
Abstract
DNA repair gene mutations are frequent in castration-resistant prostate cancer (CRPC), suggesting eligibility for poly(ADP-ribose) polymerase inhibitor (PARPi) treatment. However, therapy resistance is a major clinical challenge and genes contributing to PARPi resistance are poorly understood. Using a genome-wide CRISPR-Cas9 knockout screen, this study aimed at identifying genes involved in PARPi resistance in CRPC. Based on the screen, we identified PARP1, and six novel candidates associated with olaparib resistance upon knockout. For validation, we generated multiple knockout populations/clones per gene in C4 and/or LNCaP CRPC cells, which confirmed that loss of PARP1, ARH3, YWHAE, or UBR5 caused olaparib resistance. PARP1 or ARH3 knockout caused cross-resistance to other PARPis (veliparib and niraparib). Furthermore, PARP1 or ARH3 knockout led to reduced autophagy, while pharmacological induction of autophagy partially reverted their PARPi resistant phenotype. Tumor RNA sequencing of 126 prostate cancer patients identified low ARH3 expression as an independent predictor of recurrence. Our results advance the understanding of PARPi response by identifying four novel genes that contribute to PARPi sensitivity in CRPC and suggest a new model of PARPi resistance through decreased autophagy.
Similar content being viewed by others


Introduction
Prostate cancer (PC) is the most common non-skin cancer and the second leading cause of cancer-related death among men in western countries [1]. While early-stage localized PC may be cured, metastatic PC (mPC) is associated with high mortality rates. Conventional mPC treatment consists of androgen deprivation therapy [2] but resistance invariably develops, leading to castration-resistant prostate cancer (CRPC). Patients with CRPC have a median survival rate of only 9 to 30 months [3]. Recent studies of CRPC have revealed frequent (20–30%) deleterious mutations in DNA repair genes such as BRCA1, BRCA2 (BRCA1/2), and ATM [4,5,6], suggesting sensitivity to poly(ADP)polymerase (PARP) inhibitors (PARPi). The PARPi olaparib targets and inhibits PARP1 activity, causing double-stranded DNA break accumulation [7, 8], which in combination with certain DNA repair gene defects leads to cell death. In 2020, olaparib was granted FDA approval for CRPC patients carrying a deleterious mutation in any of 13 different homologous recombination repair genes, and who had progressed on secondary hormonal therapy [9]. However, despite initial benefits of olaparib treatment in some CRPC patients [10], therapy resistance is a major challenge [11].
Some PARPi resistance mechanisms have been described in CRPC including reversion mutations in BRCA1/2 and PALB2 in olaparib- or talazoparib-treated CRPC patients [12, 13], and an association between increased NPRL2 expression and olaparib resistance in CRPC cell line models [14]. In other cell types (breast and embryonic stem cells) PARPi resistance has been associated with loss of PARP1 [15], and loss of the deparylation enzyme poly(ADP-ribose) glycohydrolase (PARG) [16]. However, to guide treatment selection and to improve CRPC patient survival, a better understanding of the genes that control PARPi response in CRPC is needed.
The present study aimed to identify genetic modulators of PARPi response in CRPC. We performed a genome-wide knockout (KO) screen using the clustered regularly interspaced short palindromic repeats-associated protein 9 (CRISPR-Cas9) technology. From this, we identified PARP1 as well as six novel candidate genes as potential modulators of olaparib resistance. By individual gene KO, we were the first to validate that loss of PARP1, ARH3, YWHAE, and UBR5 was associated with olaparib resistance in CRPC cells. Furthermore, we show a novel model of PARPi resistance through decreased autophagy caused by PARP1 or ARH3 KO.
Results
Genome-wide CRISPR-Cas9 knockout screen and candidate gene selection
To identify novel genes contributing to resistance against the PARPi olaparib, a genome-wide CRISPR-Cas9 KO screen was performed in the human CRPC cell line C4 using the Brunello v2 library, consisting of 77 441 guide RNAs (gRNAs) targeting 19 114 genes and 1000 non-targeting controls [17]. First, stable Cas9-expressing C4 cells (C4-Cas9) were generated by lentiviral transduction with a Cas9-containing vector. Cas9 expression was validated by Western blotting (Fig. S1). Then, two pools (replicate 1 and replicate 2) of C4-Cas9 cells were transduced with the Brunello v2 library at a multiplicity of infection (MOI) of 0.5 (Fig. 1A). For each replicate, an aliquot of cells was harvested after transduction and puromycin selection (baseline). The remaining cells were treated with either: 1) DMSO (vehicle), 2) 18.5 µM olaparib (IC50), or 3) 29 µM olaparib (IC90).

A Schematic representation of experimental workflow. First, CRPC cells (C4) were transduced with the Cas9 lentiviral vector. Next, the Brunello v2 human CRISPR-Cas9 library (76 441 gRNAs) was packaged into lentiviral particles, and Cas9-expressing C4 cells were transduced at a MOI of 0.5. Library-transduced cells (in duplicate) were selected for nine days using puromycin. An aliqout of cells was harvested after puromycin selection (baseline control). The remaining cells were analyzed in three experimental groups, treated with either olaparib at IC50, olaparib at IC90, or DMSO (vehicle) for three weeks. After three weeks, the surviving cells were harvested, genomic DNA extracted, and gRNA regions were PCR amplified and sequenced. B Log2-transformed gRNA read count distributions. B/L: baseline, VEH: vehicle. C Venn diagram of top significantly enriched genes based on the criteria: called as top 500 by the MAGeCK analysis, fold change ≥1.25, and a minimum of two enriched gRNAs. IC50 (orange) and IC90 (blue). R1: replicate 1, R2: replicate 2. D Representative p-value distribution (output from MAGeCK) for genes in IC50-treated (left) and IC90-treated samples (right). E Log10-transformed read counts of individual gRNAs for PARP1, ARH3, YWHAE, and USP48 at baseline (pre-treatment) and after treatment with DMSO (vehicle) or olaparib at IC50/IC90. Black: gRNA1, green: gRNA2, orange: gRNA3, blue: gRNA4. Filled circles: replicate 1, open circles: replicate 2, line represents average of replicates.
