
Abstract
Metastatic outgrowth is supported by metabolic adaptations that may differ from the primary tumor of origin. However, it is unknown if such adaptations are therapeutically actionable. Here we report a novel aminopyridine compound that targets a unique Phosphogluconate Dehydrogenase (PGD)-dependent metabolic adaptation in distant metastases from pancreatic cancer patients. Compared to structurally similar analogs, 6-aminopicolamine (6AP) potently and selectively reversed PGD-dependent metastatic properties, including intrinsic tumorigenic capacity, excess glucose consumption, and global histone hyperacetylation. 6AP acted as a water-soluble prodrug that was converted into intracellular bioactive metabolites that inhibited PGD in vitro, and 6AP monotherapy demonstrated anti-metastatic efficacy with minimal toxicity in vivo. Collectively, these studies identify 6AP and possibly other 6-aminopyridines as well-tolerated prodrugs with selectivity for metastatic pancreatic cancers. If unique metabolic adaptations are a common feature of metastatic or otherwise aggressive human malignancies, then such dependencies could provide a largely untapped pool of druggable targets for patients with advanced cancers.
Similar content being viewed by others

Introduction
Distant metastasis is a multi-step cascade that requires malignant cells to escape the primary site of origin, disseminate in the circulation, seed foreign soils of other organs, and achieve successful metastatic outgrowth [1]. Although metastasis is the most common cause of cancer deaths, it remains one of the least understood aspects of cancer biology [2]. In patients, distant metastasis presents as either oligometastatic disease (limited tumor burden) or widely metastatic disease (heavy tumor burden) [3]. The former is often treatable and sometimes curable, whereas the latter is universally lethal.
A striking example is pancreatic ductal adenocarcinoma (PDAC) [4,5,6]. Most PDAC patients develop widely metastatic disease, which presents suddenly and progresses rapidly to diffusely fill the liver and/or lungs with hundreds to thousands of treatment-resistant metastatic tumors [7, 8]. A smaller subset of patients may also develop peritoneal carcinomatosis [7, 8], which refers to intra-abdominal metastatic implants seeded directly off the primary pancreatic tumor with secretion of malignant ascites fluid. Surprisingly, driver mutations and consequential copy number changes are shared between primary tumor and metastasis in treatment naïve patients [8,9,10], including those required for metastasis itself [11].
Barriers exist at each step of the metastatic cascade that renders the overall process inefficient [12, 13]. One barrier is the metabolic demands of metastasis. For example, metastatic dissemination within the circulation requires metabolism of reactive oxygen species [14] while metastatic outgrowth within other organs requires metabolism of nutrients that may differ substantially from those available in the primary tumor [15,16,17,18]. In the setting of shared genetic drivers, new metabolic adaptations that uniquely support metastasis may be required. If so, such adaptations would present metastasis-intrinsic therapeutic targets [19].
We recently described a nutrient-dependent metabolic adaptation that was intrinsic to PDAC distant metastasis [17, 18, 20, 21]. Well-characterized, sequence-verified clonal PDACs collected during rapid autopsies of patients with widely metastatic disease [7, 8, 22] consumed excess glucose [17], a nutrient that is depleted in stroma-rich primary PDACs [16, 17]. The excess glucose fueled abnormally high PGD catalytic activity (PGDhigh) [17, 18], which converts glucose-derived substrates [18] into pro-tumorigenic biosynthetic metabolites [20]. Because PGDhigh reciprocally stimulated glucose consumption [17], this metabolic adaptation was self-maintained through positive feedback rather than genetic mutations [9, 17, 18, 20]. The excess glucose also fueled hyperacetylation of PDAC chromatin [17] into an epigenetic state that was permissive for activation of the metastatic transcriptome [20, 23]. Thus, the rapid autopsy metastatic (sub)clones are strongly dependent on this adaptation. We therefore leveraged these unique patient samples to formally test the hypothesis that a metastasis-intrinsic metabolic adaptation (PGDhigh) can be pharmacologically targeted. This could be especially relevant for PDAC patients, since most (~70–80%) present with widespread distant metastasis [7, 8] that may favor acquisition of PGDhigh [17, 20]
Results and discussion
6-aminonicotinamide (6AN, Fig. 1A) [24] is a derivative of pyridine-3-carboxamide (nicotinamide) that is metabolized into NADP byproducts that inhibit NADP oxidoreductases, including PGD [25]. Consistent with this, we previously reported that 6AN was active against PGDhigh PDACs [17, 18, 20]. However, high concentrations were required in culture, off-target activities including non-selective inhibition of glucose-6-phosphate dehydrogenase (G6PD) are well-known [25, 26], and severe neurotoxic side-effects preclude animal experiments [24, 27]. Thus, 6AN lacks potency and selectivity in vitro and cannot be used as a therapeutic agent in vivo. We hypothesized that 6AN might instead be useful as a lead compound to identify alternative small molecules that selectively target PGDhigh.

