutation, and no toxicity) was analyzed through DMET array that interrogates 1936 variants in 231 genes of the ADME process. We further confirmed the genotype of selected variants in an extended cohort of 49 patients (the original cohort and 15 new cases, all with exon 11 primary mutation), identifying 6 SNPs— ABCB4 rs1202283, ABCC2 rs2273697, ABCG1 rs1541290, CYP11B1 rs7003319, CYP7B1 rs6987861, and NQO1 rs10517—significantly associated with response to imatinib. Three SNPs, ABCB4 rs1202283, ABCC2 rs2273697, and NQO1 rs10517, which had a significant association after adjusted multivariate analysis, were included in a genetic prediction model. We confirmed that these SNPs could stratify the cohort of 49 patients according to the risk of developing progression under imatinib treatment. In conclusion, we identified a genetic signature of response to imatinib therapy in GIST patients able to stratify patients at low and high risk to progress, according to their genotype.
Similar content being viewed by others


Introduction
Gastrointestinal stromal tumors (GIST) are rare mesenchymal diseases, with an estimated incidence of around 1–1.5/100,000 per year. Around 85–90% of GIST harbors a driver, gain-of-function mutation on KIT/PDGFRA genes [1,2,3,4]. The identification of these mutations paved the way to the successful treatment of GIST with tyrosine kinase inhibitors (TKI)—imatinib (first-line, 2000), sunitinib (second-line, 2006), and regorafenib (third-line, 2013) [5, 6]. To date, imatinib represents the sole approved drug for the first-line treatment of patients with metastatic or unresectable GIST. Imatinib achieves objective response or prolonged stable disease in about 75–90% of the patients [5]. However, patients who have a good response to imatinib quite invariably develop disease progression.
Even though in vitro data have shown an equal sensitivity to imatinib, irrespectively to mutational status, the clinical practice has highlighted a different scenario [7, 8]. The sensitivity to imatinib is strongly correlated with the KIT/PDGFRA tumor genotype and, in this regard, the example that best fits is that GIST patients with a KIT exon 11 primary mutation have a higher response rate to imatinib compared with any other GIST patient [7,8,9,10,11,12,13,14,15,16]. Despite the fact that patients with KIT exon 11 mutations appear to receive the greatest benefit from imatinib therapy, nevertheless, a wide inter-individual variability among these patients has been observed [7, 9,10,11]. In this context, differences in the absorption, distribution metabolism, and excretion (ADME) processes may be responsible for the observed variability. Indeed, it is well known that a drug, starting from its intake, undergoes a specific pharmacokinetic itinerary, determining the final intratumoral drug disposition, responsible for the final clinical response [17,18,19]. In this context, imatinib disposition might be influenced by single-nucleotide polymorphisms (SNPs) in critical genes coding for enzymes involved in the ADME processes. Besides this foreword, only a few SNPs, identified through a candidate gene approach, have been proposed as involved in imatinib response [12, 20,21,22,23,24], and no studies have investigated the relationship with imatinib-related toxicity. A multi-gene approach through the drug-metabolizing enzyme and transporter (DMET) genetic platform (Affymetrix) might represent a more complete approach to evaluate genetic variants involved in imatinib response. Indeed, this platform, successfully used in different cancer populations and other diseases [25,26,27,28,29,30,31,32,33,34,35], permits to investigate simultaneously 1936 genetic variants in 231 genes taking part in drug metabolism and transport [36]. In this context, we used the DMET genetic platform in GIST patients under imatinib treatment with the aim to identify a genetic signature predictive of clinical response and drug-related toxicity.
Materials and methods
Patients
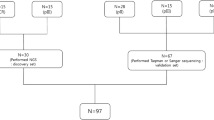
A total of 49 GIST patients, with metastatic or unresectable disease, collected between 2004 and 2014, were included in the study; all patients were of Caucasian ethnicity and treated with imatinib 400 mg daily. In particular, 38 patients, all with a KIT exon 11 primary mutation, were used as discovery set. Thirty-four patients were included in the treatment outcome analysis, while four patients were excluded due to severe imatinib-related toxicity. Subsequently, 15 GIST, all with a KIT exon 11 primary mutation, were added to extend the discovery cohort to verify the consistency of the data, and/or exclude chance findings (Fig. 1). The study was approved by the local Ethical Committee of the Sant’Orsola-Malpighi Hospital, Bologna; genomic analysis was performed after written informed consent for study participation and anonymous data publication in accordance with national legislation and the Helsinki Declaration.

