
Abstract
Despite significant progression in the study of hepatocellular carcinoma (HCC), the role of the proteasome in regulating cross talk between mTOR signaling and glycolysis in liver cancer progression is not fully understood. Here, we demonstrate that deficiency of REGγ, a proteasome activator, in mice significantly attenuates DEN-induced liver tumor formation. Ablation of REGγ increases the stability of PP2Ac (protein phosphatase 2 catalytic subunit) in vitro and in vivo, which dephosphorylates PRAS40 (AKT1 substrate 1) and stabilizes the interaction between PRAS40 and Raptor to inactive mTORC1-mediated hyper-glycolytic metabolism. In the DEN-induced animal model and clinical hepato-carcinoma samples, high levels of REGγ in HCC tumor regions contribute to reduced expression of PP2Ac, leading to accumulation of phosphorylated PRAS40 and mTORC1-mediated activation of HIF1α. Interestingly, mTORC1 enhances REGγ activity in HCC, forming a positive feedback regulatory loop. In conclusion, our study identifies REGγ-PP2Ac-PRAS40 axis as a new layer in regulating mTORC1 activity and downstream glycolytic alterations during HCC development, highlighting the REGγ-proteasome as a potential target for personalized HCC therapy.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) represents the sixth most common cancer and the third major cause of cancer-related mortality globally [1]. Although studies have reported advances in the diagnosis and treatment of HCC, the incidence and mortality still are increasing [2, 3]. To promote personalized therapy of HCC, it is urgent to understand the precise molecular mechanism for tumorigenesis and progression in HCC.
Cancer cells possess a high rate of glycolytic metabolism, called the “Warburg effect” or “aerobic glycolysis” [4]. This is to fulfill the bioenergetic and biosynthetic demands of rapid proliferation and survival of cancer cells in hypoxic environments. Upregulating glycolysis leads to an increased lactate production, which leads to acidification of the microenvironment and cancer spread [5, 6]. One of the major transcription factors regulating glycolytic response is hypoxia-inducible factor-1α (HIF-1α), which promotes the expression of a number of pivotal glycolytic enzymes and glucose transporter1 (GLUT1), playing central roles in regulation of cancer-specific glucose metabolism [7,8,9,10]. In addition to oxygen-dependent regulation, HIF-1α can also be regulated at the translational level via PI3K/Akt/mTOR pathway under normoxia [11, 12]. In human liver cancer HepG2 cells, it has been reported that activation of PI3K/Akt cascades could induce HIF-1α protein accumulation [13].
Activation of the PI3K/AKT/mTOR pathway and PTEN (a dual protein/lipid phosphatase) loss have been found in ~50% of patients with HCC [14,15,16]. In line with this, upregulation of the mTOR pathway is related to tumor recurrence and poor prognosis in HCC [14]. The AKT effector, mTOR, is a serine/threonine protein kinase and exists as mTORC1 or mTORC2 complex in organisms. The mTORC1 complex is sensitive to Rapamycin and includes mTOR, Raptor (regulatory-associated protein of mTOR), MLST8 (mammalian orthologue of LST8, also known as GpL), PRAS40 (proline-rich Akt substrate, the molecular weight is 40 kDa), and deptor (DEP domain containing mTOR-interaction protein). Non-phosphorylated PRAS40 can bind with Raptor and inhibit the activity of mTORC1. When PRAS40 is phosphorylated, it will no longer interact with Raptor and fails to inhibit the activity of mTORC1 [17]. Recently, Yiqun et al. comprehensively examined the PI3K/AKT/mTOR pathway in over 10,000 human cancers and 32 cancer types profiled by TCGA, and found many cancers exhibited high mTOR pathway activity without associated canonical genetic or genomic changes, suggesting a variety of post-transcriptional mechanisms for pathway activation and regulation [18].
REGγ (also known as PA28γ, PSME3, and Ki) is a member of the 11S family of proteasome activators. It has been found to promote degradation of numerous target proteins in an ubiquitin- and ATP-independent manner [19, 20], with functions in multiple cellular processes, including cell cycle [20, 21], autoimmunity [22], and carcinogenesis [23,24,25]. Our recent discoveries that overexpression of REGγ in HCC highly correlates with ki67 upregulated tumor samples in bioinformatics analyses prompted us to investigate the role of REGγ in hepatocellular carcinoma.
In our current study, we have discovered that REGγ boosts DEN-induced liver cancer in mice through mTORC1-HIF1α pathway, at least in part. PP2Ac is found as the bridge molecule between REGγ and mTORC1 complex, acting via dephosphorylation of PRAS40. Interestingly, a high level of REGγ is observed when mTORC1, but not mTORC2 is activated, reflecting a specific positive feedback loop in HCC. Importantly, analyses of animal tumor and clinical samples from HCC patients substantiate the tumor promoting function of REGγ, providing new insights for REGγ as a putative therapeutic target against HCC in the future.
Results
REGγ promotes DEN-induced HCC in mice via the mTORC1 pathway
To understand the role of REGγ in human HCC, we carried out bioinformatics analyses of a dataset from TCGA database. The results revealed not only an averaged overexpression of REGγ in human HCC samples, but also a positively-correlated upregulation of Ki67 in tumors, indicating a potential function of REGγ in HCC (Fig. 1a). To validate the impact of REGγ on HCC development and growth, we generated the well-established DEN-induced HCC model in REGγ+/+ and REGγ−/− mice. Histopathological data demonstrated that 5-month-old DEN-induced REGγ knockout mice had delayed tumor development (Supplementary Fig. S1a). After 9 months DEN induction, the tumor number (Figs. 1b and c) and the tumor size (Fig. 1d and Supplementary Fig. S1b) were much smaller in REGγ−/− than in WT mice. Histopathological images (Supplementary Fig. S1a) and detection of the liver cancer markers [26], including Prom1 (also known as CD133), Cd44 (Aldh1a1), and Afp (Fig. 1e) revealed that the liver tumors in DEN-induced REGγ+/+ mice mimicked poorly differentiated human HCC. Overall, these results suggest that REGγ is a tumor promoter and its deficiency restrains liver tumor growth.

