
Abstract
Due to the complexity and heterogeneity of breast cancer, the therapeutic effects of breast cancer treatment vary between subtypes. Breast cancer subtypes are classified based on the presence of molecular markers for estrogen or progesterone receptors and human epidermal growth factor 2. Thus, novel, comprehensive, and precise molecular indicators in breast carcinogenesis are urgently needed. Here, we report that ZNF133, a zinc-finger protein, is negatively associated with poor survival and advanced pathological staging of breast carcinomas. Moreover, ZNF133 is a transcription repressor physically associated with the KAP1 complex. It transcriptionally represses a cohort of genes, including L1CAM, that are critically involved in cell proliferation and motility. We also demonstrate that the ZNF133/KAP1 complex inhibits the proliferation and invasion of breast cancer cells in vitro and suppresses breast cancer growth and metastasis in vivo by dampening the transcription of L1CAM. Taken together, the findings of our study confirm the value of ZNF133 and L1CAM levels in the diagnosis and prognosis of breast cancer, contribute to a deeper understanding of the regulation mechanism of ZNF133 for the first time, and provide a new therapeutic strategy and precise intervention target for breast cancer.
Similar content being viewed by others


Introduction
Breast cancer is the most commonly diagnosed malignancy worldwide and the commonest cause of cancer-related mortality in women. Poor prognosis in breast cancer patients is generally associated with metastatic disease [1]. The risk profiles and treatment strategies for breast cancer depend mainly on three major subtypes categorized according to estrogen or progesterone receptor expression and ERBB2 gene amplification [2, 3]. However, numerous molecular players and mechanisms behind the complexity of its clinical behavior remain unknown [4]. Novel molecules are expected to be revealed through breast cancer research. Thus, in-depth molecular characterizations play critical roles in the current anticancer research era. The future of cancer diagnosis and prognosis relies on tremendously precise molecular indexing [5].
Zinc finger proteins (ZFPs) constitute the largest individual family of transcription factors in tetrapod vertebrates [6]. In humans, approximately one-third of about 800 ZFPs contain the Krüppel-associated box (KRAB). Most members of the KRAB-ZFPs family have been identified as crucial regulators of diverse cellular processes, including embryonic development, cell differentiation, cell proliferation, apoptosis, neoplastic transformation and cell cycle regulation [7]. Accordingly, dysfunction of zinc-finger protein 133 (ZNF133), a member of the KRAB-ZFPs family, is implicated in various pathological states including malignancies. ZNF133 was reported to be involved in posterior polymorphous corneal dystrophy (PPCD), inflammatory bowel diseases, and conditions related to alcoholism [8,9,10]. In addition, an aberrant expression of ZNF133 was noted in benign bone tumors, osteosarcomas, neuroblastomas, and breast cancer [11,12,13]. For example, ZNF133 was suggested as a potential protein involved in the repression of S100A4, which was associated with the increased metastatic capacity of various types of cancer cells [14]. In 2020, ZNF133 was found to be overexpressed after irradiation in patients who survived a childhood malignancy; thus, it was considered a candidate for radiation-sensitive genes in childhood tumors [15]. Furthermore, the quantity of the rare allele of ANLN:rs12535394 was reported to be positively associated with the expression of ZNF133; it was prognostic of a favorable clinical outcome in breast cancer [13]. However, the molecular mechanism underlying the role of ZNF133 in breast tumorigenesis is poorly understood.
The L1 cell adhesion molecule (L1CAM)) is a transmembrane glycoprotein. Its full-length form is composed of a long ectodomain that comprises six immunoglobulin-like (Ig) domains followed by five fibronectin type III repeats, a single transmembrane domain and a relatively short highly conserved cytoplasmic domain [16]. Initially, L1CAM was found in the neurons of the central nervous system; it is known for its role in neural development and its ability to regulate neurite outgrowth, fasciculation, cell adhesion, cell migration, myelination, and cell survival [17, 18]. Subsequent studies unveiled that L1CAM was overexpressed in primary tumors and metastases of various types of cancers, such as glioma, melanoma, ovarian, colon, breast and pancreatic cancers [19,20,21,22]. Additionally, L1CAM was associated with poor prognosis. In these cancers, tumor cells tend to use the same mechanisms involving L1CAM as neurons do to obtain increased migratory and proliferative potential. In pancreatic cancer cells, for instance, downregulation of L1CAM inhibits cell proliferation and invasion and arrests cell cycle progression l [23]. In 2018, studies from the Memorial Sloan Kettering Cancer Center (MSK) revealed that L1CAM was necessary for the metastatic colonization of multiple organs in breast cancer, such as lung, bone, brain, and liver [24]. Hence, L1CAM is a valuable diagnostic marker and an attractive target for breast cancer therapy.
The control of L1CAM expression in cancers is complex and is affected by both transcriptional and epigenetic mechanisms. Positive regulation of L1CAM was reported for the transcription factors β-catenin and SLUG in colon, endometrial, and pancreatic cancer cells [20, 25], and negative regulation of L1CAM was found for AR and REST in triple-negative breast cancers and small cell lung cancer [26, 27]. Nevertheless, the molecular mechanism underlying the regulation of L1CAM expression in breast carcinogenesis remains poorly explored.
In this study, we report that L1CAM expression is suppressed by ZNF133, a novel KRAB zinc-finger protein in breast cancer cells. The findings demonstrate that ZNF133 recruits the KAP1 complex to the L1CAM promoter and represses L1CAM transcription. Additionally, the findings show that the ZNF133 inhibits the proliferation and invasive potential of breast cancer cells in vitro and suppresses breast cancer growth and metastasis in vivo. These data support a role for ZNF133 as a novel transcription repressor and a potential tumor suppressor in breast cancer.
Results
A lower expression level of ZNF133 predicts a poorer prognosis in patients with breast cancer
To explore the novel and intricate molecular mechanisms underlying the development and progression of breast cancer and to further explore the molecular function of KRAB-ZFPs, we performed a Kaplan–Meier survival analysis (http://kmplot.com/analysis/) in public datasets. We found that higher ZNF133 expression was associated with better relapse-free survival in breast cancer patients (Fig. 1A). Analysis of the clinical specimens dataset in the Human Protein Atlas (https://www.proteinatlas.org/) also consistently showed that the higher expression of ZNF133 indicated longer overall survival time in breast cancer (Fig. 1B). In order to further explore the level of ZNF133 in each breast cancer subtype, we used the Gene expression-based Outcome for Breast Cancer Online (GOBO) tool (http://co.bmc.lu.se/gobo/gsa.pl) to examine the public dataset for ZNF133 expression. The result showed that the expression of ZNF133 was lower in each breast cancer subtype than in normal-like tissue (Fig. 1C). Moreover, we found that the level of ZNF133 expression was negatively correlated with the pathologic grade of the tumors (Fig. 1D). Next, we investigated the association between ZNF133 expression and breast cancer stage. Analysis from the TCGA database in the UALCAN portal (http://ualcan.path.uab.edu/index.html) revealed that the level of ZNF133 expression was higher in normal patients than patients in the other four individual breast cancer stages (Fig. 1E). Overall, the data above indicate the ability of ZNF133 to act as a potential suppressive marker in breast cancer. To further confirm the clinical relevance of ZNF133 expression, we assessed ZNF133 levels in other two clinical databases. The body map from Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/) showed that the median level of ZNF133 expression in breast cancer is lower than in normal samples (Fig. 1F). The dot plot showed the gene expression profile across all tumor samples and paired normal tissues (q cutoff = 0.01) (Fig. 1G). Additionally, we analyzed the mRNA expression of ZNF133 in the Oncomine database (https://www.oncomine.org/). In the TCGA breast cancer datasets, the subtypes of invasive breast carcinoma tissues had lower ZNF133 expression levels than noncancerous tissues (Fig. 1H). In particular, the male breast carcinoma or mucinous breast carcinoma samples, which had lower levels of ZNF133, statistically tended to be considered highly aggressive. Based on the above assessment of ZNF133 expression levels in breast cancer tissues versus normal tissues and correlation with subtypes and stages using publicly available datasets, we preliminary ascertained that ZNF133 may be a breast cancer suppressor.

A Kaplan–Meier survival analysis for the relationship between survival time and ZNF133 signature in breast cancer using the online tool (http://kmplot.com/analysis/). The patient samples were split into two groups according to quantile expressions of ZNF133 to analyze the prognostic value of ZNF133. The two patient cohorts were compared by a Kaplan–Meier survival plot, and the hazard ratio with 95% confidence intervals and log-rank P-value was calculated. B Kaplan–Meier analysis was performed from the online database of the Human Protein Atlas (https://www.proteinatlas.org/), and the results showed that a higher expression level of ZNF133 predicts better overall survival. C The expression of ZNF133 in different subtypes of breast cancer via the Gene expression-based Outcome for Breast Cancer Online (GOBO) analysis (http://co.bmc.lu.se/gobo/gsa.pl). The P value was calculated using one-way ANOVA for multiplegroup comparison. The number of tumor samples in each subtype is Basal (n = 304), HER2-enriched (n = 240), Luminal A (n = 465), Luminal B (n = 471), and Normal-like (n = 304). D The correlation of ZNF133 expression and histological grade using the GOBO analysis. The number of tumor samples in each grade is grade I (n = 239), grade II (n = 677), and grade III (n = 495). The P value is calculated using one-way ANOVA for multiplegroup comparison. E The mRNA expression of ZNF133 in four individual breast cancer stages compared with the normal samples (n = 114) in the UALCAN database (http://ualcan.path.uab.edu/index.html). The number of tumor samples in each stage is stage I (n = 183), stage II (n = 615), stage III (n = 247), and stage IV (n = 20). P values were analyzed by a two-tailed unpaired t-test (**P < 0.01 and ***P < 0.001). F The interactive body map of median ZNF133 expression in breast cancer (BRCA) and normal samples in the Gene Expression Profiling Interactive Analysis (GEPIA) web server (http://gepia.cancer-pku.cn/). G The dot plot of ZNF133 expression profile across BRCA (n = 1084) and paired normal tissues (n = 291) from GEPIA datasets. Each dot represents the expression of samples. H The mRNA expression of ZNF133 in different groups of breast cancer samples compared with noncancerous tissue samples from the Oncomine database (https://www.oncomine.org/resource/login.html#). P values were analyzed by a two-tailed unpaired t-test (*P < 0.05 and ***P < 0.001).