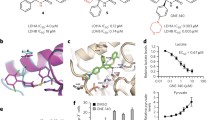
A 6AN lead compound with numbered pyridine ring and functional groups as indicated. B X-ray structure (PDB: 2JKV) showing the nicotinamide group of NADP docked within the PGD active site. Note the water molecule (red dot) situated between PGD amino acids and the pyridine C6. C Glide docking simulation showing 6AN-riboside docked within PGD. Note that the C6-amino group replaces the water and models hydrogen bonds with the same PGD amino acids. D Clonal cell lines from a test pilot PGDhigh distant metastasis (left) and a PGDlow peritoneal implant (right) from the same patient were plated into 3D suspensions and treated with the indicated analogs. Those with substitutions away from the 6-amino group selectively blocked tumoroid growth in PGDhigh. Those with substitutions of the C6-amino group or nearby atoms did not. DMSO and 6AN control treatments are the top two rows, respectively. Scale bars: 400 µm.
Comparative glide docking simulations examining the X-ray structure of NADP (Fig. 1B) or 6AN-riboside (Fig. 1C) bound within the PGD active site predicted that the amino group attached to C6 (of 6AN-riboside) was free of steric hinderance and could hypothetically displace a water molecule to form hydrogen bonds with the backbone amides of Ans103, Gly101, and Val128 of PGD. The N1-riboside exit point appeared similar in both structures, but this area was more sterically restricted due to crowding with the ribose sugar (Fig. 1B, C). In contrast, the area surrounding the C3-carboxamide group was not crowded and the group itself appeared sterically flexible in both structures (Fig. 1B, C). Because the in silico simulations predicted that the presence of a C6-amino group enhances docking stability while the C3-carboxamide group is flexible to modification, we hypothesized that aminopyridine analogs with a retained C6-amino group combined with C3-carboxamide substitutions might provide a class of 6AN-like compounds that selectively target PGD-dependent PDACs.
Cell-based assays were employed to screen a panel of pyridine analogs designed to test the docking predictions. Genetic inactivation of PGD does not significantly impair proliferative growth or viability of PGDhigh PDACs in 2D cultures [18, 20], which precludes use of traditional 2D plate screening formats. Rather, PGD inactivation strongly impairs tumoroid outgrowth of PGDhigh PDACs in 3D cultures. 3D assays were therefore selected as the screening format. Because this initial screen was designed for selectivity (rather than potency), high exposures (250 µM for 21 days) were used to identify analogs that selectively target PGDhigh without nonspecific effects on PGDlow control cells. As predicted by the glide docking simulations, analogs with a C6-amino group combined with C3-carboxamide substitutions strongly blocked 3D outgrowth of 6AN-sensitive PGDhigh test cells, which we observed in both suspension (Fig. 1D) and solid (Fig. S1A) 3D formats. As also predicted by the docking simulations, substitutions affecting the C6-amino group rendered the PGDhigh test cells resistant (Figs. 1D and S1A). Substituting nitrogen for carbon at C2 or C5 on the pyridine ring yielded similar results (Fig. 1D). In contrast, a PGDlow peritoneal implant isolated from the same rapid autopsy patient was resistant to all analogs tested, with the lone exception of a C4-fluorinated analog which could reflect nonspecific toxicity (Fig. 1D). Collectively, these experiments suggested that 6-aminopyridines represent a class of compounds with selectivity for PGDhigh PDACs.