a Bioinformatics analyzed the expression of REGγ and Ki67 in normal tissue (n = 50) and liver cancer (n = 373) samples from TCGA dataset. ***p < 0.001 (two-tailed Student’s t-test) (left panel). The expression correlation between REGγ and Ki67 was calculated by employing Spearman method (right panel). b Representative photographs showing liver tumor growth in REGγ+/+ and REGγ−/− mice 9 months after DEN treatment. c, d Average number of tumors per mouse liver from WT and REGγ−/− mice (c) and Liver tumor size (d) were quantitated (n = 12 each group). ***p < 0.001 (two-tailed Student’s t-test). e Quantitative RT-PCR analysis of Prom1, Afp, CD44 genes expression in the liver tumors. n = 5. Data represent means ± SEM. **p < 0.01 (two-tailed Student’s t-test). f DEN-induced liver tumor cells from REGγ+/+ and REGγ−/− mice were lysed and western blot was performed using indicated antibodies. Sample in each lane was from an individual mouse. g, h WT or REGγ−/− mice liver cancer cells and HepG2 cells were plated at a density of 2 × 103 cells per well in 96-well plate and treated with or without Rapamycin. Plates were read at different time points. Experiment was repeated for three times. Data represent means ± SEM. **p < 0.01, ***p < 0.001 (two-tailed Student’s t-test).
The PI3K/AKT/mTOR and MEK/ERK pathways play pivotal roles in the development of HCC [27]. To clarify the mechanism by which REGγ primarily regulates HCC, we compared the key components of these signaling pathways. We observed that mTORC1 activity, but not ERK, p38 or AKT, was obviously suppressed in DEN-induced REGγ-deficient liver tumors (Fig. 1f and Supplementary Fig. S1c), suggesting that a turning point between AKT and mTORC1 is regulated by REGγ. We obtained consistent results that REGγ promoted mTORC1 activity in HepG2 cells by regulating p-S6K and p-4EBP1 (Supplementary Fig. S1d). To further demonstrate REGγ could facilitate DEN-induced HCC growth, we isolated and immortalized DEN-induced REGγ+/+ and REGγ−/− mouse hepatoma cells that were verified for the presence of liver tumor specific markers (Supplementary Fig. S1e). Cell proliferation assays indicated that REGγ+/+ hepatoma cells grew much faster than REGγ−/− tumor cells, while the proliferation rate was nearly equal in both REGγ+/+ and REGγ−/− hepatoma cells (Fig. 1g) when mTORC1 activity was blocked by Rapamycin, a specific inhibitor of mTORC1. Similar growth effects, including growth inhibition by Rapamycin, were observed in HepG2 (shN) cells vs. HepG2 with REGγ knockdown (shR) (Fig. 1h), suggesting that the mTOR pathway is downstream of REGγ.
REGγ affects HIF-1α accumulation via mTORC1 in hepatocellular carcinoma
HIF-1α, and Myc are the leading transcription factors in response to mTORC1, promoting onset Warburg effect in cancers [28]. Western blotting experiments showed a reduced HIF-1α expression in REGγ−/− liver tumor tissues or in shR HepG2 cells (Fig. 2a). Immunohistologically, HIF-1α level was also lower in REGγ−/− normal liver tissues than in WT (Supplementary Fig. S2a). However, c-Myc protein and mRNA levels were not reduced in REGγ-silenced HepG2 cells (Supplementary Fig. S2b and S2c). Importantly, the mRNA level of HIF-1α was significantly attenuated in REGγ knock-down cells (Fig. 2b), even after the mTORC2 was blocked by silencing Rictor, which is essential to mTORC2 activation. In contrast, silencing Raptor, a component required for mTORC1 activity, led to indiscriminate expression of HIF-1α in normal and REGγ knock-down cells (Fig. 2b), demonstrating that REGγ regulates HIF-1α expression in an mTORC1-dependent manner.

a Expression of HIF-1α in liver tumor tissue from REGγ+/+ and REGγ−/− mice, and in HepG2 REGγ shN and shR cells was analyzed by western blotting. b In HepG2 REGγ shN and shR cells, HIF-1α gene’s transcription was measured by RT-qPCR technology after transfecting small interfering RNAs (siRNAs) specific for mTORC1 subunit Raptor or mTORC2 subunit Rictor for 36 hr. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). c The mRNA levels of glycolytic genes in HepG2 REGγ shN and shR cells were detected by RT-qPCR. Data are shown as mean ± SEM from three independent experiments. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). d Analysis of glycolytic gene expression in HepG2 REGγ shN and shR cells by western blot. e, f Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in HepG2 REGγ shN and shR cells were measured by Seahorse Bioscience XF96 analyzer. Data were repeated 3 times, **p < 0.01 (two-tailed Student’s t-test). g REGγ shN and shR HepG2 cells were treated with CoCl2 followed by RT-PCR analysis of glycolytic gene expression. Experiments were repeated two times. h HepG2 REGγ shN and shR cells were transfected with siRNAs for 48 h or treated with Rapamycin for 24 h. Then cells were collected for RT-qPCR analysis of HIF1α, Glut1 and HK2 gene expression. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). i, j REGγ shN and shR HepG2 cells were treated with Rapamycin or mock. Glucose uptake and lactate production were determined. Experiments were repeated three times. Data represent means ± SEM. **p < 0.01 (two-tailed Student’s t-test).
REGγ-dependent hepatoma cells proliferation and glycolysis are affected by the mTORC1-HIF-1α pathway
Next, we examined whether REGγ could regulate glycolysis and the Warburg effect to affect HCC development. We performed principal component analysis (PCA) of metabolomics and found clearly different clustering of the glycolytic profiles between 293T WT and REGγ knockout cells (Supplementary Fig. S2d). The pyruvate/lactate ratio was dramatically increased in REGγ-deficient cells comared to WT (Supplementary Fig. S2e), indicating restricted glycolysis in REGγ-depleted cells. Bioinformatics analysis of the TCGA liver cancer datasets showed that most glycolysis genes were upregulated (Supplementary Fig. S2f) and REGγ was positively correlated with the expression of these glycolysis genes, including HK2 (hexokinase 2), ALDOA (aldolase A), PKM2 (pyruvate kinase M2), GAPDH (glyceraldehyde 3-phosphate dehydrogenase), ENO1 (enolase 1), and PGM1 (phosphoglucomutase1) (Supplementary Fig. S2g).
