
Abstract
Background
Metabolic changes have been recognized as an important hallmark of cancer cells. Cancer cells can promote their own growth and proliferation through metabolic reprogramming. Particularly, serine metabolism has frequently been reported to be dysregulated in tumor cells. 3-Phosphoglycerate dehydrogenase (PHGDH) catalyzes the first step in the serine biosynthesis pathway and acts as a rate-limiting enzyme involved in metabolic reprogramming. PHGDH upregulation has been observed in many tumor types, and inhibition of PHGDH expression has been reported to inhibit the proliferation of PHGDH-overexpressing tumor cells, indicating that it may be utilized as a target for cancer treatment. Recently identified inhibitors targeting PHGDH have already shown effectiveness. A further in-depth analysis and concomitant development of PHGDH inhibitors will be of great value for the treatment of cancer.
Conclusions
In this review we describe in detail the role of PHGDH in various cancers and inhibitors that have recently been identified to highlight progression in cancer treatment. We also discuss the development of new drugs and treatment modalities based on PHGDH targets. Overexpression of PHGDH has been observed in melanoma, breast cancer, nasopharyngeal carcinoma, parathyroid adenoma, glioma, cervical cancer and others. PHGDH may serve as a molecular biomarker for the diagnosis, prognosis and treatment of these cancers. The design and development of novel PHGDH inhibitors may have broad implications for cancer treatment. Therapeutic strategies of PHGDH inhibitors in combination with traditional chemotherapeutic drugs may provide new perspectives for precision medicine and effective personalized treatment for cancer patients.
Graphical abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Metabolic changes have been recognized as hallmarks of cancer, including dysregulation of serine metabolism. In general, extracellular serine alone is sufficient to meet the need of tumor cell proliferation by entering the cells via amino acid transporters [1, 2]. Some tumor cells, however, have the ability to increase serine synthesis through glycolysis intermediates even when sufficient extracellular serine is available. Actively synthesized serine is utilized for nucleotide synthesis, redox homeostasis, amino acid transport and folic acid metabolism, thereby promoting tumor cell proliferation [3,4,5,6,7]. Serine biosynthetic fluxes derived from glycolytic intermediates are driven by amplification of the gene coding for 3-phosphoglycerate dehydrogenase (PHGDH), one of the few metabolic enzymes that are known to be dysregulated in cancer [8, 9]. PHGDH catalyzes the first step of the serine biosynthetic pathway downstream of glycolysis and is a key enzyme in serine biosynthesis. Several studies have reported that PHGDH is highly expressed in various cancers, including melanoma, glioma, and breast, cervical, rhinitis and colon cancers. Glucose catabolism in PHGDH-amplified cancer cells follows a typical glycolytic pathway, whereby 3-phosphoglycerate (3-PG) is produced through phosphoglycerate kinase (PGK). Amplified PHGDH competes with the phosphoglycerate mutant enzyme PGAM for the 3-PG substrate, thereby increasing the serine biosynthetic flux, and activation of phosphoserine aminotransferase (PSAT), phosphoserine phosphatase (PSPH) and serine hydroxy-methyltransferase (SHMT), supplying cells with 2-α-ketoglutarate and folic acid pools. 2-α-Ketoglutarate enters the tricarboxylic acid (TCA) cycle, and the folate pool is used to support nucleotide synthesis and methylation of homocysteine to form methionine [10]. In PHGDH-amplified tumor cells, carbon is transferred from the glycolytic pathway to the serine synthesis pathway (SSP), which may subsequently induce a variety of biosynthetic reactions that promote the growth and proliferation of tumor cells [10]. Interestingly, the SSP exhibits heterogeneity in different cancer cells. PHGDH-overexpressing cancer cells show high pathway activity, and its inhibition disrupts tumor cell proliferation, whereas cancer cells expressing little or no PHGDH are insensitive to inhibition of this pathway [11].
Because tumor cells have unique metabolic methods to meet their own survival needs, targeting specific enzymes such as PHGDH is considered a viable therapeutic strategy, in particular for cancers exhibiting PHGDH overexpression [12]. In a previous review the relationship between PHGDH amplification and changes in glucose metabolism in human melanoma has been explained [13], whereas Kraoua et al.. performed biochemical and genetic studies in two unrelated Tunisian families with PHGDH deficiency and expounded on clinical and biochemical analyses, imaging, electroencephalography and treatment [14]. Grant et al. described the different types, homology, structure and catalytic activity of PHGDH [15], whereas Li et al.. discussed how mammalian cells tightly control the synthesis of serine, and outlined the current status of therapeutic serine synthesis-based methods [16]. Unterlass et al. [17] discussed the role of metabolic oncogenes such as PHGDH, and the therapeutic potential that lies within targeting altered metabolic phenotypes in cancer. Here, we summarize the significance of PHGDH and the serine biosynthesis pathway in cancer, as well as the development and use of PHGDH-targeting inhibitors.
2 PHGDH and the serine biosynthesis pathway in cancer
Tumor cells are often situated in a microenvironment that is subject to stress such as hypoxia, lack of nutrition and oxidative stress. Such stressed tumor microenvironments play important roles in determining tumor cell metabolism [18]. Under nutritional stress, tumor cells can promote cell proliferation and survival through metabolic changes, which are a common feature of these cells. Elucidating the molecular mechanisms determining how tumor cells adapt to a nutritionally deficient microenvironment is essential for our understanding of the occurrence and development of tumors and for the design of targeted treatment regimens. Metabolic adaptations vary by cell type. The main metabolic route in normal cells is oxidative phosphorylation, whereas that in tumor cells is aerobic glycolysis. Glycolysis is the process of breaking down glucose in the cytoplasm to produce pyruvate, which yields two ATP molecules. Regardless of the absence or presence of oxygen, tumor cells utilize glycolysis for generating energy. This major discovery was made by the German physiologist Otto Warburg and was later called the Warburg effect. Warburg found that cancer cells produce ATP mainly through the glycolytic pathway, rather than through the tricarboxylic acid cycle. Even in the presence of sufficient oxygen, cancer cells use this less efficient route to obtain energy [19,20,21]. It has been reported that the main function of aerobic glycolysis is to maintain high levels of glycolytic intermediates (such as glyceraldehyde-3-phosphate, acetyl-CoA, glyceryl-3-phosphate, glucose-6-phosphate, dihydroxyacetone-phosphate and fructose-6-phosphate) to support intracellular anabolic responses [22]. This may be the reason why cancer cells utilize this metabolic route.
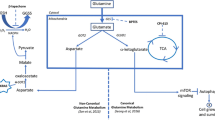
Glucose and glutamine are two important energy sources for tumor cells to survive. The growth and proliferation of tumor cells highly depends on these two substances [18, 23,24,25]. They can not only provide ATP, but also supply carbon and nitrogen sources for the biosynthesis of proteins, nucleotides and lipids [18, 20, 26, 27]. Glucose can support tumor cell proliferation either through glycolysis or the pentose phosphate pathway (PPP) [28]. Glutamine can support cells to resist oxidative stress and supplement glucose for the generation of macromolecules [26]. In addition, glucose and glutamine may serve as major sources for serine/glycine biosynthesis. Glucose and glutamine provide the precursors 3-phosphoglycerate (3-PG) and glutamate, respectively, which promote serine synthesis. Serine is a non-essential amino acid, but plays an important role in cell proliferation. Many anabolic reactions involving proteins, lipids and nucleic acids require serine [29,30,31,32]. In addition, serine is essential for the functioning of the central nervous system [30, 33]. Okabe et al.. found that the PHGDH-mediated serine biosynthesis pathway plays an important role in glucose metabolism in adipose tissues and, thus, may potentially be utilized as a target for the treatment of diabetes [34]. The synthesis of serine by PHGDH is key to heme production in endothelial cells [35]. In addition to promoting growth and proliferation through metabolic reprogramming, it can help tumor cells to overcome cellular stress [36,37,38,39]. The serine biosynthetic pathway is thought to allow tumor cells to survive, proliferate and maintain redox homeostasis [37,38,39,40,41,42,43,44]. In vivo synthesis by tumor cells of serine from 3-PG, an intermediate metabolite in the glycolytic pathway, involves several steps (Fig. 1). The first step is to synthesize 3-phosphate hydroxy-pyruvate (pPYR) under the action of phosphoglycerate dehydrogenase (PHGDH) and NAD+. Then, pPYR is transaminated by phosphoserine aminotransferase (PSAT) to form phosphoserine (pSER) and α-ketoglutarate (α-KG), thereby providing nitrogen from glutamate. Finally, pSER is dephosphorylated to form serine under the action of phosphoserine phosphatase (PSPH) [13, 40, 45]. At the same time, L-serine is produced through the non-phosphorylation pathway, instead of being produced through the phosphorylation pathway and directly through diet [46, 47]. The non-phosphorylated pathway starts with 2-phosphoglycerate, which is converted to D-glycerate. This is followed by the conversion of D-glycerate to hydroxy-pyruvate, after which serine is formed from hydroxy-pyruvate [48]. It has been reported that both pathways are important in animal systems, but that the relative contribution of each pathway to serine synthesis varies among animal species. In the livers of dogs and frogs, the main pathway for serine formation is non-phosphorylation, whereas in cattle and chicken livers the formation of serine occurs mainly through the phosphorylation pathway [49]. However, extensive data indicate that L-serine biosynthesis in mammalian tissues occurs predominantly through the phosphorylation pathway [50, 51]. Synthetic serine can provide carbon atoms to supply folate and convert serine to glycine and tetrahydrofolate to form methyl-tetraaminofolate, which subsequently enters the folate cycle [52, 53]. The serine synthesis pathway can be considered as a branch of the glycolytic pathway at 3-PG. 3-PG flows into the serine synthesis pathway, which may result in the following effects: (1) limitation of the production of ATP, (2) conversion of NAD+ to NADH to affect the redox state, (3) the products of the serine synthesis pathway (serine and glycine) may serve as precursors of the biosynthetic pathway and (4) when serine synthesizes glycine, it provides a carbon unit to the folate pool, which facilitates biosynthesis and DNA methylation [3, 23, 24, 54, 55].

The serine biosynthetic pathway. 3-PG, 3-phosphoglycerate; pPYR, 3-phosphate hydroxypyruvate; PSAT, phosphoserine aminotransferase; α-KG, α-ketoglutarate; pSER, phosphoserine; PSPH, phosphoserine phosphatase; Glu, glutamate; SER, serine; GLY, glycine
In the absence of glucose or glutamine, multiple metabolic pathways in tumor cells are altered. One of the most important changes is activation of the aforementioned serine synthesis pathway, which can support the survival of tumor cells under nutritional stress by regulating redox homeostasis and cell cycle progression. It has been shown that activation of the c-Myc-mediated serine biosynthetic pathway is essential to early cancer development under nutritional deprivation [56]. Under nutritional glucose or glutamine deficiency stress conditions, c-Myc is activated. By regulating the expression of PHGDH, PSAT, PSPH and other metabolic enzymes in the serine synthesis pathway, the remaining main energy sources glutamine or glucose are utilized to support the serine synthesis pathway for the survival of tumor cells by maintaining redox homeostasis [56].
