
Abstract
More than 25 years of research and preclinical validation have defined EphA2 receptor tyrosine kinase as a promising molecular target for clinical translation in cancer treatment. Molecular, genetic, biochemical, and pharmacological targeting strategies have been extensively tested in vitro and in vivo, and drugs like dasatinib, initially designed to target SRC family kinases, have been found to also target EphA2 activity. Other small molecules, therapeutic targeting antibodies, and peptide-drug conjugates are being tested, and more recently, approaches harnessing antitumor immunity against EphA2-expressing cancer cells have emerged as a promising strategy. This review will summarize preclinical studies supporting the oncogenic role of EphA2 in breast cancer, lung cancer, glioblastoma, and melanoma, while delineating the differing roles of canonical and noncanonical EphA2 signaling in each setting. This review also summarizes completed and ongoing clinical trials, highlighting the promise and challenges of targeting EphA2 in cancer.
Similar content being viewed by others

Introduction
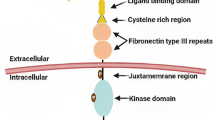
EphA2 is a member of the Eph family of receptor tyrosine kinases (RTK), the largest RTK family in the vertebrate genome. These single transmembrane (TM) receptors are characterized by structural similarities and are subdivided into two distinct classes based on sequence homology and binding to two distinct classes of membrane-bound ligands, called ephrins. Class A Eph receptors generally bind to A class ephrin ligands, which are anchored to the cell membrane by a glycosylphosphatidylinositol linkage (Fig. 1A). Class B Eph receptors generally bind to B class ephrin ligands, which contain a TM-spanning domain and an intracellular domain that includes a cytoplasmic tail with tyrosine residues and a PDZ binding motif [reviewed in [1, 2]] (Fig. 1).

A Eph receptors bind to membrane-bound ligands. Ephrin-A ligands, tethered to the membrane of adjacent cells by a GPI linkage, generally bind to Eph A class receptors, while Ephrin-B ligands, tethered by a transmembrane-spanning domain, generally bind Eph B class receptors. This enables signaling through the receptor (forward signaling) and the ligand (reverse signaling). Upon activation by ligand, Eph receptors oligomerize and are autophosphorylated at juxtamembrane tyrosine residues. B Noncanonical EphA2 signaling differs from canonical signaling in several ways. These mechanisms include signaling in the absence of ephrin ligand, heterodimerization of the EphA2 receptor with other RTKs, such as ERBB2/HER2 or EGFR, and phosphorylation of serine 897 (S897) by AKT, RSK, or PKA. C Key effects of oncogenic EphA2 signaling.
Structurally, the N-terminal extracellular portion of Eph receptors consists of a ligand-binding domain, a Sushi domain, an epidermal growth factor-like motif within a cysteine-rich domain, and two fibronectin-type III repeats (FN III1 and FN III2) followed by a single TM domain. The intracellular C-terminus contains a juxtamembrane (JM) region, followed by a kinase domain (KD), sterile α motif and a PDZ (postsynaptic density protein 95, discs large 1, and zonula occludens-1) binding motif (Fig. 1). In the case of EphA10 and EphB6, modifications within their KDs block kinase activity [3]. Ligands share a conserved extracellular, N-terminal receptor-binding domain [2].
The normal functions of EphA2 include embryonic lens and kidney development, mammary epithelial branching morphogenesis, and bone homeostasis [4]. In addition to these roles, EphA2 is a key regulator of tumorigenesis and cancer progression (Fig. 1C). The role of EphA2 in cancer is complex, with differential effects on malignant transformation and progression exerted by canonical ligand-dependent signaling vs. noncanonical ligand-independent signaling. This review will focus specifically on the oncogenic functions of EphA2 in tumor epithelium of breast and lung cancers, glioblastoma, and melanoma, preclinical studies validating these functions, and ongoing clinical testing of therapeutics that target EphA2 in these and other malignancies.
EphA2 canonical vs. noncanonical signaling in cancer
Canonical Eph receptor and ephrin ligand (Eph/ephrin) signaling is initiated upon binding of the ligand on one cell to the receptor on a neighboring cell in trans [5, 6]. Binding induces a conformational change in both receptor and ligand that allows for oligomerization with neighboring Eph/ephrin complexes and autophosphorylation of conserved tyrosine residues in the JM region [7, 8]. This then exposes the KD, rendering it active, and initiates a phosphorylation cascade along the intracellular region that allows for the recruitment and docking of downstream effector molecules with SRC homology 2 domains [8,9,10]. Because both Eph receptors and ephrin ligands are membrane-bound in this canonical pathway, ligand-dependent activation triggers a unique bidirectional signaling mechanism, with “forward signaling” in the receptor-expressing cell and “reverse signaling” in the ephrin expressing cell (Fig. 1A).
To make matters even more complicated, Eph receptors can also signal in the absence of ligand-binding and kinase activation through cross-talk with other surface receptors and interactions with intracellular kinases, broadly classified as noncanonical signaling mechanisms. For example, EphA2 has been shown to dimerize with E-Cadherin, EGFR, HER2, and integrins and alter downstream signaling in a noncanonical, ligand-independent manner [Fig. 1B; [10,11,12,13]]. EphA2 also undergoes activation through a key phosphorylation event at S897 mediated by kinases including AKT, RSK, and PKA [14,15,16]. Many of these noncanonical signaling mechanisms have been shown to generate markedly different outcomes than ligand-dependent EphA2 activation and contribute to the pro-tumor effects of EphA2 across tumor types; these are discussed in more detail in the following sections and summarized in Table 1. A more extensive review of noncanonical signaling can be found in [17]. Generally, the literature supports an oncogenic role of EphA2 noncanonical signaling in cancers of the breast and lung, glioblastoma, and melanoma, with more variable pro- and antitumor effects of canonical signaling by study and cancer type. Furthermore, some studies provide insufficient information regarding the immediate upstream and downstream events to confidently classify EphA2-mediated phenotypes as canonical or noncanonical in nature. Thus, deciphering the many different mechanisms by which Eph receptors and ephrin ligands can contribute to physiological and pathological processes remains a challenge, but also presents an opportunity to determine which targeting strategies are best suited for specific types of cancers in which EphA2 plays a tumor-promoting role.
EphA2 expression and function in human malignancies
Breast cancer
EphA2 overexpression has been reported in several malignancies, including breast cancer. Breast cancer is the most frequently diagnosed malignancy in women in the United States, and remains the second leading cause of death in women [18]. Breast cancer can be classified based on histological and molecular characteristics. Mammary carcinomas can be divided into subtypes based on cell of origin [19]. For example, ductal carcinomas arise from epithelial cells that line the mammary ducts and make up 60–80% of all mammary carcinomas. More recently, immunohistochemistry (IHC), fluorescence in situ hybridization (FISH), and gene expression profiling have led to two distinct but parallel molecular classification systems. IHC/FISH-based profiling evaluates the protein expression of estrogen (ER), progesterone (PR), and HER2 receptors, along with FISH analysis of HER2 amplification, and identifies categories based on high and low expression of these biomarkers. For example, hormone receptor positive cancers are generally ER+ and/or PR+, while triple-negative breast cancers (TNBC) have low or negative expression of all three biomarkers. In contrast, global gene expression profiling classifies breast cancers into luminal A, luminal B, basal-like, HER2-enriched, claudin-low, and normal breast-like groups [20]. While certain groups correspond with IHC/FISH-profiled categories, this classification system is not exactly aligned with receptor biomarker expression. TNBCs, which make up a high percentage of basal-like and claudin-low tumors, is an aggressive subtype of breast cancer, conferring a higher chance of metastasis, that lacks effective therapeutic options [20]. As discussed below, EphA2 is expressed in breast cancers across multiple subtypes, with enriched, high-level expression in the HER2+ subtype and the basal-like, TNBC subtype, presenting opportunities for therapeutic targeting.