To validate this regulation in REGγ-WT and REGγ-silenced HepG2 cells or REGγ−/− mouse hepatoma cells, we examined expression of glycolysis-related genes, including GLUT1 (glucose transporter 1 or SLC2A1), HK2, GPI1 (glucose-6-phosphate isomerase1), PFKL (6-phosphofructokinase, liver type), ALDOA, TPI1 (triosephosphate Isomerase 1), GAPDH, ENO1, PGM1/2, PKM2, and LDHA (lactate dehydrogenase A). Results displayed dramatically decreased mRNA (Fig. 2c and Supplementary Fig. S3a) and protein (Fig. 2d and Supplementary Fig. S3b) levels for most of the genes in REGγ-knockdown HepG2 and REGγ−/− hepatoma cells. Accordingly, we detected significantly increased basal oxygen consumption rates (OCRs, an indicator of oxidative phosphorylation) and maximal respiratory capacity in REGγ knock-down HepG2 compared to WT cells (Fig. 2e). Whereas the extracellular acidification rates (ECAR, an indicator of aerobic glycolysis) was reduced in REGγ-deficient HepG2 cells (Fig. 2f). Thus, our results indicate that REGγ may contribute to HCC metabolic adaptation by regulating glycolysis-related genes.
To validate whether REGγ regulated the glycolysis in an mTORC1- and HIF-1α-dependent manner, we treated HepG2 cells with CoCl2, which can mimic hypoxia and induce HIF-1α expression [29]. Results showed that glycolytic genes’ expression in REGγ knock-down cells was significantly elevated to comparable level with WT cells after treatment (Fig. 2g). Similar results were observed after transfecting HIF-1α constructs into cells (Supplementary Fig. S3c). Following treatment with Rapamycin, or silencing the HIF-1α, all the HIF-1α, Glut1, and HK2 transcriptions in both shN and shR cells were diminished to nearly the same level as in shR HepG2 cells (Fig. 2h). Silencing Raptor instead of Rictor reached to similar results (Supplementary Fig. S3d and S3e). In agreement, Rapamycin significantly blocked glucose uptake (Fig. 2i) and lactate production (Fig. 2j), predominantly in REGγnormal cells. Accordingly, lactate production was significantly reduced after knocking-down HIF-1α (Supplementary Fig. S3f). Overexpression of HIF-1α increased glucose uptake, lactate production, and cell proliferation to a similar level in normal and REGγ knock-down cells (Supplementary Fig. S3g–i). These data indicate that REGγ regulates glycolytic gene expression and HCC via the mTORC1-HIF-1α axis.
Ablation of REGγ inhibits mTORC1 via potentiating dephosphorylation of PRAS40 by PP2Ac
We next addressed how REGγ regulates the mTORC1 pathway. By analyzing the key components regulating mTORC1 [30], we found only phosphorylated PRAS40 was consistently decreased in REGγ-shR liver cancer cells, whereas the phosphorylation of AMPK, PTEN, PDK1, and the mTORC1 components (Raptor and mTOR) was unaltered (Fig. 3a). A general model is that PRAS40 could be phosphorylated by Akt and then dissociated from Raptor, leading to mTORC1 activation [31]. Co-immunoprecipitation analysis revealed the interaction of PRAS40 with Raptor was strengthened in REGγ knockdown cells (Fig. 3b). As expected, overexpressing or silencing PRAS40 (Fig. 3c and Supplementary Fig. S4a) eliminated the discrepancy in mTORC1 activity between REGγ shN and shR cells, indicating that REGγ modifies mTORC1 activity in a PRAS40-dependent way.

a Western blotting was performed using indicated antibodies in HepG2 REGγ shR or shN cells. Experiments were repeated at least 3 times. b HepG2 REGγ shR or shN cells were collected and lysed. The lysates were immunoprecipitated with anti-PRAS40 antibody and detected by western blot using indicated antibodies. c HepG2 REGγ shR or shN cells were transfected with PRAS40 plasmid, then the cell lysate was subjected to western blot analysis. Data shown were representative of three repeats. d PRAS40 plasmid was transfected into 293T cells with empty vector or with increasing amounts of PP2Ac plasmid. Western blot was employed for analysis. Data shown were representative of three repeats. e Co-transfecting the indicated plasmids into 293T cells for 36 h, and then cell lysates were subjected to immunoprecipitation with conjugated anti-Flag beads. Data shown are representative of three repeats. f HA-PRAS40 or Flag-PP2Ac immunoprecipitated from 293T cells were incubated as indicated, and western blotting was performed with indicated antibodies. Data shown are representative of three independent experiments. g HepG2 REGγ shR or shN cells were transfected with empty vector or PP2Ac or the mutant PP2Ac (H118N), followed by western blotting detection with indicated antibodies. Experiments were repeated three times.
Since REGγ deficiency had no influence on Akt phosphorylation (Supplementary Fig. S4b), we reasoned that REGγ may regulate PRAS40 phosphorylation by phosphatases. By screening a phosphatase library [32, 33], we identified several candidate phosphatases, including PP2A catalytic subunit PP2Ac (the protein product of PP2Aβ), PPP3CA, and PP5C (Supplementary Fig. S4c), among which, only PP2Ac was considered as a potential substrate of the REGγ-proteasome due to its faster degradation in doxycycline-inducible WT REGγ-expressing cells, but not in REGγ (N151Y)-mutant cells (Supplementary Fig. S4d). Therefore we focused on PP2Ac in subsequent studies. Dephosphorylation of PRAS40 by PP2Ac was initiated by coexpressing PRAS40 and increasing amounts of PP2Ac (Fig. 3d). Exogenously expressed PRAS40 and PP2Ac interacted with each other (Fig. 3e). Moreover, PRAS40 could only be dephosphorylated by WT PP2Ac, but not by the inactive mutation of PP2Ac (H118N) in vitro (Fig. 3f and Supplementary Fig. S4e), reinforcing that PRAS40 is a substrate of PP2Ac.
As expected, inhibition of phosphatase by okadaic acid (OA) dramatically increased phosphorylation of PRAS40 (Fig. 3f and Supplementary Fig. S4f). However, reduction of phosphorylated PRAS40 in shR-treated cells still could be detected even in the presence of MK2206, an Akt inhibitor (Supplementary Fig. S4f), suggesting that PP2Ac, but not Akt mediates REGγ-dependent regulation of PRAS40. Alternatively, overexpression of WT-PP2Ac decreased the phosphorylated PRAS40 to a similar level in REGγ shN and shR cells, while overexpression of mutant-PP2Ac (H118N) had no obvious effect (Fig. 3g). Correspondingly, mTORC1 exhibited parallel activity, indicated by phosphorylated S6K and 4EBP1 (Fig. 3g), validating the regulation of PRAS40 and mTORC1 by REGγ via PP2Ac.
