
Abstract
The protein encoded by the ephrin type-A receptor 2 (EphA2) gene is a member of the ephrin receptor subfamily of the receptor tyrosine kinase family (RTKs). Eph receptors play a significant role in various biological processes, particularly cancer progression, development, and pathogenesis. They have been observed to regulate cancer cell growth, migration, invasion, tumor development, invasiveness, angiogenesis, and metastasis. To target EphA2 activity, various molecular, genetic, biochemical, and pharmacological strategies have been extensively tested in laboratory cultures and animal models. Notably, drugs, such as dasatinib, initially designed to target the kinase family, have demonstrated an additional capability to target EphA2 activity. Additionally, a novel monoclonal antibody named EA5 has emerged as a promising option to counteract the effects of EphA2 overexpression and restore tamoxifen sensitivity in EphA2-transfected MCF-7 cells during in vitro experiments. This antibody mimicked the binding of Ephrin A to EphA2. These methods offer potential avenues for inhibiting EphA2 activity, which could significantly decelerate breast cancer progression and restore sensitivity to certain drugs. This review article comprehensively covers EphA2’s involvement in multiple malignancies, including ovarian, colorectal, breast, lung, glioma, and melanoma. Furthermore, we discuss the structure of EphA2, the Eph-Ephrin signaling pathway, various EphA2 inhibitors, and the mechanisms of EphA2 degradation. This article provides an extensive overview of EphA2’s vital role in different types of cancers and outlines potential therapeutic approaches to target EphA2, shedding light on the underlying molecular mechanisms that make it an attractive target for cancer treatment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Cancer, the deadliest disease impacting millions worldwide each year, is becoming more prevalent in developing nations due to the widespread adoption of unhealthy Western diets and sedentary lifestyles. The Eph-Ephrin system has been extensively studied in cancer, although its exact role remains unclear. Ephrin-receptors (Eph) are members of the receptor tyrosine kinase (RTKs) family, essential cell surface molecules involved in oncogenesis, embryonic development, and the growth of various malignancies [1]. Tumour suppressors and oncogenes, which govern the growth and division of normal cells, were first identified in the 1970 and 1980 s, respectively [2, 3]. The discovery of EphA1 in liver cancer cells during RTK screening in 1987 marked the first Eph receptor finding [4]. Eph-receptors are single transmembrane proteins categorized into EphA (EphA1-A8 and EphA10) and EphB (EphB1-B4 and EphB6) based on their extracellular (N-terminal) domain, responsible for ligand binding affinity. Human systems have nine EphA receptors and five EphB receptors [5]. Eph ligands, EphrinA (EphrinA1-A5) and EphrinB (EphrinB1-B3) activate EphA and EphB receptors, respectively. The ligands differ by the presence of a GPI anchor (EphrinA) and a transmembrane segment (EphrinB) [6]. Eph-receptors have dual roles in promoting and repressing tumor growth in various cancers, such as breast, lung, colorectal, prostate, and brain [1,2,3,4,5,6]. The presence of these molecules on the surfaces of two interacting cells of the same or different types is necessary for the spatial arrangements of Eph receptors and ephrin ligands. These molecules act as bridges between cells, facilitating their recognition and interaction with each other. These receptor-ligand combinations exhibit contact-mediated physiological roles in various aspects, such as axon guidance, neuronal development, vascular patterning, migration, and metastasis in many tumors. Forward signaling, which suppresses tumorigenicity, is a result of ligand binding and receptor clustering in cells that express the signal. This, in turn, prevents the phosphorylation of FAK, Akt, and ERK, thereby regulating cell survival and motility [7]. Current biochemical and mutational studies suggest that the juxtamembrane region of vertebrate RTKs may control Eph receptor activation [8]. Effective kinase activity is achieved by trans-phosphorylating clustered Eph receptors in the juxtamembrane domain [9], which leads to the suppression of cell proliferation and migration. However, Eph and Ephrin reverse signaling promotes direct cell-cell interactions, resulting in cell repulsion in ligand-expressing cells. Ephrin can concurrently reduce the amplitude of Eph receptor forward signaling in the same cell [10].
The primary source of EphA2 is the adult human epithelial cells, which are mostly active during development. Although the exact role of EphA2 in normal cells remains unclear, recent research indicates that it is involved in angiogenesis, cell proliferation, migration, and survival [11]. Elevated levels of EphA2 are commonly observed in cancer cells, and its overexpression has been associated with various types of cancer, including colon, lung, prostate, and breast cancers [12]. This suggests that EphA2 plays a significant role in the initiation, progression, and metastasis of cancer cells. Specifically, EphA2 in cancerous cells lacks tyrosine phosphorylation but retains enzyme activity [13]. Increased levels of unphosphorylated EphA2 promote tumor growth, invasion, and survival, acting as oncoproteins in malignant cells [14]. Moreover, EphA2 regulates the expression of other proteins and enzymes involved in apoptosis, angiogenesis, and metastasis, making it a crucial player in cancer development. EphA2 thus loses its ability to inhibit cell development and instead redistributes along the cell surface, where it associates with processes that promote tumor cell proliferation, invasion, and survival in a positive manner [15]. For instance, EphA2 regulates the expression of matrix metalloproteinases-9 (MMP-9), an enzyme involved in tumor growth and metastasis.
EphrinA1 and its receptors are essential for the development of ECs and the formation of vascular structures. Our primary focus has been on Eph/ephrin molecules and their function in human malignancies. Elevated levels of EphA2 have been observed in the early stages of cancer and are associated with poor patient prognosis. We are particularly interested in the EphA2 structure, signaling, and function of Eph/ephrin molecules in relation to gastric cancers, ovarian cancer, colorectal cancer, and breast cancers, as well as glioblastoma and medulloblastoma.
Structure of EphA2
EphA2 is a transmembrane glycoprotein activated upon binding with an Ephrin ligand. Initially identified as epithelial cell kinase (eck) during screening against a HeLa cell DNA library in the 1990s, EphA2 is primarily found in proliferating epithelial cells in adults [14,15,16,17]. However, its exact significance and functions in these cells still need to be better understood.
With a molecular weight of about 130 kDa and 976 amino acids, the EphA2-receptor is encoded by the EphA2 gene located on chromosome 1p36 in humans [17]. While EphA2 receptors can bind with any of the Ephrin-A ligands, they most frequently interact with EphA1 ligands [18]. The EphrinA1 gene, on the other hand, has a molecular weight of approximately 22 kDa and consists of approximately 205 amino acids. It is situated on chromosome 1p21-p22 [6].
The native structure of Ephrin comprises a ligand-binding domain, a cysteine-rich region, two fibronectin type III repeats, and a transmembrane segment, forming the extracellular domain of the receptor. In contrast, the intracellular domain contains a kinase domain (protein tyrosine kinase), a sterile alpha motif (SAM) protein-protein interaction domain, and a C-terminal PDZ binding motif (Fig. 1) [14,15,16]. The ligand of choice must bind to the ligand-binding domain of the Eph receptor, which is linked to the plasma membrane through a connecting segment.