We next investigated if any of the selective analogs possessed superior potency. Comparative dose response treatments down to 1 µM identified a previously unstudied compound, 5-(aminomethyl)pyridin-2-amine (or 6-aminopicolamine: 6AP), as the most potent inhibitor of 3D tumoroid outgrowth (Figs. 2A, S1B). This activity reflected a strong cytostatic effect, as viable cell numbers remained nearly constant throughout the entire low dose 6AP treatment period (Fig. S2A, B). As expected, 6AP was more potent than 6AN, which we replicated in both suspension (Fig. 2A) and solid (Fig. 2B) 3D formats. Despite the higher potency against PGDhigh, 6AP did not possess the same off-target activity against immortalized PGDlow human pancreatic ductal epithelial (HPDE) cells as 6AN (Fig. 2C), and a panel of PGDlow PDACs of diverse origin (primary tumor, peritoneal implant, peritoneal ascites fluid) [17, 20] were either completely insensitive to 6AP or only modestly affected at high concentrations (Fig. 2D). 6AP dose response treatments further demonstrated activity down to submicromolar (high nanomolar) concentrations against the PGDhigh pilot cells (EC50: 250 nM, Figs. 2E and S2C), and a 5 µM ceiling dose retained anti-tumorigenic activity across a larger panel of well-characterized PGDhigh PDACs [8, 17, 18, 20] (Figs. 2F and S2D).

A The PGDhigh pilot metastasis were plated into 3D suspensions and treated with 1 µM of the indicated analogs. Only 6AP impaired tumoroid growth. B PGDhigh cells were plated into solid 3D discs and treated with 1 µM of 6AN or 6AP. Only 6AP impaired tumoroid outgrowth at this concentration. C PGDlow non-malignant HPDE cells were plated into solid 3D discs and treated with 1 µM 6AN or 6AP as indicated. 6AP did not demonstrate the off-target effects of 6AN. D PGDlow PDACs from non-distant sites (as indicated) were plated into solid 3D discs and treated with 1 µM or 50 µM 6AP. 6AP demonstrated either no effect or modest effects on tumoroid outgrowth, implying selectivity for PGDhigh. E The PGDhigh pilot metastasis was plated into solid 3D discs and treated with increasing concentrations of 6AP as indicated to determine the dose-response (approximate EC50: 0.25 µM). F A panel of the indicated PGDhigh PDACs were plated into 3D suspensions and treated with 5 µM 6AP. 6AP impaired tumoroid growth of all the tested samples. Note: For all quantified 3D assays, tumoroid growth is calculated and plotted by the mean size (surface area in mm2) of twelve individual tumoroids representing 3–4 technical replicate experiments (error bars: s.d.m., p values calculated by two-tailed t tests). Scale bars: 400 µm for all panels.
We postulated that if 6AP is a potent inhibitor of PGDhigh, then it should reverse other PGD-dependent traits. The PGDhigh adaptation allows malignant cells to consume abnormally high amounts of glucose, which facilitates global hyperacetylation PDAC chromatin (Fig. S3A) [17]. We therefore tested if 6AP could reverse these traits with high potency, since both are straightforward to quantify. As expected [17], treating PGDhigh cells with high concentrations (25 µM) of 6AN slowed glucose consumption rates down to levels comparable with PGDlow cells from the same patient, whereas lower concentrations (5 µM) did not (Fig. S3B). In contrast, 6AP lowered glucose consumption rates with equal efficacy at both concentrations (Fig. S3C). Confocal immunofluorescence experiments further demonstrated that 5 µM 6AP (but not 5 µM 6AN) reversed global histone hyperacetylation (H3 lysine27 and H4 lysine16), which we replicated in both 2D (Fig. S3D) and 3D formats (Fig. S3E). Thus, 6AP potently and selectively reversed PGD-dependent malignant properties, including intrinsic tumorigenic capacity, high glucose consumption, and histone hyperacetylation.