Flowchart describing genetic markers selection
All clinical information was collected from the patients’ medical records. Tumors and patients’ characteristics are summarized in Table 1. Progression free survival (PFS) was calculated as the time elapsed between imatinib initiation and disease progression, interpreted as the escalation dose from 400 to 600 or 800 mg daily or switch to the second-line sunitinib. None of the clinical variables reported in Table 1 correlated with the PFS (Supplementary Table S1). All patients were followed up until death; none of them dropped out during surveillance.
We further evaluated the influence of genotype on toxicity in 38 patients of the discovery set, among which four had to stop imatinib due to serious adverse events (AE). In particular, the four patients experienced severe skin rash, severe diarrhea, mucositis, anasarca, pleural and pericardial effusion, and peripheral edema.
Genotype analysis
Germline DNA was isolated from whole blood using QIAamp DNA Blood Mini Kit (QIAGEN, Hilden, Germany) and quantified according to the manufacturer’s instructions. The Affymetrix DMET Plus Premier Pack (Affymetrix, CA, USA) was used to genotype DNA from 38 patients (discovery set), as described by Dumaual et al. [37]. Genotypes of all SNPs on the DMET array were reported either as “call” or as “no call” markers and call rates less than 95% were excluded from further analysis. Selected SNPs significantly associated with PFS were confirmed by real-time PCR using Taqman® Assay (Applied Biosystems - Thermo Fisher Scientific Inc. brand, Waltham, MA, USA) in 49 GIST patients, 34 from the discovery set, and 15 new cases which did not experience AE.
Statistical analysis
As shown in the flowchart in Fig. 1, from the initial 1936 variants, SNPs located on chromosome X were excluded; we then excluded variants with an identical genotype in all patients and/or with a minor allelic frequency (MAF) < 10%. Through Library R “Hardy–Weinberg” and exact test, we tested the Hardy–Weinberg equilibrium (HWE) to further eliminate SNPs deviating from the equilibrium (P-value ≤ 0.05). Variants were correlated with imatinib response and imatinib-related toxicity. Genotyping data are available at http://www.ncbi.nlm.nih.gov/geo (accession number GSE119558).
Results were analyzed using both the recessive (AA + Aa vs. aa) and dominant (AA vs. Aa + aa) models. Principal components analysis (PCA) was performed to corroborate genetic uniformity of the studied population. Univariate and multivariate analyses (adjusted for the main demographic—age and gender—and clinical parameters—tumor site and size, status at onset, and mitotic index) were performed using Cox proportional hazards models, with estimation of the hazard ratio (HR) and the corresponding two-sided 95% confidence interval (95% CI). Finally, multivariate models were chosen using a stepwise selection approach of the most significant SNPs in a univariate analysis. A predictive genetic model was generated, considering all the possible combinations of protective and risk alleles. The prognostic value and the discrimination ability (i.e., the ability to separate patients with different prognoses) of the model were evaluated with Harrell’s Concordance (C)-index. The C-index measures how well the model discriminates between different responses. The C-index (0 ≤ C ≤ 1) is a probability of concordance between predicted and observed survival, with C > 0.5 representing good prediction ability. Survival curves of the predictive genetic model were estimated and plotted with the Kaplan–Meier method. The curves were compared using the Cox model and the curve with higher number of patients was used as reference. With regard to imatinib-related toxicity, the analysis was performed in a total of 38 cases. The genotype frequencies were analyzed through DMET-Analyzer Tool, comparing patients with and without toxicity manifestation [38], by two-tailed Fisher’s exact test with FDR adjustment. In addition, haplotype blocks between SNPs were inferred using the Haploview software package and PHASE software, and their association with PFS was evaluated using a Cox regression model. Given the limited sample size of the study, probability values and additional parameter estimates were not adjusted for multiplicity. Therefore, results should be interpreted as exploratory. A power analysis for the design of future studies was also discussed. Results of potential interest were those SNPs with P-value ≤ 0.05.