After three weeks of treatment, cells were harvested for DNA extraction and gRNA regions were sequenced. Sequence data was mapped to the Brunello v2 library to count individual gRNA reads. On average, 48.6% (range 43–54%) of the reads mapped to the library (Fig. S2), while most unmapped reads aligned with vector backbone sequences (data not shown). Median gRNA read counts were comparable between all sample groups but with a slightly larger variance observed for IC90 samples likely reflecting the stronger selection pressure (Fig. 1B). Each gene was represented by at least one gRNA, and an average of 97.7% (range 95.5–98.9%) of the original library was represented in individual samples (Additional information, Table 1).
Model-based Analysis of Genome-wide CRISPR-Cas9 Knockout (MAGeCK) [18] was used to identify enriched gRNAs in olaparib (IC50 or IC90) versus vehicle-treated cells. In each comparison genes were filtered by: 1) gene ranked in top 500 by MAGeCK analysis, and 2) an average fold change ≥1.25 for gRNAs targeting that gene compared to vehicle, and 3) at least two out of four gRNAs for that gene were deemed significantly enriched by the MAGeCK analysis (based on Robust Rank Aggregation score, false discovery rate ≤ 0.05 as significance cut-off [19]). Filtered gene lists were compared between replicates. There was an overlap of 46 genes between IC50 replicates (46/787 = 5.8%), 52 genes between IC90 replicates (52/780 = 6.7%), and 13 genes were shared by all four olaparib-treated samples (Fig. 1C; Additional information Table 2). In all samples, PARP1 was the top ranked gene (Fig. 1D, Table S1) with gRNA read counts consistently enriched compared to vehicle-treated and baseline samples (Fig. 1E). This result supports the validity of our screen, as PARP1 is the main target of olaparib.
Based on a total of 469 shortlisted genes (Fig. 1C), we selected six top candidates for experimental validation: ARH3, CCND1, TEAD4, UBR5, USP48, and YWHAE (Table S1). These genes have not previously been described in relation to PARPi resistance, but have been linked to various DNA repair pathways (ARH3: base excision repair [20]; CCND1, TEAD4, UBR5, and YWHAE: homologous recombination [21,22,23,24]; USP48: Fanconi’s Anemia pathway [25]). Of these, ARH3, USP48, and YWHAE were among the 13 top enriched genes shared by all four olaparib-treated samples (Fig. 1C, D). For ARH3 and USP48, gRNA read counts were consistently enriched as compared to baseline and vehicle-treated cells, which was also true for YWHAE except gRNA4 (Fig. 1E). We further included PARP1 as this was the top hit.
Generation of KO cell populations and clones for individual candidate genes
To validate the relationship between olaparib resistance and KO of ARH3, CCND1, PARP1, TEAD4, UBR5, USP48, and YWHAE observed in the screen, we generated single-gene KO cell populations and clones by CRISPR-Cas9 editing in C4 CRPC cells. We selected the two best performing gRNAs for each gene and a non-targeting gRNA (Control_237) from the Brunello v2 library as a control (Table S2).
We successfully cloned one gRNA for YWHAE and two gRNAs for each of the remaining six genes into the GFP-encoding plasmid PX458 (Ref. [26]; Table S2). C4-Cas9 cells were transfected with the plasmids and FACS sorted based on GFP expression, generating a total of 13 KO populations for the seven candidate genes (Table S3). Indel frequencies were determined by Tracking of Indels by DEcomposition (TIDE) [27] analysis in 11/13 KO populations and ranged from 16.5% (ΔCCND1_4) to 82.9% (ΔARH3_2) (Fig. 2A, Table S2).

A, B Estimation of genomic CRISPR-Cas9 KO editing efficiency by TIDE analysis. ND: not determined. A Indel frequencies in KO cell populations. B Indel frequencies in KO clones. C Representative olaparib dose-response curves of C4 KO cell lines: (first) ΔPARP1_2 and ΔPARP1_4, (second) ΔARH3_2 and ΔARH3_3, (third) ΔYWHAE_2, (fourth) ΔUBR5_3, and (fifth) KO clones ΔUBR5_2_7B, ΔUBR5_3_9C, and ΔUBR5_3_10C. Dose-response curves show viability normalized to the corresponding cell line treated with DMSO. Assays were performed three times, each including technical triplicates. WT: wildtype C4-Cas9. *p-value < 0.05, **p-value < 0.001 (Multiple Student’s t-test). D Representative IC50 fold change compared to WT of Control_237, ΔCCND1_2, ΔCCND1_4, ΔTEAD4_2, ΔTEAD4_4, ΔUBR5_2, ΔUSP48_3, and ΔUSP48_1. E Representative olaparib dose-response curves of LNCaP KO cell lines: (left) ΔPARP1_2(LNCaP) and ΔPARP1_4(LNCaP), (middle) ΔARH3_2(LNCaP) and ΔARH3_3(LNCaP), (right) ΔUBR5_2(LNCaP) and ΔUBR5_3(LNCaP).
For four genes (UBR5, CCND1, TEAD4, and USP48), we also generated clonal KO cell lines by single cell sorting (FACS) of transfected C4-Cas9 cells followed by expansion of individual clones. In total, eight KO clones were established: one for CCND1, two for TEAD4, two for USP48, and three for UBR5 (Table S2). Indel frequencies ranged from 77.5% (ΔTEAD4_4_2D) to 98.1% (ΔUSP48_1_7A) and were in all cases higher and more homogenous than for the corresponding KO population (Fig. 2B, Table S2).
Phenotypic validation - Knockout of PARP1, ARH3, YWHAE or UBR5 causes olaparib resistance
To validate the phenotypic effect of KO cell lines, we measured the viability of each KO cell population/clone after six days exposure to a serial dilution of olaparib, as compared to wildtype C4-Cas9 cells (WT).