PHGDH belongs to the D-hydroxylate dehydrogenase family and is common in prokaryotes and eukaryotes [57, 58]. It can catalyze the de novo synthesis of serine and is the key enzyme of the serine biosynthetic pathway [48, 59]. PHGDH is highly homologous to glycerol dehydrogenase (GDH) and formate dehydrogenase (FDH) [60, 61]. Ichihara and Greenberg first reported the presence of this enzyme in mammalian systems [62, 63]. Subsequently, it has been found that PHGDH is abundantly expressed in adult and fetal brain tissues [64,65,66] and that PHGDH deficiency generally leads to bradykinesia, epilepsy and congenital microcephaly [65]. Thus, PHGDH plays an important role in the development and functioning of the central nervous system [67, 68].
Achouri et al. cloned the first mammalian PHGDH gene from rat liver, reveaing an open reading frame of 533 amino acids [69]. Its 5’-end and 3’-end non-coding regions are composed of 68-bp and 113-bp sequences, respectively. The PHGDH sequence of rat liver is similar to that of Escherichia coli, Haemophilus influenzae, Saccharomyces cerevisiae and Bacillus subtilis [70,71,72]. Rat PHGDH shows a high similarity to that of Bacillus subtilis, and is about 120 residues longer than that of Escherichia coli or Haemophilus influenzae [69]. Cho et al. determined the nucleotide sequence of the human PHGDH gene [73], which is located on the long arm of human chromosome 1, has 12 exons and is translated into a protein with a molecular mass of 56.8 kDa. The 5′-end non-coding region is 692 bp in length and the 3′-end non-coding region is 187 bp in length, including an 11-bp poly (A) tail [73]. The similarity of PHGDH between human and rat is 94.0 %, indicating that the gene is highly conserved among mammals [69]. In addition, the human PHGDH gene shows 33 %, 25 % and 24 % similarity to that of Caenorhabditis elegans [74], Schizosaccharomyces pombe and Saccharomyces cerevisiae, respectively. Human PHGDH also shows 37 %, 28 %, 26 % and 26 % similarity to that of gram-positive and -negative bacteria, i.e., Bacillus subtilis, Hemophilus influenzae, Bordetella pertussis and Escherichia coli, respectively [70,71,72]. The reported differences in the PHGDH gene mainly involve the C-terminal regulatory region [73]. The C-terminal region of PHGDH from Caenorhabditis elegans is very short and has no regulatory region, whereas that from mammals and Bacillus subtilis is about 200 residues long, 120 residues more than PHGDH of yeast and other bacteria [73].
Cho et al. analyzed the tissue distribution of PHGDH mRNA expression and detected two different transcripts, i.e., a 2.1-kb transcript (predominating) and a 710-bp transcript. They found that the 2.1-kb transcript was highly expressed in brain, kidney, liver, pancreas, ovary, testis and prostate, and exhibited a low expression in colon (mucosal lining), a very low expression in thymus, small intestine and heart, and no expression in spleen, placenta, skeletal muscle, peripheral blood leukocytes and lungs [73]. The 710-bp transcript was also expressed in tissues in which the 2.1-kb transcript was expressed, and its expression in heart and skeletal muscle was higher than that of the 2.1-kb transcript [73]. These data indicate that PHGDH exhibits different expression patterns in different tissues, i.e., exhibits tissue specificity.
Mitoma et al. cloned the mouse PHGDH gene, which is located in the F2-F3 region of chromosome 3, has a total length of ~ 27 kb, and consists of 12 exons. All exon and intron boundaries comply with the GT/AG splicing rules [75]. Also, the homologies of the N-terminal sequences of mouse and rat PHGDH are 97.0 % and of similar size [76]. The typical TATA-box near the transcription start site is missing in the mouse PHGDH gene, but there are several GC-rich regions in the first 200 bp of the 5’ flanking region. The GC-box contains a recognition sequence for Spl and plays a vital role in the basic promoter activity of the mouse PHGDH gene [75].
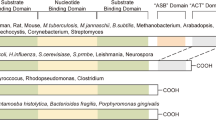
PHGDH exists in at least three different basic structural forms, called type I, type II and type III [77]. The PHGDH gene of human and Mycobacterium tuberculosis is of type I, which has the most complex structure [78]. The three types of PHGDH all contain two common domains, i.e., a cofactor-binding domain and a substrate-binding domain [78]. According to the Protein Data Bank (PDB), several PHGDH crystal structures have been analyzed, mostly from Homo sapiens. In Fig. 2 we present three representative cartoon models of PHGDH crystal structures from different sources. The crystal structure of PHGDH from Mycobacterium tuberculosis is a tetramer [79]. Although the tetramer is made up of the same subunits, obvious asymmetry is found in the tertiary structure of the subunits [79]. The crystal structure of PHGDH of Escherichia coli is also a tetramer [80]. The crystal structure of human PHGDH is a dimer [78]. Unfortunately, the full-length crystal structure of mammalian PHGDH still remains to be resolved.

Crystal structures of PHGDH derived from different sources. These pictures are plotted with pymol in cartoon models. a Crystal structure of PHGDH derived from Mycobacterium tuberculosis (PDB code: 1YGY). b Crystal structure of PHGDH derived from Escherichia coli (PDB code: 1PSD). C. Crystal structure of PHGDH derived from Homo sapiens (PDB code: 2G76)
Modern targeted drug design methods depend to a large extent on a thorough understanding of the target structures at the molecular level. Developments in the field of structural biology have facilitated the generation of protein structure information and has been applied to various stages of drug research. As such, the three-dimensional structure of PHGDH may be utilized to guide structure-based functional research, to reveal the relationship between structure and function and to elucidate its mechanism of action, thereby providing a structural basis for PHGDH-targeted drug discovery. Currently, all crystal structures of PHGDH that have been submitted to PDB have been obtained by X-ray diffraction. It is anticipated that with the development of three-dimensional reconstruction technologies using cryo-electron microscopy, more detailed structures will be determined in the future.
As mentioned above, the serine biosynthesis pathway mediated by PHGDH plays an important role in glucose metabolism in adipose tissues and heme production in endothelial cells [34, 35]. In addition, it has been reported that blood triglyceride levels may be related to PHGDH DNA methylation [81], that PHGDH mutations cause Neu-Laxova syndrome (NLS), a rare autosomal recessive disease [82] and that anti-PHGDH antibodies can be used for the diagnosis of autoimmune hepatitis (AIH) disorder [83]. In addition, several studies have shown that PHGDH is highly expressed in various tumors, including breast cancer [84], melanoma [13], pulmonary fibrosis [85], kidney cancer [86], lung adenocarcinoma [87], glioblastoma [88] and colorectal cancer [89].
3 PHGDH and cancer development, diagnosis and prognosis
Snell et al. showed, using rat liver cancer as a model, that PHGDH has a relatively high activity in tissues with a strong cell renewal ability and an increased activity in neonatal and regenerating livers. Serine methyltransferase activity has also been reported in liver cancer tissues [90]. Later, Snell et al. used human colon cancer and transplantable rat sarcomas as models to measure the activities of PHGDH, serine hydroxy-methyltransferase, serine dehydratase and serine aminotransferase, and found that serine dehydratase and serine aminotransferase activities were absent in these two tumor models, whereas PHGDH and serine hydroxy-methyltransferase activities were high [91]. PHGDH is a key enzyme for the de novo synthesis of serine, whereas serine hydroxy-methyltransferase catalyzes the synthesis of glycine and methyl-tetrahydrofolate, suggesting that serine hydroxy-methyltransferase may serve as a potential target for tumor therapy. Snell et al. did, however, not examine the role of PHGDH at that time. In a RNA interference (RNAi)-based loss of function screen to identify new tumor suppressor genes, Possemato et al. used human breast cancer xenograft models involving orthotopic transplantation in mice [41]. They found that the PHGDH gene is in a genomic region of recurrent copy number gain in breast cancer, and that the PHGDH protein level was up-regulated in 70 % of estrogen receptor (ER)-negative breast cancers. In addition, they found that the serine synthesis flux of breast cancer cells with a high PHGDH expression was increased. Also, PHGDH activity was found to be inhibited in cell lines overexpressing PHGDH, but not in cell lines that did not express PHGDH, resulting in a significant decrease in cell proliferation and serine synthesis. In addition, they found that inhibition of PHGDH activity did not affect intracellular serine levels, but instead decreased α-ketoglutarate levels, which is another product of this pathway, as well as tricarboxylic acid cycle intermediates [41]. Based on their results, the authors suggested that targeting PHGDH may serve as a novel treatment approach for breast cancer. In another study, RNAi was used to silence the expression of PHGDH in estrogen receptor-positive and -negative breast cancer cells, leading to disruption of mitochondrial redox homeostasis and increased apoptosis, thereby abrogating breast cancer stem cell (BCSC) enrichment under hypoxic conditions [42]. In addition, they found that PHGDH silencing decreased the metastatic ability of tumor cells. Based on genome-wide expression profiling of the metastatic variant breast cancer cell line MDA-MB-231(SA) and its isogenic parental cell line MDA-MB-231, it was found that the PHGDH expression level was upregulated in the metastatic variant [43]. Their research confirmed that PHGDH overexpression was significantly associated with decreased relapse-free and overall survival rates of patients as well as malignant phenotypic features of breast cancer [43]. In addition, it has been reported that a high expression of PHGDH is correlated with triple-negative (PR/ER/Her2) and basal subtypes of breast cancer, confirming that the expression of PHGDH is subtype-specific [40, 41, 92]. Inhibition of PHGDH may make triple-negative breast cancers (TNBC) sensitive to doxorubicin and, thus, PHGDH may be a promising target for improving the effectiveness of chemotherapy in this subgroup of cancer patients [93]. A relationship between cancer and D-2-hydroxyglutarate (D-2HG) was first noted through high levels of D-2HG resulting from mutations in isocitrate dehydrogenase. Additional studies have, however, reported increases in D-2HG in breast cancer cells without isocitrate dehydrogenase mutations. It has also been reported that, in addition to catalyzing the oxidation of 3-phosphoglycerate, PHGDH can reduce NADH-dependent α-ketoglutarate to D-2HG [94]. So, PHGDH appears to play a critical role in breast cancer and, as a consequence, has been studied extensively in this cancer type.