Results
From the initial 1936 variants, 1138 SNPs were excluded due to homozygosity for the wild-type allele, 249 SNPs showed MAF < 10%, 46 SNPs were located on chromosome X, and 21 were not in HWE. The variants which fulfilled the criteria were 482; from them 64 haploblocks containing 199 haplotypes were generated. With regard to PCA analysis, according to the number of SNPs analyzed we did not observe genetic structure differences (data not shown).
Clinical outcome according to the analyzed polymorphisms
Single-locus analysis
The 482 SNPs were tested for association with PFS in 34 GIST patients. The analysis highlighted that 26 SNPs, in 19 different genes, showed a significant association with PFS (Table 2). Among the 26 significant SNPs, 12 SNPs in the recessive model and 14 in the dominant one were found significantly associated with the PFS. In both models, SNPs were mainly located in metabolizing genes. Given the exploratory nature of the study and to limit false-positive results, we extended the discovery set adding 15 new cases. We selected 12 SNPs, among the 15 with a P-value ≤ 0.030, for confirmation by Taqman assays. With regard to the 15 SNPs, 3 in CYP11B1 gene were in complete linkage—rs7003319, rs1134095, and rs4736312, as a result, only rs7003319 was selected for confirmation; CYP4Z1 rs4926802, was excluded from further analysis due to uncertainty associated with the low call rate. Amongst the 12 remaining SNPs, only 6 maintained statistical significance: 3 in the recessive model (ABCB4 rs1202283, ABCC2 rs2273697, and ABCG1 rs1541290) and 3 in the dominant model (CYP11B1 rs7003319, CYP7B1 rs6987861, and NQO1 rs10517), while 1 SNP (CYP11B1 rs5303, dominant model) reached only a borderline significance; the details of the results are reported in Table 3. Considering the discovery set only, we observed 100% concordance between genotyping by DMET array and Taqman Assays, excluding any methodological issue, associated with the different chemistry used for genotyping.
Survival curves of the significant genetic models were analyzed, results are shown in Fig. 2. With regard to ABCB4 (rs1202283; Fig. 2a), ABCC2 (rs2273697; Fig. 2b), and ABCG1 (rs1541290; Fig. 2c), the presence of at least one major allele was significantly associated with a prolonged PFS (P = 0.0003, P = 0.031, and P = 0.016, respectively). Similarly, the presence of at least one minor allele in CYP7B1 (rs6987861; Fig. 2d), CYP11B1 (rs7003319; Fig. 2e), and NQO1 (rs10517; Fig. 2f) was significantly associated with a prolonged PFS (P = 0.045, P = 0.047, and P = 0.011, respectively). In the multivariate stepwise Cox regression analysis, three SNPs, ABCB4 (rs1202283), ABCC2 (rs2273697), and NQO1 (10517), maintained the statistical significance (rs1202283: HR = 0.25, 95% CI 0.12–0.56, P = 0.0001; rs2273697: HR = 2.16, 95% CI 1.09–4.27, P = 0.027; and rs10517: HR = 2.56, 95% CI 1.14–5.26, P = 0.022).

Kaplan–Meier PFS estimates for ABCB4 (rs1202283, a); ABCC2 (rs2273697, b); ABCG1 (rs1541290, c); CYP7B1 (rs6987861, d); CYP11B1 (rs7003319, e); and NQO1 (rs10517, f)
Haplotype analysis
We evaluated the association of 64 haplotype blocks with imatinib clinical outcome. Cox analysis revealed that three haplotypes were significantly associated with PFS: block 3 on chromosome 8 (Fig. 3a), formed by four polymorphisms of the CYP11B1 gene, and block 4 on chromosome 19 (Fig. 3b), formed by three polymorphisms of the CYP4F11 gene. In particular, carriers of the CYP11B1–TCAT haplotype (rs4736312, rs1134095, rs7003319, and rs5303) had a lower risk to progress compared with the most frequent GTGC haplotype (HR = 0.43, 95% CI 0.22–0.85, P = 0.015; Fig. 4a). On the contrary, the CYP4F11–GCT haplotype (rs1060463, rs8104361, and rs3765070) was associated with a higher risk of progression, with respect to ACC haplotype (HR = 2.3, 95% CI 1.05–5.07; P = 0.037; Fig. 4b).