Although Control_237 cells seemed to have slightly higher viability at 10 µM and slightly lower viability above 35 µM of olaparib, no significant differences (Student’s t-test) were observed at remaining concentration points (Fig. 2C), indicating similar olaparib sensitivity as compared to WT.
In contrast, PARP1 KO populations (ΔPARP1_2 and ΔPARP1_4) had significantly higher viability compared to WT (Multiple student’s t-test, p-value < 0.05; Fig. 2C). This confirms the results of our genome-wide KO screen, where PARP1 was the top hit (Fig. 1C, D).
Of the remaining six candidates, olaparib resistance upon gene KO was successfully validated for the three genes: ARH3, YWHAE, and UBR5. Compared to WT cells, the two ARH3 KO populations (ΔARH3_2 and ΔARH3_3), the YWHAE KO population (ΔYWHAE_2), one UBR5 KO population (ΔUBR5_3) and three UBR5 KO clones showed significantly increased survival upon olaparib treatment (Fig. 2C). The remaining UBR5 KO population (ΔUBR5_2) did not show olaparib resistance (Fig. 2D), likely explained by low indel frequency in these cells (25.5%; Table S2).
KO of ARH3, YWHAE, and UBR5 had little to no effect on proliferation rates of untreated cells (Fig. S3A), indicating that the observed olaparib resistance was not simply an indirect effect of reduced cell division rates upon gene KO. ARH3, YWHAE, and UBR5 gene KO was further validated at protein level by Western blotting (Fig. S3D, E) and at RNA level by RT-qPCR (Fig. S3F).
For the remaining three candidates (CCND1, TEAD4, and USP48), we were unable to validate a significant association between gene KO and olaparib resistance in C4 cells based on analyses of six KO populations (Fig. 2D) and five KO clones (Fig. S4). Hence, CCND1, TEAD4, and USP48 were excluded from further analysis.
To further confirm that PARP1, ARH3, YWHAE, or UBR5 KO caused olaparib resistance in PC cells, we aimed to generate gene KO models in the androgen sensitive PC cell line LNCaP, using the same two gRNAs per gene as used for C4 cell lines (Table S3). Successful PARP1, ARH3, and UBR5 KO was confirmed by Sanger sequencing or RT-qPCR in the LNCaP KO populations: ΔPARP1_2(LNCaP), ΔPARP1_4(LNCaP), ΔARH3_2(LNCaP), ΔARH3_3(LNCaP), and ΔUBR5_3(LNCaP) (Table S3), while low editing efficacy was observed for ΔUBR5_2(LNCaP) (Table S3). Despite numerous attempts, we were not able to produce an LNCaP YWHAE KO model. Dose response experiments showed increased olaparib resistance in LNCaP cell lines with KO of either PARP1, ARH3, or UBR5 (Fig. 2E). This confirmed our results from C4 cells (Fig. 2C) and demonstrated that these phenotypes were not restricted to androgen insensitive CRPC cells.
Cross-resistance to other PARPis and carboplatin
As KO of PARP1, ARH3, YWHAE, or UBR5 caused olaparib resistance, we wanted to evaluate possible cross-resistance to additional PARPis (niraparib and veliparib) and to the DNA cross-linking agent carboplatin in C4 cells.
Dose-response curves for the Control_237 cell line were slightly right-skewed for niraparib and veliparib compared to WT (Fig. 3A, B). Hence, KO cell lines were only deemed resistant if their dose-response curves were clearly different from both WT and Control_237 cell lines.

Representative niraparib (A) and veliparib (B) dose-response curves of C4 KO cell lines: (left) ΔPARP1_2 and ΔPARP1_4, (middle) ΔARH3_2 and ΔARH3_3, (right) ΔUBR5_3, ΔUBR5_2_7B, ΔUBR5_3_9C, and ΔUBR5_3_10C. C LNCaP KO viability after treatment with (left) 4 µM niraparib or (right) 60 µM veliparib.
The two PARP1 KO populations (ΔPARP1_2 and ΔPARP1_4) and the two ARH3 KO populations (ΔARH3_2 and ΔARH3_3) showed clear cross-resistance to both niraparib and veliparib (Fig. 3A, B), but not to carboplatin (Fig. S5).
For UBR5 KOs, we did not find robust evidence for cross-resistance to niraparib. Although two UBR5 KO clones (ΔUBR5_3_9C and ΔUBR5_3_10C) displayed increased viability upon niraparib (Fig. 3A) and veliparib treatment (Fig. 3B), no clear effect was seen for the third KO clone (ΔUBR5_2_7B) nor the KO population (ΔUBR5_3) (Fig. 3A, B). Likewise, we did not find clear evidence for cross-resistance to carboplatin upon UBR5 KO (Fig. S5).
The YWHAE KO population did not exhibit cross-resistance to niraparib nor veliparib (Fig. S6) but showed significant resistance to carboplatin in C4 cells (Fig. S5).
KO of PARP1 and ARH3 also caused cross-resistance to niraparib and veliparib in LNCaP cells (Fig. 3C), while UBR5 KO in LNCaP cells seemed to be associated with moderate cross-resistance to veliparib, but not to niraparib (Fig. 3C). This supports our findings from C4 cells (Fig. 3A, B).