EphA2 has emerged as a major regulator of breast tumorigenesis and malignant progression, and derangement of its signaling—particularly that which favors a ligand-independent signature—is sufficient to induce transformation in some models [21, 22]. Colocalizing with ephrin-A1, EphA2 regulates normal postnatal mammary epithelial branching morphogenesis during puberty via its traditional, canonical signaling route [23]. However, an imbalance of receptor and ligand favors oncogenic signaling in the breast; we reported a correlation between elevated expression of EphA2 and decreased overall and/or recurrence-free survival across multiple breast cancer subtypes, as well as protein expression in both tumor epithelium and vascular endothelium in human breast cancer tissue microarrays [24]. More recently, we found the greatest enrichment of EphA2 overexpression in the basal-like TNBC subtype in data curated from TCGA. In addition, EphA2 loss-of-function in transgenic (C3-TAg) and xenograft (MDA-MB-231, BT549, PDX) mouse models of TNBC consistently reduced tumorigenesis and tumor growth [25]. In both studies, EphA2 protein expression inversely correlated with the expression of its primary ligand, ephrin-A1, in breast tumor cells [24, 25]. Furthermore, restoration of ephrin-A1 signaling decreased cancer cell viability and tumor growth [26]. These data support the hypothesis that loss of ligand expression alleviates tumor-suppressive signaling to promote oncogenic ligand-independent activation and signaling by EphA2 [14].
In addition to TNBC, EphA2 overexpression in the HER2-positive breast cancer subtype [25] is associated with increased tumorigenesis, growth, and metastasis in the MMTV-Neu transgenic mouse model of HER2+ disease and in human HER2+ xenografts via ligand-independent effects [10]. Mechanistically, EphA2 is able to physically and functionally interact with ErbB2/HER2, activating ligand-independent, tumor-promoting signaling pathways like those driven by MAPK and Rho [10]. These pathways are also activated under ephrin-A1 host deficiency in a spontaneous murine breast tumor model overexpressing activated Neu, the rat homolog of the human HER2 oncogene. Deficiency of ephrin-A1 in this model elevated ligand-independent EphA2 activity, which resulted in the loss of ligand-mediated tumor-suppressive signaling [27]. Alleviation of ligand-mediated tumor suppression is supported by a correlation between high levels of EphA2 phosphorylation at S897, a target of Akt and marker for ligand-independent signaling [14], and low levels of expression of ephrin-A1 ligand in human breast tumor cells in tissue microarrays [27]. These data strongly support a ligand-independent, pro-oncogenic role of EphA2 in HER2+ cancer.
EphA2 has also been recently implicated as a key mediator of breast cancer EMT and metastasis in response to high extracellular matrix stiffness; this involves recruitment of LYN kinase to S897-phosphorylated EphA2 and is ligand-independent [28]. Recently, we also uncovered a role for EphA2 in promoting breast-to-bone metastasis and osteolytic disease by inducing osteoclast differentiation. This correlated with enrichment of EphA2 in breast-to-bone metastasis relative to levels in breast-to-brain and breast-to-lung metastasis in human metastatic cancer samples (Vaught et al. [23], submitted). Though the role of ligand-dependent vs. independent signaling in this process remains unclear, no correlation between A class ephrin ligands and breast-to-bone metastasis were detected. We are currently investigating the role of canonical vs. noncanonical signaling in our models. Thus, EphA2 regulates tumorigenesis and progression in multiple models of breast cancer, which is also reflected in EphA2 expression profiles and survival trends in breast cancer patients.
In addition to its primary roles in malignant transformation and growth, preclinical studies have linked elevated EphA2 expression with resistance to trastuzumab in human HER2-dependent cell lines through activation of Src and subsequent amplification of the PI3K/Akt and MAPK signaling pathways [29], which provides rationale to investigate EphA2-targeting agents in trastuzumab-resistant human breast cancer. In preclinical models of tamoxifen-resistant ER+ breast cancer, dual targeting of EphA2 and ER receptor through tamoxifen restored tamoxifen sensitivity [30].
In summary, EphA2 is overexpressed in several breast cancer subtypes, where it functions to regulate tumor growth, invasion, metastatic progression, and drug resistance.
Lung cancer
Lung cancer is the second most frequently diagnosed malignancy in American men and women, and remains the leading cause of death for both sexes [18]. Lung cancer is histologically divided into small cell lung cancer (SCLC), which comprise 15% of lung cancers, and non-SCLC (NSCLC), which make up the majority of cases [31]. SCLC is neuroendocrine in origin and typically responds well to chemotherapy regimens. NSCLC can be further categorized based on cell of origin, for example, squamous cell carcinoma, adenocarcinoma, and large cell carcinoma, which are identified by morphological features and IHC staining. With advances in genome sequencing and the advent of the precision medicine era, NSCLC is now more practically categorized into molecular subtypes [32]. For example, over half of all metastatic lung adenocarcinomas are driven by either EGFR or KRAS aberrations [32, 33]. Monoclonal antibodies and small-molecule inhibitors against epidermal growth factor receptor (EGFR), the protein encoded by EGFR, have shown superior therapeutic benefit in patients with mutations in this gene, compared to chemotherapy agents [34]. KRAS mutant lung cancer has been notably more difficult to target. Despite the development of a specific inhibitor against the KRAS K12C mutation [35], there is still no effective targeted therapy for a majority of KRAS mutant cancers.
EphA2 regulates tumor growth and survival in lung cancer, both in KRAS and EGFR-dependent disease models. While one study did not find a prognostic role for EphA2 in overall survival of human lung cancer [36] and another found that higher levels of EphA2 correlated with better overall survival in early-stage NSCLC [37], others reported a significant correlation between EphA2 overexpression and poor survival in NSCLC [38]. These discrepancies could be due to differences in patient stratification, since Brannan et al. also found a positive association between EphA2 and a history of smoking, activated but not mutated EGFR, and KRAS mutational status. This is consistent with the reported predictive value of high EphA2 levels when determining survival, overall recurrence, and site of recurrence, particularly brain metastases [39]. High co-positivity for RNA-dependent protein kinase and EphA2 was significantly associated with poorer overall survival relative to low co-positivity in NSCLC [40]. While EphA2 mutations are rare, a form mutated in the extracellular FN III domain (G391R) has been found in squamous cell lung carcinoma patients [41], and others have been found in both squamous cell lung carcinoma and malignant pleural mesothelioma [42]. Although functional studies demonstrate that these mutations can increase cell proliferation or invasion, the structural changes induced by these mutations are less well-studied. The fact that these mutations are not clustered in “hotspot” regions nor induce constitutively active kinase activity suggests that structural changes in various different domains can lead to ligand-independent oncogenic signaling. In addition, most preclinical models support tumor-promoting roles for EphA2 in lung cancer. Genetic and pharmacologic inhibition of EphA2 promotes apoptosis in a genetically engineered mouse model of KRAS-dependent NSCLC in vivo [43]. Moreover, EphA2 expression levels are elevated in EGFR tyrosine kinase inhibitor (TKI) resistant human lung cancers post-treatment, as determined by IHC staining of tumor samples pre- and post-treatment [44]. In a genetically engineered mouse model of erlotinib-resistant NSCLC, inducible loss of EphA2 increased tumor cell apoptosis and decreased tumor cell proliferation in vivo, supporting the putative role of EphA2 in EGFR TKI drug resistance [44]. This is also supported by proteomic analysis showing a tenfold increase in EphA2 levels in gefitinib-resistant HCC827, as well as the ability of dasatinib, for which EphA2 is one of the targets, to restore sensitivity to gefitinib [45].