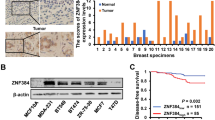
Next, we performed immunohistochemical staining of human tissue arrays containing 40 breast carcinoma samples paired with normal mammary tissues. We found that ZNF133 was mainly detected in the nucleus and significantly downregulated in breast carcinomas compared with adjacent noncancerous tissues. Moreover, ZNF133 expression level was negatively correlated with the breast cancer stage (Fig. 2A), and low ZNF133 expression significantly correlated with advanced pathological grade (p < 0.001) (Fig. 2B). Likewise, we also observed that ZNF133 displayed a positively strong expression in normal breast tissues, where ZNF133 was mostly specific for the epithelium glands and myoepithelial cells, and a weak expression in breast cancer (Fig. 2C), based on clinical specimens from the Human Protein Atlas. The above data indicated that ZNF133 is associated with a good prognosis and appears to have a negative effect on breast cancer progression.

A, B Immunohistochemical staining of ZNF133 in a tissue microarray (TMA) containing 40 breast cancer samples with paired adjacent normal tissues. Representative sections of normal and breast cancer tissues (A), as well as histological grades II and III of breast samples (B) that were immunostained with ZNF133 antibodies, are presented. Bar, 20 μm. The scores were determined by evaluating the extent and intensity of immunopositivity and were analyzed by a two-tailed paired t-test (***p < 0.001). C The expression of ZNF133 in breast cancer specimens and the adjacent normal breast tissues were acquired from the online database of the Human Protein Atlas. Bar, 200 μm.
ZNF133 inhibits the proliferation and invasion of breast cancer cells
To confirm this hypothesis, we first tested the effect of ZNF133 on the proliferation of breast cancer cells in vitro. To this end, MCF-7 and MDA-MB-231 cells were infected with lentiviruses carrying ZNF133 expression or ZNF133 shRNA, and the number of cells was examined. Our results showed that ZNF133 overexpression had a significant inhibitory effect on both MCF-7 and MDA-MB-231 cells, whereas the knockdown of ZNF133 promoted the proliferation of these cells (Fig. S1A). In addition, colony formation assays in MCF-7 and MDA-MB-231 cells revealed that ZNF133 overexpression was associated with a decreased colony number and ZNF133 knockdown led to an increase in colony number (Fig. S1B). These experiments support the role of ZNF133 in the inhibition of cell proliferation.
Metastases occur frequently in patients with breast cancer and are associated with significant morbidity and exceptionally poor survival. To investigate the role of ZNF133 in the invasion of breast cancer cells, we first performed a transwell cell invasion and tested the effect of ZNF133 on the invasive potential of breast cancer cells. MDA-MB-231 cells were infected with lentiviruses carrying ZNF133 or ZNF133 shRNA. We found that the overexpression of ZNF133 was associated with a decrease in the invasive potential of breast cancer cells. Furthermore, the knockdown of ZNF133 led to an increase in the invasive potential of cells (Fig. S1C). As epithelial–mesenchymal transition (EMT) is an early event in cell invasion, we then used western blotting to measure the expression of epithelial/mesenchymal markers in MCF-7 and MDA-MB-231 cells. The results showed that ZNF133 overexpression resulted in the induction of epithelial protein markers (E-cadherin) and the reduction of mesenchymal markers (N-cadherin and Fibronectin) (Fig. S1D). Conversely, the depletion of ZNF133 resulted in the reduction of epithelial markers and induction of mesenchymal markers (Fig. S1D). These results support the role of ZNF133 in the regulation of the invasive potential of breast cancer cells.
ZNF133 is a transcription repressor
ZNF133 is mapped to chromosome 20p11.23 and consists of ten exons. The predicted molecular weight of ZNF133 is 73.33 kDa. Bioinformatics analysis indicates that ZNF133 harbors an N-terminal KRAB domain and eleven C2H2 zinc fingers (Fig. S2A).
Amino-acid sequence alignment has confirmed that the similarity of human ZNF133 with homologs in other organisms is 99.2% in Pan troglodytes, 96% in Macaca mulatta, 86.8% in Canis lupus familiaris, and 78.7% in Rattus norvegicus (Fig. S2B). The percentage identity of ZNF133 from the organisms is shown in Fig. S2C. In addition, phylogenetic analysis indicates that ZNF133 is an evolutionarily well-conserved gene (Fig. S2D), thus suggesting its crucial physiological role of ZNF133.
Immunofluorescent imaging of ZNF133 in MCF-7, T47D and MDA-MB-231 cells indicated that ZNF133 is primarily localized in the nucleus (Fig. 3A). To confirm the expression of the full-length ZNF133 protein, FLAG-tagged ZNF133 (FLAG-ZNF133) expression plasmids were transfected into MCF-7 and MDA-MB-231 cells . They were then analyzed by western blotting with a monoclonal antibody against FLAG. The results showed that ZNF133 is a protein with a molecular weight of ~75 kDa (Fig. 3B).

A Subcellular localization of ZNF133 protein. Bar, 10 μm. B Western blotting analysis of ZNF133 protein expression. MCF-7 or MDA-MB-231 cells were transfected with empty vector or FLAG-ZNF133. Cellular proteins were extracted from indicated cell lines and western blotting was performed with antibodies against ZNF133, FLAG, or GAPDH. C The schematic diagram showing the Gal4-luciferase reporters. D Transcription repression by ZNF133. HEK293T or MCF-7 cells were transfected with different amounts of Gal4-ZNF133 expression plasmids, together with the indicated Gal4-luciferase reporter. Forty-eight hours later, luciferase activity was measured. Relative luciferase activity was calculated as firefly luciferase activity divided by renilla luciferase activity and shown relative to the control (transfected with Gal4-DBD vector). The expression of Gal4-ZNF133 was shown by western blotting. Error bars represent mean ± SD for three independent experiments. P values were determined by Student’s t-test. *P < 0.05.
As stated above, the KRAB-ZFP family proteins have been implicated in transcriptional regulation. To explore the transcriptional activity of ZNF133, we fused the full length of ZNF133 to the C-terminus of the Gal4 DNA-binding domain and analyzed the transcriptional activity of the fused construct in HEK293T and MCF-7 cells. We utilized three different Gal4-driven luciferase reporter systems, which differed in basal promoter elements (Fig. 3C). Based on the results, ZNF133 significantly inhibited the reporter activity in a dose-dependent manner in all three reporter systems in both HEK293T and MCF-7 cells (Fig. 3D), suggesting that ZNF133 is involved in transcription repression. Meanwhile, overexpression of FLAG-ZNF133 did not affect the activity of Gal4-driven reporter in MCF-7 cells (Fig. 3E); therefore, ZNF133 must physically bind to DNA to exert its transcriptional activity.
ZNF133 is physically associated with the KAP1 complex
To further elucidate the molecular mechanism underlying ZNF133-mediated transcription repression, we interrogated the ZNF133 interactome by affinity purification-coupled mass spectrometry. FLAG-ZNF133 was stably expressed in HEK293T cells, and the whole-cell extracts were prepared and subjected to affinity purification using an anti-FLAG affinity gel. Mass spectrometric analysis indicated that ZNF133 was co-purified with components of the KAP1 complex, i.e., KAP1, HDAC1/2, RBBP4/7, and HP1 (Fig. 4A).

A Mass spectrometry analysis of ZNF133-associated proteins. Cellular extracts from HEK293T cells stably expressing FLAG-ZNF133 were immunopurified with an anti-FLAG affinity column and eluted with FLAG peptides. The eluates were resolved by SDS-PAGE and silver-stained. The protein bands were retrieved and analyzed by mass spectrometry. B Interaction of ZNF133 with the components of the KAP1 complex. Whole-cell lysates from MCF-7, MDA-MB-231, or MCF-10A cells transfected with FLAG-ZNF133 were prepared. Iimmunoprecipitation was performed with anti-FLAG followed by immunoblotting with antibodies against indicated proteins. C Whole-cell lysates from MCF-7 or MDA-MB-231 cells transfected with FLAG-ZNF133 were immunoprecipitated with antibodies against indicated proteins or IgG followed by immunoblotting with the antibodies against FLAG. D Co-fractionation of ZNF133 and KAP1 complex by FPLC. Proteins of MCF-7 cells transfected with FLAG-ZNF133 were extracted, concentrated, and then fractionated on Superose 6 size exclusion columns. Chromatographic elution profiles and immunoblotting analysis of the chromatographic fractions are shown. The elution positions of calibration proteins with known molecular masses are indicated, and an equal volume from each fraction was analyzed. E Western blotting analysis of ZNF133 protein expression in MCF-7, MDA-MB-231, or MCF-10A cells. Cellular proteins were extracted from indicated cell lines, and western blotting was performed with antibodies against ZNF133 or GAPDH. F Bacterially expressed GST or GST-fused indicated proteins were performed in GST pull-down experiments with in vitro transcribed/translated FLAG-ZNF133. Error bars represent mean ± SD for three independent experiments.
