
Abstract
DNA repair pathways play an essential role in cancer susceptibility by maintaining genomic integrity. This led us to investigate the influence of polymorphisms in the genes coding repair pathway enzymes on gastrointestinal stromal tumours (GIST) susceptibility, tumour characteristics and clinical outcome. We investigated a panel of 20 polymorphisms in 11 genes in 81 cases and 147 controls. The XPD rs13181 wild-type allele and hOGG1 rs1052133 and XPF rs1800067 minor alleles were significantly associated with disease susceptibility. XPA rs1800975 and rs2808668 were associated with tumour size (P = 0.018), metastatic status at onset (P = 0.035) and mitotic index (P = 0.002). With regards to outcome treatment, the XPD rs50872 minor allele had a significant favourable impact on time to progression (TTP). Similarly, the XPC rs2228000 minor allele was correlated with a longer TTP (P = 0.03). On the contrary, the XPC rs2228001 and hOGG1 rs1052133 minor alleles were associated with a diminished TTP (P = 0.005 and P = 0.01, respectively). Regarding OS, we found the presence of at least one hOGG1 (rs1052133) minor allele that had a 60 % lower risk to die compared to the wild-type carriers (P = 0.04). Furthermore, the XRCC3 rs861539 variant allele is associated with a hazard of early death compared with the wild-type genotype (P = 0.04). To the best of our knowledge, this is the first study on polymorphisms in DNA repair genes, belonging to the different pathways, extensively evaluated in GIST patients. Through this multiple candidate gene approach, we report for the first time the significant associations between polymorphisms in DNA repair genes, susceptibility, clinical pathological features and clinical outcome in GIST.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Gastrointestinal stromal tumours (GISTs) are the most common mesenchymal tumours of the gastrointestinal tract. Classically, more than 80 % of cases harbour activating mutations in the proto-oncogene receptor tyrosine kinase (KIT) or platelet-derived growth factor receptor, alpha polypeptide (PDGFRA) gene. About 10 % of GIST do not present any mutation in these receptors and are defined as KIT/PDGFRA wild type (WT), a heterogeneous family of tumours with distinct molecular hallmarks, including defects in the succinate dehydrogenase (SDH) complex and mutations of neurofibromatosis type 1 (NF1), BRAF or KRAS genes [1–4]. KIT and PDGFRA mutations, somatic and mutually exclusive, cause a constitutional, ligand-independent, activation of the receptor, leading to activation of the downstream signalling cascade [5]. Primary mutations in KIT mainly affect exon 11 (65–70 %), 9 (10–20 %), 13 (1–4 %) and 17 ( ̴ 1 %), whereas in PDGFRA, mutations are mainly found in exon 18 (6–7 %), 12 (<1 %) and 14 (<0.5 %). The discovery of these tyrosine kinase (TK) receptor mutations has paved the way for the introduction into clinical practise of TK inhibitors (TKIs). The TKI imatinib represents the gold standard therapy for GIST. Of patients, 80–90 % respond well to imatinib, with a median response rate of 24 months, though the majority of them experience disease progression [1–6]. Prognostic factors, including age at diagnosis, tumour size, histologic grade and subtype, tumour depth, margin status and tumour site, have long been used to assess the risk of tumour relapse and disease-related death [5]. However, additional features would represent a novel strategy for stratifying GIST patients for tumour recurrence risk, which, in turn, would allow treatment options tailoring to the individual [6]. In the last decade, many evidences have highlighted the importance of the DNA repair pathways in the cancerogenetic process. Indeed, deficient DNA repair may lead to deregulated cell growth, imbalance of cell cycle control and development of many diseases, including cancer [6]. Overall, the types of DNA lesions are multiple, different repair systems are involved, and the concerted action of many enzymes is required [6]. These include XPA, XPC, XPD, XPF, XPG, RAD23B (belonging to the nucleotide excision repair (NER) system), APE1, hOGG1, XRCC1 (members of the base excision repair (BER) system), NBS1 and XRCC3 (participants of the homologous repair (HR) system). The highly conserved NER pathway is the main path responsible for the excision of a large variety of bulky DNA lesions; sporadic mutations in NER genes across different cancer types have been reported [7, 8]. However, to date, no NER deficiencies have been reported in GIST. Similarly, no BER defects have been described in GIST; indeed, somatic mutations in BER genes are uncommon, and no human disorders linked with inherited BER deficiencies are known [9]. The BER path deals with the repair of modest, non-bulky, DNA lesions caused by oxidative base lesions or endogenous metabolic processes [10]. Another important repair path is the HR system, representing the main repair mode for double-strand breaks (DSBs), arising from ionizing radiations, chemicals, free radicals or during replication. DSBs are the most severe lesions and bear high risk for cancer [11]. Several genetic disorders have been associated with the lack of an effective HR pathway, and somatic mutations in HR genes have been linked to cancer, such as the BRCA gene mutations in ovarian cancer [12]. Beyond these considerations, DNA repair genes have repeatedly attracted the research interest as they are characterized by functional genetic polymorphisms that have been associated with decreased or increased enzymatic activities, with a plausible alteration in the DNA repair machinery. The hypothesis of altered DNA repair pathways—due to genetic polymorphisms—could explain the association with increased DNA damage found in vitro [13, 14] or ex vivo after genotoxic exposure [15–17] cancer risk [18, 19], treatment outcome [20, 21] and mutational status [22, 23]. All these studies highlighted the importance of effective DNA repair machinery and prompted us to analyse the frequencies of 18 genetic polymorphisms in 10 genes belonging to the different DNA repair pathways, in GIST patients and controls. To this purpose, we selected XPD and RAD23B polymorphisms on the basis of the work by O’Brien and coworkers, who indicated these NER-belonging genes as potential risk variants for GIST. Given the high complexity of the DNA repair pathways, we investigated additional genes involved in NER, BER and HR paths, including XPA, XPC, XPF, XPG, APE1, hOGG1, XRCC1, XRCC3 and NBS1. In particular, we selected genes with a central role in the specific pathway of belonging, and polymorphisms reported as associated with other cancer type and clinical outcome [18–23]. We investigated two additional polymorphisms in the CYP1B1 gene, which were found to be associated with KIT exon 11 mutations in GIST patients [22]. Specific aims of this study were to test if the analysed polymorphisms (i) influence the risk of developing GIST; (ii) are associated with specific patients’ characteristics, tumour genotype and clinical features and (iii) influence treatment response. This last aim arises from the importance of the DNA repair machinery in genomic stability maintenance, along with the fact that the appearance of secondary point mutations in KIT or PDGFRA genes is one of the major mechanisms of acquired resistance in GIST patients undergoing imatinib therapy.