Overview of EphA2 structure featuring Ephrin-binding, extracellular fibronectin type-III repeats with EGF-like motif, juxtamembrane, intracellular kinase, SAM, and PDZ-binding domains
EphA2-Ephrin signaling: a crucial pathway in cancer progression
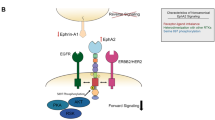
EphA2 binds to the Ephrin ligand in a healthy state, leading to Eph-Ephrin signaling when cells come into contact [19]. This interaction increases forward signaling (trans-interaction between the Eph-receptor and Eph-ligand) and reverses signaling. However, Ephrin cis-interaction can suppress trans-interaction signaling [5]. Upon EphA2 oligomerization and phosphorylation, forward signaling promotes enhanced kinase activity, leading to cell repulsion. Phosphorylated EphA2 reduces cell-extracellular matrix (ECM) attachment, and the association of Ephrin with EphA2 inhibits various kinase activities, including focal adhesion kinase (FAK), extracellular regulated protein kinase (ERK), and AKT phosphorylation, all of which play critical roles in regulating motility, viability, and proliferation in different types of cancer (Fig. 2) [20, 21].
Reverse signaling, in contrast, lacks enzyme activity and is referred to as kinase-independent signaling [20]. However, the mechanism of reverse signaling by EphrinA1 needs to be better understood. EphA2 is overexpressed in various cancers, such as breast, lung, prostate, colorectal, ovarian, and brain cancer, and its signaling with EphrinA1 regulates multiple cellular processes, including adhesion, survival, migration, proliferation, and cell-to-cell repulsion [22]. The Eph-receptor mediates the forward signaling process involving trans-activation between the Eph-receptor and Ephrin-ligand. In contrast, the reverse signaling is mediated by the Ephrin-ligand respectively [25].

EphA2 signaling in cancer: EphA2-Ephrin interaction initiates signaling, promoting cell repulsion and reducing cell-ECM attachment. Phosphorylated EphA2 inhibits FAK, ERK, and AKT phosphorylation, impacting cancer motility and viability
.
EphA2 in breast cancer: implications and therapeutic opportunities
The involvement of EphA2 in breast cancer has revealed its significant role within the Eph kinase family, which has been extensively researched. Breast cancer, which is the second leading cause of cancer-related deaths in women globally and ranks following lung cancer, frequently exhibits overexpression of EphA2 in tumors, with 40% of cases showing this occurrence. This overexpression is often associated with unfavorable prognosis, as demonstrated in studies [23,24,25]. This receptor exhibits both pro-oncogenic and anti-oncogenic properties, governing cancer initiation and metastasis through various signaling pathways. Phosphorylation of EphA2 via PI3K activation and AKT phosphorylation, independent of Ephrin, triggers an oncogenic signal that enhances EphA2-dependent cell migration and invasion [26]. EphA2 is a critical component in cellular processes, as it interacts with the Ras/ERK pathway and exerts both positive and negative modulation on ERK. Furthermore, its involvement extends to various proteins associated with cell migration, such as Src, FAK, GTPase, and AKT, thereby influencing cancer progression [27]. Additionally, EphA2 engages in molecular networks within cells, including the EGFR, FAK, and VEGF pathways, underscoring its significant role in angiogenesis, invasion, and metastasis. Notably, EphA2’s impact on angiogenesis is essential for tumor survival and growth because it intricately regulates the PI3K signaling pathway. Recent studies have highlighted the association between elevated EphA2 levels and enhanced angiogenesis, leading to an increase in microvessel counts in breast cancer [27, 28]. Prolonged trastuzumab treatment leads to EphA2 phosphorylation by Src kinase in vivo, activating the PI3K/AKT and MAPK pathways (Table 1) and causing trastuzumab resistance [28]. Interestingly, knockdowns of EphA2 in human breast cancer cells have been observed to reduce tumorigenicity [29]. Furthermore, the low-molecular-weight phosphotyrosine phosphatase (LMW-PTP) can dephosphorylate EphA2 forward signaling, transforming mammary epithelial cells into cancerous breast cancer cells [20]. At the cellular level, the phosphoprotein Anks1 promotes tumorigenesis by facilitating the trafficking of EphA2/Erbb2 complexes into COPII.
EphA2 in lung cancer: significance and therapeutic implications
Lung cancer, compared to breast, colorectal, and prostate cancer, stands as one of the most lethal malignancies globally. Non-small cell lung cancer (NSCLC), an aggressive and highly invasive carcinoma, exhibits a dismal 5-year survival rate of less than 15% [30], making up about 80% of all lung cancer cases. Eph receptors, discovered in the 1990s, have gained increasing recognition for their involvement in normal development and disease. Among the approximately 14 known Eph-receptor members in humans, EphA2 is notably more prevalent in lung cancer. According to genomic analyses, it is overexpressed in lung cancer and is linked to poor patient survival, smoking history, brain metastases, and disease relapse [30,31,32]. High EphA2 expression has been associated with developing brain metastases and advanced stages of NSCLC [33]. Notably, the EphA2 G391R mutation has been associated with the phosphorylation of a serine residue in mTOR, suggesting its potential role in EphA2 invasion. In lung cancer cells, EphA2 has been shown to regulate tumor cell motility and proliferation through the JNK-c-JUN pathway (Table 1) [33]. However, the exact signaling mechanism that activates JNK in lung cancer cells is not fully understood.
EphA2 is involved in various intracellular signaling pathways in cancer cells, and EphA2 ligand activation can suppress ERK and AKT phosphorylation, tumor cell proliferation, and motility in malignant glioma, prostate cancer, and lung cancer. In NSCLC, EphA2 is overexpressed in adherent cells, dependent on endothelial growth factor (EGFR) activation. Enhanced cell adhesion via EGFR leads to increased transcription of EphA2 mRNA through downstream activation of the MP1 and SRC signaling pathways. Additionally, EphA2 significantly inhibits hedgehog signaling-mediated cell proliferation and metastasis in lung adenocarcinoma [1].
The role of EphA2 in prostate cancer: implications for pathogenesis and therapeutics
Like breast cancer, prostate cancer (PCa) exhibits an overexpression of EphA2, making it the third most common cancer among men. PSA levels in the blood serve as a biomarker for PCa screening and clinical management [34]. Early studies have shown that PCa cell lines with high metastatic potential also display elevated levels of the EphA2-receptor protein. Distinguishing aggressive from non-aggressive tumors in PCa poses a challenge. The upregulation of EphA2 expression in prostate epithelial cells is associated with a more aggressive nature [35].