To confirm the interaction between ZNF133 and the KAP1 complex in breast cancer cells, protein extracts from MCF-7, MDA-MB-231, and MCF-10A cells overexpressing FLAG-ZNF133 were immunoprecipitated with the anti-FLAG followed by immunoblotting with antibodies against KAP1, HDAC1/2, RBBP4/7, and HP1. The results showed that components of the KAP1 complex could be efficiently co-immunoprecipitated by ZNF133 (Fig. 4B). Reciprocally, IP with antibodies against components of the KAP1 complex followed by IB with antibodies against FLAG also confirmed the interaction of ZNF133 with the KAP1 complex (Fig. 4C). Next, analysis of FLAG-ZNF133 affinity eluate by fast protein liquid chromatography (FPLC) with Superose 6 columns and a high salt extraction and size exclusion approach in MCF-7 cells stably expressing FLAG-ZNF133 was performed. We found a multiprotein complex containing ZNF133, KAP1, HDAC1/2, RBBP4/7 and HP1 (Fig. 4D). However, we compared the ZNF133 protein levels in the three cell lines and noted that ZNF133 expression was higher in MCF-10A than in MCF-7 or MDA-MB-231 cells (Fig. 4E). The differential levels of ZNF133 expression in different breast cancer cells might result in varying degrees of suppressive action on cancer cell proliferation and motility.
Next, we employed glutathione S-transferase (GST) pull-down assays to investigate the molecular details involved in this interaction.. In the experiment, recombinant GST-tagged KAP1, HDAC1/2, RBBP4/7 proteins, or GST proteins were induced by isopropyl-1-thiob-d-galactopyranoside (IPTG) and purified by Glutathione-Sepharose 4B from bacterial supernatants. Then, they were incubated with in vitro transcribed/translated FALG-ZNF133 in a binding reaction. The final pull-down products by Glutathione-Sepharose 4B were washed and collected for immunoblotting. The results showed that ZNF133 was detected in the pull-down products with GST-tagged KAP1, but not in the GST control reaction or other components of the KAP1 complex reaction (Fig. 4F). These results indicate that ZNF133 can interact with KAP1 specifically and directly in vitro. Taken together, these findings support the specific interaction between ZNF133 and KAP1 complex.
Identification of genome-wide transcriptional targets for the ZNF133
To explore the biological significance of the physical interaction of ZNF133, we performed chromatin immunoprecipitation-based deep sequencing (ChIP-seq) that analyzed the genome-wide transcriptional targets of ZNF133. In these experiments, ChIP experiments were performed in MCF-7 cells stably expressing FLAG-ZNF133. Then, ZNF133-associated DNAs were amplified using nonbiased conditions, labeled, and then sequenced using HiSeq2500. Using MACS (version2) with a p-value cutoff of 10−3, we identified 2,907 ZNF133-specific binding peaks, and a total of 1122 genes in the promoter cluster targeted by ZNF133 were identified. The distribution of specific binding peaks was 42.98% promoter, 29.73% intron, 3.79% exon, 21.71% distal intergenic, 0.14% 5′ UTR5, 1.10% 3′ UTR3, and 0.55% downstream (≤3 kb) (Fig. 5A). The chromosome distribution of the peaks is also shown in Fig. 5B. The 1122 genes targeted by ZNF133 were then classified into various cellular biological processes using the Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/). These biological processes include the development of dendrite, perikaryon and neuronal cell bodies, as well as cell proliferation and cycle pathways (Fig. 5C). Furthermore, we analyzed the genomic binding signatures for ZNF133 and identified the most significant binding motif CTGTCCATGGTCCTGA (Fig. 5D).

A ChIP-seq analysis was performed in MCF-7 cells transfected with FLAG-ZNF133 using a specific antibody against FLAG, and the peaks’ distribution of ZNF133 was determined. B The relative peaks’ chromosome distribution is shown. C KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway database was used to identify the pathways that the ZNF133 target genes were involved in. D The analysis of ZNF133-bound motifs using MEME suite. E Verification of the ChIP-seq results by qChIP analysis of the indicated genes. MCF-7 or MDA-MB-231 cells transfected with FLAG-ZNF133 were collected and qChIP experiments were performed with FLAG or KAP1 antibodies. The results are represented as fold change over control IgG with GAPDH as a negative control. F Total cellular RNAs were prepared from MCF-7 or MDA-MB-231 cells with ZNF133 overexpression for quantitative real-time RT-PCR analysis of the indicated genes. Error bars represent mean ± SD for three independent experiments. P values were determined by Student’s t-test. *P < 0.05.
By using specific antibodies against FLAG, KAP1, or isotypic normal IgG, quantitative ChIP (qChIP) analysis in MCF-7 and MDA-MB-231 cells stably expressing FLAG-ZNF133 showed a strong enrichment of ZNF133 and KAP1 on the promoters of selected genes including L1CAM, NPAS4, VGF, SYNDIG1L, CADM3, SYP, CNTNAP2, PRKD1, PCNA that represent each of the classified pathways of these genes, (Fig. 5E), thus validating the ChIP-seq results. Meanwhile, measurement of the mRNA expression of the selected genes by real-time RT-PCR in MCF-7 and MDA-MB-231 cells indicates that the expression of these genes decreased when ZNF133 was overexpressed (Fig. 5F). The above results suggest that ZNF133 recruited KAP1 on target gene promoters in breast cancer cells to repress the gene transcription.
Transcription repression of L1CAM by ZNF133/KAP1 complex
Among the identified genes, L1CAM has been reported to correlate with aggressive tumor progression in patients with breast cancer; it is also present at increased levels in primary tumors and metastases of various types of cancer [19, 28,29,30] . This indicates the importance of transcriptional regulation of L1CAM in breast carcinogenesis and its potential as a therapeutic target against metastatic breast cancer. We found that ZNF133, HDAC1 (GSM3499149), and HDAC2 (ENCFF000QOA) had similar peak locations on the promoter of L1CAM (Fig. 6A), supporting the notion that ZNF133, HDAC1, and HDAC2 physically interact and are functionally linked. Furthermore, we searched the proximal promoter of the human L1CAM gene and identified one site in the promoter region that matched the consensus ZNF133-binding element (Fig. 6A), further proving that L1CAM is a potential target of ZNF133.

A The ChIP-seq track of ZNF133, HDAC1 (GSM3499149), and HDAC2 (ENCFF000QOA) at L1CAM promoter. The red line indicates the binding site of ZNF133 on the L1CAM promoter. The sequence of the binding peak containing the significant motif (pink underlined) of ZNF133 is shown below. B MCF-7 cells were co-transfected with L1CAM promoter-Luc wild-type or mutants and expression construct for ZNF133. Luciferase activity was measured 48 h after transfection. Relative luciferase activity was calculated as firefly luciferase activity divided by renilla luciferase activity and shown relative to the control. C MCF-7 cells were transfected control or KAP1 shRNA together with L1CAM promoter-Luc construct and ZNF133 expression constructs. Luciferase activity was measured 48 hours after transfection. D Total cellular proteins were prepared from MCF-7 cells with ZNF133 overexpression or knockdown and analyzed for L1CAM protein expression by western blotting. E MCF-7 cells were transfected with shRNA of control or KAP1 together with empty vector or ZNF133 expression constructs. The mRNA or protein level of L1CAM was measured by real-time RT-PCR. Error bars represent mean ± SD for three independent experiments. P values were determined by Student’s t-test. *P < 0.05. F Analysis of the public dataset for the correlations in mRNA expression between ZNF133 and L1CAM, or KAP1 (GSE31192). The relative level of L1CAM or KAP1 was plotted against that of ZNF133. G Kaplan–Meier survival analysis of the TCGA data set for the relationship between survival time and ZNF133/L1CAM or ZNF133/KAP1 signature in breast cancer.
We then cloned the 384 bp peak fragment containing the ZNF133-binding element from the L1CAM promoter to construct a luciferase reporter to test ZNF133’s capacity to repress the L1CAM promoter-driven luciferase reporter (L1CAM-Luc). Next, MCF-7 cells were co-transfected with L1CAM-Luc and ZNF133 expression plasmid. The results showed that ZNF133 repressed the activity of the L1CAM promoter only when the CTGTCCATGGTCCTGA motif existed, but not the mutant L1CAM luciferase reporter (Fig. 6B). Additionally, ZNF133 was no longer able to repress L1CAM-Luc activity when KAP1 was knocked down (Fig. 6C), further supporting the targeting of the L1CAM gene by the ZNF133/KAP1 complex.
To further confirm the transcription repression of L1CAM by the ZNF133, ZNF133 was either overexpressed or knocked down in MCF-7 cells, and western blotting analysis was performed. The results showed that ZNF133 upregulation was associated with a reduced expression of L1CAM, whereas ZNF133 downregulation was accompanied by an elevated expression of L1CAM (Fig. 6D). As the subsequent extracellular signal-related kinase 1/2 (ERK1/2) activation was implicated in L1CAM-mediated regulation, we analyzed the phosphorylation of ERK and found that ZNF133 inhibited the intrinsic activation of ERK1/2 simultaneously (Fig. 6D), indicating that ZNF133 may restrain cancer cell progression via the L1CAM/ERK1/2 signaling pathway. Real-time RT-PCR assay consistently showed that the reduced level of L1CAM mRNA responded to ZNF133 overexpression, which could be reversed when KAP1 was depleted in MCF-7 cells (Fig. 6E).