Knockout of ARH3 decreases autophagy
As both C4 ARH3 KO populations (ΔARH3_2 and ΔARH3_3) showed clear olaparib resistance, we wanted to further characterize these cells. First, we investigated transcriptomic changes by RNA sequencing. ARH3 KO populations and WT C4-Cas9 cells were treated with either olaparib or vehicle (DMSO) for six days, followed by RNA sequencing (all experiments in duplicate). Differentially expressed genes were identified using EdgeR [28]. The analysis revealed similar patterns in differentially expressed genes between olaparib- vs. vehicle-treated cells for both ARH3 KOs and for WT (Additional information, Table 3), indicating similar transcriptional changes in olaparib treated cells, irrespective of ARH3 gene KO status. However, comparing vehicle-treated ARH3 KO cells to vehicle-treated WTs, we identified 61 differentially expressed genes shared by ΔARH3_2 and ΔARH3_3 (Additional information, Table 4). Gene-set enrichment analysis (EnrichR [29, 30]) of these genes revealed pathways involved in oxidative phosphorylation (OXPHOS) (Fig. S7) with enrichment and increased expression of three genes encoding key subunits of mitochondrial complex I: NDUFV1, NDUFA3, and NDUFB7 in both olaparib- and vehicle-treated ARH3 KO populations (Fig. 4A).

A Normalized log2 counts of the NADH:ubiquinone oxidoreductases NDUFV1, NDUFA3, and NDUFB7 and EIF5A. Expression is shown for ΔARH3 and WT C4-Cas9 cells. Filled circles: olaparib-treated, open circles vehicle-treated. B Seahorse metabolic analysis: Top: basal oxygen consumption rate (OCR), bottom: maximal respiration rate of WT C4 cells, ARH3 KO cells (ARH3_2 and ARH3_3), and PARP1 KO cells (PARP1_2 and PARP1_4) untreated or treated with olaparib for 72 h. Graphs show data from two pooled experiments. Statistics compared untreated KOs to untreated WT cells or treated KOs to treated WT cells. C LC3II/I ratios after 50 µM chloroquine treatment of WT C4 cells, ARH3 KO cells (ARH3_2 and ARH3_3), and PARP1 KO cells (PARP1_2 and PARP1_4) based on western blots quantified by densitometric analysis. Statistics compared KOs to WT. D Viability of WT C4 cells, ARH3 KO cells (ARH3_2 and ARH3_3), and PARP1 KO cells (PARP1_2 and PARP1_4) after treatment with (top) 10 µM olaparib or 10 µM olaparib+10 nM rapamycin (two pooled experiments, n = 6), (bottom) 10 µM olaparib or 10 µM olaparib+5 µM chloroquine (two pooled experiments, n = 6). All: Error bars show standard deviation. Differences between groups were tested with unpaired Student’s t-tests, p-values < 0.05 were considered significant, *p-value < 0.05, **p-value < 0.01.
Based on this, we investigated the oxygen consumption rates as a measure of OXPHOS in ARH3 KO and WT C4-Cas9 cell lines using the Seahorse assay. As ARH3 works downstream of PARP1, we included PARP1 KOs in the following analysis to investigate whether ARH3 and PARP1 KOs showed similar biological changes.
Oxygen consumption rates increased significantly upon olaparib treatment in KO and WT cells (Figs. 4B, S8), but no consistent differences between KO and WT cells were observed either with or without olaparib treatment (Figs. 4B, S8). Similarly, there were no significant differences in glycolysis, determined by extracellular acidification rate, between KOs and WT cells (Fig. S8). We further assessed NAD+ and NADH levels by liquid chromatography-mass spectrometry. In accordance with oxygen consumption rate data, we did not discover consistent differences in NAD+ or NADH levels between KOs and WT (Table S4). Thus, we did not find clear evidence for altered OXPHOS in ARH3 KO cell lines, despite increased expression of NDUFV1, NDUFA3, and NDUFB7 (Fig. 4A). Consistent with this, none of the other ~37 known genes encoding key complex I subunits [31] were deregulated in the RNA-seq data for ARH3 KO vs. WT cells (data not shown).
OXPHOS has been linked to drug resistance through autophagy activation [32] and we noted that ARH3 KOs displayed upregulation of EIF5A (Fig. 4A), which encodes a translation initiation factor for the key autophagy protein ATG3. Thus, to investigate possible changes in autophagy in ARH3 cells compared to WT, we measured the basal conversion of LC3-I to LC3-II after addition of the autophagic flux inhibitor chloroquine. Strikingly, we observed a marked decrease in basal autophagic flux, demonstrated by approximately 50% lowered LC3II/I ratios in KO cells compared to WT (Figs. 4C, S9). While this may seem to contradict the observed increase in EIF5A expression (Fig. 4A), we speculate that the latter could serve as a compensatory mechanism aiming to increase basal autophagy, which was inhibited upon ARH3 KO. We performed the same analysis on PARP1 KOs and also found decreased autophagy as compared to WT (Fig. 4C).
A fundamental pathway responsible for regulating autophagy involves the AMP activated protein kinase (AMPK), which is activated by elevated levels of AMP. Following its activation, AMPK activates autophagy through inhibition of mTOR [33]. To further elucidate the mechanism for reduced autophagy upon ARH3 and PARP1 KO, we measured AMP levels by liquid chromatography-mass spectrometry and found a 60% decrease in AMP levels in both ARH3 and PARP1 KO cells compared to WT (Table S4). Together, this suggests that KO of ARH3 or PARP1 reduces cellular AMP levels, which in turn would decrease AMPK activation (and reduce mTOR inhibition) and hence decrease autophagy.
To further elucidate this, we investigated whether pharmacological inhibition (chloroquine) or activation (mTOR inhibitor rapamycin) of autophagy would alter olaparib sensitivity of ARH3 KO, PARP1 KO, or WT cells. Notably, activation of autophagy lead to increased olaparib sensitivity in ARH3 and PARP1 KO cells but not in WT (Fig. 4D). Conversely, inhibition of autophagy lead to decreased olaparib sensitivity in ARH3 and PARP1 KO cells but not in WT (Fig. 4D).
Together, this indicates that ARH3 and PARP1 KO leads to decreased autophagy, which in turn might explain the PARPi resistant phenotype observed for PC cells with ARH3 or PARP1 KO.