Ligand-independent signaling appears to mediate many pro-tumor effects of EphA2 in lung cancer. This has been supported by studies demonstrating that either stimulation with ephrin-A1-Fc or knockdown of EphA2 in NSCLC cells decreases cell proliferation and migration in vitro primarily via decreased phosphorylation of S897 and downregulation of ERK1/2 activity [38, 44, 46]. Similarly, in KRAS mutant lung adenocarcinoma ligand-dependent EphA2 stimulation is tumor suppressive, while its loss promotes cancer cell proliferation [47]. Activation of P-EphA2 S897 by RSK has been reported in metastatic cancers, and co-localization of P-EphA2 S897 and the phosphorylated, active form of RSK was detected in human cancer tissue microarrays that included cancers from colon, stomach, liver, thyroid, ovary, uterine corpus, and lung squamous and adenocarcinoma [15, 48]. For lung cancer, double positivity staining correlated with poor survival, especially in those with a smoking history [15]. However, some literature conversely supports a ligand-dependent role for EphA2 in lung cancer. Furthermore, downstream signaling through Jnk/c-Jun and PLCγ link EphA2 to lung tumor growth and motility, but through mechanisms not yet fully elucidated [49, 50]. In summary, EphA2 is overexpressed in several lung cancer subtypes, where it functions to regulate growth, survival, invasion, metastatic progression, and drug resistance, often, but not always, through confirmed ligand-independent interactions.
Glioblastoma
Representatives of both A- and B-type Eph receptors and ephrins play essential roles in central nervous system developmental processes including neurogenesis, neuronal migration and organization, and synapse formation [reviewed in [51]]. Eph RTKs and their ligands continue to be expressed in areas of the mammalian brain retaining stem-like properties into maturity, i.e., the subventricular zone [52], where ligand-receptor interaction negatively regulates further neural outgrowth [53] and directs cell migration [54]. In primary CNS malignancies, however, this role of Eph RTKs/ephrins becomes dysregulated, and EphA2 emerges as a negative prognostic indicator and major driver of the cancer stem cell (CSC) phenotype.
EphA2 has repeatedly been shown to be highly overexpressed in primary and recurrent samples of glioblastoma multiforme (GBM), the most common and aggressive primary CNS malignancy of adults, as compared to its relatively low levels in normal brain tissue [55,56,57,58]. This overexpression is correlated with greater tumor vascularity and reduced survival [55, 57]. One study demonstrated an association of ephrin-A1 stimulation of EphA2-overexpressing GBMs with decreased anchorage-independent growth and invasiveness in GBM [56], while another conversely demonstrated ligand-dependent EphA2 activation increased GBM cell proliferation through activation of the MAPK pathway [57]. These conflicting studies differed in duration of stimulation and observed baseline levels of ephrin-A1, revealing that restoration of canonical signaling in a relatively ephrin-deficient system suppresses tumorigenic properties [56], while the addition of ephrin-A1-Fc to a system with pre-existing overexpression does not produce the same benefit and may indeed even be detrimental [57]. This once again highlights the diverse and context-dependent roles of Eph RTK signaling, which must be carefully considered when selecting agonistic or antagonistic targeting strategies.
Reflecting their physiologic roles in early neural development and tissue organization, one explanation for how EphA2 drives malignant behavior in GBM is its role in the maintenance of CSCs [59]. In glioblastoma, as in other malignancies, CSCs promote tumor heterogeneity, recurrence, and resistance to therapy [60, 61]. Enrichment of EphA2 is a common feature of GBM stem-like tumor-propagating cells and is associated with increased self-renewal and tumorigenicity compared to cells with low EphA2 expression [62]. As suggested above, this imbalance of ligand to receptor may favor ligand-independent signaling. Indeed, restoring the balance by treating these cells with either EphA2-targeting siRNA or ephrin-A1-Fc induced astroglial differentiation and suppressed xenograft tumor growth, suggesting EphA2 is not merely a passenger but rather a key driver of the stem cell phenotype. More recent studies have supported this conclusion, as RNA aptamers targeting EphA2 were shown to localize specifically to the GBM stem cell subpopulation [63], where they reduced cell viability and migration, downregulated transcription of stem cell markers, and increased expression of differentiation markers [64]. Reported decreases in cancer cell stemness and/or phenotypic aggressiveness subsequent to EphA2 silencing has been associated with ERK and AKT activation [62], suggesting that suppression of these signaling pathways by aberrant EphA2 expression promotes the stem-like phenotype. However, additional investigation supports a more complicated cross-talk, with AKT-mediated EphA2 S897 phosphorylation required to facilitate the increased invasiveness of EphA2-overexpressing glioblastoma stem cells, as well as opposing pro- and anti-oncogenic roles of ligand-independent and -dependent signaling, respectively [14, 65]. Another recent study has demonstrated phosphorylation of EphA2 S897 by ERK/RSK downstream of EGF in GBM, which was not suppressed by PI3K or AKT inhibition [66], suggesting ligand-independent, pro-oncogenic signaling may be promoted by multiple upstream mediators in GBM. Expression of EGFR vIII, a constitutively active EGFR mutant found commonly in GBM, also increases EphA2 transcript in vitro and is associated with higher EPHA2 protein levels in human GBM samples [67]. Although the involved proteins vary, these data imply a complex feedback loop amongst Eph surface receptors and intracellular kinases which function as both effectors and regulators of the former, emphasizing the need for further investigation into these intricate relationships and how they may be leveraged to mitigate disease.
Whether downstream signaling is ligand-dependent or -independent, the frequently elevated expression of EphA2 in GBM cells coupled with its low to undetectable levels in normal brain tissue mark it as a tumor-associated antigen and strong candidate for immunotherapeutic targeting in both adult and pediatric malignant gliomas [56, 58, 68]. Indeed, peripheral blood mononuclear cells stimulated with an EphA2 peptide resulted in specific cytotoxic T-lymphocyte (CTL) responses against EphA2-presenting GBM cells in vitro, and mice immunized with the peptide similarly demonstrated specific anti-EphA2 CTL activity in splenocytes [58]. This same peptide was administered along with peptide epitopes of two other glioma-associated antigens as a course of intramuscular injections to 12 pediatric patients with recurrent malignant gliomas; nine of ten participants assayed showed evidence of T-cell response against at least one of the three antigens, with all nine responding to EphA2, and minimal toxicity was noted [69]. EphA2-specific CAR-T cell therapies have also shown promise, with strong induction of IFN-γ upon exposure of CAR-T cells to EphA2, specific tumor cell killing in vitro, and inhibition or regression of GBM xenografts in vivo [70, 71]. EphA2, alongside other Eph receptors, has also proven an effective target for the delivery of cytotoxins [72]. From a mechanistically driven standpoint, small-molecule inhibition of EphA2 blocking both key serine and tyrosine phosphorylation events has also shown promise, although further research is needed to shed light on the relative benefit of agonistic vs. antagonistic or nonselective targeting strategies [73]. Overall, these data suggest EphA2 as a highly relevant target for GBM, both as a tumor-associated antigen and an important functional driver of GBM in its own right.