Moreover, after investigating the public dataset for ZNF133, KAP1, and L1CAM expression, we found a negative correlation between the mRNA levels of ZNF133 and L1CAM, as well as a positive correlation between ZNF133 and KAP1 (Fig. 6F). To further extend our observations to a clinicopathologically relevant setting, we stratified patient groups based on the inverse expression of ZNF133 and L1CAM or the co-expression of ZNF133 and KAP1 in the TCGA database, indicating a better survival of breast cancer patients in high ZNF133 and low L1CAM levels or high ZNF133 and KAP1 levels (though not significant) (Fig. 6G). Collectively, these results clearly showed that the ZNF133/KAP1 complex potentially represses the transcription of L1CAM in breast cancer.
Role of L1CAM in promoting breast cancer progression
To consolidate the significant role of L1CAM in breast cancer development and progression [19, 24], we first analyzed the overall survival time of patients suffering from breast cancer with the online clinical Human Protein Atlas dataset and confirmed that higher L1CAM expression levels indicated shorter overall survival times (Fig. S3A). In addition, in the GOBO dataset, the level of L1CAM expression was positively associated with tumor histological staging (Fig. S3B) and the highest in the basal subtype of breast cancer with the strongest metastasis potential (Fig. S3C).
To verify the potential effect of L1CAM on the proliferation and invasion of breast cancer cells, MCF-7 or MDA-MB-231 cells were infected with lentivirus carrying L1CAM expression vector or L1CAM shRNA. Cell proliferation and invasion were analyzed using cell counting, colony formation, and transwell assays, respectively. The results showed that L1CAM overexpression promoted cell proliferation and increased the number of invasive cells, while L1CAM knockdown significantly inhibited cell proliferation and invasion (Fig. S3D–F). What’s more, the results of western blotting showed that the E-cadherin protein (epithelial marker) was inhibited but N-cadherin and fibronectin protein (mesenchymal marker) was heightened by L1CAM in MCF-7 and MDA-MB-231 cells; inversely, the knockdown of L1CAM inhibited the progress of EMT (Fig. S3G). With all of the above results, we confirmed that L1CAM promoted proliferation, invasion, and EMT in breast cancer cells.
ZNF133/KAP1 complex suppresses the proliferation and invasion of breast cancer cells through inhibition of L1CAM in vitro
To investigate the role of the ZNF133/KAP1-L1CAM axis in the development and progression of breast cancer, we infected MCF-7 and MDA-MB-231 cells with lentiviruses carrying ZNF133 and/or L1CAM or infected with lentiviruses carrying ZNF133 shRNA and/or L1CAM shRNA. Then, cell proliferation was examined by cell counting assays and colony formation assays. Our results showed that the proliferation inhibitory effect of ZNF133 overexpression was rescued by simultaneous overexpression of L1CAM to the original level, and the promoted breast cancer cell proliferation by depletion of ZNF133 could be abrogated by co-knockdown of L1CAM (Figs. 7A, B, and S4A, B). Furthermore, lentiviruses carrying ZNF133 or KAP1 shRNA concurrently were infected in MCF-7 and MDA-MB cells and a colony formation experiment was performed. The results showed that the effect of ZNF133 overexpression on colony number could be offset by the knockdown of KAP1 in both cell lines (Figs. 7C, D and S4C, D), supporting that ZNF133 inhibited cell proliferation via association with KAP1 complex. Taken together, these results indicated the role played by the ZNF133/KAP1 complex in the inhibition of cell proliferation through the downregulation of L1CAM expression.

A Cell counting assay. MCF-7 cells were infected with lentiviruses carrying FLAG-tagged ZNF133 or/and L1CAM expression vector, or lentiviruses carrying ZNF133 or/and L1CAM shRNA. Cell counting assay was performed on the indicated day. B MCF-7 cells stably infected with indicated lentiviruses were maintained for 14 days before staining with crystal violet and counting for colony numbers in colony formation assay. C MCF-7 cells were infected with the control or KAP1 shRNA together with expression constructs for ZNF133 and maintained for 14 days before staining with crystal violet and counting for colony numbers in colony formation assay. Error bars represent mean ± SD for three independent experiments. P values were determined by Student’s t-test. *P < 0.05. D Western blotting analysis for KAP1 knockdown or ZNF133 overexpression in MCF-7 cells. GAPDH was used as a loading control. E MCF-7 cells were infected with lentiviruses carrying FLAG-tagged ZNF133 or/and L1CAM, or with lentiviruses carrying ZNF133 shRNA or/and L1CAM shRNA, and the expressions of epithelial and mesenchymal protein markers were tested by western blotting. F MDA-MB-231 cells stably infected with indicated lentiviruses were starved for 18 h before cell invasion assays were performed using Matrigel transwell filters. The invaded cells were stained and counted. G MCF-7 and MDA-MB-231 cells were infected with the control or KAP1 shRNA together with expression constructs for ZNF133, and a transwell assay was performed. Error bars represent mean ± SD for three independent experiments. P values were determined by Student’s t-test. *P < 0.05.
To investigate whether or not the ZNF133/KAP1-L1CAM axis played a crucial role in regulating the invasive potential of breast cancer cells in vitro, we measured the expression of epithelial/mesenchymal markers in MCF-7 and MDA-MB-231 cells. Based on alteration in the EMT marker protein levels, the results showed that the inhibitory effect on EMT by ZNF133 was rescued by recovery of L1CAM to the original level, while the positive effects for EMT progress by knockdown of ZNF133 were attenuated by co-knockdown of L1CAM (Figs. 7E and S4E). What’s more, transwell cell invasion assay proved that the reduced invasion ability of MDA-MB-231 cells associated with ZNF133 overexpression was offset by the overexpression of L1CAM, whereas the increase in the invasive potential by ZNF133 knockdown was partially offset by co-knockdown of L1CAM (Fig. 7F). Meanwhile, the effect on the decreased invasive potential by ZNF133 overexpression was attenuated by the simultaneous knockdown of KAP1 (Fig. 7G). Collectively, these results indicated that ZNF133 inhibited the invasion of breast cancer cells in vitro through its interaction with the KAP1 complex and repression of target genes including L1CAM.
ZNF133 restrains the growth and metastasis of breast cancer in vivo
To investigate the in vivo role of ZNF133 against the growth and metastasis of breast cancer, we infected MDA-MB-231 cells stably expressing firefly luciferase (MDA-MB-231-Luc-D3H2LN) with lentiviruses carrying ZNF133 or/and L1CAM or carrying ZNF133 shRNA or/and L1CAM shRNA. These cells were then orthotopically implanted onto the abdominal mammary fat pad (n = 6) of 6-week-old female mice with severe immunodeficiency NCG (NOD/ShiLtJGpt-Prkdcem26Cd52Il2rgem26Cd22/Gpt). The growth/dissemination of tumors was monitored weekly by bioluminescence imaging with the IVIS imaging system. Tumor metastasis was measured by quantitative bioluminescence imaging after 8 weeks. Metastatic events were defined as any detectable luciferase signal above the background and away from the primary tumor site. The results showed that ZNF133 overexpression was inhibited while ZNF133 knockdown promoted the growth of the primary tumor and metastasis of the MDA-MB-231-Luc-D3H2LN tumors to the lung, liver, intestine, or spleen (Fig. 8A–C). Remarkably, simultaneous overexpression of L1CAM counteracted the ZNF133 overexpression-associated suppression of primary tumor growth and metastases. Additionally, growth and metastases were significantly inhibited in mice carrying shZNF133 + shL1CAM tumors compared with shZNF133 (Fig. 8A–C). Above all, these results supported that ZNF133 suppressed the growth of breast cancer by inhibiting L1CAM and thus may be a promising approach in the treatment of breast cancer (Fig. 8D).

A, B MDA-MB-231-Luc-D3H2LN cells infected with lentiviruses carrying indicated plasmids were inoculated into the left abdominal mammary fat pad (5 × 106 cells) of 6-week-old immunocompromised female SCID beige mice. Tumor size was measured after 6 weeks (mammary tumors, n = 6). Lung, liver, spleen or intestine metastases were quantified using bioluminescence imaging after 8 weeks of initial implantation, and representative in vivo bioluminescent images are shown. Error bars represent mean ± S.D. for three independent experiments. P values were determined by Student’s t-test. *P < 0.05. C Representative images of lung, liver, spleen, or intestine sections stained with H&E are shown. Scale bar 200 μm. D Proposed model of the ZNF133/KAP1 complex repressing L1CAM to inhibit the proliferation and invasion of breast cancer cells.
Discussion
In this study, we revealed that the ZNF133 zinc-finger protein, which is a repressive transcription factor, interacts with the KAP1 complex to inhibit the expression of L1CAM, an oncogene in breast cancer. Therefore, it suppresses breast cancer development and progression in vitro and in vivo.
ZNF133 is an evolutionarily conserved gene and can be detected in all tissues. Several studies have suggested the diverse functions of ZNF133 in various malignancies. ZNF133 has been identified as a novel amplified gene in the neuroblastoma cell line [11] and an up-regulated protein in osteosarcoma tissues [12]. However, studies on several cell lines, including breast cancer cells, revealed that ZNF133 was implicated in the repression of the S100A4 protein, which is associated with bad prognostic outcomes for cancer [14]. In the current study, we show that the expression of ZNF133 is progressively reduced during breast cancer progression, and it represses the proliferation and invasive potential of breast cancer cells in vitro and in vivo. We speculated three reasons for the inconsistent role of ZNF133 in cancers: (1) The different levels of ZNF133 in different cancer tissues may be due to the dynamic regulation of tissue-specific upstream regulators binding on the znf133 gene or the unique degradation mechanism of the ZNF133 protein. (2) Different effects of ZNF133 on cell behavior in different cell lines may result from different downstream target genes. Alternatively, they may exert varying functions in osteosarcoma or breast cancer tissues despite having the same target genes. (3) The variety and uniqueness of the ZNF133- interacting complex in certain cancer cell lines may also result in different degrees of impact on target genes and eventually lead to different cell behavior. Although the genetic background of the aberrant downregulation of ZNF133 in breast cancer development is currently unknown, it is clear that ZNF133 is a potent biomarker for the diagnosis and prognosis of breast cancer, and a potential therapeutic point for breast cancer.