Haplotypes block 3 on chromosome 8 (a) and block 4 on chromosome 19 (b) significantly associated with PFS

Kaplan–Meier PFS estimates for haplotypes block 3 (CYP11B1) on chromosome 8 (a) and block 4 (CYP4F11) on chromosome 19 (b)
Assessment of a predictive genetic model
The three SNPs (ABCB4 rs1202283, ABCC2 rs2273697, and NQO1 rs10517), which maintained a significant association after multivariate analysis, constitute the response predictive model. To generate the model, we grouped the patients according to the number of protective and/or risk alleles and the product of their HR. In particular, group 1 included patients with protective genotype for ABCB4 rs1202283 (TT or Tc) and zero or one risk genotype for ABCC2 rs2273697 (AA or Ag) and NQO1 rs10517 (CC); group 2 included patients with ABCB4 protective genotype and both risk genotypes for ABCC2 rs2273697 and NQO1 rs10517, whereas group 3 consisted of GIST carriers of the ABCB4 risk genotype (cc) and only one risk genotype for ABCC2 rs2273697 and NQO1 rs10517. Finally, group 4 contained patients carrying the three risk genotypes. We applied the generated genetic model to the extended cohort of 49 GIST patients with KIT exon 11 primary mutation, representing a homogeneous group of patients. The resulting model, with a Harrell’s C = 0.714, is represented in Fig. 5. We calculated the risk of each group to experience progression; all the groups had a significantly higher risk to progress compared with group 1 (group 2: HR = 2.36, 95% CI 1.05–5.30, P = 0.037; group 3: HR = 3.19, 95% CI 1.15–9.17, P = 0.031; and group 4: HR = 25.78, 95% CI 6.86–96.77, P < 0.0001).

Predictive genetic model. Kaplan–Meier PFS estimates in 49 patients grouped on the basis of the number of protective and/or risk alleles for ABCB4 rs1202283, ABCC2 rs2273697, and NQO1 rs10517
Imatinib-related toxicity according to genotype
To evaluate the genotype influence on imatinib-related toxicity, we compared 34 GIST cases who did not experience AE with four patients forced to stop imatinib due to occurrence of severe AE; significant results are summarized in Table 4. We identified six SNPs in five genes (CYP2F1 rs305968, CHST1 rs9787901, SLC19A1 rs12659, and rs1051266, SLC22A1 rs628031, and UGT2A1 rs11249454) significantly associated with imatinib-related toxicity. In particular, with regard to the polymorphisms in the SLC19A1 (rs12659 and rs1051266), presence of at least one major allele prevents the development of AE. On the contrary, patients with at least one variant allele for CHST1 rs9787901 or carriers of the SLC22A1 rs628031 major alleles experienced toxicity.
Discussion
Imatinib represents the only gold standard therapy in metastatic GIST treatment. About 75–80% of the patients reach an objective response or experience a prolonged stable disease. The main factor influencing imatinib response is the KIT/PDGFRA genotype. However, even among the patients with a genotype favorable to imatinib response, as GIST with a KIT exon 11 primary mutation, a wide inter-individual variability in treatment response has been observed. This knowledge prompted us to launch a pharmacogenetic study, as genetic variability might be plausibly responsible for the observed clinical outcome heterogeneity. In particular with this study, we moved, for the first time in GIST, from testing a few SNPs in a limited number of genes [12, 20,21,22,23,24] to the application of a pharmacogenetic platform, such as the Affymetrix DMET array. This array permits the simultaneous analysis of a wide panel of genetic variants in genes involved in the ADME processes. Indeed, the published literature on pharmacogenetics in GIST used a multiple gene candidate approach, which is focused on specific polymorphisms but, as a downside, is likely to miss important significant results.