REGγ promotes PP2Ac degradation
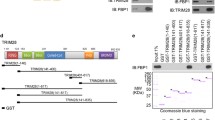
To elucidate how REGγ regulates PP2Ac, we analyzed PP2Ac protein expression profiles and found markedly increased PP2Ac in DEN-treated REGγ−/− mouse liver and in REGγ-knockdown HepG2 cells (Fig. 4a and Supplementary Fig. S5a). We also obtained the results of inverse correlation between REGγ and PP2Ac in 293 doxycycline-inducible REGγ-expressing cells (Supplementary Fig. S5b). Immunofluorescence analysis displayed nuclear accumulation of PP2Ac in DEN-induced REGγ−/− mouse liver cancer cells (Supplementary Fig. S5c). Nevertheless, the PP2Ac mRNA level was not changed in WT and REGγ−/− cells (Fig. 4b). PP2Ac degraded much faster in REGγ shN than in REGγ shR HepG2 cells (Fig. 4c). Similar dynamic changes were observed in CHX-treated doxycycline-inducible REGγ expression cells, but not in cells expressing a dominant-negative mutation (N151Y) REGγ (Supplementary Fig. S5d and S5e). Reciprocal co-IP analysis and GST pulldown assays disclosed physical interactions between REGγ and PP2Ac (Fig. 4d and e). Finally, in vitro cell-free proteolytic analysis showed REGγ and 20S-mediated degradation of PP2Ac (Fig. 4f), substantiating that PP2Ac is a direct target of REGγ.

a DEN-induced WT or REGγ−/− mice liver tissues were lysed and subjected to western blotting analysis using indicated antibodies. Experiments were repeated at least 3 times. b Total RNA was extracted from WT or REGγ−/− mice and PP2Ac mRNA was analyzed by real-time RT-PCR. Experiments were repeated three times or more. c HepG2 REGγ shN or shR cells were treated with CHX (cycloheximide, 100 μg/mL) for indicated time and followed by western blotting analysis. Representative images were from three repeats (left panel). Statistical analysis results are shown on the right panel. *p < 0.05 (two-tailed Student’s t-test). d 293T cells were transiently transfected with indicated plasmids for 36 h, and then cell lysates were subjected to immunoprecipitation with conjugated anti-Flag beads and detected by indicated antibodies. Representative images were from one of three repeats. e GST or GST-PP2Ac protein was pre-incubated with GST beads for 2 h, then purified REGγ was added for binding overnight at 4 °C. The samples were loaded for western blot analysis. f In vitro-translated PP2Ac, or p21 (as positive control) were incubated with purified REGγ and 20S proteasome alone or combined as indicated, followed by western blotting analysis.
REGγ accelerates liver tumorigenesis primarily via a PP2Ac-PRAS40/mTORC1-HIF1α axis
To determine the role of the REGγ-PP2Ac-PRAS40/mTORC1-HIF-1α regulatory axis in liver tumorigenesis, we inspected the expression of the key molecules in mouse liver cancer cells and in DEN-induced liver cancer tissues. In REGγ-deficient cells (Fig. 5a) and tissues (Supplementary Fig. S6a), higher PP2Ac was associated with reduced phosphor-PRAS40 and suppressed mTORC1 activity, along with attenuated expression of HIF-1α and glycolytic genes. Importantly, silencing PP2Ac in REGγ−/− mouse liver cancer cells significantly ablated the OCR rates and increased the ECAR rates, results comparable to those in REGγ+/+ cells (Fig. 5b and c). In agreement, knocking down of PP2Ac narrowed the proliferation gap between REGγ−/− and REGγ+/+ mouse liver cancer cells (Fig. 5d). Silencing PP2Ac rescued, although partially, the proliferation in REGγ-deficient cells (Fig. 5d). These results verify that PP2Ac indeed mediates REGγ action on the mTOR pathway in liver cancer cells to affect metabolic adaptation and cell growth.

a Western blotting detected the indicated proteins in WT or REGγ−/− mice liver cancer cells. Representative results were from two repeats. b, c In WT or REGγ−/− mice liver cancer cells, the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by Seahorse Bioscience XF96 analyzer after transfection with indicated siRNA. Experiments were repeated 3 times. Statistical results are shown on the bottom panel. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). d WT or REGγ−/− mice liver cancer cells were plated at a density of 2 × 103 cells per well in 96-well plate after transfection with indicated siRNA. Plates were read at different time points. Experiments were repeated for three times. Data represent means ± SEM. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). e HepG2 REGγ shN or shR cells were transfected with PRAS40 plasmids, or PRAS40-specific siRNA, then cells were plated in 96-well plate and measured the proliferation at indicated time points. Data shown are representative of two independent experiments. Data represent means ± SEM. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). f HepG2 REGγ shR or shN cells were transfected with PRAS40, or PRAS40 mutant plasmids, followed by analysis of indicated glycolytic gene expression. g, h IHC analysis of indicated proteins in DEN-induced liver tumors from REGγ+/+ or REGγ−/− mice (n = 8 pairs) (g) or in specimens from liver cancer patients (n = 21) (h) (Magnification: ×40. Bar = 50 μm, T: tumor region, P: paracancerous region).
In line with the impact of PP2Ac on cell growth, overexpression of PRAS40, significantly restricted proliferation in REGγ-shN cells to levels equivalent to REGγ-shR cells (Fig. 5e). In contrast, knocking down PRAS40 in REGγ-shR cells led to increased expression of glycolytic genes and cell proliferation compared with control shR cells, which was greater than in REGγ-shN cells (Fig. 5e and Supplementary Fig. S6b). Given the report that Thr 246 and Ser 183 in PRAS40 are the key phosphorylation sites required for association with Raptor to regulate mTORC1 activity [34], we constructed the PRAS40 mutations (T246A/S183A and T246D/S183D) to determine their action in the context of REGγ manipulation. WT PRAS40 and PRAS40 (T246A/S183A) indeed interacted with Raptor better than PRAS40 (T246D/S183D) mutations (Supplementary Fig. S6c). In agreement, overexpression of WT PRAS40 or PRAS40 (T246A/S183A), but not PRAS40 (T246D/S183D), markedly suppressed mRNA levels of HIF-1α and other glycolysis-related genes to comparable levels in both REGγ shN and shR cells (Fig. 5f).