Material and methods
Study population
A total of 81 unresectable/metastatic GIST patients were retrospectively enrolled at the Sant’Orsola-Malpighi Hospital, Bologna. Clinical information was collected retrospectively from the patients’ medical records. Patients’ characteristics and clinical features of their tumours are summarized in Table 1. With regards to tumour features, mitotic index was expressed as the number of mitoses per 50 high-power field, and the value were stratified into three subgroups (<5, 6–10 and >10 cm); KIT/PDGFRA mutational status was investigated through Sanger sequencing. Overall survival (OS) was defined as the time from the first day of treatment to death from disease. Dates of death were obtained and cross checked using the inpatient medical records. If a patient was alive at the time of the analysis, OS was censored at the time of the last follow-up. For the 81 patients on standard first-line imatinib therapy, time to progression (TTP) was calculated from the start of imatinib therapy to the date of disease progression documented by the CT scan performed approximately every 3–4 months. Data for patients who did not progress at the last follow-up, TTP evaluation was censored at that time. In order to exclude disease susceptibility, we also genotyped 147 controls, anonymous blood donors from the Centro Trasfusionale, Sant’Orsola-Malpighi Hospital, Bologna (Table 1). The analysis, approved by the hospital Ethics Committee, was done after written informed consent for study participation and anonymous data publication in accordance with national legislation and the Helsinki Declaration.
Genotype analysis
We selected 20 common [minor allele frequency (MAF) > 0.05 in Caucasian], well-studied functional variants—located in regulatory regions, causing non-synonymous amino acid changes and/or repeatedly associated with cancer risk, OS or treatment response. Patients with available peripheral blood were eligible for this retrospective study. Genomic DNA was extracted from fresh or frozen whole blood using a DNA isolation kit from Qiagen (QIAamp® DNA Mini Kit, Qiagen, Hilden, Germany). Characteristics of the studied polymorphisms are described in supplementary table S1. Genotypes were performed by real-time PCR using Taqman® Assay (Applied Biosystems—Thermo Fisher Scientific Inc. brand, Waltham, MA, USA) as recommended by the manufacturer (Table S1). Positive and negative controls were included in each reaction as quality control. In addition, for internal quality control (accuracy of genotyping), 90 % of the samples were repeated. The concordance between the original and the duplicate samples for all the analysed polymorphisms was 100 %.
Statistical analysis
The distribution of genotypes was tested for departures from the Hardy-Weinberg equilibrium using the χ 2 test. The frequency distributions of categorical variables were compared using Pearson’s chi-squared and Fisher’s exact test as appropriate. Survival analysis methods were used to examine the relationship between genotypes [homozygous wild-type (WT), heterozygous and homozygous for the variant allele] and TTP. In univariate analysis, the survival curves were estimated and plotted with the Kaplan-Meier method. The curves were compared with the log-rank test of equality of survivor functions (statistical significance defined as P < 0.05). In multivariate analysis, hazard ratios (HRs) and 95 % confidence interval (95 % CI) were estimated by Cox proportional hazard models, using gender and age at diagnosis, as covariates in addition to the genotype. The proportional hazard assumption was tested using Schoenfeld residuals. Multiple logistic regression was used to assess the relation between individual polymorphisms and primary resistance. Given the limited small size of the present study, probability values and additional parameter estimates were not adjusted for multiplicity. Results should be interpreted as exploratory. Linkage disequilibrium (LD) and haplotype blocks between markers in DNA repair genes were analysed using Haploview software package [24]. LD was measured using r2, and haplotype blocks were generated taking in consideration default algorithm from Gabriel et al., Science, 2002. After that, PHASE software v 2.1 [25] was used to infer haplotype data of significant block regions for each patient in analysis. Only common haplotypes with a frequency ≥5 % were used for further analysis, and the most common haplotype was used as the reference. Statistical analysis was conducted using Stata Intercooled version 12.0 [26].