Interestingly, 6AP itself failed to directly inhibit recombinant PGD in vitro (Fig. 3A). Because 6AN incorporates into NADP to produce 6ANADP [25], we hypothesized that 6AP was a prodrug that must be metabolized into similar metabolites (for example, 6APADP) with inhibitory bioactivity against PGD. We therefore devised an experimental strategy to recover intracellular metabolite pools containing 6APADP (Fig. 3B), which would allow us to formally test whether 6AP-derived metabolites could directly inhibit recombinant PGD. Liquid chromatography followed by mass spectrometry (LC-MS) on metabolites extracted from PGDhigh cells first confirmed that metabolites with mass-to-charge ratios (m/z) corresponding to 6APADP were enriched in 6AP-treated cells and absent from DMSO-treated controls (Fig. 3C). High performance liquid chromatography (HPLC) separation of the metabolite extracts further revealed a uniquely migrating peak in 6AP-treated cells that was also absent from DMSO controls and migrated differently than the known 6ANADP-containing peak [25] from 6AN-treated cells (Fig. 3D). To further remove non-nucleotide impurities, this unique peak was subjected to a second HPLC which produced two closely migrating 6APADP-containing fractions with partially overlapping retention times of 26 and 28 m (Fig. 3E). Both fractions were therefore subjected to downstream bioactivity testing against recombinant PGD.

A Recombinant PGD catalytic rates were measured in the presence of DMSO, 5 µM 6AP, or 10 µM 6AP as indicated. Consistent with a prodrug, 6AP failed to directly inhibit recombinant PGD activity (n = 3 technical replicates, error bars: s.d.m., p values calculated by two-tailed t tests). B HPLC-based strategy to recover intracellular metabolite pools containing bioactive 6APADP. The LC-MS mass-to-charge ratio (m/z) in positive ion mode for 6APADP is indicated at the bottom. C Extracted ion chromatograms (m/z: 745.1107–745.1181) of metabolites from PGDhigh cells treated with either DMSO (top) or 6AP (bottom). Closely migrating signals within this range (corresponding to 6APADP) were enriched in 6AP-treated cells. D Representative time resolved HPLC separation of metabolites extracted from PGDhigh cells treated as indicated (λ: 260 nm). 6AP shows a unique peak (arrow) that is not present in DMSO and distinct from 6AN (arrow). LC-MS confirmed this unique peak contained 6APADP (not shown). E The unique 6APADP peak was further separated into two major peaks (26 and 28 m, λ: 256 nm) that both contained 6APADP (LC-MS, not shown). F Recombinant PGD was incubated with increasing volumes of the 6APADP-containing metabolite fractions as indicated, producing a dose-response inhibition and approximate volumetric IC50 values (n = 3 biological replicates, error bars: s.e.m.). G Recombinant PGD (left) and G6PD (right) were incubated with the IC50 volumes of the indicated 6APADP fractions. Both fractions selectively inhibited PGD but not G6PD (n = 3 biological replicates, error bars: s.e.m., p values calculated by two-tailed t tests).
The unavoidable partial degradation of temperature sensitive 6APADP compounds during the complex recovery procedure limited yields and concentration estimates of full-length 6APADP in the collected peak fractions. Because of this, input volumes used for recombinant enzyme activity assays were determined empirically. Remarkably, identical input volumes from both peak fractions inhibited recombinant PGD in a linear, dose-responsive range with comparable volumetric IC50 estimates (Fig. 3F). Importantly, inhibition was selective for PGD as neither fraction inhibited G6PD (Fig. 3G). Although we cannot exclude the possibility that other metabolites might have contributed to results (including 6APADP breakdown products), these labor-intensive experiments confirmed that the PGDhigh cells manufacture 6AP-derivative metabolite pools that inhibit PGD, as expected for a prodrug. This also implies plausible mechanisms for how 6AP might discriminate PGDhigh from PGDlow PDACs, including inhibition of abnormally high PGD catalytic rates and/or preferential incorporation into differentially activated NAD(P) biosynthetic pathways.
We next investigated if 6AP monotherapy possessed selective anti-metastatic efficacy against PGDhigh in animal models of metastasis. To first determine if 6AP is safe to administer, we compared it against 6AN which causes debilitating peripheral neuropathy with acute weight loss [24, 27]. Unlike 6AN, mice treated with 6AP showed little to no clinical symptoms of acute neuropathy (hind limb spasticity, Fig. S4A) and weight loss was accordingly minimal (Fig. S4B). This prompted us to simulate the dosing requirements of a longer-term therapeutic efficacy study (saline vehicle verses 20 mg/kg 6AP administered every four days for twenty-one weeks). This dosing scheme was very well-tolerated, as mice remained clinically asymptomatic with similar weights throughout the entire course of the trial (Fig. S4C, D). Anti-metastatic efficacy trials were therefore conducted using this treatment schedule, which may be conservative given the low dose and spaced dosing frequency.