Locasale et al. used mass spectrometry, nuclear magnetic resonance and stable isotope labeling to study alternative pathways of glucose metabolism in cancer cells, and found that a large amount of glycolytic carbon was transferred to serine and glycine biosynthesis through PHGDH in at least some cancer cell types [40]. Thus, cancer cells can adjust their metabolic processes to promote the biosynthesis of large molecules such as serine and glycine, allowing cells to rapidly grow and proliferate. Through bioinformatics analysis of 3,131 human cancers, the authors found that the PHGDH gene was recurrently amplified in melanoma, and that the amplification was related to increased protein expression. RNA interference was used to reduce the expression of PHGDH, thereby affecting the growth of PHGDH-amplified cells and the flux of serine metabolism [40]. Their findings show that PHGDH can induce glycolytic fluxes and promote tumorigenesis. Although increased PHGDH levels promote abnormal accumulation of melanin and affect melanocyte biology, PHGDH expression alone is not sufficient to cause melanoma [95]. It has also been found that PHGDH may serve as a target of p53 in melanoma cells, and that p53 can inhibit serine biosynthesis by inhibiting the expression of PHGDH. Furthermore, it was found that increased PHGDH expression significantly inhibited apoptosis and that RNAi-mediated knock down of endogenous PHGDH expression led to increased apoptosis [96].
PHGDH expression levels are relatively high in astrocytic tumors, and increase further in more aggressive brain tumors [97]. Inhibiting the expression of PHGDH in glioma cells has been found to result in reduction in the expression levels of MMP-2, cyclin D1, VEGF and CHK2, a decrease in proliferation and invasion of glioma cells in vivo and in vitro, and a reduction in expression of the oncogenic transcription factor FOXMI [97]. It was also found that PHGDH can interact with and stabilize FOXM1, thereby promoting the proliferation and invasion of glial cells. This work suggests that PHGDH may serve as a prognostic marker for glial brain tumors, and that targeting the PHGDH-FOXM1 axis may provide a means for brain tumor treatment.
PHGDH also plays a role in human cervical adenocarcinoma [98]. Expression of PHGDH was measured in 54 cervical adenocarcinoma samples, and its relationship with clinical pathological parameters and prognosis was evaluated. The results showed that PHGDH is highly expressed in cervical adenocarcinoma and is related to tumor size and prognosis [99]. PHGDH silencing led to decreased tumor growth and a reduction in the expression of the anti-apoptotic protein Bcl-2, and an increase the expression of caspase-3 [99]. Therefore, PHGDH may serve as a target for cervical adenocarcinoma treatment.
qRT-PCR and tissue array-based immunohistochemical methods were used to evaluate PHGDH expression in non-small cell lung cancer (NSCLC) samples, and it was found that both the mRNA and protein levels were significantly upregulated [100]. More importantly, patients with low PHGDH levels exhibited higher five-year overall survival rates than those with high PHGDH levels [100]. This study thus suggests a clinical significance of PHGDH in NSCLC, and that its targeting may be employed for its treatment. Others found that in NSCLC the transcription factor NRF2 may regulate the expression of PHGDH, PSAT and SHMT2 through ATF4 and, thereby, affect the serine/glycine biosynthetic pathway [44]. The expression of these serine/glycine biosynthetic enzymes was found to be associated with a poor prognosis [44].
It has been reported that PANC-1 pancreatic cancer cells consume glucose at a high rate during de novo serine and glycine synthesis. Downregulation of PHGDH resulted in a significant reduction in PANC-1 tumor growth in xenografted mice, thereby extending their survival time [101]. The authors also found that pancreatic cancer patients with a better overall survival exhibited lower PHGDH expression levels. In addition to catalyzing the synthesis of serine, the authors found that PHGDH also promotes mRNA translation initiation by interacting with elF4A1 and elF4E, thereby accelerating the occurrence and development of pancreatic cancer [101]. So, inhibiting the interaction of PHGDH with eIF4A1/eIF4E may be employed for the treatment of pancreatic cancer. Another study using human pancreatic cancer BxPC-3 and SW1990 cells as in vivo models showed that the expression of PHGDH was significantly upregulated compared to that in its adjacent normal tissues and, in addition, that PHGDH upregulation in patients with pancreatic cancer was correlated with TNM, tumor size and lymph node metastasis status [102]. Knocking down PHGDH in pancreatic cancer cells resulted in inhibition of their proliferation, invasion and migration by reducing the expression of cyclin D1, cyclin B1, MMP-9 and MMP-2 [102]. Therefore, PHGDH may serve as a prognostic biomarker for patients with pancreatic cancer and as a potential target for treatment.
Cho et al. observed PHGDH expression in cervical carcinoma HeLa S3 cells, colon adenocarcinoma COLO 320DM cells, murine lymphoma BW5147.G.1.4 cells and human lymphoblastic lymphoma Sup-T1 cells, but not in human leukemia K562 cells [73]. They found that the human PHGDH gene is tissue-specifically regulated at the transcriptional level and that increases in PHGDH activity are caused by mRNA expression upregulation rather than by changes in enzyme activity. As such, its transcriptional regulatory mechanism may be used as a potential therapeutic target. PHGDH has also been found to serve as a prognostic indicator for patients with gastric cancer (GC). Compared with benign tissues, both PHGDH mRNA and protein were found to be preferentially expressed in GC tissues [103]. PHGDH has also been suggested as a key to targeting the hypoxia-inducible factor HIF in renal cell carcinoma (RCC) [104]. Expression profiling of EBV-miR-BART1 expressing nasopharyngeal carcinoma (NPC) cells revealed numerous genes that are related to metabolism [105]. Among these genes, the expression levels of PHGDH and PSAT were found to be significantly increased [105]. A high expression of PHGDH was also found in two thyroid cancer subtypes, i.e., papillary thyroid cancer (PTC) and poorly differentiated cancer (PDC) [106]. Using a cSNP chip technology in 10 cases of hepatocellular carcinoma (HCC), it was found that six specimens carried a polymorphism in PHGDH (rs1801955) T->A, suggesting a relationship between PHGDH and HCC [107]. By studying sorafenib resistance in advanced HCC, PHGDH was found to play a key role. Sorafenib activates the serine synthesis pathway by inducing PHGDH expression, whereas subsequent inactivation of PHGDH can lead to elevated reactive oxygen species (ROS) levels and the induction of apoptosis [108]. Surprisingly, it was found that the PHGDH inhibitor NCT-503 was able to synergize with sorafenib and inhibit the growth of HCC cells. Other FDA-approved tyrosine kinase inhibitors (TKIs) yielded similar results [108]. This latter study suggests that inhibiting PHGDH may be an effective strategy to resolve TKI resistance in HCC. Others found that in lung adenocarcinomas with EGFR activating mutations, inhibition of PHGDH can alleviate resistance to EGFR tyrosine kinase inhibitors (EGFR-TKIs) [109]. Using IHC to study the expression of serine synthesis-related enzymes in colorectal cancer (CRC) tissues, it was found that the expression level of PHGDH was significantly increased [110]. Concordantly, others found that the PHGDH mRNA and protein expression levels were upregulated in CRC and led to a poor prognosis [89]. Studies on the effect of glutamine metabolism on leukemia cells revealed that glutamine insufficiency inhibits the growth of leukemia cells, but promotes the serine biosynthetic pathway, and that lack of glutamine results in upregulation of PHGDH and PSAT [111]. When PHGDH was silenced without controlled use of serine, the growth of leukemia cells was inhibited, and this effect was further enhanced when glutamine metabolism was inhibited [111]. Another recent study showed that PHGDH expression may be increased in CD138+ cells of patients with multiple myeloma (MM) relapse, and to correlate with a short survival time [112]. Moreover, the authors found that overexpression of PHGDH in MM cells resulted in increased proliferation, tumor formation and resistance to bortezomib (BTZ), both in vitro and in vivo. The specific PHGDH inhibitor NCT-503 inhibited the growth and BTZ resistance of MM cells [112]. Thus, PHGDH may also serve as a therapeutic target for MM. It has been reported that Raman spectroscopy can discriminate healthy parathyroid glands from parathyroid adenoma based on specific molecular fingerprints [113]. A further analysis of available Raman data to assess metabolic differences between healthy parathyroid glands and parathyroid adenomas revealed an increase in the expression of PHGDH and glucose-6-phosphate dehydrogenase (G6PD) in parathyroid adenomas [114]. Therefore, PHGDH may serve as a target for the treatment of parathyroid adenoma. PHGDH may also produce metabolites other than serine, which may be important for tumor cell growth, proliferation and invasion. In addition, PHGDH may enhance the resistance of tumor cells to oxidative stress, which in turn may be related to enhanced metastatic properties. PHGDH expression has also been found to be upregulated in triple-negative breast cancer patients and to be an inherent feature of the ER-PgR-CK5 positive subpopulation of breast epithelial cells [115]. In other tissues and cancer types, PHGDH upregulation appears to be related to the CK5-positive cell lineage. In general, it can be concluded that targeting PHGDH may be of significance for the treatment of different types of cancer and, thus, that the development of PHGDH inhibitors should be a priority. Such inhibitors may also be employed to further clarify the biological role and pathological functions of PHGDH.
4 PHGDH-targeting inhibitors
An in vitro enzymatic assay was used to screen 800,000 drug-like small molecules, after which 408 potential PHGDH inhibitors were identified. Next, these were analyzed for NAD(P)-dependent dehydrogenases to screen for specific inhibitors [116]. 13C6-glucose was used to assess the inhibitor’s ability to deprive cells from de novo serine synthesis in an acute environment. Finally, compound CBR-5884 was identified as the most effective inhibitor (Table 1). It selectively blocks the synthesis of serine without affecting other glycolytic intermediates [11, 116]. In addition, using a breast cancer cell line model, CBR-5884 was found to be selectively toxic to PHGDH-overexpressing cells, but not to cells with little or no PHGDH expression. Concerning the two substrates of PHGDH, i.e., NADH and 3-PG, CBR-5884 was found to inhibit PHGDH in a non-competitive manner. CBR-5884 causes instability of the tetrameric form of PHGDH, resulting in its conversion into dimers. Unfortunately, however, CBR-5884 has been found to be unstable in plasma, thus urging further investigations into its use as a potential drug [116].