At present, we have only defined the repressive relationship between ZNF133 and the malignant progression of breast cancer, even though the ZNF133 protein level in MDA-MB-231 cells was confirmed to be lower than that in MCF-7 and MCF-10A cells (Fig. 4E). The exact values of ZNF133, ER, PR, and HER2 levels from the clinicopathologic analysis are still needed to distinguish the molecular subtypes and to determine the relationship between ZNF133 and the above three indicators. It is also necessary to identify and validate the specific differences in ZNF133 protein levels in different molecular subtypes from a large number of clinical samples. Thus, ZNF133 has not been fully confirmed to be indicative of better patient stratification for BRCA molecular subtypes. This will be the future focus of our research.
L1CAM was reported to be associated with a shorter disease-free interval, overall survival and impaired prognosis in breast cancer. This was confirmed by serious analysis of L1CAM expression in primary breast cancers using a combination of western blotting, DNA-microarray analysis, and immunohistochemistry [19, 24, 28, 31, 32]. Therefore, understanding the regulation of L1CAM expression is of great importance in breast cancer prevention and intervention. However, there is limited information about the negative regulation of L1CAM expression in breast cancer. It has just been reported that L1CAM could be repressed by androgen receptor (AR) in triple-negative breast cancer (TNBC). Overexpression of AR leads to the loss of L1CAM expression and concomitant binding of AR to binding sites in the L1CAM promoter region in TNBC cells [27]. Here, we report that L1CAM is transrepressed by ZNF133 in MCF-7, MDA-MB-231, and MCF-10A cells. Additionally, we confirm that ZNF133 does so by recognizing a CTGTCCATGGTCCTGA motif in the promoter of L1CAM with the KAP1 repressive complex, adding to our knowledge of molecular mechanisms on L1CAM expression regulation. However, the existence of other transcriptional regulators on the L1CAM promoter in breast cancer cells remains to be investigated. It is also still unknown how different regulators might be coordinated in the regulation of L1CAM expression in a specific cell lineage or subtype. Therefore, it is crucial to further investigate the genetic or/and epigenetic abnormalities of transcriptional regulators associated with L1CAM expression to control breast carcinogenesis.
Physiologically, the tissue expression for ZNF133 is ubiquitous and specially enhanced in the nervous systems, such as the cerebellar hemisphere, neurons, and oligodendrocytes (Bgee, https://bgee.org/; tissue-specific cell type information in Human Protein Atlas, https://www.proteinatlas.org/humanproteome/tissue+cell+type). L1CAM is also involved in the mammalian nervous system where it regulates diverse aspects of brain development. L1CAM plays a crucial role in the regulation of synaptic development and plasticity, as well as axon-axonic innervation[17, 18]. The expression distributions of ZNF133 and L1CAM have suggested a distinctive association between them in different tissues, which is possibly influenced by unique upstream regulators on the two genes from certain conditions or factors especially combined with ZNF133. In our studies, we have revealed the repressive effect of the ZNF133/KAP1 complex on L1CAM expression in breast cancer, in which ZNF133 is gradually reduced in breast cancer and L1CAM plays an important role in breast tumorigenesis.
Additionally, as we know, there exists intricate crosstalk among the components of neurodevelopment and cancer metastasis. For example, whole-genome sequencing of gastric cancer revealed that the axon guidance pathway (netrins, ephrins, semaphorins and slit-Robo) is mutated in 59% of gastric tumors, demonstrating that the axon guidance pathway may be involved in the progression of cancer and might be a promising therapeutic target [33, 34]. Netrins were first identified as axon guidance molecules that guide nerve cells and axons migration during neural development. Increasing evidence has addressed the important role of Netrin-1 in non-neural systems, including various types of cancers [35,36,37]. Thus, it is reasonable to reinforce the role of L1CAM, the nervous system protein, in breast cancer metastasis and support the involvement of ZNF133 in target L1CAM to suppress the progression of breast cancer.
The KAP repressive complex consists of HDAC1, HDAC2, HP1, RBBP4, RBBP7, and SetDB1 [38]. KAP1 acts as an interaction partner of members of the KRAB domain-containing zinc-finger transcription factors family, which is also supported by our current study that the KRAB domain is involved in ZNF133. The actions of KAP1 depend on its recruitment to chromatin by various transcription factors capable of DNA binding. Thus, the recruitment of KAP1 via its RBCC domain by different transcription factors could elicit different cellular outputs; this could possibly explain the discrepancy between the effects of KAP1 on EMT and survival observed in our experiments versus the experiments reported by others [39]. We found that the ZNF133/KAP1 complex targets the transcription of a cohort of genes and regulates several cellular key biological processes including dendrite, perikaryon, and cell proliferation. Clearly, the cellular functions of the ZNF133 are numerous, far beyond what we investigated in the current study. However, by no means do we exclude the involvement and importance of other downstream target genes besides L1CAM. Nevertheless, our results indicate that ZNF133 is a transcription repressor and a potential suppressor of L1CAM. The findings add to the understanding of L1CAM-related breast carcinogenesis and support the pursuit of ZNF133 as a novel focus point for breast cancer.
Conclusion
Traditional histopathological classification aims to categorize breast cancer into subgroups to inform clinical management decisions, but there remains considerable diversity within these subgroups. Complexity pervades breast tumors at every level, from (epi)genomic, transcriptomic and proteomic landscapes, to cellular composition and clinical behaviors. This poses great challenges in predicting the development of primary tumors and treating metastatic disease. We discovered that ZNF133 worked as a novel effective biomarker to reflect a good prognosis with less metastasis in breast cancer patients. The expression of ZNF133 was lower in invasive breast carcinoma tissue subtypes and negatively correlated with advanced pathological grades. Moreover, ZNF133 was effective against L1CAM expression in breast cancer cells. Here, using the murine models of breast cancer, we reported that exogenous ZNF133 could suppress breast cancer growth and metastasis in vivo through the inhibition of L1CAM. These data suggest that ZNF133 has the potential to act as not only a good prognosis indicator for breast cancer but also a focal point for clinical trial testing of the therapeutic potential of ZNF133-based gene therapy.
Materials and methods
Tissue microarray (TMA) and immunohistochemistry
The clinical significance of ZNF133 expression in breast cancer patients was analyzed using TMAs obtained from Shanghai Outdo Biotech Co., Ltd (China) that contained 40 breast tumor tissues and adjacent normal tissues. Shanghai Outdo Biotech Co., Ltd also provided patients’ information, including gender, age, pathological grade, and TNM stage. The use of the TMAs complied with relevant regulations and was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital. Antigen retrieval was performed by incubating the samples in sodium citrate solution (0.01 mol/L, pH 6.0) buffer for 15 min. Subsequently, endogenous peroxidase activity was blocked using 3% hydrogen peroxide, the TMAs were incubated with anti-ZNF133 antibody (1:200) at 4 °C overnight and then incubated with secondary antibodies for 1 h at room temperature. For immunohistochemistry quantification, all samples were blind scored by three independent observers. The average score was calculated for each tumor sample.
Gene construction and lentivirus production
The cDNAs for ZNF133 were purchased from WZ Biosciences. The cDNAs for L1CAM were purchased from Sino Biological. The cDNA for ZNF133 was amplified by PCR and ligated into XbaI/SwaI sites of a pCDHO-CMV-FLAG vector, NOTI/EcoRV sites of a pCDNA3.1 with one tag of FLAG, or KpnI/SmaI sites of a Gal4-DBD-3XFLAG vector. All clones were confirmed by DNA sequencing. The Gal4-DBD-3XFLAG, Gal4-SV40-LUC, Gal4-TK-LUC, Gal4-AdML-LUC, and renilla plasmids were from Dr. Yongjie Ma (Tianjin Medical University Cancer Institute and Hospital, China). The pGEX-4T-1-KAP1 was from Dr. Fanbiao Meng (Tianjin Medical University Cancer Institute and Hospital, China). The pGEX-4T-1-HDAC1/HDAC2/RBBP4/RBBP7 were from Dr. Lin Shan (Capital Medical University, China). The 384 bp fragment from the L1CAM promoter and its motif mutant were generated from Azenta Life Sciences. The pLKO.1 lentiviral RNA interference (RNAi) expression system was used to construct lentiviral short hairpin RNA (shRNA) for genes. The sequences of shRNA used in this study included the following: shSCR: 5’-CCTAAGGTTAAGTCGCCCTCG-3’, shZNF133-1: 5’-GCAACCTTCGGATTCCTTATA-3’, shZNF133-2: 5’-CACCTCACCTTACATCAAATG-3’, shKAP1: 5’-GAGAATTATTTCATGCGTGAT-3’, shL1CAM: 5’-CCACTTGTTTAAGGAGAGGAT-3’. For viral packaging, the lentiviral plasmid pCDHO-CMV-ZNF133, pCDHO-CMV-L1CAM, pLKO.1-shZNF133, pLKO.1-shKAP1 or pLKO.1-shL1CAM, together with psPAX2 and pMD2.G were co-transfected into the packaging cell line HEK293T using PEI co-precipitation at 10:5:5 mg (for a 10-cm dish). The transfection medium containing PEI and plasmid mixture was replaced with a fresh complete medium after incubation for 6 h. Viral supernatants were collected 48 h later, clarified by filtration, and concentrated by ultracentrifugation. The concentrated virus was used to infect 5 × 105 cells (20–30% confluent) in a 60 mm dish with 5 μg/ml polybrene. Infected cells were selected with puromycin and/or neomycin. For the re-expressing or re-silencing L1CAM experiments, the level of L1CAM expression was controlled by creating stable clones of cells that were expressing different levels of L1CAM, and the clones with L1CAM levels close to the original L1CAM level were chosen for phenotype experiments.