To determine the pathogenic impact of REGγ in DEN-induced HCC, and clinical relevance of our discoveries, we analyzed the correlation among REGγ, PP2Ac, PRAS40/mTORC1 and HIF-1α in DEN-induced REGγ+/+ and REGγ−/− mouse liver cancer tissues (Fig. 5g and Supplementary Fig. S6d) as well as in specimens from human liver cancers (Fig. 5h and Supplementary Fig. S6e and S6f). As predicted, in REGγ-enriched region, PP2Ac was reduced, while expression of phosphorylated PRAS40 and HIF1α were potentiated, implicating regulatory control by a REGγ-PP2Ac-PRAS40/mTORC1-HIF-1α axis in HCC.
Reciprocal positive regulation between REGγ and mTORC1
Lastly, mTORC1 was found to promote the expression of REGγ (Fig. 6a, b). Since TSC2 is a negative regulator of mTORC1 [35], we determined REGγ expression in TSC2−/− MEF cells and found REGγ was largely enhanced, and could be repressed in a dose-dependent manner by Rapamycin (Fig. 6a). Glucose starvation led to suppressed mTORC1 activity and low expression of REGγ in mouse liver cancer cells (Fig. 6b). CHX-treated TSC2−/− cells not only displayed a higher level of REGγ than in WT cells, but also demonstrated a faster decay of PP2Ac and an increased level of p-S6K (Fig. 6c and d) in TSC2−/− cells, reflecting a mTORC1-REGγ interplay. Rapamycin also inhibited REGγ expression in a time-dependent manner (Fig. 6e). Only when mTORC1, but not mTORC2, activity was blocked, REGγ expression would be attenuated (Fig. 6f). Alternatively, overexpressing PRAS40 (Figs. 6g and 3c) or knocking down PRAS40 (Fig. 6h and Supplementary Fig. S6b) in hepatic cancer cells also suppressed or triggered REGγ expression. Altogether, our data establish a REGγ-PP2Ac-PRAS40/mTORC1-HIF1α regulatory axis and reinforce a positive feedback loop between REGγ and mTORC1 in liver cancers.

a Cell lysates from WT or TSC2−/− MEF cells were subjected to Western blotting analysis using indicated antibodies. Representative results were one of three repeats. b Mice liver cancer cells were cultured with normal or low glucose for 24 h. Western blotting analysis was performed to measure indicated protein expression. Experiments were repeated 3 times. c Western blotting analysis of the indicated proteins in TSC2−/− and WT MEF cells treated with mock or CHX for indicated time. Representative results were from three repeats. d Quantitative analysis of PP2Ac degradation and p-p70S6K expression in TSC2−/− and WT MEF cells. Data represent means ± SEM. *p < 0.05, **p < 0.01 (two-tailed Student’s t-test). e HepG2 cells were treated with Rapamycin for indicated time. Western blotting analysis was performed to measure indicated protein expression. f HepG2 cells were transfected with siRNA specific-formTORC1 subunit Raptor or for mTORC2 subunit Rictor. Then REGγ expression were analyzed by Western blot. g HepG2 REGγ shR or shN cells were transfected with Pras40 plasmid and analyzed by Western blotting. Data shown are representative of three independent experiments. h HepG2 cells were transfected with control siRNA, or si-PRAS40 48 h, then REGγ protein level was inspected by western blot.
Discussion
REGγ has been reported to promote cancers [36] via distinct mechanisms including cell cycle [20, 21], survival [37], and genomic instability [38]. In the present study, we demonstrate the function and molecular mechanisms of REGγ in DEN-induced HCC to potentiate glycolytic metabolism and cancer growth. We propose a working model, in which activation of mTORC1 signaling promotes increased expression of REGγ, that in turn expedites PP2Ac decay in an ATP- and Ubiquitin-independent manner, as well as accumulation of phosphorylated PRAS40. Releasing the inhibitory p-PRAS40’s association from Raptor leads to activation of mTORC1, and subsequently induces elevated expression of glycolysis-related genes that ultimately accelerates cell proliferation and tumorigenesis. Conversely, lack of REGγ stabilizes PP2Ac protein, leading to dephosphorylation of PRAS40 that fails to dissociate from mTORC1 complex. Ultimately, attenuated mTORC1 activity results in inhibition of glycolysis and tumor progression in DEN-treated hepatocytes (Fig. 7). Thus, our results establish a positive feedback regulatory loop between REGγ and mTORC1 signaling pathway in DEN-induced hepatocellular carcinoma.

REGγ+/+ liver cancer cells exhibit higher glucose uptake, with initial activation of mTORC1 facilitating expression of REGγ. Elevated REGγ accelerate the degradation of PP2Ac protein in an ubiquitin-independent manner. Less PP2Ac leads to enrichment of phosphorylated PRAS40, which does not associate with Raptor, resulting in activation of mTORC1, overexpression of glycolytic genes, and accelerating cell proliferation and tumorigenesis. In contrast, lack of REGγ leads to accumulation of PP2Ac protein, so that PRAS40 is dephosphorylated and interacts with the mTORC1 complex, ultimately mTORC1 activity, glycolysis and tumor progression are inhibited.
Besides PI3K/Akt/mTOR and HIF-1α, Myc and MEK/ERK are also involved in the regulation of glucose metabolism in tumor cells to satisfy bioenergetic and biosynthetic requirements [28, 39]. We demonstrated that REGγ enhanced glycolysis through facilitating PP2Ac degradation (Figs. 4 and 5). Although PP2A was known to dephosphorylate Akt, MEK, and ERK1/2, to blunt their activity [40], we didn’t observe any reduction of phosphorylated Akt, MEK or ERK1/2 in REGγ-deficient hepatocellular carcinoma cells, perhaps due to loss of cross-talks between mTORC1 and mTORC2 (the latter contributes to phosphorylating some AGC kinases like Akt and SGK). Intriguingly, phosphorylation of PRAS40 was diminished in REGγ-deficient cells (Fig. 3). Our studies reinforce an anti-tumor function for PP2A, reminiscent of PTEN as a tumor suppressor to inhibit the canonical PI3K/Akt/mTOR signaling cascade. How PP2A regulation affects cross-talks between mTORC1 and other pathways remains to be further determined.