Pacold et al. indicated that new PHGDH inhibitors may selectively prevent the growth of PHGDH-dependent cancer cells and may be used in serine biosynthesis through one-carbon unit metabolism. Based on this presumption, they performed a four-point dose-response screening of the 400,000-compound NIH Molecular Library Small Molecule Repository (MLSMR) for potential PHGDH inhibitors [117]. After continuous optimization, two powerful piperazine-1-thioamide PHGDH inhibitors (NCT-502 and NCT-503) were identified (Table 1) [117]. The IC50 values of NCT-502 and NCT-503 were found to be 3.7 ± 1.0 µM and 2.5 ± 0.6 µM, respectively. In addition, NCT-503 showed a non-competitive inhibition mode on the substrates 3-PG and NAD+. When tested in cancer cell lines and xenograft models, the PHGDH inhibitors showed potent anticancer activities against PHGDH-dependent cancer cells, but not against non-PHGDH-dependent cancer cells [117]. In addition to serine synthesis inhibition, these PHGDH inhibitors did not disrupt the synthesis of other amino acids, with the exception of aspartic acid. Furthermore, the authors found that the PHGDH inhibitors not only affect the pathway of glucose to serine, but also reduce carbon incorporation of intracellular and extracellular serine into nucleotides [117]. This led to new insights into serine metabolism in cancer cells, although some unresolved issues remain, such as uncertainty about the mechanism of action of the PHGDH inhibitors. In 2018, Rohde et al. reported on the progress made on NCT-502 and NCT-503, and described the discovery and optimization of piperazine-1-thiourea-based human PHGDH inhibitors [118].
A library containing 600 fragments was screened for binding to PHGDH by differential scanning fluorimetry (DSF) [119]. Next, the selected fragments were identified using thermal shift assays, and later verified by X-ray crystallography and ITC experiments, showing that they can bind to PHGDH, specifically to the adenine subsite. Among these fragments, compound fragment 5 was found to be most effective and to bind deeper into the adenine-binding pocket (Table 1). The hit fragment proposed in this study competes with NAD at the cofactor binding site of PHGDH [119]. This work may provide a basis for the development of competitive inhibitors.
AstraZeneca used a method of fragment-based lead compound design to identify inhibitor 1 that targets PHGDH, with an IC50 of 0.26 µM (Table 1) by binding to the adenine region of NAD+ [120, 121]. In addition, approximately 336 molecules were screened using a fluorescence-based enzymatic assay, of which 279 were from the fragment library and 57 from an internal compound collection [122]. All of these compounds have thionyl moieties and common structural elements such as amine or acetophenone. Based on their findings, the authors synthesized α-ketothioamides using a convergent pharmacophore approach (Table 1), allowing the identification of PHGDH inhibitors that selectively slow down the proliferation of tumor cells with a high PHGDH expression [122]. These results may pave the way for the development of PHGDH inhibitors based on the α-ketothioamide backbone.
Two potential allosteric sites (I and II) in PHGDH were identified based on calculations. Among these, site I was found to be closer to the active site and the NAD+/NADH cofactor binding site, whereas site II was found to be located in the substrate-binding region [123]. Subsequent virtual screening of compounds capable of binding to the two sites revealed that PKUMDL-WQ-2101 and PKUMDL-WQ-2201 can bind to sites I and II, respectively (Table 1). Both compounds were found to inhibit de novo serine synthesis and metabolism in tumor cells, as well as tumor cell growth, at the micromolar level by targeting PHGDH. PKUMDL-WQ-2101 and PKUMDL-WQ-2201 showed good selectivity and activity on human breast cancer cells overexpressing PHGDH. In addition, in vivo analysis showed that these compounds can also inhibit the growth of PHGDH-amplified mouse breast cancer cells [123]. This study was the first to identify a specific, allosteric inhibitor that directly targets PHGDH using structure-based drug design methods. Allosteric inhibitors are superior to substrate-competitive inhibitors in providing new binding sites, increasing selectivity and improving the effects of drug-resistant mutations. This notion may be a basis for future research on and the development of PHGDH inhibitors.
We screened an internal database of natural products (NPs) containing approximately 600 compounds using an enzyme-based analysis method. By doing so, Azacoccones C and E, both azaepicoccone derivatives from Aspergillus flavus, were identified as PHGDH inhibitors (Table 1) [124]. Azacoccones C and E significantly inhibited the activity of PHGDH, with IC50 values of 11.71 ± 2.65 and 9.76 ± 4.32 µM, respectively. Compared with the IC50 value of the PHGDH inhibitor CBR-5884 (31.58 ± 7.18 µM), Azacoccones C and E showed better inhibitory activities. At the same concentration, the two compounds showed no difference in inhibitory effects on another NAD(P)+-dependent dehydrogenase. Besides, the inhibitory effects of Azacoccone E were found to be time-dependent and non-competitive [124]. Using microscale thermophoresis (MST) and cell thermal displacement (CETSA) methods, as well as molecular docking experiments, we found that Azacoccone E directly binds to PHGDH. It coordinated at the allosteric site of PHGDH and was far from the active site, which is crucial to reducing enzyme activity. The most encouraging result was that Azacoccone E selectively inhibited the proliferation of PHGDH-dependent cancer cells and induced their apoptosis, thereby underscoring their potential significance for cancer treatment [124].
Ixocarpalactone A is a natural compound derived from Physalis ixocarpa (Table 1) that can significantly inhibit the activity of PHGDH, with an IC50 value of 1.66 ± 0.28 µΜ [125]. MST and molecular docking experiments confirmed that Ixocarpalactone A can directly bind to PHGDH [125]. It was found that Ixocarpalactone A can specifically inhibit the proliferation of cancer cells with a high PHGDH expression (such as HeLa, SW1990 and MCF-7), without causing significant toxicities to normal cells (such as HPDE6-C7, LO2 and L929) [125]. In addition, Ixocarpalactone A is coordinated at the allosteric site of PHGDH, i.e., it is non-competitive to the NAD co-enzyme, thereby reducing its toxicity [125]. Compared with other previously discovered inhibitors targeting PHGDH, Ixocarpalactone A has unique advantages, i.e., it has a high specificity and low toxicity for the treatment of pancreatic cancer and can easily be transformed into clinical applications [126].
Mullarky et al. used inhibitor 1 reported by AstraZeneca as a lead compound to synthesize a more potent PHGDH inhibitor and to explore its mode of action [127]. They first synthesized compound 1 and found that it significantly inhibits PHGDH, with an IC50 value of 0.238 ± 0.035 µM. Due to its relatively poor permeability through the cell membrane, compound 1 lacks efficacious cellular activity. Therefore, structure-based transformation of compound 1 was performed to obtain a series of indole amide compounds (Table 1). The authors found that these compounds can significantly inhibit the activity of PHGDH and serine synthesis, as well as the growth of Carney cancer cells in serine-free media. The mechanism of action involves binding to the adenine sites of PHGDH in a non-covalent and reversible manner [127]. Here, we show the crystal structure of the complex of another compound (15) with human PHGDH (Fig. 3). Also, the researchers designed prodrugs of compound 1 such as compound 18, which is more effective than its parent compound 15 and almost completely disrupts the proliferation of Carney cancer cells [127]. However, the synthetic ester prodrugs are easily hydrolyzed in plasma, which in turn hampers their in vivo use. Further structural optimization is thus warranted.

X-Ray co-crystal structure of compound 15 and PHGDH (PDB code: 6PLG). These pictures were plotted with pymol. The overall structure is displayed as cartoon mode in blue and those binding amino acids are displayed in stick mode; red sticks represent oxygen atoms, blue sticks represent nitrogen atoms, and the red dotted line represents a hydrogen bond. Brown sticks represent carbon atoms in compound 15, and yellow sticks indicate chlorine atoms
Weinstabl et al. found that a NADH/NAD+ competitive PHGDH inhibitor, BI-4924, and its prodrug BI-4916 (Table 1) exhibit a high selectivity [128]. Through high-throughput screening, BI-4924 was defined as a PHGDH inhibitor with lipophilicity and high binding to plasma protein [128]. The crystal structure of the complex of PHGDH and BI-4924 is also shown (Fig. 4). BI-4916 is an ester prodrug of BI-4924 that increases cell penetration. The intracellular lactone cleavage of the ester prodrug BI-4916 results in intracellular enrichment of the actual carboxylic acid-based drug BI-4924, thereby overcoming the effect of high concentrations of NADH/NAD+ in the cytoplasm [128]. However, the instability of ester prodrugs has limited the in vivo use of BI-4916. Therefore, BI-4924 and its prodrug BI-4916 require further optimization.

X-Ray co-crystal structure of BI-4924 and PHGDH (PDB code: 6RJ6); These pictures were plotted with pymol. The overall structure is displayed as cartoon mode in blue, and the binding amino acids are depicted in a stick mode. Red sticks represent oxygen atoms, blue sticks represent nitrogen atoms, and red dotted lines represent hydrogen bonds. Brown sticks represent carbon atoms in BI-4924, and yellow sticks depict sulfur atoms
In 2016 a patent was assigned to a series of small molecule PHGDH inhibitors. Although the patent does not indicate how these compounds were designed, the backbone of these compounds and their corresponding activities were provided. Among these inhibitors, compound 19 is the most active, with an IC50 value less than 50 µM [129]. A recent study additionally reported that DSF (Table 1), a drug for treating chronic alcoholism, can destroy the PHGDH active tetramer form via special cysteine oxidation, thereby inhibiting human PHGDH activity and exhibiting anticancer activity [130]. Thus, repurposing existing drugs may also be an attractive strategy.
Several important issues should be considered when designing PHGDH inhibitors. First, the source of serine in the central nervous system mainly involves resynthesis in vivo. If a designed PHGDH inhibitor enters the central nervous system, it will affect serine synthesis by inhibiting PHGDH and, thereby, affect the synthesis of other biomolecules, resulting in undesired side effects. Therefore, the currently developed PHGDH inhibitors cannot cross the blood-brain barrier. Second, if the designed inhibitors compete with substrates or coenzymes and have little effects on other NAD-dependent dehydrogenases, the molecules preferably contain a carboxyl group or a negatively charged functional group to specifically interact with the alkaline side chain of the binding pocket of the substrate. If it is designed to compete with the substrate, the molecule should be as small as possible, because the size of the substrate pocket is only 10–12 Å. The molecule can also stretch into the NAD binding pocket to compete with NAD+ and, thereby, interfere with the activity of PHGDH.