Because the liver is the most common site of PDAC distant metastasis [7], efficacy trials were conducted in mice using a well-established model of experimental hepatic metastasis (splenic implantation) [17, 28]. Two widely used strains of immunodeficient mice (Nu/J and NSG) were employed because they are differentially permissive for metastatic outgrowth, they facilitate investigation of cell-autonomous sensitivities since confounding tumor-immune interactions are minimized, and because they can host human cells. A PGDhigh primary tumor subclone that seeded distant metastasis [8, 20] in a treatment naive patient from the rapid autopsy cohort was chosen for experiments. Unlike the other rapid autopsy samples collected from fully established metastatic tumors, this subclone (13Pr) retains a fuller spectrum of the metastatic cascade which allows 13Pr cells to efficiently seed hepatic metastasis in immunodeficient mice [17]. As might be expected, 13Pr is also closely related to distant metastases from the same patient based on phylogenetic relationships [8], epigenomic state [20], and metabolic dependencies [17, 18, 20] including PGDhigh. Thus, 13Pr was ideal to test if 6AP monotherapy could target PGD-dependent metastasis in vivo.
In the less permissive Nu/J strain, metastatic outgrowth of 13Pr cells is stochastic and manifests as low tumor burden after a latency of at least 12–20 weeks. This model therefore allowed us to test if 6AP qualitatively influenced the probability of developing metastatic outgrowth in the liver. As expected, saline-treated control mice developed hepatic metastasis in a stochastic manner (4/8 mice, Fig. 4A top panels). 6AP-treated mice were less prone to develop metastasis (1/8 mice, Fig. 4A top panels), and metastatic tumor that did develop was highly necrotic relative to controls (Fig. 4A bottom panels). Thus, although 6AP did not prevent stochastic outgrowth with 100% efficacy, it did lower the overall frequency (saline: 50%, 6AP: 12.5%, Fig. 4A top graph) with impaired PDAC viability when metastasis did occur (saline: 72%, 6AP: 30%, Fig. 4A bottom graph).

A Top panels: Fewer 6AP-treated Nu/J mice developed PGDhigh (13Pr cells) hepatic metastasis than saline controls after 21 weeks of treatment, as shown in the gross images (scale bars: 1 cm) and the plots quantifying percent of mice that developed metastasis (n = 8 mice per group, two-tailed p-values are calculated from the chi-square analysis of observed metastases versus the 50% expected by chance in this stochastic model). Bottom panels: Metastasis that did develop during 6AP treatments was largely necrotic, as shown by representative H&E stains (circled, scale bars: 400 µm) and plots quantifying percent viable tumor, stroma, and necrosis (n = 4 saline mice, n = 1 6AP mouse). B Only a subset of 6AP-treated NSG mice developed grossly diffuse PGDhigh (13Pr cells) hepatic metastasis as shown in the gross images (scale bars: 1 cm). Individual 6AP-treated mice also developed less bulky intrahepatic metastasis with more tumoral necrosis than saline-treated mice as shown by representative H&E stains (top panels: metastases circled, scale bars: 1 mm; bottom panels: necrosis circled, scale bars: 400 µm) and plots quantifying percent of hepatic parenchyma involved by metastasis and percent of necrotic tumor (n = 6 mice per group, error bars: s.e.m., p values calculated by two tailed t tests). C 6AP did not impair pancreatic tumor growth of PGDlow 38Per cells orthotopically implanted into NSG mice as shown in the gross images (scale bars: 1 cm), representative H&E images (top panel scale bars: 1 mm, bottom panel scale bars 400 µm, T: tumor, P: pancreas), and the plots quantifying gross tumor size measurements (top plot, n = 6 mice per group, error bars: s.d.m., p values calculated by two-tailed t tests) and histologic percent tumor necrosis (bottom plots, n = 6 mice per group, error bars: s.e.m., p values calculated by two-tailed t tests).