Genetic and transcriptional alterations of ARH3, YWHAE, and UBR5 in prostate cancer
The clinical relevance of ARH3, UBR5, and YWHAE was evaluated using tumor genome sequencing data from cBioPortal [34] including 2 625 patients with localized PC (LPC) and 638 patients with mPC. In addition, we used in-house RNA sequencing data from two cohorts. Cohort 1 included 126 LPC, 17 mPC, and 38 non-malignant (NM) prostate tissue samples and clinical follow-up data. Cohort 2 included 41 LPC, 72 mPC, and 45 NM samples.
For ARH3, genomic alterations (amplifications, deletions, or mutations) were detected in 0.46% (12/2 625) of LPC and 1.9% (12/638) of mPC tumors in the cBioPortal data (Fig. 5A), indicating enrichment for ARH3 alterations in metastatic vs. localized PC (Fisher’s exact test, p = 0.0008). In cohort 1, ARH3 expression was downregulated in mPC vs. NM and LPC samples, respectively (Mann Whitney U Test, p = 0.04 and 0.02), but no significant differences were seen in cohort 2 (Fig. 5B). Furthermore, in cohort 1, BCR-free survival analysis showed that high expression of ARH3 was associated with significantly better outcome in multivariate cox regression (HR = 0.42; p = 0.041; Fig. 5C), although only borderline significant in univariate analysis (Table S5). These results indicate that loss/downregulation of ARH3 is associated with more aggressive PC. Based on our in vitro data these tumors might also be resistant to PARPi treatment.

A Stacked alteration frequencies for ARH3, UBR5, and YWHAE in localized prostate cancer (LPC) and metastatic prostate cancer (mPC), extracted from cBioPortal. B Log2-normalized expression of ARH3, YWHAE, and UBR5 in (left) patient cohort 1 and (right) patient cohort 2, comparing expression values between normal and adjacent-normal (NM), LPC, and mPC. Error bars show SD. Differences between groups were tested with either Mann-Whitney U test or unpaired Student’s t-tests, p-values<0.05 were considered significant, *p-value < 0.05, **p-value < 0.01, ***p-value < 0.001, ****: p-value < 0.0001. C Forest plot of Cox multivariate regression analysis, p-values < 0.05 were considered significant, *p-value < 0.05, ***p-value < 0.001.
For YWHAE, genomic alterations were found in 0.69% (18/2 625) of LPCs and in 2.19% (14/638) of mPCs in the cBioPortal data (Fisher’s exact test, p = 0.0024) (Fig. 5A). The majority of YWHAE alterations in LPC were deep deletions (89%; 16/18), whereas a larger proportion of amplifications were found in mPC (43%; 6/14; Fig. 5A). In both cohort 1 and 2, YWHAE expression was significantly upregulated in LPC and mPC, compared to NM prostate tissue samples (p < 0.05) (Fig. 5B). There were no significant associations between YWHAE expression and BCR-free survival in either univariate (Table S5) or multivariate analysis (Fig. 5C) of cohort 1.
For UBR5, genomic alterations (mainly amplifications) were present in 4.65% (122/2 625) of LPC and 24.0% (158/638) of mPC tumors (Fisher’s exact test, p < 0.0001) (Fig. 5A). In cohort 1, UBR5 expression was significantly upregulated in LPC vs. NM samples (p = 0.01), and further upregulated in mPC vs. LPC (p = 0.002) (Fig. 5B), supported by cohort 2 (NM vs. mPC, p = 0.003; Fig. 5B). Together, results indicate that UBR5 and YWHAE are overexpressed in some PCs, which might be favorable for olaparib response as our in vitro data showed that KO of UBR5 or YWHAE caused olaparib resistance in CRPC cells.
For the remaining candidates, CCND1, TEAD, USP48, or PARP1, we observed no clear association between gene expression levels in PC tissue and metastasis status in cohorts 1 and 2 (Fig. S10), nor with recurrence-free survival (Table S5). Still, genomic alterations affecting CCND1, TEAD, USP48, or PARP1 were significantly more common in metastatic (SU2C) than in localized PC (TCGA) patients, as also seen for ARH3, UBR5, and YWHAE (Fig. S11, Table S6). Genomic alterations in neither of the seven genes were mutually exclusive (Fig. S12), which at least in part may reflect their association with distinct DNA repair pathways and/or molecular redundancy in DNA repair pathways.
Discussion
This study is the first to perform an unbiased genome-wide CRISPR-Cas9 KO screen in the C4 CRPC cell line to identify genes involved in olaparib resistance. We identified and validated PARP1, ARH3, UBR5, and YWHAE as novel genes associated with olaparib resistance in CRPC.
PARP1 was the top hit in both treatment conditions in the CRISPR screen, in line with previous (non-PC related) genome-wide CRISPR studies [16, 35]. As PARP1 is the main target of olaparib, this emphasizes the capacity of our screen to identify biologically relevant olaparib resistance genes. This is the first study to demonstrate an association between PARP1 loss and PARPi resistance in CRPC.
ARH3 KO induced PARPi (olaparib, niraparib, and veliparib) resistance in androgen sensitive (C4) and insensitive (LNCaP) CRPC cells. This is consistent with a previous report [16] identifying PARG depletion as a PARPi resistance mechanism. ARH3 and PARG belong to the family of (ADP-ribosyl) hydrolases, which degrade large poly(ADP-ribose) (PAR) structures and ADPr mono- and oligomers at DNA-damaged sites regulating PARylation levels [36]. While PARG has been studied in PC [37], no PC related studies on ARH3 were identified.
Our RNA sequencing analyses of ARH3 KO CRPC cells revealed upregulation of EIF5A compared to WT cells, indicating induction of autophagy. However, further analyses showed decreased autophagy in ARH3 KO cells compared to WT cells. Although increased autophagy has been associated with drug resistance [38] it can also increase apoptotic cell death of prostate cancer cells [39]. Olaparib has previously been shown to increase autophagy in breast cancer cell lines, where inhibition of autophagy caused a significant decrease in olaparib sensitivity [40]. In line with this, our study showed that inhibition of autophagy decreased olaparib sensitivity of ARH3 KO cell lines while activation of autophagy increased their sensitivity. Furthermore we showed that ARH3 KO cells had decreased AMP levels compared to WT in agreement with a previous study showing that PARG silencing reduced AMP levels as PAR catabolism contributes to the AMP pool [41]. Based on the observed reduction of AMP levels in ARH3 KO cells, our results indicate that autophagy might be lowered due to a hampered AMPK-mTOR pathway, ultimately protecting ARH3 KO cells from olaparib induced cell death.