Cell culture and transfection
All cells were obtained from the American Type Culture Collection (ATCC) and maintained according to ATCC recommendations. MCF-7 and HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS). MCF-10A cells were cultured in Dulbecco′s modified Eagle′s medium/Nutrient Mixture F-12 Ham supplemented with 100 ng/ml cholera toxin, 20 ng/ml epidermal growth factor (EGF), 10 μg/ml insulin, 500 ng/ml hydrocortisone and 5% heat-inactivated horse serum. Cells were maintained in a humidified incubator equilibrated with 5% CO2 at 37 °C. MDA-MB-231 cells were cultured in L-15 medium supplemented with 10% FBS and without CO2. Transient transfections of expression plasmids in MCF-7, and HEK293T cells were carried out using poly (ethylene imine) (PEI) (Sigma) according to the manufacturer’s recommendations. Transfections of expression plasmids in MDA-MB-231 and MCF-10A cells were carried out using Lipofectamine 3000 (Invitrogen) as the recommendations. All of the cells were authenticated by examination of morphology and growth characteristics, and were confirmed to be mycoplasma-free.
Reagents and antibodies
Anti-FLAG M2 affinity gel (A2220), and FLAG peptide (F3290) were purchased from Sigma. Protein A/G PLUS-agarose (sc-2003) was purchased from Santa Cruz. Puromycin was purchased from Invitrogen, and neomycin was from Gibco. Crystal violet was purchased from Solarbio. d-luciferin was from Genomeditech. Antibodies used were as follows: anti-ZNF133 (Abcam, ab237736, 1:200 for IHC, 1:100 for IF and 1:1000 for WB); anti-FLAG (MBL, M185-3L, IP and 1:8000 for western blot); anti-fibronectin (Proteintech, 15613-1-AP, 1:1000 for WB); anti-E-cadherin (Proteintech, 20874-1-AP, 1:1000 for WB); anti-N-cadherin (Proteintech, 22018-1-AP, 1:1000 for WB); anti-GAPDH (Proteintech, 60004-1-Ig, 1:8000 for WB); anti-KAP1 (Proteintech, 66630-1-Ig, ChIP and 1:1000 for WB)/(Proteintech, 15202-1-AP, IP and 1:1000 for WB); anti-HDAC1 (Proteintech, 10197-1-AP, IP and 1:1000 for WB); anti-HDAC2 (Proteintech, 12922-3-AP, IP and 1:1,000 for WB); anti-RBBP4 (Proteintech, 20364-1-AP, IP and 1:1000 for WB); anti-RBBP7 (Proteintech, 20365-1-AP, 1:1000 for WB); anti-HP1 (Proteintech, 11831-1-AP, IP and 1:1000 for WB); anti-L1CAM (Abcam, ab270455, 1:1000 for WB); anti-ERK (Proteintech, 11257-1-AP, 1:1000 for WB); anti-p-ERK (Proteintech, 28733-1-AP, 1:1000 for WB); anti-β-actin (Sigma, A5316, 1:5000 for western blot).
Fluorescence confocal microscopy
MCF-7, T47D, or MDA-MB-231 cells growing on six-well chamber slides were washed with PBS, fixed in 4% (w/v) paraformaldehyde for 20 min, permeabilized with 0.1% (v/v) Triton X-100 in PBS for 10 min, blocked with 3% BSA, and incubated with appropriate primary antibodies followed by staining with Alexa Fluor 488-coupled secondary antibodies (Life Technologies, 1:150). The cells were washed four times, and a final concentration of 0.1 mg/mL DAPI (Sigma) was included in the final washing to stain nuclei. Images were visualized with a fluorescence microscope (Zeiss Imager Z2).
Luciferase reporter assay
HEK293T and MCF-7 cells in 24-well plates were transfected with luciferase reporter, renilla, and indicated expression constructs. The amount of DNA in each transfection was kept constant by addition of empty vector. Thirty-six hours after transfection, the firefly and renilla luciferase were assayed according to the manufacturer’s protocol (Promega), and the firefly luciferase activity was normalized to that of renilla luciferase. Each experiment was performed in triplicate and repeated at last three times.
Immunopurification and mass spectrometry
HEK293T cells stably expressing FLAG-ZNF133 were washed twice with cold PBS, scraped, and collected by centrifugation at 1500 × g for 5 min. Cellular extracts were prepared by incubating the cells in lysis buffer containing protease inhibitor cocktail (Roche). Anti-FLAG immunoaffinity columns were prepared using anti-FLAG M2 affinity gel (Sigma) following the manufacturer’s suggestions. Cell lysates were obtained from about 5 × 108 cells and applied to an equilibrated FLAG column of 1-ml bed volume to allow for adsorption of the protein complex to the column resin. After binding, the column was washed with cold PBS plus 0.1% Nonidet P-40. FLAG peptide (Sigma) was applied to the column to elute the FLAG protein complex as described by the vendor. Fractions of the bed volume were collected and resolved on NuPAGE 4–12% Bis-Tris gel (Invitrogen), silver-stained using Pierce silver stain kit, and subjected to LC–MS/MS (Q-Exactive HF X, BGI) sequencing.
Co-immunoprecipitation and western blotting
Cellular lysates were prepared by incubating the cells in lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.3% NP-40, 2 mM EDTA) containing protease inhibitor cocktail (Roche) for 40 min at 4 °C, followed by centrifugation at 12,000 × g for 15 min at 4 °C. The protein concentration of the lysates was determined by the BCA protein assay kit (Pierce) according to the manufacturer’s protocol. For immunoprecipitation, 500 μg of protein was incubated with 2 μg specific antibodies for 12 h at 4 °C with constant rotation; 50 μl of 50% protein A or G agarose beads was added and incubated for an additional 3 h at 4 °C. Beads were then washed five times using the lysis buffer. Between washes, the beads were collected by centrifugation at 1000 × g for 3 min at 4 °C. The precipitated proteins were eluted from the beads by resuspending the beads in 2 × SDS-PAGE loading buffer and boiling for 10 min. The resultant materials from immunoprecipitation or cell lysates were resolved using SDS-PAGE gels and transferred onto nitrocellulose membranes. For routine western blotting, cells were washed twice with cold PBS, then lysed in buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycer-ophosphate, 1 mM sodium vanadate, 1 mg/ml leupeptin, 1 mM phenylmethyl-sulfonylfluoride). Equal amounts of protein were loaded onto SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) blotting membranes. Membranes were incubated with appropriate antibodies for 1 h at room temperature or overnight at 4 °C followed by incubation with a secondary antibody. Immunoreactive bands were visualized using western blotting Luminol reagent (Santa Cruz) according to the manufacturer’s recommendation.
GST pull-down assay
GST-tagged KAP1, HDAC1/2, or RBBP4/7 was expressed by inserting cDNA segments into the pGEX 4T-1 vector. The plasmid was transformed into Escherichia coli strain BL21 (DE3) and induced by IPTG to express GST fusion protein. As the GST, GST-KAP1, HDAC1/2, or RBBP4/7 proteins were expressed, the crude bacterial lysates were prepared by sonication in TEDGN (50mM Tris-HCl, pH 7.4, 1.5mM EDTA, 1mM dithiothreitol, 10% (v/v) glycerol, 0.4M NaCl) in the presence of the protease inhibitor mixture. In vitro transcription and translation experiments were done with rabbit reticulocyte lysate (TNT systems, Promega) according to the manufacturer’s recommendation. Then, 50 μl Glutathione-Sepharose 4B slurries beads (Amersham Biosciences) were pre-washed and equal amounts (20 μg) of GST fusion proteins were immobilized on 50 μl of 50% glutathione-Sepharose 4B slurry beads in 0.5 ml of GST pull-down binding buffer (10 mM HEPES, pH 7.6, 3 mM MgCl2, 100 mM KCl, 5 mM EDTA, 5% glycerol, 0.5% CA630). After incubation for 1 h at 4 °C with rotation, beads were washed three times with GST pull-down binding buffer and resuspended in 0.5 ml of GST pull-down binding buffer before adding 10 μl of in vitro transcribed/translated FLAG-ZNF133 proteins for 2 h at 4 °C with gentle rotation. After incubation, the beads were then washed three times with binding buffer. The bound proteins were eluted by boiling in 30 μl of 2 × sample loading buffer and subjected to western blotting and detected with an anti-FLAG antibody.
Fast protein liquid chromatography
Cellular lysates of MCF-7 stably expressing FLAG-ZNF133 were prepared by incubating the cells in lysis buffer containing protease inhibitor cocktail (Roche). Lysates were then applied to an 850 × 20 mm Superose 6 size exclusion column (Amersham Biosciences) that was equilibrated with PBS and calibrated with protein standards (blue dextran, 2000 kDa; thyroglobulin, 669 kDa; ferritin, 440 kDa; aldolase, 158 kDa; all from Amersham Biosciences). The column was eluted at a flow rate of 0.5 ml/min and fractions were separately collected.