HIF1α executes a multitude of functions in HCC. For example, it can upregulate several glycolytic genes to help produce ATP and provide an energy source for the growth of HCC. It also upregulates growth factors, such as VEGF to promote tumor proliferation [41]. In our present study, we established the correlation between REGγ-HIF1a-glycolysis and hepatocellular carcinoma cell proliferation. More evidence is needed to verify that downstream changes in glycolysis are responsible for the hepatocellular carcinoma cell proliferation or tumor growth. Recently, Guo et al. have reported that REGγ plays oncogenic roles in pancreatic cancer by inhibiting the degradation of c-Myc and enhancing glycolysis [25]. However, Li et al. demonstrated that REGγ could proteolyze c-Myc in HeLa and MCF cells as well as in Drosophilia [42]. In our study, we did not observe significant changes in c-Myc between REGγ WT and REGγ-depleted HCC cells (Supplementary. Fig. S2b and S2c), suggesting that the interplay between REGγ and c-Myc may be context-dependent.
The molecular mechanisms for the development of HCC are complicated. Major alterations in HCC include upregulation of Ras/Raf/MEK/ERK and PI3K/Akt/mTOR signaling, aberrant expression of RTK (such as VEGFR, PDGFR, and MET), and reprogrammed energy metabolism [43]. Although sorafenib, an inhibitor both for Raf/MEK/ERK and VEGFR/PDGFR, has been approved as one of the few available anti-HCC drugs [44], the improvement of overall survival rates in HCC patients is still limited [45]. In recent years, several drugs including everolimus, an mTOR inhibitor, have failed in the third phase clinical trials [45, 46], possibly due to the heterogeneity of HCC. Previous study displayed that REGγ is required for the virulence of HCV core protein. HCV core protein transgenic mice with REGγ ablation are not susceptible to core protein-induced diseases, including HCC [47, 48]. Our results highlight new functions of the REGγ-proteasome in glycolytic metabolism and decipher molecular pathogenic mechanism of REGγ in DEN-induced HCC, providing novel insights to development of new biomarkers and targets for personalized therapy against HCC.
Materials and methods
Mice and HCC induction
The origin and feeding of REGγ−/− mice with C57BL/6 genetic background were described previously [22]. Age and gender-matched mice were randomly allocated to experimental groups without blinding. A single intra-peritoneal injection of DEN (diethylnitrosamine) to 15-day-old mice at a dose of 10 mg/kg body weight was given to induce liver tumors in male mice. Mice were sacrificed 8–9 months later, and tumors bigger than 1 mm in diameter on the liver surface were counted. Tumors bigger than 5 mm across were dissected for biochemical and molecular analyses. The sample size (at least 5) of our experiments was determined by our previous experience.
Cell culture
293T and HepG2 cell lines were obtained from the ATCC. 293T REGγ knockout cells, 293 inducible REGγ (WT) or REGγ (N151Y) expressing cells were established in our laboratory [33]. TSC2−/− MEF cells were kindly provided by Dr. Wei Liu. Cells were authenticated by a short tandem repeat (STR) profiling and routinely tested for Mycoplasma contamination. Cells were cultured in DMEM (GIBCO) containing 10% FBS and 50 mg/ml penicillin/streptomycin (Invitrogen), 37 °C with 5% CO2.
Bioinformatics analysis
We downloaded the expression matrixes and related clinical information of liver cancer in TCGA (The Cancer Genome Atlas) database from GDC (https://portal.gdc.cancer.gov/). The dataset contained a total of 373 liver cancer samples and 50 normal tissues. Differential expression analysis of interested genes was conducted using Student’s t-test. We calculated the expression correlation between two genes by Spearman method.
Extracellular acidification rate and oxygen consumption rate assays
ECAR and OCR were measured using Glycolysis Stress Test Kit and Cell Mito Stress Test Kit (Agilent Technologies), respectively. Briefly, cells were plated in appropriate growth medium the day prior to the assay and allowed to adhere overnight. For ECAR, the glycolytic inhibitor 20 nM 2-DG were sequentially added into each well at the indicated time points, and for OCR, Oligo (oligomycin), the reversible inhibitor of oxidative phosphorylation, FCCP (p-trifluoromethoxy carbonyl cyanide phenylhydrazone), a potent uncoupler of oxidative phosphorylation, and the mitochondrial complex I inhibitor rotenone plus the mitochondrial complex III inhibitor AA (Antimycn A) were sequentially injected. Data were analyzed by Seahorse XF-96 Wave software. ECAR is reported in mpH/minute and OCR in pmols/minute. ECAR and OCR were normalized to cell count following the assay.
Glucose uptake and lactate assay
Cells were treated as described in the figure legends. For glucose uptake, 1 × 104 cells were plated into 96-well plate and incubated for 12 h. Then cells were washed three times with PBS and starved for glucose by preincubating with 100 ml Krebs-Ringer-Phosphate-HEPES buffer containing 2% BSA for 40 min. 10 mM 2-DG was added and incubated for 20 min. Cells were lysed with 90 μl of extraction buffer, frozen/thawed once, heated at 85 °C for 40 min and added 10 μl of neutralization buffer. Then lysates were centrifugated at 12,000 rpm for 3 min, the supernatant was used for determination of glucose uptake at 412 nm in a reader. Values were normalized to cell number. For lactate production measurement, 1 × 105 cells were seeded into plate and incubated in full medium for 12 h. The media were removed and cells were incubated in DMEM without FBS. After incubation for 1 h, the supernatant was collected. The reaction mixture was incubated for 30 min at RT in dark and measured at 450 nm in a reader. Results were normalized to cell number.
In vitro phosphatase assay
GST-PRAS40, GST-PP2Ac and GST-PP2Ac (H118N) were expressed and purified from Escherichia coli. GST-PP2Ac and GST-PP2Ac (H118N) with or without ATP and AKT were incubated with GST-PRAS40 in buffer (20 mM HEPES, 20 mM MgCl2, 0.03% β-mercaptoethanol) for 30 min at 30 °C with every five minutes gentle shaking. The reactions were stopped by adding 2× protein loading and followed by boiling for 5 min. Flag-PP2CB and PRAS40 were expressed in HEK293T cell followed by OA (okadaic acid) treatment. Then Flag-PP2Ac protein was immunoprecipitated using anti-Flag beads from cell lysates and eluted with 3x Flag peptide. PRAS40 was immunoprecipitated by PRAS40 antibody-coupled beads and washed three times with 1× phosphatase buffer (50 mM HEPES (pH 7.5), 100 mM NaCl, 2 mM dithiothreitol, 0.01% Brij 35,1 mM MnCl2). The elute was divided into equal parts. One aliquot was treated with OA (okadaic acid), the others were mock treated. After incubation with PP2Ac at 30 °C for 10, 20, and 30 min with gentle shaking, the reactions were stopped and boiled for 5 min.