Our study focused on the identification of a predictive genetic signature of imatinib response and toxicity in GIST patients. For this purpose, we initially selected a homogeneous cohort of GIST patients, with a KIT exon 11 primary mutation, under imatinib treatment, and divided them in patients with progressive or non-progressive disease and patients with or without imatinib-related toxicity. Overall, after a confirmation step in an extended cohort of patients, we identified six SNPs in six genes—ABCB4 rs1202283, ABCC2 rs2273697, ABCG1 rs1541290, CYP11B1 rs7003319, CYP7B1 rs6987861, and NQO1 rs10517—significantly associated with response to imatinib. Deepening the investigation through a multivariate analysis, adjusted for ties, the three SNPs ABCB4 rs1202283, ABCC2 rs2273697, and NQO1 rs10517 maintained their statistical significance. To test if the three SNPs could represent genetic biomarkers with a predictive role, we included them in a genetic prediction model. We confirmed they were able to stratify the cohort of patients with KIT exon 11 primary mutations, according to the risk of developing progression during imatinib treatment. The three SNPs have not been previously reported as associated with imatinib treatment in GIST, however, some of the data might be supported by findings in chronic myeloid leukemia cells or patients, which share with GIST the gold standard treatment (imatinib). With regard to ABCC2, a study has reported the association of a haplotype with imatinib resistance in chronic myeloid leukemia patients [39]. ABCC2 and ABCB4 are both efflux transporters [40], and it is reasonable to assume that SNPs in these genes may affect the pharmacokinetics and, therefore, treatment response. With regard to NQO1, no evidence of a correlation with imatinib response has been so far described. A study on leukemia cells identified NQO2 as imatinib off-target protein, while binding to the related NQO1 was prevented by steric hindrance [41]. Interestingly, a study on imatinib-resistant leukemia cells showed that drug sensitivity was enhanced through the downregulation of NRF2 and genes downstream, as NQO1 [42]. No other evidence, both in vitro or in vivo, have been reported in the literature, and to the best of our knowledge, our study provides the first evidence of a possible involvement of this enzyme, in particular of a SNP in its 3′UTR region, in imatinib clinical outcome. UTR regions are involved in regulating RNA stability and translational efficiency; therefore we speculate that the presence of the SNP may favorably impact the PFS, reducing NQO1 antioxidant activity. This is intriguing, as in leukemia cells imatinib resistance has been associated with resistance to oxidative stress [43, 44]. Besides this, a further interesting point concerns the recent demonstration that NQO1 significantly influenced the autophagy process [45]. Autophagy is attracting the research interest in both tumor development and mechanisms of imatinib resistance in GIST [46]. Overall, given the small sample size, we cannot rule out that the significances found are a chance. However, given their function as transporters (ABCC2 and ABCB4) and in oxidative stress (NQO1), SNPs in their coding genes might show a plausible role in imatinib response. In view of these considerations, in the near future, it would be important to verify in cellular models the implication of ABCC2 and ABCB4 in imatinib transport and oxidative stress as mechanisms involved in imatinib response; once established their involvement, functional studies on these SNPs will be desirable.
With regard to imatinib toxicity, the small number of patients forced to discontinue the therapy due to AE precludes any definitive conclusion. Indeed, even we observed interesting associations between SNPs in CHST1, CYP2F1, SLC19A1, SLC22A1, and UGT2A1, the present study should be considered as hypothesis generating. Therefore, a follow-up validation study involving a large number of GIST patients experiencing AE is strongly warranted. Certainly, a robust validation of this finding could help clinicians, leading to a better management and monitoring of patients at risk of AE.
Despite the enthusiasm about the intriguing result, we are aware of important limitations, including the lack of adequate statistical power of the analysis, which might be affected by the small population size. In this context, future studies should be large enough for attaining necessary statistical power, taking into consideration the observed associations; ideally a sample size of 130 patients should give 80% power at an alpha value of 0.0001, which will also include provision for correction for multiple comparisons. However, we would like to stress out that GIST is a rare entity, with an estimated incidence of around 1–1.5/100,000 per year, which makes difficult enrolling large sample size of patients with homogeneous characteristics. Specifically, to minimize the confounding factors, we limited the study to patients with a KIT exon 11 primary mutation, as these patients are the most common GIST and the ones receiving the greatest benefit from imatinib. This aspect represents certainly a strength, however, we are aware that besides the potential contribution to the clinical outcome of germline DNA, tumor heterogeneity, which characterizes advanced diseases, may play an additional role in the phenotypic drug response. Consequently, it would be interesting to apply the genetic model to an extended heterogeneous GIST population. Overall the result is certainly interesting, as the identification of a potential biomarker of disease progression could be advantageous in the clinical approach for a better surveillance and follow-up of these patients. However, before its application in a real clinical setting, further investigations in independent and larger cohort of patients are mandatory.
In conclusion, to the best of our knowledge, this study represents the first example of DMET array application in GIST patients. The most eye-catching result is the establishment of a genetic model, able to stratify patients at high and low risk to progress under imatinib, according to their genotype. Despite the enthusiasm, in light of limitations and strengths, this represents a hypothesis-generating study that should prompt additional prospective validation studies.