Similar results were observed in our analyses of PARP1 KO cells, suggesting that olaparib resistance in ARH3 and PARP1 KO are mediated – at least partly - by a similar mechanism that involves reduced autophagy. In line with this, ARH3 is known to be involved in dePARrylation, which is required for PARP1 activity [42], further supporting our findings.
Furthermore, our clinical data showed decreased ARH3 expression in mPC compared to normal and localized PC tissue samples. Thus, loss of ARH3 might also be associated with PC aggressiveness.
We also validated a correlation between KO of YWHAE or UBR5 and olaparib resistance. However, KO of YWHAE or UBR5 did not show significant cross resistance to other PARPis (veliparib/niraparib). As PARPis differ regarding PARP trapping ability and metabolization [43, 44], KO of some genes might cause olaparib resistance while not affecting sensitivity to other PARPis.
For YWHAE and UBR5, data from the cBioportal revealed amplifications as being the most common alteration in mPC (Fig. 5A). Similarly, our RNA sequencing data revealed a significant increase in YWHAE and UBR5 expression from NM to mPC (Fig. 5B, C). Thus, increased expression of these genes might be beneficial for olaparib treatment. However, further investigations are needed to fully understand the role of YWHAE and UBR5 in PC and PARPi response.
We did not validate the phenotypic effects of CCND1, TEAD1, and USP48 KO indicated by the screen data, emphasizing the importance of functional validation of CRISPR screen results through individual KO of genes.
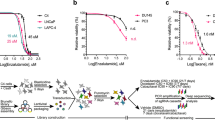
This study sheds light on novel genes contributing to PARPi resistance in CRPC and propose a novel mode of olaparib resistance through increased autophagy (Fig. 6). Further investigations are needed to clarify whether in vitro findings can be translated to clinical practice. Thus, future research should include comprehensive clinical studies to elucidate the potential of ARH3, UBR5, and YWHAE as biomarkers for PARPi treatment response in CRPC.

A Overview of results from the genome-wide loss-of-function screen, performed to identify genes causing olaparib resistance. First, the Brunello v2 library was packed into lentiviral particles and transduced into the C4 prostate cancer cell line. Next, cells were selected by PARP inhibition (olaparib) and surviving clones were identified. Four genes (ARH3, PARP1, YWHAE, and UBR5) were individually validated as causing olaparib resistance. B Proposed mechanism for PARP inhibitor resistance in ARH3 deficient prostate cancer cells, based on functional experiments. In ARH3 wildtype cells (top), PARP1 adds poly-(ADP-ribose) subunits (PARylation) to proteins. ARH3 breaks down the PARylation to PAR and ADPr, which increases AMP. Increased AMP activates AMP activated protein kinase (AMPK), resulting in inhibition of mTOR and increased autophagy. This increased autophagy sensitizes cells to PARP inhibition. In ARH3 knockout cells (bottom), PARP is inhibited by accumulation of PARylation. The lack of dePARylation results in decreased AMP, which decreases AMPK activity, increases mTOR activity, and ultimately decreases autophagy. Decreased autophagy causes cells to survive PARP inhibitor treatment. C Overview of results from clinical data analyses, which showed that patients with metastatic prostate cancer have lowered ARH3 expression compared to patients with localized prostate cancer. Furthermore, decreased expression of ARH3 is a marker of biochemical recurrence.
Materials and methods
Cell cultivation
All CRPC cell lines (C4 and LNCaP) were grown in Roswell Park Memorial Institute (RPMI)-medium (Lonza, Walkersville, MD, USA) and HEK293T cells were grown in Dulbecco’s Modified Eagle’s (DMEM)-medium (Lonza). Medium was supplemented with 10% Fetal Bovine Serum (FBS, Gibco), and 1% penicillin-streptomycin (10000 U/mL) (Gibco). Cells were cultured in incubators at 37 °C and 5% CO2 and, authenticated by short tandem repeat profiling, and tested regularly for mycoplasma infection.
CRISPR KO screen
CRISPR-Cas9 KO screen
The Human Brunello v2 CRISPR KO pooled library (76 441 gRNAs, targeting 19 114 genes, including 1 000 controls) was a generous gift from David Root and John Doench (Addgene, Watertown, MA, USA, Addgene ID: 73178) [17].
Viral particles were produced as described by Ryø et al. [45] (plasmids listed in Supplementary Methods). SpCas9 was introduced into C4 cells using the lentiCas9-Blast vector. In duplicates, Cas9-expressing C4 cells were screened using Brunello v2 in lentiGuide-Puro vectors at an MOI of 0.5 (based on a viral titer experiment; Table S7). Transduced cells were selected by nine days of puromycin (3 µg/mL). Following, a minimum of 40 million cells from each replicate were harvested to establish a baseline group. Of the remaining cells, cells were treated with either 18.5 µM olaparib (IC50), 29 µM olaparib (IC90), or DMSO (vehicle). During the screen, cells were passaged at 80–90% confluency, while maintaining a minimum coverage of 500 cells per gRNA. Medium was replaced every third day. After 21 days, cells were collected and extracted for genomic DNA by salting out. By PCR, gRNA regions were amplified using staggered primers (Table S8). Adaptors for Illumina Sequencing and indices were applied using the KAPA Hyper Prep Kit (KAPABiosystems, Wilmington, MA, USA). Products were purified on SPRI beads (Agencourt AMPure XP, Beckman Coulter, Brea, Ca, USA) and sequenced on Illumina NexSeq500 with a coverage of 500 cells per gRNA and aiming at a read depth of 40 million reads per sample.