Histologic analysis and immunohistochemistry
The liver tissues were fixed in 4% neutral formalin overnight and then were dehydrated and embedded in paraffin before cutting into different sections (4–5 μm thick). The sections were subsequently de-paraffined and stained with hematoxylin and eosin (H&E) or analyzed by immunohistochemistry (IHC) [22]. After incubation with anti-REGγ (1:800), anti-Ki67 (1:100), anti-PP2Ac (1:100), anti-PRAS40 T246 (1:100) and anti-HIF-1α (1:200) overnight at 4 °C, sections were incubated with biotinylated secondary antibody for 30 min at RT and then were rinsed and added streptavidin–horseradish peroxidase conjugates for 10 min. After staining with DAB solution, the slides were counterstained with hematoxylin and were dehydrated for mounting. 21 paraffin-embedded human liver cancer specimens from Changzheng Hospital and 56 human liver tumor samples in a tissue microarray from Shanghai Cancer Institute were included in our clinical related analysis.
Statistical analysis
All in vitro experiments were repeated at least three times. Prism software (GraphPad Software) was used for statistical analysis. The data were shown as either a scatter plots or bar graphs with mean ± S.E.M. Statistical significance between two samples was determined when the P value was less than 0.05 with two-tailed Student’s t-test. The variance between the groups was similar.
Ethical statement
All animal experiments complied with the National Institutes of Health guide for the care and use of Laboratory animals. Research involving human material, or human data were performed in accordance with the ethical principles of the World Medical Association (Declaration of Helsinki). The participants signed appropriate informed consent forms before the study to collect samples. All studies were reviewed and approved by the Ethics Committee of East China Normal University.
The information for reagents used in this paper was listed in Table S1.
The siRNA sequences for specific gene were shown in Table S2.
The primers used in this manuscript were listed in Table S3 (for human genes) and Table S4 (for mice genes).
References
Laursen L. A preventable cancer. Nature. 2014;516:S2–3.
Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA Cancer J Clin. 2014;64:9–29.
Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, et al. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–88.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519–30.
Alfarouk KO, Muddathir AK, Shayoub ME. Tumor acidity as evolutionary spite. Cancers (Basel). 2011;3:408–14.
Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–9.
Gill KS, Fernandes P, O’Donovan TR, McKenna SL, Doddakula KK, Power DG, et al. Glycolysis inhibition as a cancer treatment and its role in an anti-tumour immune response. Biochim Biophys Acta. 2016;1866:87–105.
Harris AL. Hypoxia–a key regulatory factor in tumour growth. Nat Rev Cancer. 2002;2:38–47.
Karim S, Adams DH, Lalor PF. Hepatic expression and cellular distribution of the glucose transporter family. World J Gastroenterol. 2012;18:6771–81.
Mendez-Blanco C, Fondevila F, Garcia-Palomo A, Gonzalez-Gallego J, Mauriz JL. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50:134.
Agani F, Jiang BH. Oxygen-independent regulation of HIF-1: novel involvement of PI3K/AKT/mTOR pathway in cancer. Curr Cancer Drug Targets. 2013;13:245–51.
Duvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–83.
Han X, Sun S, Zhao M, Cheng X, Chen G, Lin S, et al. Celastrol stimulates hypoxia-inducible factor-1 activity in tumor cells by initiating the ROS/Akt/p70S6K signaling pathway and enhancing hypoxia-inducible factor-1alpha protein synthesis. PLoS ONE. 2014;9:e112470.
Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: current state and future trends. J Hepatol. 2014;60:855–65.
Kunter I, Erdal E, Nart D, Yilmaz F, Karademir S, Sagol O, et al. Active form of AKT controls cell proliferation and response to apoptosis in hepatocellular carcinoma. Oncol Rep. 2014;31:573–80.
Khemlina G, Ikeda S, Kurzrock R. The biology of hepatocellular carcinoma: implications for genomic and immune therapies. Mol Cancer. 2017;16:149.
Bhat M, Sonenberg N, Gores GJ. The mTOR pathway in hepatic malignancies. Hepatology. 2013;58:810–8.
Zhang Y, Kwok-Shing NgP, Kucherlapati M, Chen F, Liu Y, Tsang YH. et al. A pan-cancer proteogenomic atlas of PI3K/AKT/mTOR pathway alterations. Cancer Cell. 2017;31:820–32. e3.
Li X, Lonard DM, Jung SY, Malovannaya A, Feng Q, Qin J, et al. The SRC-3/AIB1 coactivator is degraded in a ubiquitin- and ATP-independent manner by the REGgamma proteasome. Cell 2006;124:381–92.
Chen X, Barton LF, Chi Y, Clurman BE, Roberts JM. Ubiquitin-independent degradation of cell-cycle inhibitors by the REGgamma proteasome. Mol Cell. 2007;26:843–52.
Li X, Amazit L, Long W, Lonard DM, Monaco JJ, O’Malley BW. Ubiquitin- and ATP-independent proteolytic turnover of p21 by the REGgamma-proteasome pathway. Mol Cell. 2007;26:831–42.
Yao L, Zhou L, Xuan Y, Zhang P, Wang X, Wang T, et al. The proteasome activator REGgamma counteracts immunoproteasome expression and autoimmunity. J Autoimmun. 2019;103:102282.
Xiong S, Zheng Y, Jiang P, Liu R, Liu X, Qian J, et al. PA28gamma emerges as a novel functional target of tumour suppressor microRNA-7 in non-small-cell lung cancer. Br J Cancer. 2014;110:353–62.
Wang Q, Gao X, Yu T, Yuan L, Dai J, Wang W, et al. REGgamma controls hippo signaling and reciprocal NF-kappaB-YAP regulation to promote colon cancer. Clin Cancer Res. 2018;24:2015–25.