Similarly, EphA3 is also overexpressed in the more invasive PCa cell lines [36]. In PCa cells, tyrosine phosphorylation of EphA2 plays a crucial role in cell motility, adhesion, and invasion. Recent research indicates that EphA2 activation or expression in PC3 cell lines does not significantly affect cell proliferation regulation. However, EphA2 activation in PC3 prostate cancer cells triggers cell motility by activating the SRC/FAK complex (Table 1) [37]. According to recent research, transfection of cancer cells with EphA2 mutants, designed to inhibit EphA2 dimerization and activation, enhances their pro-migratory capabilities. To investigate the possibility of efficiently reversing EphA2 pro-migratory qualities in cell culture and metastasis in vivo, we used EphA2 dimerizing drugs, such as ephrinA1-Fc or 135H12. Over time, they observed that both drugs resulted in a sustained receptor decrease; however, both drugs considerably reduced the ability of PC-3 prostate cancer cells to migrate, with ephrinA1-Fc having a marginally higher effect. In contrast to ephrinA1-Fc (1 µg/mL, or approximately 22 nM of the dimeric macromolecule), 135H12 requires a comparatively high dosage (10 µM) to produce a considerable reduction in migration [38]. These findings suggest that agonistic drugs targeting EphA2 in prostate cancer have substantial therapeutic potential.
Additionally, EphA2 activation inhibits integrin-mediated cell attachment and spreads into the extracellular matrix (ECM) [39]. The phosphorylation of the EphA2 receptor in tumor cells is controlled by cell density, and mutants with faulty phosphorylation can inhibit EphA2 kinase in PCa. As discussed earlier, phosphorylated EphA2 plays a pivotal role in prostate cancer cells, affecting various clinical functions, including inhibiting cytoskeleton spreading, retraction fiber formation, and control of cell rounding [40].
EphA2 in brain cancer: implications in glioblastoma and medulloblastoma pathogenesis
EphA2 is known to be overexpressed in two of the most aggressive brain cancers: glioblastoma multiform (GBM) and medulloblastoma [41]. In GBM, EphA2 overexpression has been associated with a poor prognosis, as it is not observed in healthy brain tissue [41]. Upon binding of epidermal growth factor (EGF), EphA2 undergoes serine phosphorylation, which plays a crucial role in promoting tumor activities through the MEK/ERK and PI3K/AKT pathways, leading to enhanced cell migration and invasion [21, 41]. EphA2’s molecular actions in GBM also involve decreased ERK phosphorylation, altered Akt interaction, and modified stem cell invasiveness. It suggests that EphA2 regulates stemness and contributes to the glioma phenotype, with these effects being suppressed upon siRNA-mediated knockdown of EphA2. Upon binding with its ligand, EphA2 internalizes, leading to alterations in GBM cell morphology, migration, and adhesion [42].
Medulloblastoma, a subtype of glioma and a pediatric brain tumor, is also influenced by Eph receptors. EphA2 expression in medulloblastoma is associated with vasculogenic mimicry (VM), invasion, migration, and signaling pathways through metalloproteinase-2 (MMP-2) and PI3K (Table 1) [7]. On the other hand, EphA3 overexpression has been linked to glioblastoma, where it prevents the activation of the MAKP pathway, maintaining tumor cells in a dedifferentiated tumorigenicity state [1].
EphA2 in colorectal cancer: emerging insights and therapeutic implications
Colorectal cancer (CRC) is the most commonly diagnosed disease in both men and women, ranking as the fourth leading cause of cancer-related death worldwide. Approximately 30% of CRC cases contribute significantly to CRC-related mortality [43]. Over the past few decades, numerous studies have investigated the role of the Eph-Ephrin system in CRC, but its complete functionality remains unclear. Among the Eph receptors studied in CRC, EphA1 and EphA2 have been the focus of research. Recent studies have associated EphA2 overexpression with poor patient survival, particularly in the early stages of the disease. It suggests a pro-angiogenic effect, as the elevated EphA2 expression also correlates with increased microvessel formation [44]. Carcino Embryonic Antigen Cell Adhesion Molecule-1 (CEACAM1), a cell adhesion molecule, has been linked to the growth and spread of CRC tumors [45]. As a member of the multifunctional CEA family, CEACAM functions as a negative regulator of various signaling pathways. The signaling role of EphA2 in CRC can be either anti-oncogenic or pro-oncogenic, depending on the presence of ligands [46]. In a ligand-dependent manner, EphA2 can suppress ERK/MAPK and PI3K/AKT/mTOR signaling pathways activation through autophosphorylation, displaying an anti-oncogenic function.
Conversely, serine residue phosphorylation can activate these pathways, leading to cell migration and metastasis, representing a pro-oncogenic function. CEACAM’s role in tumor growth is contradictory, and its downregulation has been associated with the early stages of CRC tumor development (Table 1) [45, 47]. As a co-receptor, CEACAM is believed to modulate EphA2 signaling, and the inhibition of EphA2 has been shown to significantly reduce levels of phosphorylated CEACAM [47].
EphA2 expression in ovarian cancer: clinical significance and prognostic implications
Ovarian cancer is a relatively uncommon but hazardous form of cancer, often referred to as the “silent killer” due to its difficulty in early detection. In recent research, the receptor tyrosine kinase (RTK) receptor EphA2 has emerged as a significant player in the development and progression of ovarian and uterine cancer tumors. Studies have shown that approximately 75% of invasive tumors express EphA2, while non-invasive tumors do not exhibit this expression. It suggests that EphA2 expression may be associated with the aggressiveness of ovarian carcinoma, indicating its potential as a prognostic marker [48]. Moreover, investigations have revealed that EphA2 overexpression contributes to various cancer-promoting activities. It has been found that elevated EphA2 levels enhance cellular invasiveness, meaning cancer cells become more adept at penetrating surrounding tissues and spreading to distant sites. Additionally, in experimental settings, ovarian cancer cells with higher levels of EphA2 exhibit increased tumor growth, both in laboratory cultures and in vivo, when tested in animal models. These findings indicate that EphA2 is crucial in fostering tumor progression and metastasis in ovarian cancer [49]. The growing understanding of EphA2’s involvement in ovarian cancer presents new opportunities for targeted therapeutic strategies. By targeting EphA2, researchers aim to develop novel treatments that could specifically inhibit the cancer-promoting activities of this receptor, potentially leading to improved outcomes for patients with ovarian cancer. However, further research is required to fully elucidate the molecular mechanisms underlying EphA2’s actions in ovarian cancer and to develop effective and safe therapeutic interventions.
EphA2 expression in gastric cancer: clinical relevance and prognostic significance
Gastric cancer is the third leading cause of cancer-related deaths worldwide and the fifth most common malignant tumor. Various receptor tyrosine kinases (RTKs) such as EGFR, HER2, and MET have been implicated in gastric cancer progression due to alterations in their activity [50,51,52]. Among the RTK family, Eph-receptors, including EphA and EphB receptors, are frequently overexpressed in human cancers.