Melanoma
EphA2 has long been implicated in the progression of melanoma, the deadliest cancer of the skin and one of the most commonly diagnosed malignancies for US adults overall. Early data by Easty et al. provided the evidence of ectopic or enhanced EphA2 expression in a subset of melanomas and its positive correlation with advanced and metastatic tumor status [74, 75]. These data have been borne out by more recent studies demonstrating similarly high levels of expression across multiple melanoma cell lines, particularly those deriving from metastatic disease or tumors with more aggressive behavior [76, 77]. Although it is unclear whether canonical or noncanonical signaling is at play in these studies, they support EphA2 as a key driver of oncogenic behavior in melanoma, as its depletion decreases cancer cell proliferation and migration and induces apoptosis, while overexpression in cell lines with low endogenous levels enhances their proliferation. For example, downregulation of EphA2 in one highly-expressing metastatic melanoma-derived cell line promoted phosphorylation of several growth-inhibiting and pro-apoptotic kinases including P53, CHK2, and AMPKα-1 [77].
Other studies have demonstrated that EphA2 expression is induced by ultraviolet radiation in melanocytes via a p53-independent, MAPK-dependent mechanism [78]. However, in contrast to the anti-apoptotic role of EphA2 in melanoma cells described above, overexpression of EphA2 in normal melanocytes promotes apoptosis via a caspase-8-dependent mechanism, once again implying a complicated balance of pro- and anti-oncogenic signaling. It is possible that the pro-apoptotic effects of EphA2 may be neutralized by other mutations in melanoma, thus tipping the balance in favor of its oncogenic signaling properties, or that precancerous melanocytes which are able to survive despite escalating levels of pro-apoptotic stimuli may select for a more aggressive tumor phenotype dependent on that oncogenic signal, as described in the “oncogene overdose-addiction” model [76].
Noncanonical EphA2 RTK signaling is associated with diverse components of melanoma progression, including motility and invasion, metastasis, and treatment resistance. EphA2 upregulation and S897 phosphorylation occur in BRAF(V600E) inhibitor (BRAFi) vemurafenib-resistant cells, while restoration of canonical signaling via ephrin-A1-Fc or inhibition of noncanonical signaling can restore vemurafenib sensitivity [79, 80]. Noncanonical EphA2 signaling can also promote metastasis in response to BRAFi-driven selective pressure [81]. Independent of BRAFi exposure, induction of ectopic EphA2 expression favors its ligand-independent activation and triggers transition from an MMP-dependent mesenchymal to a nonproteolytic ameboid motility phenotype, which is associated with increased cell migration in vitro and metastasis in vivo [82]. It is important to note that the pro-oncogenic behavior of EphA2 RTK signaling in melanoma is likely modulated by intrinsic tumor ephrin-A1 levels, with higher ephrin-A1 expression negatively correlated with metastasis [83], suggesting a favorable effect of ligand-dependent signaling. Although the literature generally favors pro-oncogenic effects of noncanonical EphA2 signaling in melanoma, some evidence fails to support this assumption. For example, early reports support a role for ephrin-A1 as a promoter of cancer cell growth and melanoma progression [84, 85] while several other studies provide insufficient data to conclusively classify their demonstrated phenotypes as either canonical or noncanonical in nature. These data, summarized in Table 1, emphasize the importance of continued investigation of the events surrounding EphA2 activation in melanoma in order to determine how to most effectively target it in tumors.
Favorable effects of inhibiting EphA2 in melanoma have been borne out in preclinical investigation. For example, small-molecule inhibitors of EphA2 suppress proliferation and induces apoptosis of melanoma cells [79]. A monoclonal antibody to EphA2 reduces melanoma cell migration similarly to stimulation with ephrin-A1, consistent with pro-oncogenic ligand-independent signaling and anti-oncogenic ligand-dependent signaling in this system. Pairing this primary antibody with a cytotoxin-conjugated secondary antibody dramatically induced melanoma cell toxicity [86]. Inhibiting EphA2 directly or indirectly stimulating its dephosphorylation at S897 by targeting transcription factor FLI1 or aminopeptidase CD13/ANEP at least partially restores vemurafenib sensitivity in resistant lines [80]. These data mark EphA2 as a promising target candidate for decreasing aggressive characteristics of melanomas including invasiveness and plasticity, as well as restoring BRAF inhibitor sensitivity.
EphA2 therapeutics in clinical trials: outcomes
EphA2-targeting compounds have appeared in clinical trials for multiple types and stages of malignancy, wherein EphA2 has functioned as a cell surface marker for specific delivery of cytotoxins, an epitope for immune targeting, and the primary mechanistic target of interest (Fig. 2). With respect to the former, binding of an anti-EphA2 monoclonal antibody, IC1, to the receptor’s extracellular domain resulted in its phosphorylation, internalization, and degradation. IC1 alone did not impact in vitro cancer cell growth; however, when linked to cytotoxic agent monomethyl auristatin phenylalanine (MMAP), the antibody-drug conjugate demonstrated promising antitumor efficacy in in vitro and in vivo preclinical studies [87]. This conjugate, MEDI-547, progressed to a phase 1 dose escalation trial in patients with relapsed and refractory solid tumors of types known to overexpress EphA2 [88]. The study was discontinued after the initial cohort of six patients demonstrated high rates of severe coagulopathy, with adverse events including hemorrhage (conjunctival, mouth, and other sites), clotting/bleeding-related liver disorder, and epistaxis. Five of six patients showed disease progression while on study. These results suggest substantial off-target cross-reactivity of IC1 antibody with other antigens or undesirable on-target effects of EphA2 inhibition on EphA2-bearing vascular endothelium, as IC1-MMAP conjugate reduced HUVEC cell viability with IC50 comparable to other cell lines and was noted to induce cell death in activated endothelial cells in preclinical analysis [87].

Strategies to inhibit EphA2 in the clinic include EphA2-targeting antibody-cytotoxic drug conjugates (ADC) or peptide-drug conjugates (PDC), tyrosine kinase inhibitors (TKI) like dasatinib, CAR-T cells engineered to recognize and target EphA2 antigen, and nanocarriers designed to deliver siRNAs targeting EPHA2 to tumor cells. Potential future strategies for suppression of noncanonical signaling might also include canonical EphA2 agonists, such as soluble ephrin-A1 (A1-Fc), or other small-molecule inhibitors to block EphA2 phosphorylation at S897 (AKTi/RSKi/PKAi).
Subsequently, an afucosylated anti-EphA2 antibody against the JM domain of EphA2 was developed, which binds both wild type (intact) EphA2 and pro-oncogenic truncated EphA2 generated by proteolytic cleavage in some tumors [89]. In contrast to the agonistic effects of IC1, this antibody, DS-8895a, antagonizes EphA2 tyrosine phosphorylation upon stimulation with Ephrin-A1 and induces antibody-dependent cellular cytotoxicity. DS-8895a demonstrated anticancer activity across in vitro and in vivo models of gastric and breast adenocarcinomas and improved tumor response to cisplatin [90]. In subsequent clinical trials, tumor uptake of the compound was confirmed in 100% of imaged patients [91]. Fourteen of thirty-seven patients treated with DS-8895a in a phase 1 trial achieved stable disease or partial response with five reports of grade ≥3 cytopenia but no thrombotic or hemorrhagic events described, an improvement compared to results with MEDI-547 [92]. Bicycle Therapeutics has recently described a high-affinity EphA2-targeting peptide-cytotoxin conjugate with low affinity for other Eph proteins which dramatically reduces in vivo tumor volume in an EphA2 expression-dependent manner without hematologic adverse effects [93]. This compound, BT5528, is currently recruiting participants in a phase 1/2 trial [94].