Most PDAC patients develop widely metastatic disease that diffusely involves the liver [7]. In the more permissive NSG strain, 13Pr metastatic outgrowth consistently manifests as diffuse hepatic involvement. This model therefore allowed us to test if 6AP might quantitatively influence the kinetics of (widely) metastatic outgrowth. As expected, all saline-treated mice implanted with 13Pr developed diffuse metastasis in the liver (Fig. 4B top panels). 6AP-treated mice implanted with 13Pr also developed hepatic metastasis (Fig. 4B top panels), which was not unexpected given the highly permissive strain of the host. However, unlike saline-treated mice (grossly diffuse disease in 6/6 mice), grossly evident metastatic burden was more variable in 6AP-treated mice (grossly diffuse disease in 2/6 mice). We therefore conducted comprehensive histopathology measurements to precisely quantify the extent of metastatic outgrowth. The microscopic analysis demonstrated an overall 55% mean reduction in metastatic tumor burden (saline: 66%, 6AP: 32%, Fig. 4B top plot). Although tumor necrosis was modest in all NSG animals, a significant increase was nevertheless present in 6AP-treated mice implanted with PGDhigh (saline: 3.7%, 6AP: 8.7%, Fig. 4B bottom plot). Thus, while not qualitatively preventative, 6AP did quantitatively slow PGDhigh metastatic outgrowth by 50% in the NSG background (assuming a linear doubling time, saline: 8% mean outgrowth per week, 6AP: 4% mean outgrowth per week, p = 0.0008 by two-sided t tests).
Because 13Pr is strongly hepatotropic [17], control experiments using PGDlow PDACs were also conducted to evaluate if the in vivo effects of 6AP against PGDhigh metastatic outgrowth were selective. To that end, PGDlow cells isolated from a metastatic peritoneal implant and a primary tumor (of a different patient) were orthotopically implanted into the pancreas of NSG mice to test the effects of 6AP against PGDlow primary tumor growth. In contrast to the sensitivity of PGDhigh hepatic metastasis (Fig. 4A, B), both models of PGDlow primary tumor growth were resistant to 6AP (Figs. 4C and S5A). The frequency of stochastic peritoneal spread of the PGDlow peritoneal implant also appeared resistant to 6AP (saline: 50%, 6AP: 50%, Fig. S5B). As expected [17], no mice orthotopically implanted with PGDlow cells developed grossly evident hepatic metastasis (Fig. S5A, C). Thus, 6AP was selectively active against PGDhigh PDACs in experimental 3D formats in vitro (Figs. 1 and 2) and experimental mouse models in vivo (Figs. 4 and S5).
Here we provide proof-of-principle evidence that a metastasis-intrinsic metabolic adaptation can present druggable therapeutic target(s). Our previous studies detailed one such adaptation that is heavily dependent on PGDhigh. Based on the results presented here, we propose that 6AP and possibly other 6-aminopyridines can be further developed into possible therapies for patients with metastatic pancreatic cancer. To that end, crystal structures of 6APADP bound within PGD could clarify selective inhibitory mechanism(s) and guide design of second generation 6-aminopyridine analogs with superior properties.
There are nuances regarding the use of 6-aminopyridines that require further investigation beyond the scope of this study. First, 6-aminopyridine prodrugs must enter the NAD(P) salvage pathway for bioactive transformation. Although human PDACs reportedly utilize this pathway for NADP biosynthesis [29], subsets that do not would possess intrinsic resistance and the pathway itself offers potential routes to acquired resistance. Synthesis or purification of 6-aminopyridine riboside, mononucleotide, or dinucleotide formulations could yield direct PGD inhibitors (Fig. 3) that bypass prodrug metabolism. Second, despite high catalytic activity of the encoded protein, the PGD gene itself is not mutated or recurrently over-expressed in PGDhigh PDACs [20]. Thus, neither sequence nor expression can serve as biomarkers of PGDhigh status. However, PGDhigh strongly correlates with widely metastatic PDAC [8, 20], high glucose consumption rates [17], global histone hyperacetylation [17, 20], reduced TXNIP expression [17], and a distinctive stromal fibrosis pattern [17]. If properly developed as biomarkers, such surrogates could predict responses to 6-aminopyridines. Third, this study focused on cellular properties intrinsic to low passage PDACs collected from patients. Genetically engineered mouse models [30, 31] could inform whether 6-aminopyridines influence the PDAC immune microenvironment [32]. Finally, although 6-aminopyridine prodrugs may share similar metabolic fates [33], comprehensive drug metabolism and pharmacokinetics studies are required to formally document bioactive half-lives and tissue distributions, oral bioavailability and water solubilities may vary, and minimal tolerated dose(s) must be determined empirically.