EphA2, a member of the Eph-receptor family, has been identified to play a significant role in facilitating the epithelial-mesenchymal transition (EMT) in gastric cancer cells. Blocking EphA2 has been shown to inhibit the proliferation and invasion of gastric cancer cells [53]. However, the exact mechanisms regulating EphA2 expression remain uncertain. Recent research has identified EphA2 overexpression as a prognostic marker in gastric cancer. Studies have also demonstrated that inhibiting EphA2 in gastric cancer cell lines can significantly impact various biological processes, including cell cycle progression, proliferation, and invasiveness [53]. Another noteworthy finding is that the microRNA (miR-125a-5p) targets EphA2 to downregulate its expression and prevent gastric cancer growth. When combined with trastuzumab, miR-125a-5p inhibits the AKT downstream signaling of ERBB2, effectively curbing gastric cancer cell growth.
Furthermore, the knockdown of EphA2 using siRNA has shown promise in reducing gastric cancer cells’ invasiveness and metastatic potential in vitro [53]. These insights into EphA2’s involvement in gastric cancer provide potential avenues for targeted therapies. By focusing on EphA2 and its regulatory mechanisms, researchers aim to develop novel strategies to inhibit the growth and spread of gastric cancer, offering new hope for improved treatment options for patients with this devastating disease.
EphA2 expression in melanoma: implications for tumor progression and prognosis
Melanoma, the most dangerous form of skin cancer, originates from melanocyte cells responsible for producing melanin, the pigment that gives skin its color. In India, approximately 10 cases of melanoma are reported each year. Emerging research has unveiled the significance of EphA2 in melanoma progression, as it is overexpressed in melanoma cells.
EphA2 overexpression has been implicated in promoting several critical aspects of melanoma malignancy. It is crucial in encouraging migration, colony formation, and proliferation, ultimately leading to a malignant phenotype [77]. Furthermore, EphA2 is notably upregulated in aggressive melanoma tumor cells, a finding consistently observed by different researchers. This heightened expression has been correlated with increased invasion, proliferation, and vasculogenic mimicry (VM), all hallmarks of a metastatic tumor cell phenotype [78]. Interestingly, experimental studies have demonstrated that down-regulation of EphA2 significantly impacts melanoma behavior. Inhibition of EphA2 expression leads to diminished tumorigenic potential in in vivo models and a concomitant decrease in invasion, proliferation, and colony formation in vitro [78]. These findings underscore the critical role EphA2 plays in driving melanoma growth and metastasis. Despite these significant observations, the precise molecular mechanisms EphA2 influences melanoma proliferation are yet to be fully elucidated. Unraveling the intricate signaling pathways and molecular interactions involving EphA2 in melanoma could open new avenues for targeted therapeutic interventions. Developing strategies to inhibit EphA2’s pro-oncogenic activities may hold promise in curbing melanoma’s aggressive behavior, offering hope for more effective treatments for this deadly disease. Continued research in this area is vital to advance our understanding and improve clinical outcomes for patients battling melanoma.
Pharmaceutical approaches to combating and safeguarding against cancer
There are two strategies for suppressing the carcinogenic effects of EphA2: Eph-Ephrin interaction disruption and inhibition of EpA2 kinase activity. Activating the constitutive RAF-MAPK or MEK1 pathway may prevent AKT activation and cellular arrest. Table 2 displays a compilation of inhibitors acknowledged for their effectiveness in blocking EphA2.
Dasatinib
Dasatinib is recognized as an inhibitor of both the Brc/Abl and Src kinase families, and recent studies have revealed its ability to inhibit EphA2, a member of the receptor tyrosine kinase family [54]. This kinase inhibitor, Dasatinib, effectively hinders the phosphorylation of EphA2, as depicted in Fig. 3, and its regulatory function has been observed in cases of pancreatic and melanoma cancer. According to existing literature, the application of Dasatinib to EphA2 has been linked to the suppression of cellular proliferation, migration, and invasion [55, 56]. Furthermore, Dasatinib has been associated with promoting EphA2 internalization and degradation. Recent research on glioblastoma cells has demonstrated that the novel EphA2 inhibitor 4a, derived from Dasatinib, retains its inhibitory actions against EphA2 [79].
ALW-11-41-27
ALW-11-41-27 has been identified as an effective inhibitor of EphA2 kinase activities in non-small cell lung and lung cancer. Notably, this compound exhibits structural similarities to Dasatinib. Recent research has unveiled that ALW-11-41-27 successfully hampers the phosphorylation of Tyr-588 on EphA2. Many studies have consistently demonstrated that ALW-11-41-27 can impede tumor growth in vivo by effectively restraining cell survival, proliferation, and migration [57].
Moreover, it has come to light that the EphA2 inhibitor ALW-11-41-27 exerts a regulatory influence on PI-IBS (post-inflammatory irritable bowel syndrome), both in experimental models and in laboratory settings. It suggests its potential as a therapeutic agent for treating and preventing PI-IBS.
Cholanic acid, GW4064, 76D10 and Uni-PR1331
In the context of prostate cancer, specific compounds have exhibited the ability to inhibit EphA2 phosphorylation and induce cell retraction, as evidenced by studies [58,59,60]. Notably, Cholanic acid, GW4064, and 76D10 have all shown efficacy. Additionally, Uni-PR1331 has demonstrated its capacity to hinder EphA phosphorylation and deactivate the process by impairing angiogenesis [61].
GW4064, classified as a stilbene carboxylic acid, disrupts the EphA2-Ephrin A1 complex in a dose-dependent manner. This disruption leads to the phosphorylation of Ephrin A1 and simultaneously blocks EphA2 activation. On the other hand, 76D10, a derivative of salicylic acid with a furanyl structure, acts by inhibiting Eph-Ephrin interactions and, consequently, EphA2-mediated cell retraction (Table 2) [60].
Doxazosin
A noteworthy small-molecule antagonist targeting EphA2 is Doxazosin. Interestingly, Doxazosin induces the catalytic activation of EphA2, which occurs independently of its antagonistic effects on alpha-1 adrenoreceptors. Furthermore, Doxazosin facilitates the internalization of EphA2 receptors and effectively inhibits ERK/AKT kinase activity, mirroring the cellular responses seen with Ephrin-A1-induced cell rounding.
In in vivo experiments, the impact of Doxazosin is quite remarkable. It significantly reduces prostate cancer cell migration and suppresses the occurrence of distant metastases, as summarized in Table 2 [62, 63].
GLPG1790
There is a promising orally administered small drug capable of inhibiting Eph-receptor activities. Both in vitro and in vivo investigations have demonstrated the significant potential of GLPG790 in curbing the progression of tumor cells. It achieves this by promoting a state of dormancy in HCT15 cells and reducing the phosphorylation and activation of EphA2. Moreover, GLPG1790 has been shown to induce G1/S cell cycle growth arrest, specifically in colorectal cancer [64].