ChIP sequencing
MCF-7 cells stably expressing FLAG-ZNF133 were maintained in DMEM supplemented with 10% FBS. Approximately 5 × 107 cells were used for each ChIP-seq assay. The ChIP procedure was performed as described in the qChIP below. The chromatin DNA was precipitated by monoclonal antibodies against FLAG. The DNA was purified with the Qiagen PCR purification kit. In-depth whole-genome DNA sequencing was performed by the BGI Corporation, China.
qChIP
MCF-7 cells were crosslinked using 1% formaldehyde for 10 min at room temperature and quenched by the addition of glycine to a final concentration of 125 mM for 5 min. The fixed cells were resuspended in lysis buffer (1% SDS, 5 mM EDTA and 50 mM Tris-HCl, pH 8.1) in the presence of protease inhibitors, then subjected to 30 cycles (30-s on and off) of sonication (SCIENTZ-II D) to generate chromatin fragments of 300–500 bp in length. Lysates were diluted in buffer containing 1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl (pH 8.1) and protease inhibitors. For IP, the diluted chromatin was incubated with specific antibodies for 12 h at 4 °C with constant rotation, 50 μL of 50% (vol/vol) protein A/G Sepharose beads were then added, and the incubation was continued for an additional 2 h. Beads were washed with the following buffers: TSE I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 150 mM NaCl and 20 mM Tris-HCl, pH 8.0); TSE II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 500 mM NaCl and 20 mM Tris-HCl, pH 8.0); TSE III (0.25 M LiCl, 1% Nonidet P-40, 1% sodium deoxycholate, 1 mM EDTA and 10 mM Tris-HCl, pH 8.0) and TE (1 mM EDTA and 10 mM Tris-HCl, pH 8.0). The pulled-down chromatin complex and input were de-crosslinked at 55 °C for 12 h in elution buffer (1% SDS and 0.1 M NaHCO3). The eluted DNA was purified with the QIAquick PCR Purification Kit. qChIPs were performed using Power SYBR Green PCR Master Mix and ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA). The sequences of the primers used are listed as following: L1CAM, 5′-TCTACCCTGCCCTTCTCTCC-3′ (forward) and 5′-CTCACCTTCTCCCTGTTCCG-3′ (reverse); NPAS4, 5′-CTGTCGCATGTCTAGGGCAG-3′ (forward) and 5′-CACCGTCCAGGCTCTCTAAC-3′ (reverse); VGF, 5′-CTAGCTCGCTCCGGCTTCAG-3′ (forward) and 5′-CAATCGTCGGGCGTCCTT-3′ (reverse); SYNDIG1L, 5′-TTGACCCCAGTTCATCAGCC-3′ (forward) and 5′-TTGCACTTCGTCCCTCCCTA-3′ (reverse); CADM3, 5′-TTGAGAGCTGGCTGCCTTAC-3′ (forward) and 5′-TGGAGAGCTCAAAGCACAGG-3′ (reverse); SYP, 5′-GCGCGTACCTGATTCACCAC-3′ (forward) and 5′-CCTGGGCTGTTCCAACGA-3′ (reverse); CNTNAP2, 5′-AATCCCCAACTTCAGCACCG-3′ (forward) and 5′-AAAGGGAGACTGAGCCGAC-3′ (reverse); PRKD1, 5′-TGCTGAATGACGCCATCTGT-3′ (forward) and 5′-GTCCACACCCAGTACTGCTC-3′ (reverse); PCNA, 5′-GAGCAAAGAGCCCTGGAACA-3′ (forward) and 5′-TTCGCGCCAAAGTCACAAAG -3′ (reverse).
Quantitative RT-PCR analysis
Total cellular RNAs were isolated from samples with the Trizol reagent (Invitrogen). First strand cDNA synthesis with the Reverse Transcription System (TransGen Biotech). Quantitation of all gene transcripts was done by qPCR using Power SYBR Green PCR Master Mix and an ABI PRISM 7500 sequence detection system (Applied Biosystems, Foster City, CA) with the expression of GAPDH as the internal control. The primer pairs used were: L1CAM, 5′-TGCTCATCCTCTGCTTCATC-3′ (forward) and 5′-TCCTCGTTGTCACTCTCCA-3′ (reverse); NPAS4, 5′-CCCAGCACTGCCACGTTCCC-3′ (forward) and 5′-CCCGGAGTTGAGCTAGGGCCA-3′ (reverse); VGF, 5′-AAGGATGACGGCGTACCAGA-3′ (forward) and 5′-TGCCTGCAACAGTACCGAG-3′ (reverse); SYNDIG1L, 5′- GGCGGGACAGGCTCTGA-3′ (forward) and 5′-AGCAGCGGGTTCTGTAGTTC-3′ (reverse); CADM3, 5′-CGGAGAGGGTGAGAAATGG-3′ (forward) and 5′-CGAGAGTAAATCTGTTGGGTTG-3′ (reverse); SYP, 5′-TCTGGTGATGTTTTACGCTAC-3′ (forward) and 5′-AATCCTCAGGCTTAACTTGC-3′ (reverse); CNTNAP2, 5′- CAAGTGTTCAGGTTGTGTCAAGAA-3′ (forward) and 5′-AGCAGGGTATAATGTGGTGAGGA-3′ (reverse); PRKD1, 5′-CGCACATCATCTGCTGAACT-3′ (forward) and 5′-CTTTCGGTGCACAACGTTTA-3′ (reverse); PCNA, 5′-AAAAGCCACTCCACTGTCT-3′ (forward) and 5′-CTCTACAACAAGGGGCACAT-3′ (reverse); GAPDH, 5′-TCCTCCTGTTTCATCCAAGC-3′ (forward) and 5′-TAGTAGCCGGGCCCTACTTT-3′ (reverse).
Proliferation assay
Cells were plated in triplicate in 12-well plates at 1 × 105 cells per well in 1.5 ml of medium. After days, as indicated in experiments, wells were washed twice with PBS to remove dead cells, and then the entire contents of the well were trypsinized. Cell number was determined using a hemocytometer. Each experiment was performed in triplicate and repeated at last three times.
Colony formation assay
MCF-7 (1 × 103/well) or MDA-MB-231 (500/well) cells were maintained in culture media in 6 or 12-well plate for 14–20 days, fixed with 4% paraformaldehyde, stained with 0.1% crystal violet for colony observation, and counted using light microscope. Each experiment was performed in triplicate and repeated at last three times.
Cell invasion assay
The transwell invasion assay was performed using the transwell chamber (BD biosciences). Stably infected MDA-MB-231 cells were cultured in medium with 10% FBS at 37 °C without CO2. Cells were deprived in serum-free medium for 18 h, and harvested. These cells were washed three times in PBS and resuspended in serum-free culture medium. Afterwards, 1 × 105 of these cells in 0.3 ml of serum-free media were plated onto the matrigel-coated upper chamber of the transwell. The upper chamber was then transferred into a well containing 0.5 ml of media supplemented with 10% FBS and incubated for 5 h. Cells may actively migrate from the upper to the lower side of the filter due to FBS as attractant. Cells on the upside were removed using cotton swabs, and the invasive cells on the lower side were fixed, stained with 0.1% crystal violet solution, and counted using light microscope. Each experiment was performed in triplicate and repeated at last three times.
In vivo metastasis
The MDA-MB-231-Luc-D3H2LN cell line (MDA-MB-231 cell line engineered to stably express firefly luciferase) (Xenogen Corporation) was infected with lentiviruses carrying ZNF133 or/and L1CAM, or lentiviruses carrying ZNF133 shRNA, or/and L1CAM shRNA. These cells were inoculated into the left abdominal mammary fat pad (2.5 × 106 cells) of 6-week-old severe immunodeficient female NCG mice (T001475, GemPharmatech). Mice, according to body equal completely randomized design, were divided into groups each with six (n = 6) before injected. Sample size estimate was based on xenograft assays from literatures. Mice in which tumors did not form would be removed from the study. Tumors were observed in all the mice injected with cells. For bioluminescence imaging, mice were anesthetized and given 200 μg/g of D-luciferin in PBS by intraperitoneal injection. Fifteen minutes after injection, bioluminescence images were obtained with a charge-coupled device camera (IVIS SPECTRUM; Caliper life science). Bioluminescence from relative optical intensity was defined manually, and data was expressed as photon flux (photons/s/cm2/Steradian) and normalized to background photon flux which was defined from a relative optical intensity drawn over a mouse that was not given an injection of D-luciferin. The measurement and data processing were done by an investigator blinded to the treatment procedure. All animals were killed at the end of the experiment. All animal handling and procedures were approved by the Animal Ethical and Welfare Committee (AEWC) of Tianjin Medical University Cancer Institute and Hospital (NSFC-AE-2022199).
Statistical analysis
Data from biological triplicate experiments are presented with an error bar as mean ± SD. Before statistical analysis, variation within each group of data and the assumptions of the tests were checked. Two-tailed unpaired Student’s t-test was used for comparing two groups of data unless otherwise noted. Statistical significance was considered at a value of P < 0.05.
Data availability
ChIP-seq data for ZNF133 has been deposited in the Gene Expression Omnibus (GEO) with an accession number GSE226190. Data for Kaplan–Meier survival analysis in breast cancer patients are publicly available online at http://kmplot.com/analysis/index.php?p=background. Data for Gene expression-based Outcome for Breast cancer Online (GOBO) analysis are available from http://co.bmc.lu.se/gobo/gsa.pl. Data for Gene Expression Profiling Interactive Analysis (GEPIA) analysis are available from http://gepia.cancer-pku.cn/. Data for mRNA expression analysis in breast cancer patients are available in the Oncomine database at https://www.oncomine.org/. Access to publicly available cancer TCGA data is provided in the UALCAN web resource at http://ualcan.path.uab.edu/index.html. The open reading frame, conserved domains, and chromosomal location of ZNF133 were analyzed using the Uniprot (https://www.uniprot.org/) and NCBI (www.ncbi.nlm.nih.gov) databases. The theoretical molecular weight of ZNF133 was calculated using http://www.detaibio.com/sms2/protein_mw.html tools. The homologous alignment and phylogenetic analysis were performed using the MegAlign with Clustal V method.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Waks AG, Winer EP. Breast cancer treatment: a review. JAMA. 2019;321:288–300.