Guo J, Hao J, Jiang H, Jin J, Wu H, Jin Z, et al. Proteasome activator subunit 3 promotes pancreatic cancer growth via c-Myc-glycolysis signaling axis. Cancer Lett. 2017;386:161–7.
Hindupur SK, Colombi M, Fuhs SR, Matter MS, Guri Y, Adam K, et al. The protein histidine phosphatase LHPP is a tumour suppressor. Nature. 2018;555:678–82.
Alqahtani A, Khan Z, Alloghbi A, Said Ahmed TS, Ashraf M, Hammouda DM. Hepatocellular carcinoma: molecular mechanisms and targeted therapies. Medicina (Kaunas). 2019;55:526.
Obre E, Rossignol R. Emerging concepts in bioenergetics and cancer research: metabolic flexibility, coupling, symbiosis, switch, oxidative tumors, metabolic remodeling, signaling and bioenergetic therapy. Int J Biochem Cell Biol. 2015;59:167–81.
Munoz-Sanchez J, Chanez-Cardenas ME. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol. 2019;39:556–70.
Ding M, Bruick RK, Yu Y. Secreted IGFBP5 mediates mTORC1-dependent feedback inhibition of IGF-1 signalling. Nat Cell Biol. 2016;18:319–27.
Chong ZZ. Targeting PRAS40 for multiple diseases. Drug Disco Today. 2016;21:1222–31.
Sun W, Yu Y, Dotti G, Shen T, Tan X, Savoldo B, et al. PPM1A and PPM1B act as IKKbeta phosphatases to terminate TNFalpha-induced IKKbeta-NF-kappaB activation. Cell Signal. 2009;21:95–102.
Gao X, Wang Q, Wang Y, Liu J, Liu S, Liu J, et al. The REGgamma inhibitor NIP30 increases sensitivity to chemotherapy in p53-deficient tumor cells. Nat Commun. 2020;11:3904.
Mi W, Ye Q, Liu S, She QB. AKT inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E-BP1 axis. Oncotarget. 2015;6:13962–77.
Manning BD, Cantley LC. Rheb fills a GAP between TSC and TOR. Trends Biochem Sci. 2003;28:573–6.
He J, Cui L, Zeng Y, Wang G, Zhou P, Yang Y, et al. REGgamma is associated with multiple oncogenic pathways in human cancers. BMC Cancer. 2012;12:75.
Moncsek A, Gruner M, Meyer H, Lehmann A, Kloetzel PM, Stohwasser R. Evidence for anti-apoptotic roles of proteasome activator 28gamma via inhibiting caspase activity. Apoptosis 2015;20:1211–28.
Zannini L, Lecis D, Buscemi G, Carlessi L, Gasparini P, Fontanella E, et al. REGgamma proteasome activator is involved in the maintenance of chromosomal stability. Cell Cycle. 2008;7:504–12.
DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB. The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab. 2008;7:11–20.
Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353:417–39.
Lin D, Wu J. Hypoxia inducible factor in hepatocellular carcinoma: a therapeutic target. World J Gastroenterol. 2015;21:12171–8.
Li S, Jiang C, Pan J, Wang X, Jin J, Zhao L, et al. Regulation of c-Myc protein stability by proteasome activator REGgamma. Cell Death Differ. 2015;22:1000–11.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74.
European Association For The Study Of The L, European Organisation For R, and Treatment Of C. EASL-EORTC clinical practice guidelines: management of hepatocellular carcinoma. J Hepatol. 2012;56:908–43.
Dhanasekaran R, Venkatesh SK, Torbenson MS, Roberts LR. Clinical implications of basic research in hepatocellular carcinoma. J Hepatol. 2016;64:736–45.
Zhu AX, Kudo M, Assenat E, Cattan S, Kang YK, Lim HY, et al. Effect of everolimus on survival in advanced hepatocellular carcinoma after failure of sorafenib: the EVOLVE-1 randomized clinical trial. JAMA. 2014;312:57–67.
Miyamoto H, Moriishi K, Moriya K, Murata S, Tanaka K, Suzuki T, et al. Involvement of the PA28gamma-dependent pathway in insulin resistance induced by hepatitis C virus core protein. J Virol. 2007;81:1727–35.
Moriishi K, Mochizuki R, Moriya K, Miyamoto H, Mori Y, Abe T, et al. Critical role of PA28gamma in hepatitis C virus-associated steatogenesis and hepatocarcinogenesis. Proc Natl Acad Sci USA. 2007;104:1661–6.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31730017, 31670882, 81672883), the Science and Technology Commission of Shanghai Municipality (19JC1411900, 16ZR1410000, 16QA1401500). We thank Dr. Wei Liu from Zhejiang University for providing TSC2−/− cell line. We also thank the ECNU Multifunctional Platform for Innovation (011) for keeping and raising mice.
Author information
Authors and Affiliations
Contributions
X.T.L., B.H.Z., J.B.L., and L.F.Y. designed research. L.F.Y. and Y.X. performed the molecular and cell biology, metabolic, data analysis and animal experiments, respectively. X.L.M., T.Y.M., and X.Q.M. were involved in the molecular and cell biology study. H.Y.Z., T.Z.W., W.S.S., and H.B.W. contributed to the animal work. B.Y. contributed to metabolomics analysis. J.R.X., J.J.L., and C.G. provided clinical samples. L.L. and P.Z. contributed to material support. X.T.L., B.H.Z., R.M., and L.F.Y. wrote the paper.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yao, L., Xuan, Y., Zhang, H. et al. Reciprocal REGγ-mTORC1 regulation promotes glycolytic metabolism in hepatocellular carcinoma. Oncogene 40, 677–692 (2021). https://doi.org/10.1038/s41388-020-01558-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-020-01558-8
- Springer Nature Limited
This article is cited by
-
B3galt5 deficiency attenuates hepatocellular carcinoma by suppressing mTOR/p70s6k-mediated glycolysis
Cellular and Molecular Life Sciences (2023)
-
Identification of a BRAF/PA28γ/MEK1 signaling axis and its role in epithelial-mesenchymal transition in oral submucous fibrosis
Cell Death & Disease (2022)
-
RETRACTED ARTICLE: LNCAROD enhances hepatocellular carcinoma malignancy by activating glycolysis through induction of pyruvate kinase isoform PKM2
Journal of Experimental & Clinical Cancer Research (2021)