In studies involving tumors originating from HCT15 or HCT116, GLPG1790 has exhibited robust inhibitory effects on EphA2 phosphorylation [65].
SWL dimer
Among the EphA2 antagonists under consideration, the SWL dimer stands out as a fascinating therapeutic candidate. It possesses the unique quality of inducing EphA2 tyrosine phosphorylation in in vitro studies [66]. This distinctive characteristic sets it apart. SWL is a 12-mer peptide that interacts with the LBD epitope of EphA2.
MGCD516 (sitravatinib)
Sitravinib is an innovative multi-targeted kinase inhibitor designed to address a range of sarcoma-related kinases. Its targets include PDFGR, VEGER, and several Eph-receptors like EphA2, EphA3, EphA4, and EphB2 receptors. Notably, Sitravinib exhibits a favorable safety profile, with most gastrointestinal side effects, such as nausea and diarrhea.
MGCD16, a compound related to Sitravinib, has demonstrated a significant reduction in cell proliferation [67]. Moreover, in vivo studies have suggested that MSCD516, another derivative, outperforms crizotinib and imatinib in inhibiting tumor growth more effectively [67].
Lithocholic acid
Lithocholic acid (LCA), identified as an EphA2 antagonist, is generated through the prokaryotic transformation of chenodeoxycholic acid. As reported, LCA competitively and reversibly hinders the interaction between EphA2 and EphrinA1 while leaving EphA2 kinase activity unaffected. Additionally, when LCA is coupled with an amino acid, it interferes with the binding between EphrinA1 and Eph2, suppressing Eph2 phosphorylation [4, 68].
WW437
WW437 exhibits potent anticancer properties, effectively impeding tumorigenesis, invasion, proliferation, and various hallmark cancer characteristics, such as the epithelial-to-mesenchymal transition. In breast cancer, WW437 significantly suppresses EphA2 signaling and disrupts the HDAC2/4-EphA2 axis. Furthermore, it has been observed to substantially reduce both tumor growth and metastasis in breast cancer cell models [69].
Sorafenib
Sorafenib is an antineoplastic medication employed in treating advanced liver cancer and other malignancies that are not amenable to surgical intervention. This oral multikinase inhibitor is proficient at triggering cancer cell apoptosis while concurrently curbing tumor cell proliferation and angiogenesis. Critical pathways like EGFR and VEGFR are pivotal in driving the progression of various epithelial cancers, including non-small cell lung cancer (NSCLC) and colorectal cancer (CRC). Sorafenib, however, exerts its effects by targeting numerous receptor tyrosine kinases (RTKs) implicated in the formation of new blood vessels, including VEGFR-2 and VEGFR-3 (as illustrated in Fig. 3) [81].
Additionally, the PI3K/AKT pathway, which governs cell apoptosis, is of paramount importance. Recent research has demonstrated that sorafenib can inhibit AKT by impeding the JAK/STAT pathway (Fig. 3) [79, 80].
Lapatinib
Lapatinib is another notable kinase inhibitor employed in cancer treatment, functioning to halt or decelerate the spread of cancer cells by obstructing the aberrant protein signals that drive cancer cell multiplication. Among the inhibitors mentioned earlier, Lapatinib is the sole small molecule HER2-targeting tyrosine kinase inhibitor (TKI) approved for breast cancer treatment. This dual-action inhibitor competes with ATP binding and effectively targets the tyrosine kinase domains of both EGFR and HER2 (Fig. 3) [82].
Through interaction with these receptors, Lapatinib curtails phosphorylation, interrupting their ability to transmit signals via the PI3K/Akt and MAPK pathways (Fig. 3). Consequently, cell proliferation is suppressed, and apoptosis is initiated [83].

Cancer treatment agents, including Dasatinib, ALW, GLPG1790, Lithocholic acid, and MGCD 516, target distinct cancer-related pathways
EphA2 degradation
Synthetic ligands or antibodies can inhibit EphA2 signaling by facilitating the internalization or degradation of EphA2.
Ephrin-A1-Fc and Ephrin-A1-Soluble
Depending on the nature of cell-cell interactions, Ephrin A1 is typically found at deficient levels within cells and possesses tumor-suppressive attributes. Notably, the degradation of Ephrin A2 can be significantly expedited using soluble Ephrin A1, achieved through artificial dimerization and antibody targeting of the ECD epitope [70].
Utilizing recombinant Ephrin A1 human IgG-Fc for clustering, soluble Ephrin A1 and Ephrin A1-Fc are generated, conferring an Ephrin-like property. This process enhances EphA2 phosphorylation and reduces EphA2 protein production, collectively restraining gastric cancer progression [71].
Antibodies
Monoclonal antibodies (mAbs) designed to act as agonists against EphA2 exhibit the ability to counteract its internalization and degradation, thereby thwarting the suppression of EphA2’s oncogenic characteristics. One such mAb, EA5, effectively impedes EphA2 receptor actions independent of ligands, thus preventing receptor degradation. EA5 mAb treatment has been demonstrated to halt the growth of specific tumor cell lines [72]. Additionally, research reveals that mAb EA5 therapy reduces EphA2 expression in ovarian cancer by more than 90% in the cases studied [73].
Furthermore, another monoclonal antibody called EA2, developed through immunizing mice with the EphA2 protein, demonstrates the ability to induce cell-cell adhesion and exhibits selective binding properties [74]. In a related vein, B233, another recently discovered anti-EphA2 mAb, prompts the phosphorylation and degradation of EphA2 (as outlined in Table 3) [74].
Collectively, these scientific studies underscore the potential of monoclonal antibodies with agonistic properties against the EphA2 receptor to mitigate its internalization, degradation, and dampening of its proliferative activity [72]. Notably, mAb EA5 is a potent therapeutic candidate for curbing EphA2 expression in ovarian cancer tumors [73, 74].
Conclusion
Our current understanding of EphA2’s intricate workings reveals a dependency on a complex interplay between the receptor, its ligands, various signaling pathways, adaptor proteins, and potential drug targets. Notably, the expression of EphA2 receptors has been detected at different stages of multiple diseases, with the type of malignancy influencing the distinct patterns of receptor expression. Furthermore, Eph receptors and their ligands have emerged as influential factors in angiogenesis, tumorigenesis, and tumor angiogenesis processes.
Prospects for the future: unleashing the therapeutic potential of eph receptors and Ephrin ligands
In the past decade, exploring Eph receptors and Ephrin ligands has led to remarkable advancements in antitumor and antiangiogenic therapies. These breakthroughs have cast Eph receptors, also called Ephrins, into the limelight of numerous therapeutic approaches. As we delve deeper into understanding EphA2’s role in cancer and angiogenesis, we can envision a promising future where targeted therapies utilizing these molecular components will play a pivotal role in improving the effectiveness of cancer treatments. This journey can potentially open new horizons for personalized medicine and enhance the outcomes for patients grappling with various types of cancer.
Data availability
There is no supplementary data available.