5 Conclusions and perspectives
Under normal physiological conditions, cells rely on oxidative phosphorylation of glucose to provide energy, but in the absence of oxygen, cells will switch to the glycolytic pathway. Warburg’s groundbreaking research made scientists realize that even in an oxygen-rich environment, tumor cells continue to utilize aerobic glycolysis as a source of energy for their growth. In addition, tumor cells will use glutamine as the second largest energy source, and the intermediate products of the glutamine decomposition process can participate in the tricarboxylic acid cycle to provide energy [131]. Thus, tumor cells will reprogram to meet the large amount of energy and biosynthetic materials required for their rapid growth and proliferation. Numerous studies have confirmed the crucial role that metabolic reprogramming plays in cancer. Given that metabolic reprogramming of tumor cells is closely related to specific characteristics such as malignancy, invasion and metastasis, targeting abnormally active metabolic pathways has become a strategy for the design of new anti-tumor drugs. It has been found that serine metabolism is often dysregulated in tumor cells. Through in-depth analysis of the serine biosynthetic pathway, the rate-limiting enzyme PHGDH of this pathway was found to be overexpressed in a variety of tumors, including melanomas and estrogen receptor-deficient breast cancers. More recently, overexpression of PHGDH has also been observed in nasopharyngeal carcinoma, parathyroid adenoma, glioma, cervical cancer and others. Therefore, PHGDH may serve as a molecular biomarker for the diagnosis, prognosis and treatment of various cancers.
Breast cancer is the most common malignant tumor in women, and triple-negative breast cancer (TNBC) is a specific subtype with a high risk of recurrence and metastasis, poor therapeutic effects and a poor prognosis. At present, a variety of preclinical and clinical trials is under way to better define the biological characteristics of TNBC, to find effective therapeutic targets and to improve prognosis. In ~ 70 % of TNBC cell lines PHGDH has been found to be up-regulated, and inhibition of PHGDH can make TNBC cells sensitive to doxorubicin [93]. The development of PHGDH as a biomarker for a more accurate TNBC subtyping is of great significance for increasing the clinical treatment efficacy and improving the prognosis. Increased PHGDH expression is associated with a poor overall survival of patients with lung adenocarcinoma. The use of erlotinib for the treatment of EGFR mutation-positive lung adenocarcinoma may result in drug resistance, but this resistance can be overcome by inhibiting PHGDH [109]. Therefore, PHGDH may serve as a target to reverse erlotinib resistance in this type of cancer. Increased expression of PHGDH has been found to be related to tumor size, TNM status and lymph node metastasis in patients with pancreatic cancer. In addition, PHGDH has been found to control pancreatic cancer cell proliferation, migration and invasion [102]. Therefore, PHGDH may also serve as a prognostic indicator and therapeutic target for pancreatic cancer. Bortezomib (BTZ) is being used for the treatment of multiple myeloma (MM), but its efficacy is limited due to the occurrence of drug resistance. It has been reported that resistance to BTZ in MM is related to an increase in serine synthesis, and that PHGDH is up-regulated in BTZ-resistant MM cells [132]. Therefore, interference with serine metabolism may be a strategy to improve the efficacy of bortezomib, and PHGDH may serve as a biomarker of BTZ resistance. High PHGDH levels and advanced TNM (III + IV) stages are independent prognostic markers of non-small cell lung cancer [100]. Through genome-wide CRISPR/Cas9 library screening, PHGDH has also been found to be a key driving factor for sorafenib resistance in gastric cancer [108]. The expression of PHGDH was found to be negatively correlated with the 5-year survival rate of gastric cancer patients, and PHGDH was found to serve as a predictive factor for a poor prognosis of gastric cancer [103]. Ample evidence indicates that PHGDH inhibition can lead to a decreased proliferation of PHGDH-dependent tumor cells. Therefore, it may be utilized as a target for cancer treatment [133]. The design of PHGDH inhibitors is expected to become one of the strategies for the treatment of cancer.
Although numerous reports support contribution of the serine synthesis pathway to tumor growth and proliferation, especially the role of the rate-limiting enzyme PHGDH, a detailed explanation of the mode of action of PHGDH in tumor cells has only been recently reported. Specifically, inhibition of PHGDH has been found to alter the metabolism of nucleotides in a manner independent of the maintenance of redox homeostasis and serine utilization. Instead, inhibition of PHGDH disrupts the balance in central carbon metabolism, thereby affecting the tricarboxylic acid cycle and the pentose phosphate pathway [134].
Currently, several PHGDH inhibitors have been reported and these can be divided into two categories. One type comprises allosteric inhibitors such as CBR-5884, NCT-503 and PKUMDL-WQ-2201. These inhibitors all show good PHGDH inhibitory activities. Another category comprises orthostatic inhibitors such as compounds identified by RAZE Pharmaceuticals. As yet, however, no PHGDH inhibitors have entered clinical trials. Although PHGDH as potential target for anti-cancer drug development has gained widespread attention, the design of drugs targeting PHGDH has slowly progressed due to the following reasons: (1) the complete crystal structure of PHGDH has not been elucidated yet, (2) the prosthetic NAD+ is widely used in living organisms and the concentration is as high as 0.3 mM and (3) the size of the active pocket of the catalytic site of PHGDH is relatively small.
In summary, despite a deeper understanding of the mode of action of PHGDH, several key issues remain to be resolved, as most studies were conducted in vitro using cell line models or in vivo using animal models or, alternatively, using clinical research data. It is of translational interest to further explore PHGDH and promote the development of new drugs based on its targeting. Also, the development of new drug delivery modalities combined with traditional chemotherapeutic drugs will provide new perspectives for precision medicine and the efficacious personalized treatment of cancer patients.
Data availability
Not applicable.
Code availability
Not applicable.
References
G.A. Barker, J.C. Ellory, The identification of neutral amino acid transport systems. Exp. Physiol. 75, 3–26 (1990)
M. Palacín, R. Estévez, J. Bertran, A. Zorzano, Molecular biology of mammalian plasma membrane amino acid transporters. Physiol. Rev. 78, 969–1054 (1998)
C.F. Labuschagne, N.J. van den Broek, G.M. Mackay, K.H. Vousden, O.D. Maddocks, Serine, but not glycine, supports one-carbon metabolism and proliferation of cancer cells. Cell Rep. 7(4), 1248–1258 (2014)
K.R. Mattaini, M.R. Sullivan, M.G. Vander, Heiden, The importance of serine metabolism in cancer. J. Cell Biol. 214, 249–257 (2016)
M.R. Sullivan, K.R. Mattaini, E.A. Dennstedt, A.A. Nguyen, S. Sivanand, M.F. Reilly, K. Meeth, A. Muir, A.M. Darnell, M.W. Bosenberg, C.A. Lewis, M.G. Vander Heiden, Increased serine synthesis provides an advantage for tumors arising in tissues where serine levels are limiting. Cell Metab. 29, 1410–1421 (2019)
C. Frezza, Cancer metabolism: addicted to serine. Nat. Chem. Biol. 12, 389–390 (2016)
K. Snell, Y. Natsumeda, G. Weber, The modulation of serine metabolism in hepatoma 3924A during different phases of cellular proliferation in culture. Biochem. J. 245, 609–612 (1987)
A.R. Mullen, R.J. DeBerardinis, Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol. Metab. 23(11), 552–559 (2012)
E. Gottlieb, I.P. Tomlinson, Mitochondrial tumour suppressors: a genetic and biochemical update. Nat. Rev. Cancer 5(11), 857–866 (2005)
S.G. Dann, R.T. Abraham, Serine biosynthesis: fuel for the melanoma cell growth engine. Pigment Cell Melanoma Res. 24, 875–877 (2011)
E. Mullarky, L.L. Lairson, L.C. Cantley, C.A. Lyssiotis, A novel small-molecule inhibitor of 3-phosphoglycerate dehydrogenase. Mol. Cell. Oncol. 3, e1164280 (2016)
J. Luo, Cancer’s sweet tooth for serine. Breast Cancer Res. 13, 317 (2011)
E. Mullarky, K.R. Mattaini, M.G. Vander Heiden, L.C. Cantley, J.W. Locasale, PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 24, 1112–1115 (2011)
I. Kraoua, E. Wiame, L. Kraoua, F. Nasrallah, H. Benrhouma, A. Rouissi, I. Turki, H. Chaabouni, G. Briand, N. Kaabachi, E. Van Schaftingen, N. Gouider-Khouja, 3-Phosphoglycerate dehydrogenase deficiency: description of two new cases in Tunisia and review of the literature. Neuropediatrics 44(5), 281–285 (2013)