Results
Genotype distribution in the two studied populations
Genotype frequencies of the 20 polymorphisms were in Hardy-Weinberg equilibrium in both GIST patients and controls. MAF and Hardy-Weinberg equilibrium P value are presented in supplementary Table S2. In addition, genotype distributions were similar to those reported in the publicly available database NCBI (dbSNP) for Caucasians (CEU samples; Supp. Table S2). The results of the most significant relationships between genetic polymorphisms and GIST susceptibility are presented in Table 2. In particular, the most significant results, after adjustment by gender and age, were found for hOGG1 (rs1052133, C>G), XPD (rs13181, A>C), XPF (rs1800067, G>A) and XPC (rs2228000, C>T). With regards to hOGG1, the presence of at least one G variant allele was associated with a 2.3 higher GIST susceptibility with respect to the WT genotype (OR 2.32, 95 % CI 1.26–4.3; P = 0.007). Regarding the XPD polymorphism, the presence of at least one WT allele was significantly more common in cases compared to controls (75.0 vs 59.9 %, respectively; OR 2.23, 95 % CI 1.15–4.2; P = 0.017). Similarly, the presence of at least one XPF minor allele was associated with a major GIST susceptibility compared to the WT genotype (OR 2.38, 95 % CI 1.15–4.9; P = 0.019). A borderline association was also observed for XPC rs2228000, with the presence of at least one variant allele associated with increased risk of GIST (OR 1.87, 95 % CI 1.02–3.4; P = 0.042). None of the other investigated polymorphisms had significant association with GIST susceptibility.
Mutational status and genotypes in GIST population
Mutational status of the studied population is shown in Table 1. In the wake of the work of O’Brien and colleagues, we tested if primary mutations in KIT or PDGFRA genes were correlated with specific genotype. In order to achieve this aim, the 81 patients were grouped into two groups: KIT/PDGFRA mutants and KIT/PDGFRA wt-SDH-mutated GIST. The most significant results have been found for hOGG1 rs1052133 and XPC rs2228000. With regards to hOGG1, having the minor allele in homozygous, or harbouring at least one minor allele, was correlated with a lower risk (97 and 82 %, respectively) to have a KIT or PDGFRA mutation (OR 0.03, 95 % CI 0.0018–0.65, P = 0.02 and OR 0.18, 95 % CI 0.031–0.96, P = 0.04 respectively) compared to the homozygous WT genotype. Similarly, the presence of at least one minor XPC rs2228000 allele was associated with a higher risk to have a KIT or PDGFRA mutation (OR 6.5, 95 % CI 1.15–36.8; P = 0.03) compared to the XPC WT genotype. The majority of GIST patients (54.4 %) harboured a KIT exon 11 mutation (61.9 % involving the codons 557–558 and 36.6 % other codons). None of the analysed polymorphisms was associated with a specific KIT mutation.
Clinical features and genotypes in GIST population
We analysed the association of polymorphisms with tumour site, tumour size, status at onset and mitotic index. The most significant results are reported in Table 3. Univariate analysis showed significant associations between XPA polymorphisms and size, status at onset and mitotic index. With regards to tumour size, patients were divided in four groups (≤2, 2–5, 5–10 and ≥10). The XPA variant alleles were associated with smaller tumour size; on the contrary, the WT allele was predominant in patients (60 %) with tumour size >10 cm (P = 0.018). The same variant alleles were also associated with metastatic status at onset (P = 0.035). Similarly, the analysis showed an excess of WT XPA genotype compared to the presence of at least one variant allele in patients with a mitotic index lower than 6 (76.5 vs 23.5 %; P = 0.002). Further significant associations were found for XPC (rs2228000) and XPF (rs3136155). An excess of carriers of XPC variant allele was observed in patients with a mitotic index lower than 6, compared to patients with higher mitotic index (70.6 vs 29.4 %, P = 0.022). With regards to XPF, an association with tumour site has been observed. In particular, 74.4 % of patients with at least one XPF variant allele had a tumour localized in the small intestine, while in contrast, only 25.6 % of patients had a WT genotype (P = 0.004).
Treatment outcome of imatinib therapy and genotypes
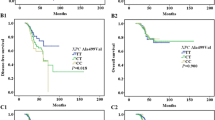
Sixty-three patients received standard first-line imatinib 400 mg daily and were evaluated for imatinib response; the median OS was 71.15 months (range 8.57–146.63); progression was observed in 58 cases (71.6 %), with a median TTP of 30.88 months (range 1.23–128.33); 12 had stable disease (14.8 %), whereas in 11 patients, the status was unknown (13.6 %). During the follow-up, 28 GIST died due to the tumour (34.6 %) and 1 patient died due to disease-unrelated causes. With regards to the association between selected polymorphisms and TTP, the most significant results are reported in Table 4. In the multivariate analysis, the presence of at least one minor allele in XPD rs50872 had a significant favourable impact on TTP (HR 0.42, 95 % CI 0.20–0.84, P = 0.015). Similarly, the presence of at least one XPC (rs2228000) minor allele was correlated with a longer TTP (HR 0.55, 95 % CI 0.32–0.96, P = 0.03). On the contrary, the XPC rs2228001 minor allele in homozygous was associated with a diminished TTP (HR 2.9, 95 % CI 1.38–6.1, P = 0.005). The hOGG1 (rs1052133) minor allele in homozygous was also associated with a shorter TTP compared to the presence of at least one WT allele (HR 17.1, 95 % CI 3.1–92.7, P = 0.01); however, caution should be used in interpreting this result, as it is based on two homozygous subjects only. None of the other analysed polymorphisms correlated with the TTP. Alleles were also correlated with TTP based on the Kaplan-Meier method. In particular, the presence of the XPC (rs2228001) minor allele in homozygous was associated with a reduced TTP (P = 0.014; Fig. 1). Similarly, the hOGG1 (rs1052133) minor allele in homozygous was associated with reduced TTP compared to the presence of at least one WT allele (P < 0.0001), though the result is based on two homozygous subjects only; we should consider it with criticism. Regarding OS, in a multivariate analysis, we found that the presence of at least one hOGG1 (rs1052133) minor allele had a 60 % lower risk to die compared to the WT carriers (HR 0.41, 95 % CI 0.16–0.99, P = 0.04). Furthermore, patients carrying the XPF rs3136155 in heterozygosis had a reduced risk to die than patients carrying the major allele TT (HR 0.36, 95 % CI 0.13–0.97, P = 0.04). Finally, we observed an association between the XRCC3 genotype and OS (rs861539). In particular, the variant allele in homozygosis is associated with a hazard of early death compared with the homozygous WT genotype (HR 2.8, 95 % CI 1.03–7.71, P = 0.04). Alleles were also correlated with OS based on the Kaplan-Meier method. In particular, the presence of the minor allele in hOGG1 (rs1052133) was associated with a longer survival (P = 0.0272, Fig. 2). In addition, carriers of the XPF rs3136155 in heterozygosis had a longer survival compared to patients homozygous for the WT allele (P = 0.01).