References
Chow AY (2010) Cell cycle control by oncogenes and Tumor suppressors: driving the Transformation of normal cells into cancerous cells. Nat Educ. 3(9).
Brantley-Sieders DM (2012) Clinical relevance of ephs and ephrins in cancer: lessons from breast, colorectal, and lung cancer profiling. Semin Cell Dev Biol 23(1):102–108
Xiao T, Xiao Y, Wang W, Tang YY, Xiao Z, Su M (2020) Targeting EphA2 in cancer. J Hematol Oncol 13(1):114
Barquilla A, Pasquale EB (2015) Eph receptors and ephrins: Therapeutic opportunities. Annu Rev Pharmacol Toxicol 55(1):465–487
Wykosky J, Debinski W (2008) The EphA2 receptor and EphrinA1 ligand in solid tumors: function and therapeutic targeting. Mol Cancer Res 6(12):1795–1806
Adu-Gyamfi EA, Czika A, Liu T-H, Gorleku PN, Fondjo LA, Djankpa FT et al (2021) Ephrin and eph receptor signaling in female reproductive physiology and pathology†. Biol Reprod 104(1):71–82
Zhou Y, Sakurai H (2017) Emerging and diverse functions of the EphA2 noncanonical pathway in Cancer Progression. Biol Pharm Bull 40(10):1616–1624. https://doi.org/10.1248/bpb.b17-00446
Barton WA, Dalton AC, Seegar TCM, Himanen JP, Nikolov DB (2014) Tie2 and eph receptor tyrosine kinase activation and signaling. Cold Spring Harb Perspect Biol 6(3):a009142–a009142. https://doi.org/10.1101/cshperspect.a009142
Wang X, Yennawar N, Hankey PA (2014) Autoinhibition of the Ron receptor tyrosine kinase by the juxtamembrane domain. Cell Communication Signaling: CCS 12(1):28. https://doi.org/10.1186/1478-811X-12-28
Falivelli G, Lisabeth EM, de la Torre ER, Perez-Tenorio G, Tosato G, Salvucci O, Pasquale EB (2013) Attenuation of eph receptor kinase activation in Cancer cells by Coexpressed Ephrin Ligands. PLoS ONE 8(11):e81445. https://doi.org/10.1371/journal.pone.0081445
Herath NI, Spanevello MD, Doecke JD, Smith FM, Pouponnot C, Boyd AW (2012) Complex expression patterns of eph receptor tyrosine kinases and their ephrin ligands in colorectal carcinogenesis. Eur J Cancer 48(5):753–762. https://doi.org/10.1016/j.ejca.2011.07.003
Van Landeghem L, Mahé MM, Teusan R, Léger J, Guisle I, Houlgatte R, Neunlist M (2009) Regulation of intestinal epithelial cells transcriptome by enteric glial cells: impact on intestinal epithelial barrier functions. BMC Genomics 10(1):507. https://doi.org/10.1186/1471-2164-10-507
Plum PS, Warnecke-Eberz U, Dhaouadi O, Alakus H, Drebber U, Metzger R, Prenzel KL, Hölscher AH, Bollschweiler E (2015) Molecular markers predicting lymph node metastasis in early esophageal cancer. Histol Histopathol 30(10):1193–1202. https://doi.org/10.14670/HH-11-618
Kurose h, ueda (2019) k., kondo, r., ogasawara, s., kusano, h., sanada, s., naito, y., nakiri, m., nishihara, k., kakuma, t., akiba, j., igawa, t., & yano, h. Elevated expression of epha2 is associated with poor prognosis after radical prostatectomy in prostate cancer. Anticancer research 39(11):6249–6257. https://doi.org/10.21873/anticanres.13834
Zhou L, Lu X, Zhang B, Shi Y, Li Z (2021) EphA2 as a new target for breast cancer and its potential clinical application. Int J Clin Exp Pathol, 14(4)
Lindberg RA, Hunter T (1990) cDNA cloning and characterization of eck, an epithelial cell receptor protein-tyrosine kinase in the eph/elk family of protein kinases. Mol Cell Biol 10(12):6316–6324
Gomez-Soler M, Pasquale EB (2021) Eph receptors and Ephrins. Encyclopedia of Molecular Pharmacology. Springer International Publishing, Cham, pp 615–628
Signaling Crosstalk. In (2013) Encyclopedia of systems Biology. Springer New York, New York, NY, pp 1938–1938
Miao H, Li D-Q, Mukherjee A, Guo H, Petty A, Cutter J et al (2009) EphA2 mediates ligand-dependent inhibition and ligand-independent Promotion of Cell Migration and Invasion via a reciprocal Regulatory Loop with Akt. Cancer Cell 16(1):9–20
Zhou Y, Sakurai H (2017) Emerging and diverse functions of the EphA2 noncanonical pathway in Cancer Progression. Biol Pharm Bull 40(10):1616–1624
Nakamoto M, Bergemann AD (2002) Diverse roles for the Eph family of receptor tyrosine kinases in carcinogenesis. Microsc Res Tech 59(1):58–67
Brantley-Sieders DM, Zhuang G, Hicks D, Fang W, Bin, Hwang Y, Cates JMM et al (2008) The receptor tyrosine kinase EphA2 promotes mammary adenocarcinoma tumorigenesis and metastatic progression in mice by amplifying ErbB2 signaling. J Clin Invest 118(1):64–78
Xu N-J, Henkemeyer M (2012) Ephrin reverse signaling in axon guidance and synaptogenesis. Semin Cell Dev Biol 23(1):58–64
Brantley-Sieders DM, Jiang A, Sarma K, Badu-Nkansah A, Walter DL, Shyr Y et al (2011) Eph/Ephrin Profiling in Human Breast Cancer Reveals Significant Associations between Expression Level and Clinical Outcome. Zhang L, editor. PLoS One. 6(9):e24426. A
Carles-Kinch K, Kilpatrick KE, Stewart JC, Kinch MS (2002) Antibody targeting of the EphA2 tyrosine kinase inhibits malignant cell behavior. Cancer Res. 62(10)
Pasquale EB (2008) Eph-ephrin bidirectional signaling in physiology and disease. Cell 133(1):38–52
De Robertis M, Loiacono L, Fusilli C, Poeta ML, Mazza T, Sanchez M, Marchionni L, Signori E, Lamorte G, Vescovi AL, Garcia-Foncillas J, Fazio VM (2017) Dysregulation of EGFR pathway in EphA2 cell subpopulation significantly associates with poor prognosis in Colorectal Cancer. Clin Cancer Res 23(1):159–170. https://doi.org/10.1158/1078-0432.CCR-16-0709
Shi X, Lingrak R, Cuizon C, Toth P, Zheng J, Smith A et al (2020) Functional oligomerization of the EphA2 receptor tyrosine kinase. Biophys J 118(3):97a
D’Addario G, Felip E (2009) Non-small-cell lung cancer: ESMO Clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol 20(SUPPL 4):iv68–70