References
Antonescu CR. The GIST paradigm: lessons for other kinase-driven cancers. J Pathol. 2011;223:251–61.
Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11:865–78.
Heinrich MC, Corless CL, Duensing A, McGreevey L, Joseph N, Singer S, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–10.
Angelini S, Ravegnini G, Fletcher JA, Maffei F, Hrelia P. Clinical relevance of pharmacogenetics in gastrointestinal stromal tumor treatment in the era of personalized therapy. Pharmacogenomics. 2013;14:941–56.
Demetri GD, von Mehren M, Blanke CD, Van den Abbeele AD, Eisenberg B, Roberts PJ, et al. Efficacy and safety of imatinib mesylate in advanced gastrointestinal stromal tumors. N Engl J Med. 2002;347:472–80.
Demetri GD, van Oosterom AT, Garrett CR, Blachstein ME, Shah MH, Verweij J, et al. Efficacy and safety of sunitinib in patients with advanced gastrointestinal stromal tumour after failure of imatinib: a randomised controlled trial. Lancet. 2006;368:1329–38.
Heinrich MC, Corless CL, Demetri GD, Blanke CD, von Mehren M, Joensuu H, et al. Kinase mutations and imatinib response in patients with metastatic gastrointestinal stromal tumor. J Clin Oncol. 2003;21:4342–9.
Maleddu A, Pantaleo MA, Nannini M, Biasco G. The role of mutational analysis of KIT and PDGFRA in gastrointestinal stromal tumors in a clinical setting. J Transl Med. 2011;9:75.
Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup Phase III Trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 Study by Cancer and Leukemia Group B and Southwest Oncology Group. J Clin Oncol. 2008;26:5360–7.
Debiec-Rychter M, Dumez H, Judson I, Wasag B, Verweij J, Brown M, et al. Use of c-KIT/PDGFRA mutational analysis to predict the clinical response to imatinib in patients with advanced gastrointestinal stromal tumours entered on phase I and II studies of the EORTC Soft Tissue and Bone Sarcoma Group. Eur J Cancer. 2004;40:689–95.
Gastrointestinal Stromal Tumor Meta-Analysis Group (MetaGIST). Comparison of two doses of imatinib for the treatment of unresectable or metastatic gastrointestinal stromal tumors: a meta-analysis of 1,640 patients. J Clin Oncol. 2010;28:1247–53.
Angelini S, Pantaleo MA, Ravegnini G, Zenesini C, Cavrini G, Nannini M, et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumours treated with imatinib therapy. Pharmacol Res. 2013;68:1–6.
Serrano C, Wang Y, Mariño-Enríquez A, Lee JC, Ravegnini G, Morgan JA, et al. KRAS and KIT Gatekeeper Mutations Confer Polyclonal Primary Imatinib Resistance in GI Stromal Tumors: Relevance of Concomitant Phosphatidylinositol 3-Kinase/AKT Dysregulation. J Clin Oncol. 2015;33:e93–6.
Ravegnini G, Sammarini G, Angelini S, Hrelia P. Pharmacogenetics of tyrosine kinase inhibitors in gastrointestinal stromal tumor and chronic myeloid leukemia. Expert Opin Drug Metab Toxicol. 2016;12:733–42.
Debiec-Rychter M, Sciot R, Le Cesne A, Schlemmer M, Hohenberger P, van Oosterom AT, et al. KIT mutations and dose selection for imatinib in patients with advanced gastrointestinal stromal tumours. Eur J Cancer. 2006;42:1093–103.
Duensing A, Medeiros F, McConarty B, Joseph NE, Panigrahy D, Singer S, et al. Mechanisms of oncogenic KIT signal transduction in primary gastrointestinal stromal tumours (GISTs). Oncogene. 2004;23:3999–4006.
Ravegnini G, Nannini M, Sammarini G, Astolfi A, Biasco G, Pantaleo MA, et al. Personalized Medicine in Gastrointestinal Stromal Tumor (GIST): Clinical Implications of the Somatic and Germline DNA Analysis. Int J Mol Sci. 2015;16:15592–608.