While the trials above aim to exploit EphA2 as a tumor-associated protein for drug delivery purposes, several completed and ongoing trials employ compounds targeting EphA2 protein function as their primary mechanism of interest using small-molecule inhibitors. The most common of these is dasatinib, a TKI with activity against BCR-ABL and SRC family kinases that is FDA approved for use in myeloid leukemias. Dasatinib is one of more than 20 small-molecule inhibitors currently approved or in clinical trials with sub-micromolar binding affinity for EphA2; others include ponatinib and bosutinib [95]. As such, in clinical trials, dasatinib has shown benefit against EphA2-associated cancer types, such as breast cancer and NSCLC [96, 97]. One recently completed phase 2 trial combined dasatinib with the intradermal injection of dendritic cells presenting tumor-associated peptides including EphA2 with promising results; patients initiating both treatments in cycle 1 showed an objective response rate of 67% [98]. However, other studies have been less favorable, with limited single-agent efficacy of dasatinib in phase 2 trials of locally advanced/metastatic breast carcinoma [99], TNBC [100], advanced melanoma [101], and pancreatic adenocarcinoma [102]. Careful selection of patients whose tumors are most likely to respond to EphA2 inhibition may yield better results, as many of these trials featured relatively unselective patient populations. However, as an ATP-competitive kinase inhibitor [103], dasatinib may be intrinsically limited in its ability to antagonize the ligand-independent oncogenic effects of EphA2, which can occur via alternate mechanisms not dependent on EphA2’s catalytic activity [14]. Trials of dasatinib and other EphA2-inhibiting compounds are ongoing in triple-negative breast cancer and other advanced or relapsed/refractory solid tumors, including some with a precision medicine element to allocate patients to treatment arms through genetic and molecular profiling of tumor samples [104,105,106]. Selected clinical trials of EphA2-inhibiting or -targeting therapeutics are listed in Table 2.
Conclusions and future directions
This review has summarized the evidence that EphA2 is a key regulator of tumorigenesis and progression in multiple malignancies, including breast cancer, lung cancer, glioblastoma, and melanoma. In addition to promoting tumor cell proliferation and invasion, EphA2 regulates host–tumor interactions including tumor cell-endothelial cell and tumor cell-immune cell communication that facilitate malignant progression. This has made EphA2 a molecule of great interest for targeting with novel experimental therapeutics, many of which have been or are currently being tested in clinical trials.
In spite of extensive data to support its relevance, promising preclinical validation studies, and ongoing clinical trials testing novel and repurposed agents targeting EphA2, several challenges persist. EphA2’s broad expression in multiple cell and tissue types represents both a challenge and an opportunity. For example, targeting EphA2 in malignancies with a high microvascular density in which both tumor cells and tumor-associated endothelium overexpress EphA2 has the potential to disrupt tumor growth and neovascularization simultaneously with a single agent, but may also lead to unintended, toxic side effects due to the disruption of EphA2 function in normal host tissue. The multiple signaling modes of the EphA2 receptor also complicate targeting strategies, as there is no consensus on the relative efficacy of agonistic vs. antagonistic agents, and indeed the most appropriate approach is likely to vary by context. In addition, traditional RTK small-molecule inhibitors blocking the kinase activity of the receptor may fail to inhibit the oncogenic, ligand-independent effects of EphA2 which may be kinase-independent. In spite of these challenges, efforts to target EphA2 with repurposed drugs like dasatinib as well as next-generation targeting peptide-toxin conjugates under development [107] are ongoing. It will also be of great interest to see if EphA2 can be exploited as a tumor antigen for antitumor immunity successfully in broader clinical trials.
Success of one or more EphA2-targeted therapies in the clinic could also provide more options for drug-resistant disease (e.g., trastuzumab-resistant HER2+ breast cancer). Moreover, this would open the door for combination therapies using agents that might synergize with anti-EphA2 therapies. For example, co-targeting TNBC with a combination of EphA2 and an agent inducing apoptosis could improve patient responses over those achieved by either agent alone. Finally, given the role of EphA2 in tumor invasion and metastasis, targeting EphA2 in metastatic malignancies may provide additional therapeutic options for advanced cancers, including breast cancer-to-bone metastatic disease (Vaught et al. [23], submitted). Overall, the body of evidence shows EphA2 to be a promising molecular target for many different human malignancies, and ongoing efforts to target EphA2 in the clinic may provide a valuable new weapon in the arsenal for fighting cancer.
References
Perez White BE, Getsios S. Eph receptor and ephrin function in breast, gut, and skin epithelia. Cell Adh Migr. 2014;8:327–38.
Liang LY, Patel O, Janes PW, Murphy JM, Lucet IS. Eph receptor signalling: from catalytic to non-catalytic functions. Oncogene. 2019;38:6567–84.
Lisabeth EM, Falivelli G, Pasquale EB. Eph receptor signaling and ephrins. Cold Spring Harb Perspect Biol. 2013;5:1–20.
Park JE, Son AI, Zhou R. Roles of EphA2 in development and disease. Genes. 2013;4:334–57.
Himanen JP, Rajashankar KR, Lackmann M, Cowan CA, Henkemeyer M, Nikolov DB. Crystal structure of an Eph receptor-ephrin complex. Nature. 2001;414:933–8.
Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, et al. Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex. EMBO Rep. 2009;10:722–8.
Wybenga-Groot LE, Baskin B, Ong SH, Tong J, Pawson T, Sicheri F. Structural basis for autoinhibition of the Ephb2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell. 2001;106:745–57.
Kullander K, Klein R. Mechanisms and functions of Eph and ephrin signalling. Nat Rev Mol Cell Biol. 2002;3:475–86.
Zisch AH, Pazzagli C, Freeman AL, Schneller M, Hadman M, Smith JW, et al. Replacing two conserved tyrosines of the EphB2 receptor with glutamic acid prevents binding of SH2 domains without abrogating kinase activity and biological responses. Oncogene. 2000;19:177–87.
Brantley-Sieders DM, Zhuang G, Hicks D, Fang WB, Hwang Y, Cates JM, et al. The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Investig. 2008;118:64–78.
Zantek ND, Azimi M, Fedor-Chaiken M, Wang B, Brackenbury R, Kinch MS. E-cadherin regulates the function of the EphA2 receptor tyrosine kinase. Cell Growth Differ. 1999;10:629–38.
Miao H, Burnett E, Kinch M, Simon E, Wang B. Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation. Nat Cell Biol. 2000;2:62–69.
Larsen AB, Pedersen MW, Stockhausen MT, Grandal MV, van Deurs B, Poulsen HS. Activation of the EGFR gene target EphA2 inhibits epidermal growth factor-induced cancer cell motility. Mol Cancer Res. 2007;5:283–93.
Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, et al. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20.
Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, et al. Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2. Nat Commun. 2015;6:7679.
Barquilla A, Lamberto I, Noberini R, Heynen-Genel S, Brill LM, Pasquale EB. Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation. Mol Biol Cell. 2016;27:2757–70.
Zhou Y, Sakurai H. Emerging and diverse functions of the EphA2 noncanonical pathway in cancer progression. Biol Pharm Bull. 2017;40:1616–24.