G.A. Grant, D-3-phosphoglycerate dehydrogenase. Front. Mol. Biosci. 5, 110 (2018)
A.M. Li, J.B. Ye, The PHGDH enigma: do cancer cells only need serine or also a redox modulator? Cancer Lett. 476, 97–105 (2020)
J.E. Unterlass, N.J. Curtin, Warburg and Krebs and related effects in cancer. Expert Rev. Mol. Med. 21, e4 (2019)
R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism. Nat. Rev. Cancer 11(2), 85–95 (2011)
O. Warburg, Über den stoffwechsel der carcinom-zelle. J. Mol. Med. 4(12), 534–536 (1925)
O. Warburg, On the origin of cancer cells. Science 123, 309–314 (1956)
O. Warburg, K. Posener, E. Negelein, Ueber den stoffwechsel der tumoren. Biochem. Z. 152, 319–344 (1924)
S.Y. Lunt, M.G. Vander Heiden, Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 27, 441–464 (2011)
I. Amelio, F. Cutruzzolá, A. Antonov, M. Agostini, G. Melino, Serine and glycine metabolism in cancer. Trends Biochem. Sci. 39, 191–198 (2014)
J.W. Locasale, Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583 (2013)
P.M. Tedeschi, E.K. Markert, M. Gounder, H. Lin, D. Dvorzhinski, S.C. Dolfi, L.L. Chan, J. Qiu, R.S. DiPaola, K.M. Hirshfield, L.G. Boros, J.R. Bertino, Z.N. Oltvai, A. Vazquez, Contribution of serine, folate and glycine metabolism to the ATP, NADPH and purine requirements of cancer cells. Cell Death Dis. 4, e877 (2013)
R.J. DeBerardinis, T. Cheng, Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene 29(3), 313–324 (2010)
R.J. Shaw, Glucose metabolism and cancer. Curr. Opin. Cell Biol. 18(6), 598–608 (2006)
T. Soga, Cancer metabolism: key players in metabolic reprogramming. Cancer Sci. 104, 275–281 (2013)
O. Kuge, K. Hasegawa, K. Saito, M. Nishijima, Control of phosphatidylserine biosynthesis through phosphatidylserine-mediated inhibition of phosphatidylserine synthase I in Chinese hamster ovary cells. Proc. Natl. Acad. Sci. U. S. A. 95, 4199–4203 (1998)
T.J. de Koning, K. Snell, M. Duran, R. Berger, B.T. Poll-The, R. Surtees, L-serine in disease and development. Biochem. J. 371, 653–661 (2003)
A.H. Futerman, H. Riezman, The ins and outs of sphingolipid synthesis. Trends Cell Biol. 15, 312–318 (2005)
M. Yang, K.H. Vousden, Serine and one-carbon metabolism in cancer. Nat. Rev. Cancer 16(10), 650–662 (2016)
J.S. Metcalf, R.A. Dunlop, J.T. Powell, S.A. Banack, P.A. Cox, L-serine: a naturally-occurring amino acid with therapeutic potential. Neurotox. Res. 33, 213–221 (2018)
K. Okabe, I. Usui, K. Yaku, Y. Hirabayashi, K. Tobe, T. Nakagawa, Deletion of PHGDH in adipocytes improves glucose intolerance in diet-induced obese mice. Biochem. Biophys. Res. Commun. 504, 309–314 (2018)
S. Vandekeere, C. Dubois, J. Kalucka, M.R. Sullivan, M. García-Caballero, J. Goveia, R. Chen, F.F. Diehl, L. Bar-Lev, J. Souffreau, A. Pircher, S. Kumar, S. Vinckier, Y. Hirabayashi, S. Furuya, L. Schoonjans, G. Eelen, B. Ghesquière, E. Keshet, X. Li, M.G. Vander Heiden, M. Dewerchin, P. Carmeliet, Serine synthesis via PHGDH is essential for heme production in endothelial cells. Cell Metab. 28, 573–587 (2018)
A. Schulze, A.L. Harris, How cancer metabolism is tuned for proliferation and vulnerable to disruption. Nature 491, 364–373 (2012)
E. Piskounova, M. Agathocleous, M.M. Murphy, Z. Hu, S.E. Huddlestun, Z. Zhao, A.M. Leitch, T.M. Johnson, R.J. DeBerardinis, S.J. Morrison, Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191 (2015)
E. Mullarky, L.C. Cantley, Diverting glycolysis to combat oxidative stress. (Springer, Tokyo, 2015), pp. 3–23
J. Ye, J. Fan, S. Venneti, Y.W. Wan, B.R. Pawel, J. Zhang, L.W. Finley, C. Lu, T. Lindsten, J.R. Cross, G. Qing, Z. Liu, M.C. Simon, J.D. Rabinowitz, C.B. Thompson, Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 4, 1406–1417 (2014)
J.W. Locasale, A.R. Grassian, T. Melman, C.A. Lyssiotis, K.R. Mattaini, A.J. Bass, G. Heffron, C.M. Metallo, T. Muranen, H. Sharfi, A.T. Sasaki, D. Anastasiou, E. Mullarky, N.I. Vokes, M. Sasaki, R. Beroukhim, G. Stephanopoulos, A.H. Ligon, M. Meyerson, A.L. Richardson, L. Chin, G. Wagner, J.M. Asara, J.S. Brugge, L.C. Cantley, M.G. Vander Heiden, Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874 (2011)
R. Possemato, K.M. Marks, Y.D. Shaul, M.E. Pacold, D. Kim, K. Birsoy, S. Sethumadhavan, H.K. Woo, H.G. Jang, A.K. Jha, W.W. Chen, F.G. Barrett, N. Stransky, Z.Y. Tsun, G.S. Cowley, J. Barretina, N.Y. Kalaany, P.P. Hsu, K. Ottina, A.M. Chan, B. Yuan, L.A. Garraway, D.E. Root, M. Mino-Kenudson, E.F. Brachtel, E.M. Driggers, D.M. Sabatini, Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350 (2011)
D. Samanta, Y. Park, S.A. Andrabi, L.M. Shelton, D.M. Gilkes, G.L. Semenza, PHGDH expression is required for mitochondrial redox homeostasis, breast cancer stem cell maintenance, and lung metastasis. Cancer Res. 76, 4430–4442 (2016)
S. Pollari, S.M. Käkönen, H. Edgren, M. Wolf, P. Kohonen, H. Sara, T. Guise, M. Nees, O. Kallioniemi, Enhanced serine production by bone metastatic breast cancer cells stimulates osteoclastogenesis. Breast Cancer Res. Treat. 125, 421–430 (2011)
G.M. DeNicola, P.H. Chen, E. Mullarky, J.A. Sudderth, Z. Hu, D. Wu, H. Tang, Y. Xie, J.M. Asara, K.E. Huffman, I.I. Wistuba, J.D. Minna, R.J. DeBerardinis, L.C. Cantley, Erratum: NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat. Genet. 48, 473 (2016)
S. Ravez, Q. Spillier, R. Marteau, O. Feron, R. Frédérick, Challenges and opportunities in the development of serine synthetic pathway inhibitors for cancer therapy. J. Med. Chem. 60, 1227–1237 (2017)
K. Snell, Enzymes of serine metabolism in normal, developing and neoplastic rat tissues. Adv. Enzyme Regul. 22, 325–400 (1984)
K. Snell, Enzymes of serine metabolism in normal and neoplastic rat tissues. Biochim. Biophys. Acta 843, 276–281 (1985)
K. Snell, The duality of pathways for serine biosynthesis is a fallacy. Trends Biochem. Sci. 11, 241–243 (1986)
D.A. Walsh, H.J. Sallach, Comparative studies on the pathways for serine biosynthesis in animal tissues. J. Biol. Chem. 241, 4068–4076 (1966)
G.P. Cheung, J. Cotropia, H.J. Sallach, The effects of dietary protein on the hepatic enzymes of serine metabolism in the rabbit. Arch. Biochem. Biophys. 129, 672–682 (1969)
E.V. Rowsell, K. Snell, J.A. Carnie, A.H. Al-Tai, Liver-L-alanine-glyoxylate and L-serine-pyruvate aminotransferase activities: an apparent association with gluconeogenesis. Biochem. J. 115, 1071–1073 (1969)
P. Stover, V. Schirch, Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methenyltetrahydrofolate to 5-formyltetrahydrofolate. J. Biol. Chem. 265, 14227–14233 (1990)
A.S. Tibbetts, D.R. Appling, R. Appling Dean, Compartmentalization of Mammalian folate-mediated one-carbon metabolism. Annu. Rev. Nutr. 30, 57–81 (2010)
M.G. Vander Heiden, J.W. Locasale, K.D. Swanson, H. Sharfi, G.J. Heffron, D. Amador-Noguez, H.R. Christofk, G. Wagner, J.D. Rabinowitz, J.M. Asara, L.C. Cantley, Evidence for an alternative glycolytic pathway in rapidly proliferating cells. Science 329, 1492–1499 (2010)
J.W. Locasale, L.C. Cantley, Altered metabolism in cancer. BMC Biol. 8, 88 (2010)
L. Sun, L. Song, Q. Wan, G. Wu, X. Li, Y. Wang, J. Wang, Z. Liu, X. Zhong, X. He, S. Shen, X. Pan, A. Li, Y. Wang, P. Gao, H. Tang, H. Zhang, cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 25(4), 429–444 (2015)
G.A. Grant, A new family of 2-hydroxyacid dehydrogenases. Biochem. Biophys. Res. Commun. 165, 1371–1374 (1989)
C. Vinals, E. Depiereux, E. Feytmans, Prediction of structurally conserved regions of D-specific hydroxy acid dehydrogenases by multiple alignment with formate dehydrogenase. Biochem. Biophys. Res. Commun. 192, 182–188 (1993)
D.M. Greenberg, A. Ichihara, Further studies on the pathway of serine formation from carbohydrate. J. Biol. Chem. 224(1), 331–340 (1957)
J.D. Goldberg, T. Yoshida, P. Brick, Crystal structure of a NAD-dependent D-glycerate dehydrogenase at 2.4 Å resolution. J. Mol. Biol. 236, 1123–1140 (1994)
V.S. Lamzin, Z. Dauter, V.O. Popov, E.H. Harutyunyan, K.S. Wilson, High resolution structures of holo and apo formate dehydrogenase. J. Mol. Biol. 236, 759–785 (1994)
A. Ichihara, D.M. Greenberg, Studies on the purification and properties of D-glyceric acid kinase of liver. J. Biol. Chem. 225, 949–958 (1957)
J.E. Willis, H.J. Sallach, The occurrence of D-3-phosphoglycerate dehydrogenase in animal tissue. Biochim. Biophys. Acta 81(1), 39–54 (1964)
M.O. Kinoshita, Y. Shinoda, K. Sakai, T. Hashikawa, M. Watanabe, T. Machida, Y. Hirabayashi, S. Furuya, Selective upregulation of 3-phosphoglycerate dehydrogenase (PHGDH) expression in adult subventricular zone neurogenic niche. Neurosci. Lett. 453, 21–26 (2009)