The presence of the XPC minor allele in homozygous is associated with a reduced TTP

The presence of at least one hOGG1 minor allele is associated with a longer OS
Haplotype analysis
Haplotype blocks were defined only for XPA (rs1800975 and rs2808668) and XPC (rs2228000 and rs2228001) gene regions (Fig. 3). Estimation of the coefficient of LD showed, as expected, that the XPA rs1800975 was in complete linkage disequilibrium with the XPA rs2808668 in both cases and controls (LD = 1.0 and LD = 0.92, respectively). The XPC polymorphisms rs2228000 and rs2228001 showed only a partial linkage in cases and controls (LD = 0.33 and LD = 0.27, respectively). We evaluated the association with XPA and XPC haplotype blocks and clinical pathological features, OS and PFS. XPA haplotype was significantly correlated with tumour site (P = 0.015), status at onset (P = 0.025) and mitotic index (P = 0.001) (Table 5). Neither OS nor PFS was correlated with XPA or XPC haplotype (data not shown).

Haplotype blocks for XPA (rs1800975 and rs2808668) and XPC (rs2228000 and rs2228001) gene regions
Discussion
To the best of our knowledge, only a case study identified several variants, including CYP1B1 and RAD23 as genetic risk factors for GIST tumour subtypes [22]. In this context, our study represents the first case-control study focusing on polymorphisms in genes of the DNA repair machinery, GIST susceptibility and clinical outcomes following imatinib therapy. In the era of personalized therapy, besides tumour genotype that clearly represents a key player in GIST management, with KIT and PDGFRA having a primary role, germline DNA and epigenetic changes could represent an important aspect impacting the susceptibility and clinical outcome in cancer patients [27, 28]. The different DNA repair pathways represent the main players acting to protect the genome from the deleterious effects on the DNA, deriving from genotoxic exposure, and to guarantee the genome stability. These mechanisms include specific and intricate networks with the aim to counteract various kinds of injuries as single-strand or double-strand breaks [29]. In the absence of this complex machinery, DNA would keep continuing to accumulate different lesions, leading to genome instability, with disease development and death as consequences [29]. The human population is characterized by a notably genetic variability, and each individual might be different from any other in the ability to repair DNA damage. Low repair efficiency has been correlated with a higher cancer risk; presumably, this depends on the fact that unrepaired somatic mutations and chromosomal aberrations contribute to cancer development and progression [30]. DNA repair genes are highly polymorphic, and the existence of these polymorphisms could impact the functionality of the proteins taking part in the DNA repair process, promoting cancer development and progression. Many findings support the hypothesis that different mechanisms could be involved in GIST carcinogenesis as supported by the vastness of different clinical behaviour of each GIST patients, regardless of the KIT/PDGFRA mutational status. In a previous study, we found significant associations between genetic polymorphisms in the key enzymes of the folate metabolic pathways and GIST susceptibility, clinical features and outcome [31]. It is widely accepted that many biological components may affect susceptibility and treatment response, including immune response, factors that regulate cell cycle and apoptosis and DNA repair enzymes. Our previous finding on folate metabolism, together with the supports given by this pathway to the DNA repair machinery, prompted us to evaluate, through a multiple candidate gene approach, 18 polymorphisms in 10 genes codifying for DNA repair enzymes. The key finding was the associations of several polymorphisms in DNA repair genes with GIST susceptibility, different clinical features and clinical outcome.