Irrespective of compound class, our data indicate that PGD inhibitors may benefit pancreatic cancer patients with PGD-dependent metastasis. We emphasize that the current study was strictly designed to evaluate if metastatic outgrowth could be prevented or slowed by PGD inhibitor monotherapies. Combining PGD inhibitors with standard chemotherapy [34], targeted therapy against genetic drivers [35], immunotherapy [32, 36], or other forms of metabolic therapy [37,38,39,40] could significantly enhance therapeutic efficacy. Beyond PGDhigh pancreatic cancers, our results indicate that it is possible to pharmacologically target a metastasis-intrinsic metabolic dependency, even if the components are not genetically altered. If such dependencies are prevalent in advanced cancers, then a largely untapped pool of druggable metabolic targets may be available for development of effective therapies for patients with late-stage disease.
Materials and methods
Reagent sources
The rapid autopsy cell lines are previously described [8, 17, 18, 20]. AsPC-1, HPAF-II, and PANC-2.03 were purchased from ATCC. 6AN was purchased from Sigma-Aldrich. Analogs were purchased from Enamine, Sigma-Aldrich, or synthesized at the Vanderbilt Institute of Chemical Biology using published protocols. All compound identities and purities were verified by LC-MS (M + H and >95% pure at λ = 215 nm and 254 nm) and 1H NMR spectroscopy prior to use. Detailed methods can be found in the Supplementary Material.
References
Vanharanta S, Massagué J. Origins of metastatic traits. Cancer Cell. 2013;24:410–21.
Lambert AW, Pattabiraman DR, Weinberg RA. Emerging biological principles of metastasis. Cell. 2017;168:670–91.
Iacobuzio-Donahue CA, Litchfield K, Swanton C. Intratumor heterogeneity reflects clinical disease course. Nat Cancer. 2020;1:3–6.
Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med. 2014;371:2140–1.
Kamisawa T, Wood LD, Itoi T, Takaori K. Pancreatic cancer. Lancet. 2016;388:73–85.
Storz P, Crawford HC. Carcinogenesis of Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2020;158:2072–81.
Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27:1806–13.
Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–7.
Makohon-Moore AP, Zhang M, Reiter JG, Bozic I, Allen B, Kundu D, et al. Limited heterogeneity of known driver gene mutations among the metastases of individual patients with pancreatic cancer. Nat Genet. 2017;49:358–66.
Reiter JG, Makohon-Moore AP, Gerold JM, Heyde A, Attiyeh MA, Kohutek ZA, et al. Minimal functional driver gene heterogeneity among untreated metastases. Science. 2018;361:1033–7.
Hayashi A, Hong J, Iacobuzio-Donahue CA. The pancreatic cancer genome revisited. Nat Rev Gastroenterol Hepatol. 2021;18:469–81.
Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, et al. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–73.
Massagué J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306.
Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–91.
Bergers G, Fendt SM. The metabolism of cancer cells during metastasis. Nat Rev Cancer. 2021;21:162–80.
Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, et al. Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res. 2015;75:544–53.
Bechard ME, Smalling R, Hayashi A, Zhong Y, Word AE, Campbell SL, et al. Pancreatic cancers suppress negative feedback of glucose transport to reprogram chromatin for metastasis. Nat Commun. 2020;11:4055.
Bechard ME, Word AE, Tran AV, Liu X, Locasale JW, McDonald OG. Pentose conversions support the tumorigenesis of pancreatic cancer distant metastases. Oncogene. 2018;37:5248–56.
Fendt SM, Frezza C, Erez A. Targeting metabolic plasticity and flexibility dynamics for cancer therapy. Cancer Disco. 2020;10:1797–807.
McDonald OG, Li X, Saunders T, Tryggvadottir R, Mentch SJ, Warmoes MO, et al. Epigenomic reprogramming during pancreatic cancer progression links anabolic glucose metabolism to distant metastasis. Nat Genet. 2017;49:367–76.
McDonald OG. Cancer metastasis: selectable traits without genetic constraints. Mol Cell Oncol. 2020;7:1825910.
Embuscado EE, Laheru D, Ricci F, Yun KJ, de Boom Witzel S, Seigel A, et al. Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther. 2005;4:548–54.
Roe JS, Hwang CI, Somerville TDD, Milazzo JP, Lee EJ, Da Silva B, et al. Enhancer Reprogramming Promotes Pancreatic Cancer Metastasis. Cell. 2017;170:875–.e820.