Enriched genes were identified using MAGeCK [18]. Restrictive criteria were imposed on the MAGeCK output for candidate gene selection. From the top 500 enriched genes in each replicate, genes with a fold change of above 1.25 (compared to vehicle), and at least two significantly enriched gRNAs per gene were identified.
Generation of KO cell lines
Cloning and transformation
Vectors were generated to contain gRNAs targeting ARH3, CCND1, PARP1, TEAD4, UBR5, USP48, or YWHAE, or a non-targeting gRNA (Table S2). Complementary gRNA sequences were phosphorylated, annealed, and cloned into the PX458 backbone vector [26]. Constructs were amplified using XL2-Blue Ultracompetent E.coli cells (Stratagene; 100 µg/mL ampicillin) and purified via the NucleoBond Xtra Midi purification kit (Macherey-Nagel, Düren, Germany). Correct gRNA sequence inserts were confirmed by Sanger Sequencing (Table S9).
C4-Cas9 and LNCaP-Cas9 cells were transfected with gRNA plasmids individually, using FuGENE HD Transfection Reagent (Promega, Madison, WI, USA). GFP positive cells were FACS sorted on the BD FACSAriaTM III Cell Sorter (FACS Core Facility, Aarhus University) either into populations or single cell suspension to establish KO clones. Genomic KO of individual genes was validated by Sanger Sequencing (primers in Table S10) and TIDE analysis [27].
Cell viability assays
Olaparib (AZD2281; KU0059436), veliparib (ABT-888), niraparib (MK-4827), and carboplatin (NSC 241240) were purchased from MedChemExpress (Shanghai, China) and reconstituted in DMSO (Sigma-Aldrich, St. Louis, MO, USA).
Cells were plated in 96-well plates at a density of 6 000 cells per well. After 24 h, cells received drug-supplemented medium at 10-point drug concentrations for each drug (Table S11) or DMSO and were cultured for six days. Cell viability was evaluated by 4 h incubation with 10% AlamarBlueTM Cell Viability Reagent (Invitrogen) followed by 530 nm/620 nm absorbance measurement (Synergy HT plate reader; BioTek).
Statistical analysis
Statistical analyses were performed using GraphPad Prism, v8.4.2. Experiments were repeated three times with technical triplicates. Viability of drug-treated cells was normalized to DMSO-treated cells. Dose-response curves were fitted using non-linear regression. Differences in viability between KO and WT cells were analyzed by pairwise t-test. A p-value < 0.05 was considered statistically significant.
References
Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30.
Cornford P, van den Bergh RCN, Briers E, Van den Broeck T, Cumberbatch MG, De Santis M, et al. EAU-EANM-ESTRO-ESUR-SIOG Guidelines on Prostate Cancer. Part II—2020 Update: Treatment of Relapsing and Metastatic Prostate Cancer. Eur Urol. 2021;79:263–82.
Kirby M, Hirst C, Crawford ED. Characterising the castration-resistant prostate cancer population: a systematic review. Int J Clin Pr. 2011;65:1180–92.
Robinson D, Van Allen EM, Wu YM, Schultz N, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–28.
Abida W, Armenia J, Gopalan A, Brennan R, Walsh M, Barron D, et al. Prospective Genomic Profiling of Prostate Cancer Across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis Oncol. 2017;1:1–16.
Tukachinsky H, Madison RW, Chung JH, Gjoerup O, Severson EA, Dennis L, et al. Genomic analysis of circulating tumor DNA in 3,334 patients with advanced prostate cancer identifies targetable BRCA alterations and AR resistance mechanisms. Clin Cancer Res. 2021;27:3094–105.
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer Res. 2012;72:5588–99.
Hopkins TA, Shi Y, Rodriguez LE, Solomon LR, Donawho CK, DiGiammarino EL, et al. Mechanistic Dissection of PARP1 Trapping and the Impact on In Vivo Tolerability and Efficacy of PARP Inhibitors. Mol Cancer Res. 2015;13:1465–77.
FDA. FDA approves olaparib for HRR gene-mutated metastatic castration-resistant prostate cancer FDA website: FDA website; 2021 [Available from: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-hrr-gene-mutated-metastatic-castration-resistant-prostate-cancer.
de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;382:2091–102.
Hussain M, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N Engl J Med. 2020;383:2345–57.
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, et al. Analysis of Circulating Cell-Free DNA Identifies Multiclonal Heterogeneity of BRCA2 Reversion Mutations Associated with Resistance to PARP Inhibitors. Cancer Disco. 2017;7:999–1005.
Goodall J, Mateo J, Yuan W, Mossop H, Porta N, Miranda S, et al. Circulating Cell-Free DNA to Guide Prostate Cancer Treatment with PARP Inhibition. Cancer Disco. 2017;7:1006–17.
Chen X, Chen Z, Zheng B, Tang W. Targeting NPRL2 to enhance the efficacy of Olaparib in castration-resistant prostate cancer. Biochem Biophys Res Commun. 2019;508:620–5.
Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nature. Communications. 2018;9:1849.
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective Loss of PARG Restores PARylation and Counteracts PARP Inhibitor-Mediated Synthetic Lethality. Cancer Cell. 2018;33:1078–93.
Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol. 2016;34:184.
Li W, Xu H, Xiao T, Cong L, Love MI, Zhang F, et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knocout screens. Genome Biol. 2014;15:554–66.
Kolde R, Laur S, Adler P, Vilo J. Robust rank aggregation for gene list integration and meta-analysis. Bioinformatics. 2012;28:573–80.
Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13:411–24.
Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, et al. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature 2011;474:230–4.
Hazan I, Monin J, Bouwman BAM, Crosetto N, Aqeilan RI. Activation of Oncogenic Super-Enhancers Is Coupled with DNA Repair by RAD51. Cell Rep. 2019;29:560–72.e4.