Polymorphisms in DNA repair genes and GIST susceptibility
With regards to GIST susceptibility, significant associations have been found for the XPD rs13181 WT allele and hOGG1 rs1052133 and XPF rs1800067 minor alleles. The most significant results have emerged for hOGG1, the main mammalian enzyme involved in removing the major adducts produced by reactive oxygen species, through the BER pathway. Interestingly, a role for hOGG1 in tumour suppression is suggested by the frequent loss of the hOGG1 chromosomal locus in human cancer [32]. With regards to the investigated hOGG1 polymorphism (rs1052133), a study performed in a cell line model has shown a two-fold reduction of its enzyme activity, which, in turn, may promote cancer initiation due to accumulation of unrepaired DNA lesions [33]. Our finding supports this hypothesis, as the variant allele is more common in GIST patients than in controls. Furthermore, this result agrees with previous studies linking the minor allele with lung, prostate, oesophageal and colorectal cancer susceptibility [34–36], even opposite results are reported in the literature [37, 38]. With regards to XPD, the enzyme has a double role, participating in both DNA repair (NER pathway) and transcription initiation [39]. In the present study, the XPD WT allele (rs13181) is associated with GIST susceptibility. Interestingly, in previous in vitro studies, the WT allele has been associated with a suboptimal DNA repair capacity and a higher cancer risk, including lung, stomach and skin cancer [40–42]. Our result is in line with these previous findings; however, we should use caution in drawing a definitive conclusion as the findings in the literature are not consistent. Indeed, the XPD (rs13181) minor allele has been associated with cancer of the upper aerodigestive tract, chronic myeloid leukaemia and glioma [43, 44]. Regarding XPF, it forms a tight complex with ERCC1 enzyme, which is a key element in the NER pathway [45]. To the best of our knowledge, no functional in vitro studies are available on XPF variants. However, we predicted the effect of the polymorphism using two different computational approaches: the structured-based method PolyPhen2 [46] and the sequence-based approach SIFT [47]. Through these in silico tools, the XPF rs1800067, an arginine-to-glutamine transition at codon 415, is predicted to be deleterious. Therefore, it could impact the formation of the XPF/ERCC1 complex, diminish its activity and, in the end, alter cancer risk [45]. This is concordant with our finding, showing that the minor allele is associated with GIST susceptibility. Previous studies reported that this polymorphism is correlated with risk of meningioma [48] and benign breast disease (precursor of breast cancer) [49]. Indeed, the data are not consistent as two meta-analyses on squamous cell carcinoma and on several human cancers did not confirm any association with cancer risk [50].
Influence of polymorphisms in DNA repair genes on KIT/PDGFRA mutational status and other tumour features
With regards to the association between DNA repair genotype and KIT/PDGFRA mutational status, we found that the hOGG1 (rs1052133) and XPC (rs2228000) variant alleles are associated respectively, with a lower and higher risk to harbour a KIT/PDGFRA mutation. Although the sample size was limited, our study suggests that hOGG1 variant allele is less likely to be involved in GIST carcinogenesis associated with KIT/PDGFRA mutation. In particular, from our analysis, it seems that hOGG1 (rs1052133) may support the appearance of mutations, typical of KIT/PDGFRA wt-SDH-mutated GIST. To our opinion, this association is intriguing as hOGG1, one of the enzymes initiating the BER process, is detected in both mitochondria and nuclei in human cells. Overall, it could be possible that the hOGG1 polymorphism rs1052133, which is associated with a lower activity, could support the appearance of SDH mutations. This hypothesis leads us to make a further reflection, on the possible involvement of the oxidative stress in KIT/PDGFRA wt-SDH-mutated GIST carcinogenesis, as mitochondria are highly exposed to reactive oxygen species as a result of their respiratory function. Unfortunately, to the best of our knowledge, no signature of oxidative stress response has been reported in both KIT/PDGFRA-mutated and KIT/PDGFRA wt-SDH-mutated GIST.
XPC has a double role in DNA repair, acting in both the DNA repair pathway, BER and NER. In BER, XPC stimulates hOGG1 DNA glycosylase activity through a direct interaction, whereas in NER, after recognition and binding of the damaged DNA, XPC needs to be stabilized and targeted by other partners, such as RAD23 [51]. In view of this, it is worthy to retain that variations on XPC, including the polymorphisms we studied, may impact these interactions and may influence the occurrence of KIT/PDGFRA mutations. In particular, the XPC rs2228000 is located in the domain interacting with RAD23B; the XPC-RAD23 complex formation is critical given that is specifically involved as the earliest damage detector in NER path initiation [52]. Although the polymorphism is predicted to be tolerated in the in silico search, we cannot exclude that the variant might interfere with the optimal XPC-RAD23 interaction. Therefore, this variant might lead to a less efficient NER path, explaining the observed higher rate of KIT/PDGFRA mutation in patients harbouring the variant allele.
In the present study, we also included two polymorphisms in the CYP1B1 gene, based on the work by O’Brien and colleagues, who found these polymorphisms associated with WT GIST and KIT exon 11 codon 557–558 deletion [22]. Due to the small size of our population, we considered only exon 11 mutations; in particular, we compared mutations affecting codons 557 and/or 558 with all other mutations. Regrettably, our study did not corroborate any of the findings by O’Brien and coworkers; indeed, we are aware of the limitation of our study, as the small sample size could have led to a low statistical power, and we cannot exclude that we missed the statistical significance.
With regards to genotype-clinical pathological features interaction, our multiple candidate gene approach highlighted that XPA polymorphisms might be significantly associated with tumour size, mitotic index and status at onset. In particular, the rs1800975 (A) and rs2808668 (C) variant alleles were significantly associated with smaller and metastatic tumours and with a mitotic index greater than 6. The association with status at onset and mitotic index was also confirmed in the haplotype analysis, which also showed a statistical significant correlation with tumour site. XPA directly binds the damaged single strand of DNA and scaffolds multiple DNA repair components of the dynamic NER complex [53]. The rs1800975, located in the proximity of the translation initiation codon, may impact the translation efficiency, and studies in vitro have shown that the variant allele is associated with an increased promoter activity [54]. In accordance with previous works [55], our results showed that rs1800975 is in strong linkage with rs2808668, an intronic polymorphism, and the interaction could act synergically in altering the regular translation efficiency. Herein, these polymorphisms seem to have a predominant correlation with features that suggest an aggressive behaviour as metastatic status and high mitotic index. The XPA gene is located on chromosome 9q, which usually is not affected by deletion/translocation in GIST allowing preserving its function during tumorigenesis. This is an interesting aspect considering that it has been proposed that XPA has a role in the cell cycle checkpoint regulation [56], and it is reasonable to suppose that the two polymorphisms together may contribute to a loss of cycle regulation and drive towards a more hostile behaviour.