American Cancer Society. Cancer Facts & Figures 2020. Atlanta: American Cancer Society; 2020.
Weigelt B, Geyer FC, Reis-Filho JS. Histological types of breast cancer: how special are they? Mol Oncol. 2010;4:192–208.
Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. 2011;5:5–23.
Kikawa KD, Vidale DR, Van Etten RL, Kinch MS. Regulation of the EphA2 kinase by the low molecular weight tyrosine phosphatase induces transformation. J Biol Chem. 2002;277:39274–9.
Zelinski DP, Zantek ND, Stewart JC, Irizarry AR, Kinch MS. EphA2 overexpression causes tumorigenesis of mammary epithelial cells. Cancer Res. 2001;61:2301–6.
Vaught D, Chen J, Brantley-Sieders DM. Regulation of mammary gland branching morphogenesis by EphA2 receptor tyrosine kinase. Mol Biol Cell. 2009;20:2572–81.
Brantley-Sieders DM, Jiang A, Sarma K, Badu-Nkansah A, Walter DL, Shyr Y, et al. Eph/ephrin profiling in human breast cancer reveals significant associations between expression level and clinical outcome. PLoS ONE. 2011;6:e24426.
Song W, Hwang Y, Youngblood VM, Cook RS, Balko JM, Chen J, et al. Targeting EphA2 impairs cell cycle progression and growth of basal-like/triple-negative breast cancers. Oncogene. 2017;36:5620–30.
Noblitt LW, Bangari DS, Shukla S, Knapp DW, Mohammed S, Kinch MS, et al. Decreased tumorigenic potential of EphA2-overexpressing breast cancer cells following treatment with adenoviral vectors that express EphrinA1. Cancer Gene Ther. 2004;11:757–66.
Youngblood VM, Kim LC, Edwards DN, Hwang Y, Santapuram PR, Stirdivant SM, et al. The Ephrin-A1/EPHA2 signaling axis regulates glutamine metabolism in HER2-positive breast cancer. Cancer Res. 2016;76:1825–36.
Fattet L, Jung HY, Matsumoto MW, Aubol BE, Kumar A, Adams JA, et al. Matrix rigidity controls epithelial-mesenchymal plasticity and tumor metastasis via a mechanoresponsive EPHA2/LYN complex. Dev Cell. 2020;54:302–16. e307
Zhuang G, Brantley-Sieders DM, Vaught D, Yu J, Xie L, Wells S, et al. Elevation of receptor tyrosine kinase EphA2 mediates resistance to trastuzumab therapy. Cancer Res. 2010;70:299–308.
Gokmen-Polar Y, Toroni RA, Hocevar BA, Badve S, Zhao Q, Shen C, et al. Dual targeting of EphA2 and ER restores tamoxifen sensitivity in ER/EphA2-positive breast cancer. Breast Cancer Res Treat. 2011;127:375–84.
Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, et al. The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol. 2015;10:1243–60.
Weinstein IB, Joe AK. Mechanisms of disease: oncogene addiction-a rationale for molecular targeting in cancer therapy. Nat Clin Pr Oncol. 2006;3:448–57.
Skoulidis F, Heymach JV. Co-occurring genomic alterations in non-small-cell lung cancer biology and therapy. Nat Rev Cancer. 2019;19:495–509.
Chan BA, Hughes BG. Targeted therapy for non-small cell lung cancer: current standards and the promise of the future. Transl Lung Cancer Res. 2015;4:36–54.
Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. 2019;575:217–23.
Shen W, Xi H, Zhang K, Cui J, Li J, Wang N, et al. Prognostic role of EphA2 in various human carcinomas: a meta-analysis of 23 related studies. Growth Factors. 2014;32:247–53.
Ishikawa M, Miyahara R, Sonobe M, Horiuchi M, Mennju T, Nakayama E, et al. Higher expression of EphA2 and ephrin-A1 is related to favorable clinicopathological features in pathological stage I non-small cell lung carcinoma. Lung Cancer. 2012;76:431–8.
Brannan JM, Sen B, Saigal B, Prudkin L, Behrens C, Solis L, et al. EphA2 in the early pathogenesis and progression of non-small cell lung cancer. Cancer Prev Res. 2009;2:1039–49.
Kinch MS, Moore MB, Harpole DH Jr. Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 2003;9:613–8.
Guo C, Shao R, Correa AM, Behrens C, Johnson FM, Raso MG, et al. Prognostic significance of combinations of RNA-dependent protein kinase and EphA2 biomarkers for NSCLC. J Thorac Oncol. 2013;8:301–8.
Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R, et al. EphA2 mutation in lung squamous cell carcinoma promotes increased cell survival, cell invasion, focal adhesions, and mammalian target of rapamycin activation. J Biol Chem. 2010;285:18575–85.
Tan YC, Srivastava S, Won BM, Kanteti R, Arif Q, Husain AN, et al. EPHA2 mutations with oncogenic characteristics in squamous cell lung cancer and malignant pleural mesothelioma. Oncogenesis. 2019;8:49.
Amato KR, Wang S, Hastings AK, Youngblood VM, Santapuram PR, Chen H, et al. Genetic and pharmacologic inhibition of EPHA2 promotes apoptosis in NSCLC. J Clin Investig. 2014;124:2037–49.
Amato KR, Wang S, Tan L, Hastings AK, Song W, Lovly CM, et al. EPHA2 Blockade Overcomes Acquired Resistance to EGFR Kinase Inhibitors in Lung Cancer. Cancer Res. 2016;76:305–18.
Koch H, Busto ME, Kramer K, Médard G, Kuster B. Chemical proteomics uncovers EPHA2 as a mechanism of acquired resistance to small molecule EGFR kinase inhibition. J Proteome Res. 2015;14:2617–25.
Larsen AB, Stockhausen MT, Poulsen HS. Cell adhesion and EGFR activation regulate EphA2 expression in cancer. Cell Signal. 2010;22:636–44.
Yeddula N, Xia Y, Ke E, Beumer J, Verma IM. Screening for tumor suppressors: Loss of ephrin receptor A2 cooperates with oncogenic KRas in promoting lung adenocarcinoma. Proc Natl Acad Sci USA. 2015;112:E6476–6485.
Volz C, Breid S, Selenz C, Zaplatina A, Golfmann K, Meder L, et al. Inhibition of tumor VEGFR2 induces serine 897 EphA2-dependent tumor cell invasion and metastasis in NSCLC. Cell Rep. 2020;31:107568.
Song W, Ma Y, Wang J, Brantley-Sieders D, Chen J. JNK signaling mediates EPHA2-dependent tumor cell proliferation, motility, and cancer stem cell-like properties in non-small cell lung cancer. Cancer Res. 2014;74:2444–54.
Song W, Kim LC, Han W, Hou Y, Edwards D, Wang S. et al. Phosphorylation of PLCγ1 by EphA2 receptor tyrosine kinase promotes tumor growth in lung cancer. Mol Cancer Res. 2020;18:1735–43.
North HA, Clifford MA, Donoghue MJ. ‘Til Eph do us part’: intercellular signaling via Eph receptors and ephrin ligands guides cerebral cortical development from birth through maturation. Cereb Cortex. 2013;23:1765–73.
Lim DA, Alvarez-Buylla A. The Adult Ventricular-Subventricular Zone (V-SVZ) and olfactory bulb (OB) neurogenesis. Cold Spring Harb Perspect Biol. 2016;8:1–33.