Li CG, Mahon C, Sweeney NM, Verschueren E, Kantamani V, Li D, et al. PPARγ Interaction with UBR5/ATMIN Promotes DNA Repair to Maintain Endothelial Homeostasis. Cell Rep. 2019;26:1333–43.
Pennington KL, Chan TY, Torres MP, Andersen JL. The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene. 2018;37:5587–604.
Velimezi G, Robinson-Garcia L, Muñoz-Martínez F, Wiegant WW, Ferreira da Silva J, Owusu M, et al. Map of synthetic rescue interactions for the Fanconi anemia DNA repair pathway identifies USP48. Nat Commun. 2018;9:2280.
Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–308.
Steensel V TIDE: Tracking of Indels by DEcomposition [cited 2020 23/4]. Available from: https://tide.deskgen.com/.
Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40.
Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucl Acids Res. 2016;44(W1):W90–7.
Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinforma. 2013;14:128.
Rath S, Sharma R, Gupta R, Ast T, Chan C, Durham TJ, et al. MitoCarta3.0: an updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucl Acids Res. 2020;49(D1):D1541–7.
Lee JS, Lee H, Jang H, Woo SM, Park JB, Lee SH, et al. Targeting Oxidative Phosphorylation Reverses Drug Resistance in Cancer Cells by Blocking Autophagy Recycling. Cells. 2020;9:2013.
Eliopoulos AG, Havaki S, Gorgoulis VG. DNA Damage Response and Autophagy: A Meaningful Partnership. Front Genet. 2016;7:204.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Disco. 2012;2:401–4.
He YJ, Meghani K, Caron MC, Yang C, Ronato DA, Bian J, et al. DYNLL1 binds to MRE11 to limit DNA end resection in BRCA1-deficient cells. Nature. 2018;563:522–6.
Niere M, Kernstock S, Koch-Nolte F, Ziegler M. Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix. Mol Cell Biol. 2008;28:814–24.
Zhang M, Lai Y, Vasquez JL, James DI, Smith KM, Waddell ID, et al. Androgen Receptor and Poly(ADP-ribose) Glycohydrolase Inhibition Increases Efficiency of Androgen Ablation in Prostate Cancer Cells. Sci Rep. 2020;10:3836.
Cahuzac M, Langlois P, Péant B, Fleury H, Mes-Masson A-M, Saad F. Pre-activation of autophagy impacts response to olaparib in prostate cancer cells. Commun Biol. 2022;5:251.
Tai S, Sun Y, Liu N, Ding B, Hsia E, Bhuta S, et al. Combination of Rad001 (everolimus) and propachlor synergistically induces apoptosis through enhanced autophagy in prostate cancer cells. Mol Cancer Ther. 2012;11:1320–31.
Arun B, Akar U, Gutierrez-Barrera AM, Hortobagyi GN, Ozpolat B. The PARP inhibitor AZD2281 (Olaparib) induces autophagy/mitophagy in BRCA1 and BRCA2 mutant breast cancer cells. Int J Oncol. 2015;47:262–8.
Formentini L, Macchiarulo A, Cipriani G, Camaioni E, Rapizzi E, Pellicciari R, et al. Poly(ADP-ribose) Catabolism Triggers AMP-dependent Mitochondrial Energy Failure*. J Biol Chem. 2009;284:17668–76.
Virág L, Szabó C. The Therapeutic Potential of Poly(ADP-Ribose) Polymerase Inhibitors. Pharmacol Rev. 2002;54:375-429.
Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355:1152–8.
Valabrega G, Scotto G, Tuninetti V, Pani A, Scaglione F. Differences in PARP Inhibitors for the Treatment of Ovarian Cancer: Mechanisms of Action, Pharmacology, Safety, and Efficacy. Int J Mol Sci. 2021;22:4203.
Ryø LB, Thomsen EA, Mikkelsen JG. Production and Validation of Lentiviral Vectors for CRISPR/Cas9 Delivery. Methods Mol Biol. 2019;1961:93–109.
Acknowledgements
This work was supported by grants from The Novo Nordisk Foundation (KDS), Aase & Ejnar Danielsens Fond (MBI), Fabrikant Einar Willumsens Mindelegat (MBI), Tømrermester Jørgen Holm & Hustru Elisa F. Hansens Mindelegat (MBI), Direktør Emil C. Hertz og Hustru Inge Hertz Fond (MBI), Graduate School of Health (Aarhus University; MBI), Helge Peetz & Verner Peetz & Hustru Vilma Peetz Fond (MBI), Beckett Fonden (MBI), and NEYE Fonden (MBI). The Danish Cancer Biobank is acknowledged for providing patient samples and information on handling and storage.
Author information
Authors and Affiliations
Contributions
KDS, MBI, EMGS, JGM, and JH designed the study. MBI and EAT conducted the CRISPR-Cas9 screen. MBI and EMGS produced single gene knockout cell lines and performed all cell line experiments. MBI performed RNA-sequencing analyses for cell lines. AD performed western blots of LC3II/I. JP performed LC-MS experiments. PB supervised Seahorse assays. KDS, JF, MR, SK, MRJ, KB, BPU, and MB were responsible for collection and sequencing of patient samples. MBI and EMGS analyzed patient RNA-sequencing data. MBI, EMGS, SW, PB, JP, and KDS interpreted data. MBI, EMGS and KDS wrote the manuscript. KDS, SW, and JGM supervised the study. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ipsen, M.B., Sørensen, E.M.G., Thomsen, E.A. et al. A genome-wide CRISPR-Cas9 knockout screen identifies novel PARP inhibitor resistance genes in prostate cancer. Oncogene 41, 4271–4281 (2022). https://doi.org/10.1038/s41388-022-02427-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02427-2
- Springer Nature Limited
This article is cited by
-
Fit-Seq2.0: An Improved Software for High-Throughput Fitness Measurements Using Pooled Competition Assays
Journal of Molecular Evolution (2023)