An additional significant association has been observed between XPF rs3136155 minor allele and small intestine GIST, with the variant allele more common in patients with small intestine GIST. Usually, small intestine GIST behaves more aggressively than gastric tumours [15]; therefore, even at the moment, there are no reports in the literature on this intronic polymorphism that can corroborate our finding; we suggest that this polymorphism could interfere with the normal DNA repair efficiency of XPF and promote a worse prognosis in GIST patients.
Polymorphisms in DNA repair genes and clinical outcome
With regards to the clinical outcome to imatinib treatment, we analysed, for the first time, the impact of polymorphisms in DNA repair genes on TTP and OS in GIST patients. Genetic polymorphisms in drug metabolizer and transporter genes were a reasonable starting point to evaluate their relevance in treatment outcome [57–59]. Indeed, also polymorphisms in DNA repair genes have attracted the research interest as it is currently understudied whether the inherited individual capability to repair DNA damage could affect the treatment results. In the present study, TTP was affected by XPD (rs50872), XPC (rs2228000 and rs2228001) and hOGG1 (rs1052133). Regarding the XPD polymorphisms, the variant allele has been associated with increased TTP. Interestingly, this poorly studied intronic polymorphism was also associated with a prolonged PFS in non-small cell lung cancer patients, treated with first-line platinum-based chemotherapy [60]. The two polymorphisms in the XPC gene have shown an opposite behaviour, as the rs2228000 and rs2228001 minor alleles are associated with a longer and shorter TTP respectively. The XPC rs2228001 variant allele is associated with suboptimal repair efficiency [52] and may promote the acquisition of additional mutations driving to tumour progression. This finding is supported by the Kaplan-Meier model that shows the presence of at least one major allele significantly associated with a longer OS. With regards to hOGG1, the rs1052133 variant allele, linked to a defective DNA repair activity, is associated with a shorter TTP. As we have seen for the XPC (rs2228001), this finding is intriguing, as a lower DNA repair activity is reflected in a reduced time before progressing. In addition, as the main cause of imatinib resistance and progression is the emergency of secondary mutations, it is worthy to think that an inadequate DNA repair capacity may contribute to a faster progression. With regards to OS, significant associations were found for hOGG1 (rs1052133), XPF (rs3136155) and XRCC3 (rs861539) genotype. Concerning hOGG1, the variant allele had a lower risk to die due to the tumour; this result is apparently in contrast with the previous finding; indeed, cells with a reduced DNA repair capacity could have a major benefit from the treatment, irrespectively of the acquisition of secondary mutations and subsequent progression. The XRCC3 gene product is required for efficient repair of double-strand breaks and DNA cross-links via HRR pathway and correct chromosomal segregation [61]. The variant allele we studied has been linked with an increased risk of developing tetraploid cells that promotes loss of chromosomes and rearrangements, causing genetic instability [62]. In the present study, in line with the role of the variant allele, we found the XRCC3 polymorphism associated with a higher risk of death.
Concluding remarks
To the best of our knowledge, this is the first time that polymorphisms in DNA repair genes, belonging to the different pathways, have been extensively evaluated in GIST patients. Through this multiple candidate gene approach, we report the significant associations between polymorphisms in DNA repair genes, susceptibility, clinical pathological features and clinical outcome in GIST. We are aware of the limitations of this study due to the small size of the investigated population. However, we would like to point out that 81 patients represent a considerable number given the rarity of this disease, and we believe that genetic polymorphisms in DNA repair genes, as those described in the present study, are worthy of further investigations.
References
Corless CL, Fletcher JA, Heinrich MC. Biology of gastrointestinal stromal tumors. J Clin Oncol. 2004;22:3813–25.
Plaat BE, Hollema H, Molenaar WM, et al. Soft tissue leiomyosarcomas and malignant gastrointestinal stromal tumors: differences in clinical outcome and expression of multidrug resistance proteins. J Clin Oncol. 2000;18:3211–20.
Dematteo RP, Heinrich MC, El-Rifai WM, et al. Clinical management of gastrointestinal stromal tumors: before and after STI-571. Hum Pathol. 2002;33:466–77.
Nannini M, Astolfi A, Urbini M, et al. Integrated genomic study of quadruple-WT GIST (KIT/PDGRA/SDH/RAS pathway wild-type GIST). BMC Cancer. 2014;14:685.
Miettinen M, Lasota J. Gastrointestinal stromal tumors: review on morphology, molecular pathology, prognosis, and differential diagnosis. Arch Pathol Lab Med. 2006;130:1466–78.
Szkandera J, Absenger G, Liegl-Atzwanger B, et al. Common gene variants in RAD51, XRCC2 and XPD are not associated with clinical outcome in soft-tissue sarcoma patients. Cancer Epidemiol. 2013;37:1003–9.
Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8.
Kim J, Mouw KW, Polak P, et al. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet. 2016;48:600–6.
Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–74.
Maynard S, Schurman SH, Harboe C, et al. Carcinogenesis. 2009;30:2–10.
Weterings E, Chen DJ. The endless tale of non-homologous end-joining. Cell Res. 2008:114–24.
Ledermann JA, Drew Y, Kristeleit RS. Homologous recombination deficiency and ovarian cancer. Eur J Cancer. 2016;60:49–58.
Vodicka P, Stetina R, Polakova V, et al. Association of DNA repair polymorphisms with DNA repair functional outcomes in healthy human subjects. Carcinogenesis. 2007;28:657–64.
Angelini S, Kumar R, Carbone F, et al. Inherited susceptibility to bleomycin-induced micronuclei: correlating polymorphisms in GSTT1, GSTM1 and DNA repair genes with mutagen sensitivity. Mutat Res. 2008;638:90–7.
Angelini S, Kumar R, Carbone F, et al. Micronuclei in humans induced by exposure to low level of ionizing radiation: influence of polymorphisms in DNA repair genes. Mutat Res. 2005;570:105–17.
Angelini S, Maffei F, Bermejo JL, et al. Environmental exposure to benzene, micronucleus formation and polymorphisms in DNA-repair genes: a pilot study. Mutat Res. 2012;743:99–104.
Milic M, Rozgaj R, Kasuba V, et al. Polymorphisms in DNA repair genes: link with biomarkers of the CBMN cytome assay in hospital workers chronically exposed to low doses of ionizing radiation. Arh Hig rad Toksikol. 2015;66:109–20.
Yuan K, Huo M, Sun Y, et al. Association between x-ray repair cross-complementing group 3 (XRCC3) genetic polymorphisms and papillary thyroid cancer susceptibility in a Chinese Han population. Tumour Biol. 2016;37:979–87.
Ji HX, Chang WS, Tsai CW, et al. Contribution of DNA repair Xeroderma pigmentosum group D genotype to gastric cancer risk in Taiwan. Anticancer Res. 2015;35:4975–81.
Yu H, Wu X, Zhang Y, et al. Genetic variability of DNA repair mechanisms influences chemotherapy outcome of gastric cancer. Int J Clin Exp Pathol. 2015;8:4106–12.
Zhou J, Liu ZY, Li CB, et al. Genetic polymorphisms of DNA repair pathways influence the response to chemotherapy and overall survival of gastric cancer. Tumour Biol. 2015;36:3017–23.
O’Brien KM, Orlow I, Antonescu CR, et al. Gastrointestinal stromal tumors, somatic mutations and candidate genetic risk variants. PLoS One. 2013;8:e62119.
Cho S, Kim MJ, Choi YY, et al. Associations between polymorphisms in DNA repair genes and TP53 mutations in non-small cell lung cancer. Lung Cancer. 2011;73:25–31.
Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–5.
Gabriel SB, Schaffner SF, Nguyen H, et al. The structure of haplotype blocks in the human genome. Science. 2002;296:2225–9.
Stata Corporation. Stata statistical software, release 11. College Station: Stata Corporation; 2011.
Ravegnini G, Nannini M, Sammarini G, et al. Personalized medicine in gastrointestinal stromal tumor (GIST): clinical implications of the somatic and germline DNA analysis. Int J Mol Sci. 2015;16:15592–608.
Nannini M, Ravegnini G, Angelini S, et al. MicroRNA profiling in gastrointestinal stromal tumors: implication as diagnostic and prognostic markers. Epigenomics. 2015;7:1033–49.
Michalska MM, Samulak D, Romanowicz H, et al. Single nucleotide polymorphisms (SNPs) of hOGG1 and XRCC1 DNA repair genes and the risk of ovarian cancer in polish women. Tumour Biol. 2015;36:9457–63.
Frank SA. Genetic predisposition to cancer—insights from population genetics. Nat Rev Genet. 2004;5:764–72.
Angelini S, Ravegnini G, Nannini M, et al. Folate-related polymorphisms in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Eur J Hum Genet. 2015;23:817–23.
Hill JW, Evans MK. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006;34:1620–32.
Bravard A, Vacher M, Moritz E, et al. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009;69:3642–9.
Su Y, Zhang H, Xu F, et al. DNA repair Gene polymorphisms in relation to non-small cell lung cancer survival. Cell Physiol Biochem. 2015;36:1419–29.
Chen Y, Li J, Li T, Mo Z. hOGG1 C1245G gene polymorphism associated with prostate cancer: a meta-analysis. Int J Biol Markers. 2015;30:e161–8.
Zhang J. A meta-analysis of the association between the hOGG1 Ser326Cys polymorphism and the risk of esophageal squamous cell carcinoma. PLoS One. 2013;8:e65742.
Zhang H, Xu Y, Zhang Z, et al. The hOGG1 Ser326Cys polymorphism and prostate cancer risk: a meta-analysis of 2584 cases and 3234 controls. BMC Cancer. 2011;11:391.
Gu D, Wang M, Zhang Z, et al. Lack of association between the hOGG1 Ser326Cys polymorphism and breast cancer risk: evidence from 11 case-control studies. Breast Cancer Res Treat. 2010;122:527–31.
Zhu ML, He J, Wang M, et al. Potentially functional polymorphisms in the ERCC2 gene and risk of esophageal squamous cell carcinoma in Chinese populations. Sci Rep. 2014;4:6281.