Holmberg J, Armulik A, Senti KA, Edoff K, Spalding K, Momma S, et al. Ephrin-A2 reverse signaling negatively regulates neural progenitor proliferation and neurogenesis. Genes Dev. 2005;19:462–71.
Conover JC, Doetsch F, Garcia-Verdugo JM, Gale NW, Yancopoulos GD, Alvarez-Buylla A. Disruption of Eph/ephrin signaling affects migration and proliferation in the adult subventricular zone. Nat Neurosci. 2000;3:1091–7.
Wang LF, Fokas E, Bieker M, Rose F, Rexin P, Zhu Y, et al. Increased expression of EphA2 correlates with adverse outcome in primary and recurrent glioblastoma multiforme patients. Oncol Rep. 2008;19:151–6.
Wykosky J, Gibo DM, Stanton C, Debinski W. EphA2 as a novel molecular marker and target in glioblastoma multiforme. Mol Cancer Res. 2005;3:541–51.
Liu F, Park PJ, Lai W, Maher E, Chakravarti A, Durso L, et al. A genome-wide screen reveals functional gene clusters in the cancer genome and identifies EphA2 as a mitogen in glioblastoma. Cancer Res. 2006;66:10815–23.
Hatano M, Eguchi J, Tatsumi T, Kuwashima N, Dusak JE, Kinch MS, et al. EphA2 as a glioma-associated antigen: a novel target for glioma vaccines. Neoplasia. 2005;7:717–22.
Chen J, Song W, Amato K. Eph receptor tyrosine kinases in cancer stem cells. Cytokine Growth Factor Rev. 2015;26:1–6.
Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–6.
Lathia JD, Mack SC, Mulkearns-Hubert EE, Valentim CL, Rich JN. Cancer stem cells in glioblastoma. Genes Dev. 2015;29:1203–17.
Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL, et al. The EphA2 receptor drives self-renewal and tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell. 2012;22:765–80.
Affinito A, Quintavalle C, Esposito CL, Roscigno G, Vilardo C, Nuzzo S, et al. The discovery of RNA APtamers That Selectively Bind Glioblastoma Stem Cells. Mol Ther Nucleic Acids. 2019;18:99–109.
Affinito A, Quintavalle C, Esposito CL, Roscigno G, Giordano C, Nuzzo S, et al. Targeting Ephrin receptor tyrosine kinase A2 with a selective aptamer for glioblastoma stem cells. Mol Ther Nucleic Acids. 2020;20:176–85.
Miao H, Gale NW, Guo H, Qian J, Petty A, Kaspar J, et al. EphA2 promotes infiltrative invasion of glioma stem cells in vivo through cross-talk with Akt and regulates stem cell properties. Oncogene. 2015;34:558–67.
Hamaoka Y, Negishi M, Katoh H. EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell Signal. 2016;28:937–45.
Ramnarain DB, Park S, Lee DY, Hatanpaa KJ, Scoggin SO, Otu H, et al. Differential gene expression analysis reveals generation of an autocrine loop by a mutant epidermal growth factor receptor in glioma cells. Cancer Res. 2006;66:867–74.
Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. J Neurooncol. 2008;88:245–50.
Pollack IF, Jakacki RI, Butterfield LH, Hamilton RL, Panigrahy A, Normolle DP, et al. Antigen-specific immunoreactivity and clinical outcome following vaccination with glioma-associated antigen peptides in children with recurrent high-grade gliomas: results of a pilot study. J Neurooncol. 2016;130:517–27.
Chow KK, Naik S, Kakarla S, Brawley VS, Shaffer DR, Yi Z, et al. T cells redirected to EphA2 for the immunotherapy of glioblastoma. Mol Ther. 2013;21:629–37.
Bielamowicz K, Fousek K, Byrd TT, Samaha H, Mukherjee M, Aware N, et al. Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro Oncol. 2018;20:506–18.
Ferluga S, Tome CM, Herpai DM, D’Agostino R, Debinski W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget. 2016;7:59860–76.
Gravina GL, Mancini A, Colapietro A, Delle Monache S, Sferra R, Vitale F. et al. The small molecule ephrin receptor inhibitor, GLPG1790, reduces renewal capabilities of cancer stem cells, showing anti-tumour efficacy on preclinical glioblastoma models. Cancers. 2019;11:1–27.
Easty DJ, Ganz SE, Farr CJ, Lai C, Herlyn M, Bennett DC. Novel and known protein tyrosine kinases and their abnormal expression in human melanoma. J Invest Dermatol. 1993;101:679–84.
Easty DJ, Herlyn M, Bennett DC. Abnormal protein tyrosine kinase gene expression during melanoma progression and metastasis. Int J Cancer. 1995;60:129–36.
Udayakumar D, Zhang G, Ji Z, Njauw CN, Mroz P, Tsao H. EphA2 is a critical oncogene in melanoma. Oncogene. 2011;30:4921–9.
Margaryan NV, Strizzi L, Abbott DE, Seftor EA, Rao MS, Hendrix MJ, et al. EphA2 as a promoter of melanoma tumorigenicity. Cancer Biol Ther. 2009;8:279–88.
Zhang G, Njauw CN, Park JM, Naruse C, Asano M, Tsao H. EphA2 is an essential mediator of UV radiation-induced apoptosis. Cancer Res. 2008;68:1691–6.
Miao B, Ji Z, Tan L, Taylor M, Zhang J, Choi HG, et al. EPHA2 is a mediator of vemurafenib resistance and a novel therapeutic target in melanoma. Cancer Discov. 2015;5:274–87.
Azimi A, Tuominen R, Costa Svedman F, Caramuta S, Pernemalm M, Frostvik Stolt M, et al. Silencing FLI or targeting CD13/ANPEP lead to dephosphorylation of EPHA2, a mediator of BRAF inhibitor resistance, and induce growth arrest or apoptosis in melanoma cells. Cell Death Dis. 2017;8:e3029.
Paraiso KH, Das Thakur M, Fang B, Koomen JM, Fedorenko IV, John JK, et al. Ligand-independent EPHA2 signaling drives the adoption of a targeted therapy-mediated metastatic melanoma phenotype. Cancer Discov. 2015;5:264–73.
Parri M, Taddei ML, Bianchini F, Calorini L, Chiarugi P. EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid-like motility style. Cancer Res. 2009;69:2072–81.
Mo J, Zhao X, Dong X, Liu T, Zhao N, Zhang D. et al. Effect of EphA2 knockdown on melanoma metastasis depends on intrinsic ephrinA1 level. Cell Oncol. 2020;43:655–67.
Easty DJ, Hill SP, Hsu MY, Fallowfield ME, Florenes VA, Herlyn M, et al. Up-regulation of ephrin-A1 during melanoma progression. Int J Cancer. 1999;84:494–501.
Easty DJ, Guthrie BA, Maung K, Farr CJ, Lindberg RA, Toso RJ, et al. Protein B61 as a new growth factor: expression of B61 and up-regulation of its receptor epithelial cell kinase during melanoma progression. Cancer Res. 1995;55:2528–32.
Sakamoto A, Kato K, Hasegawa T, Ikeda S. An Agonistic Antibody to EPHA2 Exhibits Antitumor Effects on Human Melanoma Cells. Anticancer Res. 2018;38:3273–82.
Jackson D, Gooya J, Mao S, Kinneer K, Xu L, Camara M, et al. A human antibody-drug conjugate targeting EphA2 inhibits tumor growth in vivo. Cancer Res. 2008;68:9367–74.