L.W. Klomp, T.J. de Koning, H.E. Malingré, E.A. van Beurden, M. Brink, F.L. Opdam, M. Duran, J. Jaeken, M. Pineda, L. Van Maldergem, B.T. Poll-The, I.E. van den Berg, R. Berger, Molecular characterization of 3-phosphoglycerate dehydrogenase deficiency-a neurometabolic disorder associated with reduced L-serine biosynthesis. Am. J. Hum. Genet. 67, 1389–1399 (2000)
K. Yoshida, S. Furuya, S. Osuka, J. Mitoma, Y. Shinoda, M. Watanabe, N. Azuma, H. Tanaka, T. Hashikawa, S. Itohara, Y. Hirabayashi, Targeted disruption of the mouse 3-phosphoglycerate dehydrogenase gene causes severe neurodevelopmental defects and results in embryonic lethality. J. Biol. Chem. 279, 3573–3577 (2004)
S. Furuya, T. Tabata, J. Mitoma, K. Yamada, M. Yamasaki, A. Makino, T. Yamamoto, M. Watanabe, M. Kano, Y. Hirabayashi, L-serine and glycine serve as major astroglia-derived trophic factors for cerebellar purkinje neurons. Proc. Natl. Acad. Sci. U. S. A. 97, 11528–11533 (2000)
K. Yamasaki, K. Yamada, S. Furuya, J. Mitoma, Y. Hirabayashi, Y. Watanabe, 3-Phosphoglycerate dehydrogenase (3PGDH), a key enzyme for L-serine biosynthesis, is preferentially expressed in the radial glia/astrocyte lineage and olfactory ensheathing glia in the mouse brain. J. Neurosci. 21, 7691–7704 (2001)
Y. Achouri, M.H. Rider, E.V.V. Schaftingen, M. Robbi, Cloning, sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem. J. 323(2), 365–370 (1997)
K.L. Tobey, G.A. Grant, The nucleotide sequence of the serA gene of Escherichia coli and the amino acid sequence of the encoded protein, D-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 261, 12179–12183 (1986)
R.D. Fleischmann, M.D. Adams, O. White, R.A. Clayton, E.F. Kirkness, A.R. Kerlavage, C.J. Bult, J.F. Tomb, B.A. Dougherty, J.M. Merrick, Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 269, 496–512 (1995)
A. Sorokin, E. Zumstein, V. Azevedo, S.D. Ehrlich, P. Serror, The organization of the Bacillus subtilis 168 chromosome region between the spoVA and serA genetic loci, based on sequence data. Mol. Microbiol. 10, 385–395 (1993)
H.M. Cho, D.Y. Jun, M.A. Bae, Y.H. Kim, Nucleotide sequence and differential expression of the human 3-phosphoglycerate dehydrogenase gene. Gene 245, 193–201 (2000)
R. Wilson, R. Ainscough, K. Anderson, C. Baynes, M. Berks, J. Bonfield, J. Burton, M. Connell, T. Copsey, J. Cooper, 2.2 Mb of contiguous nucleotide sequence from chromosome III of C. elegans. Nature 368, 32–38 (1994)
J. Mitoma, S. Furuya, M. Shimizu, Y. Shinoda, K. Yoshida, N. Azuma, H. Tanaka, Y. Suzuki, Y. Hirabayashi, Mouse 3-phosphoglycerate dehydrogenase gene: genomic organization, chromosomal localization, and promoter analysis. Gene 334, 15–22 (2004)
M. Robbi, Y. Achouri, C. Szpirer, E. Van Schaftingen, The gene encoding rat 3-phosphoglycerate dehydrogenase. Mamm. Genome 11, 1034–1036 (2000)
S. Dey, Z. Hu, X.L. Xu, J.C. Sacchettini, G.A. Grant, D-3-Phosphoglycerate dehydrogenase from Mycobacterium tuberculosis is a link between the Escherichia coli and mammalian enzymes. J. Biol. Chem. 280, 14884–14891 (2005)
J.E. Unterlass, R.J. Wood, A. Baslé, J. Tucker, C. Cano, M.M.E. Noble, N.J. Curtin, Structural insights into the enzymatic activity and potential substrate promiscuity of human 3-phosphoglycerate dehydrogenase (PHGDH). Oncotarget 8, 104478–104491 (2017)
S. Dey, G.A. Grant, J.C. Sacchettini, Crystal structure of Mycobacterium tuberculosis D-3-phosphoglycerate dehydrogenase: extreme asymmetry in a tetramer of identical subunits. J. Biol. Chem. 280, 14892–14899 (2005)
D.J. Schuller, G.A. Grant, L.J. Banaszak, The allosteric ligand site in the Vmax-type cooperative enzyme phosphoglycerate dehydrogenase. Nat. Struct. Biol. 2, 69–76 (1995)
V. Truong, S. Huang, J. Dennis, M. Lemire, N. Zwingerman, D. Aïssi, I. Kassam, C. Perret, P. Wells, P.E. Morange, M. Wilson, D.A. Trégouët, F. Gagnon, Blood triglyceride levels are associated with DNA methylation at the serine metabolism gene PHGDH. Sci. Rep. 7, 11207 (2017)
R. Shaheen, Z. Rahbeeni, A. Alhashem, E. Faqeih, Q. Zhao, Y. Xiong, A. Almoisheer, S.M. Al-Qattan, H.A. Almadani, N. Al-Onazi, B.S. Al-Baqawi, M.A. Saleh, F.S. Alkuraya, Neu-Laxova syndrome, an inborn error of serine metabolism, is caused by mutations in PHGDH. Am. J. Hum. Genet. 94, 898–904 (2014)
D.J. Xiang, H.P. Yan, Q. Xin, F. Lu, X. Feng, Y. Zhao, Y. Liu, J.X. Yang, Cloning and expression of 3-phosphoglycerate dehydrogenase gene and its correlative antibodies in diagnosis of autoimmune hepatitis. Zhonghua Gan Zang Bing Za Zhi 17, 378–382 (2009)
J. Chen, F. Chung, G. Yang, M. Pu, H. Gao, W. Jiang, H. Yin, V. Capka, S. Kasibhatla, B. Laffitte, S. Jaeger, R. Pagliarini, Y. Chen, W. Zhou, Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 4, 2502–2511 (2013)
R.B. Hamanaka, R. Nigdelioglu, A.Y. Meliton, Y. Tian, L.J. Witt, E. O’Leary, K.A. Sun, P.S. Woods, D. Wu, B. Ansbro, S. Ard, J.M. Rohde, N.O. Dulin, R.D. Guzy, G.M. Mutlu, Inhibition of phosphoglycerate dehydrogenase attenuates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 58, 585–593 (2018)
H. Yoshino, N. Nohata, K. Miyamoto, M. Yonemori, T. Sakaguchi, S. Sugita, T. Itesako, S. Kofuji, M. Nakagawa, R. Dahiya, H. Enokida, PHGDH as a key enzyme for serine biosynthesis in HIF2α-targeting therapy for renal cell carcinoma. Cancer Res. 77, 6321–6329 (2017)
B. Zhang, A. Zheng, P. Hydbring, G. Ambroise, A.T. Ouchida, M. Goiny, H. Vakifahmetoglu-Norberg, E. Norberg, PHGDH defines a metabolic subtype in lung adenocarcinomas with poor prognosis. Cell Rep. 19, 2289–2303 (2017)
A.L. Engel, N.I. Lorenz, K. Klann, C. Münch, C. Depner, J.P. Steinbach, M.W. Ronellenfitsch, A.L. Luger, Serine-dependent redox homeostasis regulates glioblastoma cell survival. Br. J. Cancer 122(9), 1391–1398 (2020)
X.Q. Jia, S. Zhang, H.J. Zhu, W. Wang, J.H. Zhu, X.D. Wang, J.F. Qiang, Increased expression of PHGDH and prognostic significance in colorectal cancer. Transl. Oncol. 9, 191–196 (2016)
K. Snell, G. Weber, Enzymic imbalance in serine metabolism in rat hepatomas. Biochem. J. 233, 617–620 (1986)
K. Snell, Y. Natsumeda, J.N. Eble, J.L. Glover, G. Weber, Enzymic imbalance in serine metabolism in human colon carcinoma and rat sarcoma. Br. J. Cancer. 57, 87–90 (1988)
S. Noh, D.H. Kim, W.H. Jung, J.S. Koo, Expression levels of serine/glycine metabolism-related proteins in triple negative breast cancer tissues. Tumour Biol. 35, 4457–4468 (2014)
X. Zhang, W. Bai, Repression of phosphoglycerate dehydrogenase sensitizes triple-negative breast cancer to doxorubicin. Cancer Chemother. Pharmacol. 78, 655–659 (2016)
J. Fan, X. Teng, L. Liu, K.R. Mattaini, R.E. Looper, M.G. Vander Heiden, J.D. Rabinowitz, Human phosphoglycerate dehydrogenase produces the oncometabolite D-2-hydroxyglutarate. ACS Chem. Biol. 10, 510–516 (2015)
K.R. Mattaini, M.R. Sullivan, A.N. Lau, B.P. Fiske, R.T. Bronson, M.G. Vander Heiden, Increased PHGDH expression promotes aberrant melanin accumulation. BMC Cancer 19, 723 (2019)
Y. Ou, S.J. Wang, L. Jiang, B. Zheng, W. Gu, p53 protein-mediated regulation of phosphoglycerate dehydrogenase (PHGDH) is crucial for the apoptotic response upon serine starvation. J. Biol. Chem. 290, 457–466 (2015)
J. Liu, S. Guo, Q. Li, L. Yang, Z. Xia, L. Zhang, Z. Huang, N. Zhang, Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 111, 245–255 (2013)
Z. Jing, W. Heng, D. Aiping, Q. Yafei, Z. Shulan, Expression and clinical significance of phosphoglycerate dehydrogenase and squamous cell carcinoma antigen in cervical cancer. Int. J. Gynecol. Cancer 23, 1465–1469 (2013)
Z. Jing, W. Heng, L. Xia, W. Ning, Q. Yafei, Z. Yao, Z. Shulan, Downregulation of phosphoglycerate dehydrogenase inhibits proliferation and enhances cisplatin sensitivity in cervical adenocarcinoma cells by regulating Bcl-2 and caspase-3. Cancer Biol. Ther. 16, 541–548 (2015)
J. Zhu, J. Ma, X. Wang, T. Ma, S. Zhang, W. Wang, X. Zhou, J. Shi, High Expression of PHGDH predicts poor prognosis in non-small cell lung cancer. Transl. Oncol. 9, 592–599 (2016)
X. Ma, B. Li, J. Liu, Y. Fu, Y. Luo, Phosphoglycerate dehydrogenase promotes pancreatic cancer development by interacting with eIF4A1 and eIF4E. J. Exp. Clin. Cancer Res. 38, 66 (2019)
Z. Song, C. Feng, Y. Lu, Y. Lin, C. Dong, PHGDH is an independent prognosis marker and contributes cell proliferation, migration and invasion in human pancreatic cancer. Gene 642, 43–50 (2018)
Y. Xian, S. Zhang, X. Wang, J. Qin, W. Wang, H. Wu, Phosphoglycerate dehydrogenase is a novel predictor for poor prognosis in gastric cancer. Onco. Targets Ther. 9, 5553–5560 (2016)
A. Fenner, Kidney cancer: PHGDH is key for targeting HIF in RCC. Nat. Rev. Urol. 14, 702 (2017)
Y. Ye, Y. Zhou, L. Zhang, Y. Chen, X. Lyu, L. Cai, Y. Lu, Y. Deng, J. Wang, K. Yao, W. Fang, H. Cai, X. Li, EBV-miR-BART1 is involved in regulating metabolism-associated genes in nasopharyngeal carcinoma. Biochem. Biophys. Res. Commun. 436, 19–24 (2013)