Johnson WJ, McColl JD. 6-Aminonicotinamide-a potent nicotinamide antagonist. Science. 1955;122:834.
Köhler E, Barrach H, Neubert D. Inhibition of NADP dependent oxidoreductases by the 6-aminonicotinamide analogue of NADP. FEBS Lett. 1970;6:225–8.
Dietrich LS, Friedland IM, Kaplan LA. Pyridine nucleotide metabolism: mechanism of action of the niacin antagonist, 6-aminonicotinamide. J Biol Chem. 1958;233:964–8.
Herken H, Meyer-Estorf G, Halbhübner K, Loos D. Spastic paresis after 6-aminonicotinamide: metabolic disorders in the spinal cord and electromyographically recorded changes in the hind limbs of rats. Naunyn Schmiedebergs Arch Pharm. 1976;293:245–55.
Soares KC, Foley K, Olino K, Leubner A, Mayo SC, Jain A. et al. A preclinical murine model of hepatic metastases. J Vis Exp. 2014;91:e51677.
Ju HQ, Zhuang ZN, Li H, Tian T, Lu YX, Fan XQ, et al. Regulation of the Nampt-mediated NAD salvage pathway and its therapeutic implications in pancreatic cancer. Cancer Lett. 2016;379:1–11.
Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469–83.
Rhim AD, Oberstein PE, Thomas DH, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 2014;25:735–47.
Daneshmandi S, Cassel T, Lin P, Higashi RM, Wulf GM, Boussiotis VA, et al. Blockade of 6-phosphogluconate dehydrogenase generates CD8(+) effector T cells with enhanced anti-tumor function. Cell Rep. 2021;34:108831.
Walker DL, Reid JM, Svingen PA, Rios R, Covey JM, Alley MC, et al. Murine pharmacokinetics of 6-aminonicotinamide (NSC 21206), a novel biochemical modulating agent. Biochem Pharm. 1999;58:1057–66.
Conroy T, Desseigne F, Ychou M, Bouché O, Guimbaud R, Bécouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364:1817–25.
Golan T, Hammel P, Reni M, Van Cutsem E, Macarulla T, Hall MJ, et al. Maintenance olaparib for germline BRCA-mutated metastatic pancreatic cancer. N Engl J Med. 2019;381:317–27.
Balachandran VP, Beatty GL, Dougan SK. Broadening the impact of immunotherapy to pancreatic cancer: challenges and opportunities. Gastroenterology. 2019;156:2056–72.
Yoshida T, Yamasaki S, Kaneko O, Taoka N, Tomimoto Y, Namatame I, et al. A covalent small molecule inhibitor of glutamate-oxaloacetate transaminase 1 impairs pancreatic cancer growth. Biochem Biophys Res Commun. 2020;522:633–8.
Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med. 2019;25:628–40.
Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med. 2019;25:620–7.
Recouvreux MV, Moldenhauer MR, Galenkamp KMO, Jung M, James B, Zhang Y. et al. Glutamine depletion regulates Slug to promote EMT and metastasis in pancreatic cancer. J Exp Med. 2020;217:e20200388.
Acknowledgements
This work was supported by National Institutes of Health grants R01 CA222594 (OGM) and 5P30 DK058404 (OGM).
Author information
Authors and Affiliations
Contributions
OGM conceived the study and oversaw experiments and data analysis. SRS oversaw compound design and conducted glide docking simulations. RVS and OGM performed cell culture, metabolite extractions, and HPLC studies. PJK and LJM advised and assisted with HPLC experiments. MEB, JD, DB, ERR, and OGM performed mouse experiments. MEB conducted confocal immunofluorescent imaging. OGM performed histopathologic analysis. OGM wrote the manuscript. RVS, SRS, and PJK edited the manuscript. RVS, MEB, SRS, JD, and OGM assembled the Figures. All authors agreed to the final version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Smalling, R.V., Bechard, M.E., Duryea, J. et al. Aminopyridine analogs selectively target metastatic pancreatic cancer. Oncogene 41, 1518–1525 (2022). https://doi.org/10.1038/s41388-022-02183-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02183-3
- Springer Nature Limited
We’re sorry, something doesn't seem to be working properly.
Please try refreshing the page. If that doesn't work, please contact support so we can address the problem.