Spitz MR, Wu X, Wang Y, et al. Modulation of nucleotide excision repair capacity by XPD polymorphisms in lung cancer patients. Cancer Res. 2001;61:1354–7.
Xue MH, Li GY, XJ W, Zhang CX, Zhang CF, Zhu K, et al. Genetic variability of genes in NER pathway influences the treatment outcome of gastric cancer. Int J Clin Exp Pathol. 2015;8:5563–9.
Dong Y, Zhuang L, Ma W. Comprehensive assessment of the association of ERCC2 Lys751Gln polymorphism with susceptibility to cutaneous melanoma. Tumour Biol. 2013;34:1155–60.
Bănescu C, Trifa AP, Demian S, et al. Polymorphism of XRCC1, XRCC3, and XPD genes and risk of chronic myeloid leukemia. Biomed Res Int. 2014;2014:213790.
Adel Fahmideh M, Schwartzbaum J, Frumento P, et al. Association between DNA repair gene polymorphisms and risk of glioma: a systematic review and meta-analysis. Neuro-Oncology. 2014;16:807–14.
Tsodikov OV, Enzlin JH, Schärer OD, et al. Crystal structure and DNA binding functions of ERCC1, a subunit of the DNA structure-specific endonuclease XPF-ERCC1. Proc Natl Acad Sci U S A. 2005;102:11236–41.
Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting missense mutations. Nat Methods. 2010;7:248–9.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Rajaraman P, Hutchinson A, Wichner S, et al. DNA repair gene polymorphisms and risk of adult meningioma, glioma, and acoustic neuroma. Neuro-Oncology. 2010;12:37–48.
Jorgensen TJ, Helzlsouer KJ, Clipp SC, et al. DNA repair gene variants associated with benign breast disease in high cancer risk women. Cancer Epidemiol Biomark Prev. 2009;18:346–50.
Shi TY, He J, Qiu LX, Zhu ML, et al. Association between XPF polymorphisms and cancer risk: a meta-analysis. PLoS One. 2012;7:e38606.
Bernardes de Jesus BM, Bjørås M, Coin F, et al. Dissection of the molecular defects caused by pathogenic mutations in the DNA repair factor XPC. Mol Cell Biol. 2008;28:7225–35.
He J, Shi TY, Zhu ML, et al. Associations of Lys939Gln and Ala499Val polymorphisms of the XPC gene with cancer susceptibility: a meta-analysis. Int J Cancer. 2013;133:1765–75.
Donninger H, Clark J, Rinaldo F, et al. The RASSF1A tumor suppressor regulates XPA-mediated DNA repair. Mol Cell Biol. 2015;35:277–87.
Park JY, Park SH, Choi JE, et al. Polymorphisms of the DNA repair gene xeroderma pigmentosum group a and risk of primary lung cancer. Cancer Epidemiol Biomark Prev. 2002;11:993–7.
Han W, Kim KY, Yang SJ, et al. SNP-SNP interactions between DNA repair genes were associated with breast cancer risk in a Korean population. Cancer. 2012;118:594–602.
Li Z, Musich PR, Serrano MA, et al. XPA-mediated regulation of global nucleotide excision repair by ATR is p53-dependent and occurs primarily in S-phase. PLoS One. 2011;6:e28326.
Angelini S, Pantaleo MA, Ravegnini G, et al. Polymorphisms in OCTN1 and OCTN2 transporters genes are associated with prolonged time to progression in unresectable gastrointestinal stromal tumor s treated with imatinib therapy. Pharmacol Res. 2013;68:1–6.
Angelini S, Ravegnini G, Fletcher JA, et al. Clinical relevance of pharmacogenetics in gastrointestinal stromal tumor treatment in the era of personalized therapy. Pharmacogenomics. 2013;14:941–56.
Ravegnini G, Sammarini G, Angelini S, Hrelia P. Pharmacogenetics of tyrosine kinase inhibitors in gastrointestinal stromal tumor and chronic myeloid leukemia. Expert Opin Drug Metab Toxicol. 2016;12:733–42.
Kim SH, Lee GW, Lee MJ, et al. Clinical significance of ERCC2 haplotype-tagging single nucleotide polymorphisms in patients with unresectable non-small cell lung cancer treated with first-line platinum-based chemotherapy. Lung Cancer. 2012;77:578–84.
Vangsted A, Gimsing P, Klausen TW, et al. Polymorphisms in the genes ERCC2, XRCC3 and CD3EAP influence treatment outcome in multiple myeloma patients undergoing autologous bone marrow transplantation. Int J Cancer. 2007;120:1036–45.
Yoshihara T, Ishida M, Kinomura A, et al. XRCC3 deficiency results in a defect in recombination and increased endoreduplication in human cells. EMBO J. 2004;23:670–80.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of interest
None
Additional information
Margherita Nannini, Vittorio Simeon and Muriel Musti contributed equally.
Patrizia Hrelia and Sabrina Angelini jointly directed the work.
Electronic supplementary material
ESM 1
(DOCX 37 kb)
Rights and permissions
About this article

Cite this article
Ravegnini, G., Nannini, M., Simeon, V. et al. Polymorphisms in DNA repair genes in gastrointestinal stromal tumours: susceptibility and correlation with tumour characteristics and clinical outcome. Tumor Biol. 37, 13413–13423 (2016). https://doi.org/10.1007/s13277-016-5276-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13277-016-5276-7