Annunziata CM, Kohn EC, LoRusso P, Houston ND, Coleman RL, Buzoianu M, et al. Phase 1, open-label study of MEDI-547 in patients with relapsed or refractory solid tumors. Investig N. Drugs. 2013;31:77–84.
Koshikawa N, Hoshino D, Taniguchi H, Minegishi T, Tomari T, Nam SO, et al. Proteolysis of EphA2 Converts It from a Tumor Suppressor to an Oncoprotein. Cancer Res. 2015;75:3327–39.
Hasegawa J, Sue M, Yamato M, Ichikawa J, Ishida S, Shibutani T, et al. Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol Ther. 2016;17:1158–67.
Ludwig Institute for Cancer Research, Daiichi Sankyo Co. Ltd., Austin Health. Safety and bioimaging trial of DS-8895a in patients with advanced EphA2 positive cancers. 2014. https://ClinicalTrials.gov/show/NCT02252211.
Shitara K, Satoh T, Iwasa S, Yamaguchi K, Muro K, Komatsu Y, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the afucosylated, humanized anti-EPHA2 antibody DS-8895a: a first-in-human phase I dose escalation and dose expansion study in patients with advanced solid tumors. J Immunother Cancer. 2019;7:219.
Bennett G, Brown A, Mudd G, Huxley P, Van Rietschoten K, Pavan S, et al. MMAE Delivery Using the. Mol Cancer Ther. 2020;19:1385–94.
Bicycle Tx Limited. Study BT5528-100 in patients with advanced solid tumors associated with EphA2 expression. 2019. https://ClinicalTrials.gov/show/NCT04180371.
Heinzlmeir S, Kudlinzki D, Sreeramulu S, Klaeger S, Gande SL, Linhard V, et al. Chemical proteomics and structural biology define EPHA2 inhibition by clinical kinase drugs. ACS Chem Biol. 2016;11:3400–11.
Mitri Z, Nanda R, Blackwell K, Costelloe CM, Hood I, Wei C, et al. TBCRC-010: phase I/II study of dasatinib in combination with zoledronic acid for the treatment of breast cancer bone metastasis. Clin Cancer Res. 2016;22:5706–12.
Kim C, Liu S, Crawford J, Subramaniam D, Giaccone G. MA09.01A phase I/II trial of dasatinib and osimertinib in TKI naive patients with advanced EGFR-mutant non-small cell lung cancer (NSCLC). J Thorac Oncol. 2019;14:S281–S282.
Tarhini AA, Tawbi H, Storkus WJ. Vaccine therapy + dasatinib for the treatment of patients with stage IIIB-IV melanoma. Melanoma Manag. 2016;3:251–4.
Duke University, Bristol-Myers Squibb. Phase II Dasatinib Study in advanced breast cancer. 2007. https://ClinicalTrials.gov/show/NCT00546104.
Finn RS, Bengala C, Ibrahim N, Roché H, Sparano J, Strauss LC, et al. Dasatinib as a single agent in triple-negative breast cancer: results of an open-label phase 2 study. Clin Cancer Res. 2011;17:6905–13.
Kluger HM, Dudek AZ, McCann C, Ritacco J, Southard N, Jilaveanu LB, et al. A phase 2 trial of dasatinib in advanced melanoma. Cancer. 2011;117:2202–8.
Chee CE, Krishnamurthi S, Nock CJ, Meropol NJ, Gibbons J, Fu P, et al. Phase II study of dasatinib (BMS-354825) in patients with metastatic adenocarcinoma of the pancreas. Oncologist. 2013;18:1091–2.
Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, et al. Discovery of N-(2-chloro-6-methyl- phenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4- ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658–61.
University of Wisconsin Madison, National Cancer Institute (NCI). Window of opportunity trial of dasatinib in operable triple negative breast cancers with nEGFR. 2017. https://ClinicalTrials.gov/show/NCT02720185.
OHSU Knight Cancer Institute, Oregon Health and Science University. A personalized medicine study for patients with advanced cancer of the breast, prostate, pancreas or those with refractory acute myelogenous leukemia. 2019. https://ClinicalTrials.gov/show/NCT03878524.
National Cancer Institute (NCI). Targeted therapy directed by genetic testing in treating patients with advanced refractory solid tumors, lymphomas, or multiple myeloma (The MATCH Screening Trial). 2015. https://ClinicalTrials.gov/show/NCT02465060.
Gambini L, Salem AF, Udompholkul P, Tan XF, Baggio C, Shah N, et al. Structure-based design of novel EphA2 agonistic agents with nanomolar affinity in vitro and in cell. ACS Chem Biol. 2018;13:2633–44.
Fang WB, Brantley-Sieders DM, Parker MA, Reith AD, Chen J. A kinase-dependent role for EphA2 receptor in promoting tumor growth and metastasis. Oncogene. 2005;24:7859–68.
Sugiyama N, Gucciardo E, Tatti O, Varjosalo M, Hyytiäinen M, Gstaiger M, et al. EphA2 cleavage by MT1-MMP triggers single cancer cell invasion via homotypic cell repulsion. J Cell Biol. 2013;201:467–84.
Choi K, Creighton CJ, Stivers D, Fujimoto N, Kurie JM. Transcriptional profiling of non-small cell lung cancer cells with activating EGFR somatic mutations. PLoS ONE. 2007;2:e1226.
Hess AR, Seftor EA, Gardner LM, Carles-Kinch K, Schneider GB, Seftor RE, et al. Molecular regulation of tumor cell vasculogenic mimicry by tyrosine phosphorylation: role of epithelial cell kinase (Eck/EphA2). Cancer Res. 2001;61:3250–5.
Daiichi Sankyo Co. Ltd., Daiichi Sankyo Inc. Phase 1 study of DS-8895a in subjects with advanced solid tumors. 2013. https://ClinicalTrials.gov/show/NCT02004717.
Xuanwu Hospital, Beijing, Beijing Mario Biotech Company, Hebei Senlang BIotech Company, Beijing HuiNengAn Biotech Company. Personalized chimeric antigen receptor T Cell immunotherapy for patients with recurrent malignant gliomas. 2018. https://ClinicalTrials.gov/show/NCT03423992.
M.D. Anderson Cancer Center, National Cancer Institute (NCI). EphA2 siRNA in treating patients with advanced or recurrent solid tumors. 2015. https://ClinicalTrials.gov/show/NCT01591356.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wilson, K., Shiuan, E. & Brantley-Sieders, D.M. Oncogenic functions and therapeutic targeting of EphA2 in cancer. Oncogene 40, 2483–2495 (2021). https://doi.org/10.1038/s41388-021-01714-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-021-01714-8
- Springer Nature Limited
This article is cited by
-
Targeting the EphA2 pathway: could it be the way for bone sarcomas?
Cell Communication and Signaling (2024)
-
EphA2-specific microvesicles derived from tumor cells facilitate the targeted delivery of chemotherapeutic drugs for osteosarcoma therapy
Journal of Nanobiotechnology (2024)
-
Comprehensive analysis of Epha10 as a predictor of clinical prognosis and immune checkpoint therapy efficacy in non-small cell lung cancer
Scientific Reports (2024)
-
EphA2 promotes the transcription of KLF4 to facilitate stemness in oral squamous cell carcinoma
Cellular and Molecular Life Sciences (2024)
-
Targeting EphA2: a promising strategy to overcome chemoresistance and drug resistance in cancer
Journal of Molecular Medicine (2024)