Kinch MS, Moore MB, Harpole DH (2003) Predictive value of the EphA2 receptor tyrosine kinase in lung cancer recurrence and survival. Clin Cancer Res. 9(2)
Faoro L, Singleton PA, Cervantes GM, Lennon FE, Choong NW, Kanteti R et al (2010) EphA2 mutation in lung squamous cell Carcinoma promotes increased cell survival, Cell Invasion, focal adhesions, and mammalian target of Rapamycin activation. J Biol Chem 285(24):18575–18585
Song W, Ma Y, Wang J, Brantley-Sieders D, Chen J (2014) JNK Signaling mediates EPHA2-Dependent Tumor Cell Proliferation, Motility, and Cancer Stem cell–like properties in non–small cell Lung Cancer. Cancer Res 74(9):2444–2454
Cary KC, Cooperberg MR (2013) Biomarkers in prostate cancer surveillance and screening: past, present, and future. Ther Adv Urol 5(6):318–329
Zeng G, Hu Z, Kinch MS, Pan C-X, Flockhart DA, Kao C et al (2003) High-level expression of EphA2 receptor tyrosine kinase in Prostatic Intraepithelial Neoplasia. Am J Pathol 163(6):2271–2276
Anderton M, van der Meulen E, Blumenthal MJ, Schäfer G (2021) The role of the eph receptor family in Tumorigenesis. Cancers (Basel) 13(2):206
Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G et al (2007) EphrinA1 activates a Src/Focal Adhesion Kinase-Mediated Motility Response Leading to rho-dependent Actino/Myosin contractility. J Biol Chem 282(27):19619–19628
Buricchi F, Giannoni E, Grimaldi G, Parri M, Raugei G, Ramponi G et al (2007) Redox Regulation of Ephrin/Integrin cross-talk. Cell Adh Migr 1(1):33–42
Salem AF, Gambini L, Billet S, Sun Y, Oshiro H, Zhao M, Hoffman RM, Bhowmick NA, Pellecchia M (2020) Prostate cancer metastases are strongly inhibited by agonistic EPHA2 ligands in an orthotopic mouse model. Cancers 12(10). https://doi.org/10.3390/cancers12102854
Li J-Y, Xiao T, Yi H-M, Yi H, Feng J, Zhu J-F et al (2019) S897 phosphorylation of EphA2 is indispensable for EphA2-dependent nasopharyngeal carcinoma cell invasion, metastasis and stem properties. Cancer Lett 444:162–174
Hamaoka Y, Negishi M, Katoh H (2016) EphA2 is a key effector of the MEK/ERK/RSK pathway regulating glioblastoma cell proliferation. Cell Signal 28(8):937–945
Binda E, Visioli A, Giani F, Lamorte G, Copetti M, Pitter KL et al (2012) The EphA2 receptor drives Self-Renewal and Tumorigenicity in stem-like tumor-propagating cells from human glioblastomas. Cancer Cell 22(6):765–780
Wang S, Yu L, Ling G, Xiao S, Sun X, Song Z et al (2012) Vasculogenic mimicry and its clinical significance in medulloblastoma. Cancer Biol Ther 13(5):341–348
Allemani C, Rachet B, Weir HK, Richardson LC, Lepage C, Faivre J et al (2013) Colorectal cancer survival in the USA and Europe: a CONCORD high-resolution study. BMJ Open 3(9):e003055
Kataoka H, Igarashi H, Kanamori M, Ihara M, Wang J-D, Wang Y-J et al (2004) Correlation of EPHA2 overexpression with high microvessel count in human primary colorectal cancer. Cancer Sci 95(2):136–141
Beauchemin N, Arabzadeh A (2013) Carcinoembryonic antigen-related cell adhesion molecules (CEACAMs) in cancer progression and metastasis. Cancer Metastasis Rev 32(3–4):643–671
Kim WM, Huang Y-H, Gandhi A, Blumberg RS (2019) CEACAM1 structure and function in immunity and its therapeutic implications. Semin Immunol 42:101296
Arabzadeh A, McGregor K, Breton V, Van Der Kraak L, Akavia UD, Greenwood CMT et al (2017) EphA2 signaling is impacted by carcinoembryonic antigen cell adhesion molecule 1-L expression in colorectal cancer liver metastasis in a cell context-dependent manner. Oncotarget 8(61):104330–104346
Wu Y-M, Liu C-H, Huang M-J, Lai H-S, Lee P-H, Hu R-H et al (2013) C1GALT1 enhances proliferation of Hepatocellular Carcinoma Cells via modulating MET glycosylation and dimerization. Cancer Res 73(17):5580–5590
Lu C, Shahzad MMK, Wang H, Landen CN, Kim SW, Allen J et al (2008) EphA2 overexpression promotes ovarian cancer growth. Cancer Biol Ther 7(7):1098–1103
Liu S-Y, Shun C-T, Hung K-Y, Juan H-F, Hsu C-L, Huang M-C et al (2016) Mucin glycosylating enzyme GALNT2 suppresses malignancy in gastric adenocarcinoma by reducing MET phosphorylation. Oncotarget 7(10):11251–11262
Huang M-J, Hu R-H, Chou C-H, Hsu C-L, Liu Y-W, Huang J et al (2015) Knockdown of GALNT1 suppresses malignant phenotype of hepatocellular carcinoma by suppressing EGFR signaling. Oncotarget 6(8):5650–5665
Yuan W, Chen Z, Chen Z, Wu S, Guo J, Ge J et al (2012) Silencing of EphA2 inhibits invasion of human gastric cancer SGC-7901 cells in vitro and in vivo. Neoplasma 59(01):105–113
Harb OA, Atwa HA, Haggag R, Shorbagy S, El, Abdelaziz LA, Balata SA et al (2017) The value of HER2 neu and EphA2 expressions in gastric adenocarcinoma prognosis. J Gastrointest Dig Syst. 07(02)
Hossain A, Khan HTA (2016) Identification of genomic markers correlated with sensitivity in solid tumors to Dasatinib using sparse principal components. J Appl Stat 43(14):2538–2549
Buettner R, Mesa T, Vultur A, Lee F, Jove R (2008) Inhibition of src family kinases with Dasatinib Blocks Migration and Invasion of Human Melanoma Cells. Mol Cancer Res 6(11):1766–1774
Chang Q, Jorgensen C, Pawson T, Hedley DW (2008) Effects of Dasatinib on EphA2 receptor tyrosine kinase activity and downstream signaling in pancreatic cancer. Br J Cancer 99(7):1074–1082
Amato KR, Wang S, Tan L, Hastings AK, Song W, Lovly CM et al (2016) EPHA2 blockade overcomes Acquired Resistance to EGFR kinase inhibitors in Lung Cancer. Cancer Res 76(2):305–318
Tognolini M, Incerti M, Hassan-Mohamed I, Giorgio C, Russo S, Bruni R et al (2012) Structure-activity relationships and mechanism of action of eph-ephrin antagonists: Interaction of Cholanic Acid with the EphA2 receptor. ChemMedChem 7(6):1071–1083