W.Y. Sun, H.M. Kim, W.H. Jung, J.S. Koo, Expression of serine/glycine metabolism-related proteins is different according to the thyroid cancer subtype. J. Transl. Med. 14, 168 (2016)
J. Wang, H. Ni, L. Chen, Y.X. Liu, C.B. Chen, W.Q. Song, Preparation and analysis of cSNP chip on hepatocellular carcinoma-related genes. HBPD Int. 4, 398–402 (2005)
L. Wei, D. Lee, C.T. Law, M.S. Zhang, J. Shen, D.W. Chin, A. Zhang, F.H. Tsang, C.L. Wong, I.O. Ng, C.C. Wong, C.M. Wong, Genome-wide CRISPR/Cas9 library screening identified PHGDH as a critical driver for Sorafenib resistance in HCC. Nat. Commun. 10, 4681 (2019)
J.K. Dong, H.M. Lei, Q. Liang, Y.B. Tang, Y. Zhou, Y. Wang, S. Zhang, W.B. Li, Y. Tong, G. Zhuang, L. Zhang, H.Z. Chen, L. Zhu, Y. Shen, Overcoming erlotinib resistance in EGFR mutation-positive lung adenocarcinomas through repression of phosphoglycerate dehydrogenase. Theranostics 8, 1808–1823 (2018)
S. Yoon, J.G. Kim, A.N. Seo, S.Y. Park, H.J. Kim, J.S. Park, G.S. Choi, J.Y. Jeong, Y. Jun do, G.S. Yoon, B.W. Kang, Clinical implication of serine metabolism-associated enzymes in colon cancer. Oncology 89, 351–359 (2015)
F. Polet, C. Corbet, A. Pinto, L.I. Rubio, R. Martherus, V. Bol, X. Drozak, V. Grégoire, O. Riant, O. Feron, Reducing the serine availability complements the inhibition of the glutamine metabolism to block leukemia cell growth. Oncotarget 7, 1765–1776 (2016)
X. Wu, J. Xia, J. Zhang, Y. Zhu, Y. Wu, J. Guo, S. Chen, Q. Lei, B. Meng, C. Kuang, X. Feng, Y. He, Y. Shen, X. Li, L. Qiu, G. Li, W. Zhou, Phosphoglycerate dehydrogenase promotes proliferation and bortezomib resistance through increasing reduced glutathione synthesis in multiple myeloma. Br. J. Haematol. 190, 52–66 (2020)
A. Palermo, M. Fosca, G. Tabacco, F. Marini, V. Graziani, M.C. Santarsia, F. Longo, A. Lauria, R. Cesareo, I. Giovannoni, C. Taffon, M. Rocchia, S. Manfrini, P. Crucitti, P. Pozzilli, A. Crescenzi, J.V. Rau, Raman spectroscopy applied to parathyroid tissues: a new diagnostic tool to discriminate normal tissue from adenoma. Anal. Chem. 90(1), 847–854 (2018)
A. di Masi, L. Leboffe, A. Sodo, G. Tabacco, R. Cesareo, M. Sbroscia, I. Giovannoni, C. Taffon, P. Crucitti, F. Longo, S. Manfrini, M.A. Ricci, P. Ascenzi, A. Crescenzi, A. Palermo, Metabolic profile of human parathyroid adenoma. Endocrine 67(3), 699–707 (2020)
I. Gromova, P. Gromov, N. Honma, S. Kumar, D. Rimm, M.L. Talman, V.T. Wielenga, J.M. Moreira, High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol. Oncol. 9, 1636–1654 (2015)
E. Mullarky, N.C. Lucki, R. Beheshti Zavareh, J.L. Anglin, A.P. Gomes, B.N. Nicolay, J.C. Wong, S. Christen, H. Takahashi, P.K. Singh, J. Blenis, J.D. Warren, S.M. Fendt, J.M. Asara, G.M. DeNicola, C.A. Lyssiotis, L.L. Lairson, L.C. Cantley, Identification of a small molecule inhibitor of 3-phosphoglycerate dehydrogenase to target serine biosynthesis in cancers. Proc. Natl. Acad. Sci. U. S. A. 113, 1778–1783 (2016)
M.E. Pacold, K.R. Brimacombe, S.H. Chan, J.M. Rohde, C.A. Lewis, L.J. Swier, R. Possemato, W.W. Chen, L.B. Sullivan, B.P. Fiske, S. Cho, E. Freinkman, K. Birsoy, M. Abu-Remaileh, Y.D. Shaul, C.M. Liu, M. Zhou, M.J. Koh, H. Chung, S.M. Davidson, A. Luengo, A.Q. Wang, X. Xu, A. Yasgar, L. Liu, G. Rai, K.D. Westover, M.G. Vander Heiden, M. Shen, N.S. Gray, M.B. Boxer, D.M. Sabatini, A PHGDH inhibitor reveals coordination of serine synthesis and one-carbon unit fate. Nat. Chem. Biol. 12, 452–458 (2016)
J.M. Rohde, K.R. Brimacombe, L. Liu, M.E. Pacold, A. Yasgar, D.M. Cheff, T.D. Lee, G. Rai, B. Baljinnyam, Z. Li, A. Simeonov, M.D. Hall, M. Shen, D.M. Sabatini, M.B. Boxer, Discovery and optimization of piperazine-1-thiourea-based human phosphoglycerate dehydrogenase inhibitors. Bioorg. Med. Chem. 26, 1727–1739 (2018)
J.E. Unterlass, A. Baslé, T.J. Blackburn, J. Tucker, C. Cano, M.E.M. Noble, N.J. Curtin, Validating and enabling phosphoglycerate dehydrogenase (PHGDH) as a target for fragment-based drug discovery in PHGDH-amplified breast cancer. Oncotarget 9, 13139–13153 (2018)
The 5th RSC-BMCS fragment-based drug discovery meeting at Churchill College, Cambridge (UK) (2015)
N. Fuller, L. Spadola, S. Cowen, J. Patel, H. Schönherr, Q. Cao, A. McKenzie, F. Edfeldt, A. Rabow, R. Goodnow, An improved model for fragment-based lead generation at AstraZeneca. Drug Discov. Today 21, 1272–1283 (2016)
S. Ravez, C. Corbet, Q. Spillier, A. Dutu, A.D. Robin, E. Mullarky, L.C. Cantley, O. Feron, R. Frédérick, α-Ketothioamide derivatives: a promising tool to interrogate phosphoglycerate dehydrogenase (PHGDH). J. Med. Chem. 60, 1591–1597 (2017)
Q. Wang, M.V. Liberti, P. Liu, X. Deng, Y. Liu, J.W. Locasale, L. Lai, Rational design of selective allosteric inhibitors of PHGDH and serine synthesis with anti-tumor activity. Cell Chem. Biol. 24, 55–65 (2017)
J. Guo, X. Gu, M. Zheng, Y. Zhang, L. Chen, H. Li, Azacoccone E inhibits cancer cell growth by targeting 3-phosphoglycerate dehydrogenase. Bioorg. Chem. 87, 16–22 (2019)
M. Zheng, J. Guo, J. Xu, K. Yang, R. Tang, X. Gu, H. Li, L. Chen, Ixocarpalactone A from dietary tomatillo inhibits pancreatic cancer growth by targeting PHGDH. Food Funct. 10(6), 3386–3395 (2019)
D. González-Mendoza, D. Ascencio-Martinez, A.H. Poox, V. Mendez-Trujillo, O. Grimaldo-Juarez, J.F.S. Hernández, L.C. Diaz, S.M.A. Marin, Phenolic compounds and physiochemical analysis of Physalis ixocarpa genotypes. Sci. Res. Essays 6, 3808–3814 (2011)
E. Mullarky, J. Xu, A.D. Robin, D.J. Huggins, A. Jennings, N. Noguchi, A. Olland, D. Lakshminarasimhan, M. Miller, D. Tomita, M. Michino, T. Su, G. Zhang, A.W. Stamford, P.T. Meinke, S. Kargman, L.C. Cantley, Inhibition of 3-phosphoglycerate dehydrogenase (PHGDH) by indole amides abrogates de novo serine synthesis in cancer cells. Bioorg. Med. Chem. Lett. 29, 2503–2510 (2019)
H. Weinstabl, M. Treu, J. Rinnenthal, S.K. Zahn, P. Ettmayer, G. Bader, G. Dahmann, D. Kessler, K. Rumpel, N. Mischerikow, F. Savarese, T. Gerstberger, M. Mayer, A. Zoephel, R. Schnitzer, W. Sommergruber, P. Martinelli, H. Arnhof, B. Peric-Simov, K.S. Hofbauer, G. Garavel, Y. Scherbantin, S. Mitzner, T.N. Fett, G. Scholz, J. Bruchhaus, M. Burkard, R. Kousek, T. Ciftci, B. Sharps, A. Schrenk, C. Harrer, D. Haering, B. Wolkerstorfer, X. Zhang, X. Lv, A. Du, D. Li, Y. Li, J. Quant, M. Pearson, D.B. McConnell, Intracellular trapping of the selective phosphoglycerate dehydrogenase (PHGDH) inhibitor disrupts serine biosynthesis. J. Med. Chem. 62, 7976–7997 (2019)
E. Saiah, 3-phosphoglycerate dehydrogenase inhibitors and uses thereof. Patents. (2016)
Q. Spillier, D. Vertommen, S. Ravez, R. Marteau, Q. Thémans, C. Corbet, O. Feron, J. Wouters, R. Frédérick, Anti-alcohol abuse drug disulfiram inhibits human PHGDH via disruption of its active tetrameric form through a specific cysteine oxidation. Sci. Rep. 9, 4737 (2019)
J. Son, C.A. Lyssiotis, H. Ying, X. Wang, S. Hua, M. Ligorio, R.M. Perera, C.R. Ferrone, E. Mullarky, N. Shyh-Chang, Y. Kang, J.B. Fleming, N. Bardeesy, J.M. Asara, M.C. Haigis, R.A. DePinho, L.C. Cantley, A.C. Kimmelman, Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496, 101–105 (2013)
E.A. Zaal, W. Wu, G. Jansen, S. Zweegman, J. Cloos, C.R. Berkers, Bortezomib resistance in multiple myeloma is associated with increased serine synthesis. Cancer Metab. 5, 7 (2017)
C.K. Zogg, Phosphoglycerate dehydrogenase: potential therapeutic target and putative metabolic oncogene. J. Oncol. 2014, 524101 (2014)
M.A. Reid, A.E. Allen, S. Liu, M.V. Liberti, P. Liu, X. Liu, Z. Dai, X. Gao, Q. Wang, Y. Liu, L. Lai, J.W. Locasale, Serine synthesis through PHGDH coordinates nucleotide levels by maintaining central carbon metabolism. Nat. Commun. 9, 5442 (2018)
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (NSFC) [NOs. 81773594, U1803122, U1703111, 81473254, and 81773637], the National Mega-project for Innovative Drugs (grant number 2019ZX09721001-004-007), the National Science and Technology Major Project of the Ministry of Science and Technology of China (No. 2018ZX09735005), the Chunhui Program-Cooperative Research Project of the Ministry of Education, Liaoning Province Natural Science Foundation (NO. 2020-MZLH-31, 2019-MS-299) and the Liaoning Revitalization Talents Program (NO. XLYC1807182). We thank LetPub (www.letpub.com) for its linguistic assistance during the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Yes.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Mingxue Li and Canrong Wu contributed equally to this work.
Rights and permissions
About this article

Cite this article
Li, M., Wu, C., Yang, Y. et al. 3-Phosphoglycerate dehydrogenase: a potential target for cancer treatment. Cell Oncol. 44, 541–556 (2021). https://doi.org/10.1007/s13402-021-00599-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-021-00599-9