Peng B, Lloyd P, Schran H. Clinical pharmacokinetics of imatinibby. Clin Pharmacokinet. 2005;44:879–94.
Oostendorp RL, Buckle T, Beijnen JH, van Tellingen O, Schellens JH. The effect of P-gp (Mdr1a/1b), BCRP (Bcrp1) and P-gp/BCRP inhibitors on the in vivo absorption, distribution, metabolism and excretion of imatinib. Invest New Drugs. 2009;27:31–40.
O’Brien KM, Orlow I, Antonescu CR, Ballman K, McCall L, DeMatteo R, et al. Gastrointestinal stromal tumors, somatic mutations and candidate genetic risk variants. PLoS ONE. 2013;8:e62119.
Koo DH, Ryu MH, Ryoo BY, Beck MY, Na YS, Shin JG, et al. Association of ABCG2 polymorphism with clinical efficacy of imatinib in patients with gastrointestinal stromal tumor. Cancer Chemother Pharmacol. 2015;75:173–82.
Angelini S, Ravegnini G, Nannini M, Bermejo JL, Musti M, Pataleo MA, et al. Folate-related polymorphisms in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Eur J Hum Genet. 2015;23:817–23.
Ravegnini G, Nannini M, Simeon V, Musti M, Sammarini G, Saponare M, et al. Polymorphisms in DNA repair genes in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Tumour Biol. 2016;37:13413–23.
Ravegnini G, Nannini M, Zenesini C, Simeon V, Sammarini G, Urbini M, et al. An exploratory association of polymorphisms in angiogenesis-related genes with susceptibility, clinical response and toxicity in gastrointestinal stromal tumors receiving sunitinib after imatinib failure. Angiogenesis. 2017;20:139–48.
Deeken JF, Cormier T, Price DK, Sissung TM, Steinberg SM, Tran K, et al. A pharmacogenetic study of docetaxel and thalidomide in patients with castration-resistant prostate cancer using the DMET genotyping platform. Pharm J. 2010;10:191–9.
Di Martino MT, Arbitrio M, Guzzi PH, Leone E, Baudi F, Piro E, et al. A peroxisome proliferator-activated receptor gamma (PPARG) polymorphism is associated with zoledronic acid-related osteonecrosis of the jaw in multiple myeloma patients: analysis by DMET microarray profiling. Br J Haematol. 2011;154:529–33.
Di Martino MT, Arbitrio M, Leone E, Guzzi PH, Rotundo MS, Ciliberto D, et al. Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer Biol Ther. 2011;12:780–7.
Rumiato E, Boldrin E, Amadori A, Saggioro D. DMETTM (Drug-Metabolizing Enzymes and Transporters) microarray analysis of colorectal cancer patients with severe 5-fluorouracil-induced toxicity. Cancer Chemother Pharmacol. 2013;72:483–8.
Lombardi G, Rumiato E, Bertorelle R, Saggioro D, Farina P, Della Puppa A, et al. Clinical and genetic factors associated with severe hematological toxicity in glioblastoma patients during radiation plus temozolomide treatment: A Prospective Study. Am J Clin Oncol. 2015;38:514–9.
Nieuweboer AJ, Smid M, de Graan AJ, Elbouazzaoui S, de Bruijn P, Martens JW, et al. Predicting paclitaxel-induced neutropenia using the DMET platform. Pharmacogenomics. 2015;16:1231–41.
Arbitrio M, Di Martino MT, Barbieri V, Agapito G, Guzzi PH, Botta C, et al. Identification of polymorphic variants associated with erlotinib-related skin toxicity in advanced non-small cell lung cancer patients by DMET microarray analysis. Cancer Chemother Pharmacol. 2016;77:205–9.
Rumiato E, Boldrin E, Malacrida S, Battaglia G, Bocus P, Castoro C, et al. A germline predictive signature of response to platinum chemotherapy in esophageal cancer. Transl Res. 2016;171:29–37.e1.
Drenberg CD, Paugh SW, Pounds SB, Shi L, Orwick SJ, Li L, et al. Inherited variation in OATP1B1 is associated with treatment outcome in acute myeloid leukemia. Clin Pharmacol Ther. 2016;99:651–60.
Di Martino MT, Scionti F, Sestito S, Nicoletti A, Arbitrio M, Hiram Guzzi P, et al. Genetic variants associated with gastrointestinal symptoms in Fabry disease. Oncotarget. 2016;7:85895–904.