Tsang JYS, Tse GM. Molecular classification of breast cancer. Adv Anat Pathol. 2020;27:27–35.
Liang Y, Zhang H, Song X, Yang Q. Metastatic heterogeneity of breast cancer: Molecular mechanism and potential therapeutic targets. Semin Cancer Biol. 2020;60:14–27.
Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. 2021;397:1750–69.
Cassandri M, Smirnov A, Novelli F, Pitolli C, Agostini M, Malewicz M, et al. Zinc-finger proteins in health and disease. Cell Death Discov. 2017;3:17071.
Sun M, Ju J, Ding Y, Zhao C, Tian C. The signaling pathways regulated by KRAB zinc-finger proteins in cancer. Biochim Biophys Acta Rev Cancer. 2022;1877:188731.
Shen AL, Moran SA, Glover EA, Drinkwater NR, Swearingen RE, Teixeira LB, et al. Association of a chromosomal rearrangement event with mouse posterior polymorphous corneal dystrophy and alterations in Csrp2bp, Dzank1, and Ovol2 gene expression. PLoS ONE. 2016;11:e0157577.
Ehringer MA, Thompson J, Conroy O, Goldman D, Smith TL, Schuckit MA, et al. Human alcoholism studies of genes identified through mouse quantitative trait locus analysis. Addict Biol. 2002;7:365–71.
Jung ES, Choi KW, Kim SW, Hubenthal M, Mucha S, Park J, et al. ZNF133 is associated with infliximab responsiveness in patients with inflammatory bowel diseases. J Gastroenterol Hepatol. 2019;34:1727–35.
Heiskanen MA, Bittner ML, Chen Y, Khan J, Adler KE, Trent JM, et al. Detection of gene amplification by genomic hybridization to cDNA microarrays. Cancer Res. 2000;60:799–802.
Li Y, Liang Q, Wen YQ, Chen LL, Wang LT, Liu YL, et al. Comparative proteomics analysis of human osteosarcomas and benign tumor of bone. Cancer Genet Cytogenet. 2010;198:97–106.
Dai X, Chen X, Hakizimana O, Mei Y. Genetic interactions between ANLN and KDR are prognostic for breast cancer survival. Oncol Rep. 2019;42:2255–66.
Andersen K, Smith-Sorensen B, Pedersen KB, Hovig E, Myklebost O, Fodstad O, et al. Interferon-gamma suppresses S100A4 transcription independently of apoptosis or cell cycle arrest. Br J Cancer. 2003;88:1995–2001.
Galetzka D, Muller T, Dittrich M, Endres M, Kartal N, Sinizyn O, et al. Correction to: molecular karyotyping and gene expression analysis in childhood cancer patients. J Mol Med. 2020;98:1657.
Kleene R, Lutz D, Loers G, Bork U, Borgmeyer U, Hermans-Borgmeyer I, et al. Revisiting the proteolytic processing of cell adhesion molecule L1. J Neurochem. 2021;157:1102–17.
Duncan BW, Murphy KE, Maness PF. Molecular mechanisms of L1 and NCAM adhesion molecules in synaptic pruning, plasticity, and stabilization. Front Cell Dev Biol. 2021;9:625340.
Ango F, Gallo NB, Van Aelst L. Molecular mechanisms of axo-axonic innervation. Curr Opin Neurobiol. 2021;69:105–12.
Zhang J, Yang F, Ding Y, Zhen L, Han X, Jiao F, et al. Overexpression of L1 cell adhesion molecule correlates with aggressive tumor progression of patients with breast cancer and promotes motility of breast cancer cells. Int J Clin Exp Pathol. 2015;8:9240–7.
Saha A, Cheriyamundath S, Kumar A, Gavert N, Brabletz T, Ben-Ze'ev A. A necessary role for increased biglycan expression during L1-mediated colon cancer progression. Int J Mol Sci. 2021;23:445.
Altevogt P, Doberstein K, Fogel M. L1CAM in human cancer. Int J Cancer. 2016;138:1565–76.
Chen J, Gao F, Liu N. L1CAM promotes epithelial to mesenchymal transition and formation of cancer initiating cells in human endometrial cancer. Exp Ther Med. 2018;15:2792–7.
Ben Q, An W, Fei J, Xu M, Li G, Li Z, et al. Downregulation of L1CAM inhibits proliferation, invasion and arrests cell cycle progression in pancreatic cancer cells in vitro. Exp Ther Med. 2014;7:785–90.
Er EE, Valiente M, Ganesh K, Zou Y, Agrawal S, Hu J, et al. Pericyte-like spreading by disseminated cancer cells activates YAP and MRTF for metastatic colonization. Nat Cell Biol. 2018;20:966–78.
Geismann C, Arlt A, Bauer I, Pfeifer M, Schirmer U, Altevogt P, et al. Binding of the transcription factor Slug to the L1CAM promoter is essential for transforming growth factor-beta1 (TGF-beta)-induced L1CAM expression in human pancreatic ductal adenocarcinoma cells. Int J Oncol. 2011;38:257–66.
Kreisler A, Strissel PL, Strick R, Neumann SB, Schumacher U, Becker CM. Regulation of the NRSF/REST gene by methylation and CREB affects the cellular phenotype of small-cell lung cancer. Oncogene. 2010;29:5828–38.
Doberstein K, Milde-Langosch K, Bretz NP, Schirmer U, Harari A, Witzel I, et al. L1CAM is expressed in triple-negative breast cancers and is inversely correlated with androgen receptor. BMC Cancer. 2014;14:958.
Schroder C, Schumacher U, Fogel M, Feuerhake F, Muller V, Wirtz RM, et al. Expression and prognostic value of L1-CAM in breast cancer. Oncology Rep. 2009;22:1109–17.
Laban M, El-Swaify ST, Ali SH, Refaat MA, Sabbour M, Farrag N, et al. The prediction of recurrence in low-risk endometrial cancer: is it time for a paradigm shift in adjuvant therapy? Reprod Sci. 2022;29:1068–85.
Maten MV, Reijnen C, Pijnenborg JMA, Zegers MM. L1 cell adhesion molecule in cancer, a systematic review on domain-specific functions. Int J Mol Sci. 2019;20:4180.
Li Y, Galileo DS. Soluble L1CAM promotes breast cancer cell adhesion and migration in vitro, but not invasion. Cancer Cell Int. 2010;10:34.
Samatov TR, Wicklein D, Tonevitsky AG. L1CAM: cell adhesion and more. Prog Histochem Cytochem. 2016;51:25–32.
Wong SS, Kim KM, Ting JC, Yu K, Fu J, Liu S, et al. Genomic landscape and genetic heterogeneity in gastric adenocarcinoma revealed by whole-genome sequencing. Nat Commun. 2014;5:5477.
Wang K, Zhao XH, Liu J, Zhang R, Li JP. Nervous system and gastric cancer. Biochim Biophys Acta Rev on Cancer. 2020;1873:188313.
Delloye-Bourgeois C, Brambilla E, Coissieux MM, Guenebeaud C, Pedeux R, Firlej V, et al. Interference with netrin-1 and tumor cell death in non-small cell lung cancer. J Natl Cancer Inst. 2009;101:237–47.
Mazelin L, Bernet A, Bonod-Bidaud C, Pays L, Arnaud S, Gespach C, et al. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature. 2004;431:80–84.
Shimizu A, Nakayama H, Wang P, Konig C, Akino T, Sandlund J, et al. Netrin-1 promotes glioblastoma cell invasiveness and angiogenesis by multiple pathways including activation of RhoA, cathepsin B, and cAMP-response element-binding protein. J Biol Chem. 2013;288:2210–22.
Iyengar S, Farnham PJ. KAP1 protein: an enigmatic master regulator of the genome. J Biol Chem. 2011;286:26267–76.
Yu C, Zhan L, Jiang J, Pan Y, Zhang H, Li X, et al. KAP-1 is overexpressed and correlates with increased metastatic ability and tumorigenicity in pancreatic cancer. Med Oncol. 2014;31:25.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81902811, 82002977, 82103387 and 82203471), and Tianjin Natural Science Foundation (Nos. 20JCZDJC00030 and 19JCZDJC65800).
Author information
Authors and Affiliations
Contributions
LL, XW, and KH conceived and designed the study, performed experiments, and analyzed data; KH and YC performed animal experiments and pathologic analysis; XL analyzed the clinical datasets; LQ, BW, ZW, HW, JH, LH, MW, and ZS provided technical assistance. XL, CB, YC, RC, ML, and CX provided funding support and some ideas. LL drafted the paper; BL and LL discussed and finalized the paper. We thank Home for Researchers editorial team (www.home-for-researchers.com) for language editing service.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Animal handling and procedures were approved by the Animal Ethical and Welfare Committee (AEWC) of Tianjin Medical University Cancer Institute and Hospital (NSFC-AE-2022199). The use of the TMAs complied with relevant regulations, and was approved by the Ethics Committee of Tianjin Medical University Cancer Institute and Hospital.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, L., Wang, X., Hu, K. et al. ZNF133 is a potent suppressor in breast carcinogenesis through dampening L1CAM, a driver for tumor progression. Oncogene 42, 2166–2182 (2023). https://doi.org/10.1038/s41388-023-02731-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-023-02731-5
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.