Tognolini M, Incerti M, Pala D, Russo S, Castelli R, Hassan-Mohamed I et al (2014) Target Hopping as a useful Tool for the identification of Novel EphA2 protein-protein antagonists. ChemMedChem 9(1):67–72
Noberini R, De SK, Zhang Z, Wu B, Raveendra-Panickar D, Chen V et al (2011) A disalicylic acid-Furanyl Derivative inhibits ephrin binding to a subset of eph receptors. Chem Biol Drug Des 78(4):667–678
Festuccia C, Gravina GL, Giorgio C, Mancini A, Pellegrini C, Colapietro A et al (2018) UniPR1331, a small molecule targeting Eph/ephrin interaction, prolongs survival in glioblastoma and potentiates the effect of antiangiogenic therapy in mice. Oncotarget 9(36):24347–24363
Petty A, Myshkin E, Qin H, Guo H, Miao H, Tochtrop GP et al (2012) A Small Molecule Agonist of EphA2 Receptor Tyrosine Kinase Inhibits Tumor Cell Migration In Vitro and Prostate Cancer Metastasis In Vivo. Ling MT, editor. PLoS One. 7(8):e42120
Buckens OJ, El Hassouni B, Giovannetti E, Peters GJ (2020) The role of eph receptors in cancer and how to target them: novel approaches in cancer treatment. Expert Opin Investig Drugs 29(6):567–582
Nelson H (1997) Proliferating cell nuclear antigen expression and its relationship to malignancy potential in invasive colorectal carcinomas, vol 40. Diseases of the Colon and Rectum
Colapietro A, Gravina GL, Petragnano F, Fasciani I, Scicchitano BM, Beirinckx F et al (2020) Antitumorigenic effects of inhibiting ephrin receptor kinase signaling by GLPG1790 against Colorectal Cancer Cell Lines in Vitro and in vivo. J Oncol 2020:1–16
Duggineni S, Mitra S, Lamberto I, Han X, Xu Y, An J et al (2013) Design and synthesis of potent bivalent peptide agonists targeting the EphA2 receptor. ACS Med Chem Lett 4(3):344–348
Patwardhan PP, Ivy KS, Musi E, de Stanchina E, Schwartz GK (2016) Significant blockade of multiple receptor tyrosine kinases by MGCD516 (Sitravatinib), a novel small molecule inhibitor, shows potent antitumor activity in preclinical models of sarcoma. Oncotarget 7(4):4093–4109
Chiang JYL, Ferrell JM (2020) Bile acid receptors FXR and TGR5 signaling in fatty liver diseases and therapy. Am J Physiol Liver Physiol 318(3):G554–G573
Zhang T, Li J, Ma X, Yang Y, Sun W, Jin W et al (2018) Corrigendum to ‘Inhibition of HDACs-EphA2 signaling axis with WW437 demonstrates promising preclinical antitumor activity in breast cancer’ [EBioMedicine 31 276–286]. EBioMedicine. 2020;52:102629
Davis S, Gale NW, Aldrich TH, Maisonpierre PC, Lhotak V, Pawson T et al (1994) Ligands for EPH-Related receptor tyrosine kinases that require membrane attachment or clustering for activity. Sci (80-) 266(5186):816–819
Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE (2004) Ligation of EphA2 by ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun 320(4):1096–1102
Merritt WM, Kamat AA, Hwang J-Y, Bottsford-Miller J, Lu C, Lin YG et al (2010) Clinical and biological impact of EphA2 overexpression and angiogenesis in endometrial cancer. Cancer Biol Ther 10(12):1306–1314
Landen CN, Lu C, Han LY, Coffman KT, Bruckheimer E, Halder J et al (2006) Efficacy and Antivascular effects of EphA2 Reduction with an agonistic antibody in Ovarian Cancer. JNCI J Natl Cancer Inst 98(21):1558–1570
Coffman KT, Hu M, Carles-Kinch K, Tice D, Donacki N, Munyon K et al (2003) Differential EphA2 Epitope Display on normal versus malignant cells. Cancer Res. 63(22)
Hasegawa J, Sue M, Yamato M, Ichikawa J, Ishida S, Shibutani T et al (2016) Novel anti-EPHA2 antibody, DS-8895a for cancer treatment. Cancer Biol Ther 17(11):1158–1167
Sakamoto A, Kato K, Hasegawa T (2018) An agonistic antibody to EPHA2 exhibits Antitumor effects on Human Melanoma cells. Anticancer Res 38(6):3273–3282
Udayakumar D, Zhang G, Ji Z, Njauw C-N, Mroz P, Tsao H (2011) Epha2 is a critical oncogene in melanoma. Oncogene 30(50):4921–4929
Margaryan NV, Strizzi L, Abbott DE, Seftor EA, Rao MS, Hendrix MJC et al (2009) EphA2 as a promoter of melanoma tumorigenicity. Cancer Biol Ther. 8(3)
Zhu Y, Zheng B, Wang H, Chen L (2017) New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin 38(5):614–622
Xu X, Li Y, Wu Y, Wang M, Lu Y, Fang Z et al (2023) Increased ATF2 expression predicts poor prognosis and inhibits sorafenib-induced ferroptosis in gastric cancer. Redox Biol 59:102564
Morgillo F, Martinelli E, Troiani T, Orditura M, de Vita F, Ciardiello F (2011) Antitumor activity of sorafenib in human cancer cell lines with acquired resistance to EGFR and VEGFR tyrosine kinase inhibitors. PLoS ONE. ;6(12)
D’Amato V, Raimondo L, Formisano L, Giuliano M, De Placido S, Rosa R et al (2015) Mechanisms of lapatinib resistance in HER2-driven breast cancer. Cancer Treat Rev 41(10):877–883
PJ M, Lapatinib SG (2008) : A dual inhibitor of human epidermal growth factor receptor tyrosine kinases. Clin Ther. 30(8)
Acknowledgements
The authors would like to express their heartfelt appreciation for the generous support of Integral Information and Research Centre-7 (IIRC-7) at Integral University Lucknow (IU/R&D/2023-MCN0002255).
Funding
The authors affirm that no funds, grants, or financial support were received during the preparation of this review article.
Author information
Authors and Affiliations
Contributions
M.N. wrote the review article. J. K, S.K., and M.K.A.K. edited and corrected the manuscript, while S.A. and M.K.A.K. also proofread.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not required.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Nehal, M., Khatoon, J., Akhtar, S. et al. Exploring the potential of EphA2 receptor signaling pathway: a comprehensive review in cancer treatment. Mol Biol Rep 51, 337 (2024). https://doi.org/10.1007/s11033-024-09298-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11033-024-09298-8