Arbitrio M, Di Martino MT, Scionti F, Agapito G, Guzzi PH, Cannataro M, et al. DMETTM (Drug Metabolism Enzymes and Transporters): a pharmacogenomic platform for precision medicine. Oncotarget. 2016;7:54028–50.
Agapito G, Guzzi PH, Cannataro M. DMET-Miner: efficinet discovery of association rules from pharmacogenomic data. J Biomed Inform. 2015;56:273–83.
Dumaual C, Miao X, Daly TM, bruckner C, Njau R, Fu DJ, et al. Comprehensive assessment of metabolic enzyme and transporter genes using the Affymetrix Targeted Genotyping System. Pharmacogenomics. 2007;8:293–305.
Guzzi PH, Agapito G, Di Martino MT, Arbitrio M, Tassone P, Tagliaferri P, et al. DMET-analyzer: automatic analysis of Affymetrix DMET data. BMC Bioinforma. 2012;13:258.
Au A, Baba AA, Azlan H, Norsa’adah B, Ankathil R. Clinical impact of ABCC1 and ABCC2 genotypes and haplotypes in mediating imatinib resistance among chronic myeloid leukaemia patients. J Clin Pharm Ther. 2014;39:685–90.
Chen Z, Shi T, Zhang L, Zhu P, Deng M, Huang C, et al. Mammalian drug efflux transporters of the ATP binding cassette (ABC) family in multidrug resistance: A review of the past decade. Cancer Lett. 2016;370:153–64.
Winger JA, Hantschel O, Superti-Furga G, Kuriyan J. The structure of the leukemia drug imatinib bound to human quinone reductase 2 (NQO2). BMC Struct Bio. 2009;9:7.
Chen F, Liu Y, Wang S, Guo X, Shi P, Wang W, et al. Triptolide, a Chinese herbal extract, enhances drug sensitivity of resistant myeloid leukemia cell lines through downregulation of HIF-1α and Nrf2. Pharmacogenomics. 2013;14:1305–17.
Ma D, Fang Q, Wang P, Gao R, Wu W, Lu T, et al. Induction of heme oxygenase-1 by Na + -H + exchanger 1 protein plays a crucial role in imatinib-resistant chronic myeloid leukemia cells. J Biol Chem. 2015;290:12558–71.
Synowiec E, Hoser G, Wojcik K, Pawlowska E, Skorski T, Blasiak J. UV Differentially Induces Oxidative Stress, DNA Damage and Apoptosis in BCR-ABL1-Positive Cells Sensitive and Resistant to Imatinib. Int J Mol Sci. 2015;16:18111–28.
Kim TW, Kim YJ, Kim HT, Park SR, Lee MY, Park YD, et al. NQO1 Deficiency Leads Enhanced Autophagy in Cisplatin-Induced Acute Kidney Injury Through the AMPK/TSC2/mTOR Signaling Pathway. Antioxid Redox Signal. 2016;24:867–83.
Ravegnini G, Sammarini G, Nannini M, Pantaleo MA, Biasco G, Hrelia P, et al. Gastrointestinal stromal tumors (GIST): Facing cell death between autophagy and apoptosis. Autophagy. 2017;13:452–63.
Acknowledgements
Gloria Ravegnini is supported by a MSD Italia fellowship granted by and on behalf of Merck Sharp & Dohme Corporation.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Ravegnini, G., Urbini, M., Simeon, V. et al. An exploratory study by DMET array identifies a germline signature associated with imatinib response in gastrointestinal stromal tumor. Pharmacogenomics J 19, 390–400 (2019). https://doi.org/10.1038/s41397-018-0050-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41397-018-0050-4
- Springer Nature Limited
This article is cited by
-
The GIST of it all: management of gastrointestinal stromal tumors (GIST) from the first steps to tailored therapy. A bibliometric analysis
Langenbeck's Archives of Surgery (2024)
-
Pharmacogenomics and functional imaging to predict irinotecan pharmacokinetics and pharmacodynamics: the predict IR study
Cancer Chemotherapy and Pharmacology (2021)
-
HIF-1α regulates cellular metabolism, and Imatinib resistance by targeting phosphogluconate dehydrogenase in gastrointestinal stromal tumors
Cell Death & Disease (2020)