
Abstract
Multiple cancer signalling networks take part in regulatory crosstalks with the Hippo tumour suppressor pathway through the transcriptional cofactor Yes-associated protein (YAP). Nevertheless, how YAP is controlled by pathway crosstalks in tumourigenesis remains poorly understood. Here, we performed a targeted kinase inhibitor screen in human cancer cells to identify novel Hippo pathway regulators. Notably, we identified the nerve growth factor (NGF) receptor tyrosine kinase (NTRK1), a molecule not previously associated with Hippo signalling. NTRK1 inhibition decreased YAP-driven transcription, cancer cell proliferation and migration. Furthermore, using a complementary functional genomics approach and mouse xenograft models, we show that NTRK1 regulates YAP oncogenic activity in vivo. Mechanistically, NTRK1 inhibition was found to induce large suppressor kinase 1 (LATS1) phosphorylation and to control YAP subcellular localization. Taken together, these results provide compelling evidence of crosstalks between the NGF-NTRK1 and Hippo cancer pathways.
Similar content being viewed by others

Introduction
The Hippo signalling pathway is frequently deregulated in many types of cancer [1, 2]. The core kinase cascade of the Hippo pathway consists of large suppressor kinase 1/2 (LATS1/2), STE20-like kinase 1/2 (Mst1/2) and the RASSF family of proteins [3, 4]. Transcriptional coactivator Yes-associated protein (YAP) is a key downstream effector of the Hippo pathway and when dephosphorylated translocates to the nucleus and drives gene expression programs that control cell proliferation, cell migration and invasion. Comprehensive pan-cancer analyses of common cancer types have identified frequent and widespread YAP overexpression in colorectal, hepatocellular, lung, ovarian, pancreatic, and prostate carcinomas [5]. Consistent with these reports, we have previously shown that YAP is amplified in mouse mammary tumours [6]. Furthermore, the expression of constitutively active YAP induces epithelial-to-mesenchymal transition, apoptosis suppression and anchorage-independent growth [6, 7].
The upstream mechanisms controlling Hippo pathway activation, including post-translational YAP regulation, remain poorly characterized. Hence, a better understanding of the full topology (and identity) of pathways linked to Hippo signalling is required to decipher the oncogenic mechanisms and potential targets in a broad spectrum of cancers. Since YAP signalling depends on its phosphorylation status, we sought to identify novel signalling molecules/effectors that modulate YAP function by performing a target-specific kinase screen. Our experimental design used a cell-based reporter to quantify the effects of a panel of well-characterized kinase inhibitors on YAP activity in real time. Specifically, we used two metrics in this assay: (i) luciferase activity as assessed by using a YAP-regulated reporter [8] and (ii) cell proliferation measured by Resazurin assays. Using this approach, we identified a compound, Ro 08-2750, that robustly inhibited YAP target gene connective tissue growth factor (CTGF) promoter-driven luciferase activity. Ro 08-2750 is known to inhibit nerve growth factor (NGF) binding to neurotrophic receptor tyrosine kinase 1 (NTRK1) and p75NTR, and this binding regulates synaptic strength and plasticity in the mammalian nervous system [9, 10]. NTRK1 mediates the many effects of NGF signalling in nerve cells, e.g. promoting cell proliferation or differentiation [11], through receptor auto-phosphorylation and induction of MAPK/ERK and AKT/PKB signalling pathways [12]. Of particular interest, the overexpression of NGF and NGF receptors in cancers implicates the functional role of NGF signaling in non-neuronal carcinogenesis [13].
In the current report, we show that the inhibition of NTRK1 suppresses cancer cell proliferation and migration. This effect is mediated through the direct regulation of Hippo signalling and YAP phosphorylation. Also, we provide evidence of crosstalks between the Hippo and NGF-NTRK1 cancer pathways in vivo. Collectively, these results provide unique molecular insight into both pathways, with potential implications for identifying new targets for cancer diagnosis or treatment.
Results
Identification of Ro 08-2750 as a potent inhibitor of CTGF promoter-driven luciferase activity
Deregulation of the Hippo signalling pathway promotes tumour development and progression [4, 14]. Re-activation of Hippo pathway activity has been suggested as an anti-cancer therapy [3, 15]. To identify key regulatory kinases involved in YAP oncogenic function, we co-transfected 293AD cells with YAP and a luciferase reporter driven by the CTGF promoter [8]. CTGF is a well-known YAP target that mediates the pro-proliferative function of YAP [16, 17]. We measured the effect of 80 selective kinase inhibitors on luciferase reporter activity in transfected 293AD cells. Notably, one compound, Ro 08-2750, which blocks NGF binding to p75NTR and NTRK1 [9], inhibited CTGF promoter activity by ~ 80% (Fig. 1a) and in a dose-dependent manner (Fig. 1b); however, this compound had a negligible effect on 293AD cell proliferation (Supplementary Fig. 1A). Also, several other kinase inhibitors were identified that could inhibit CTGF promoter activity (Supplementary Fig. 1B). However, we decided to focus our efforts on Ro 08-2750, because our results implicated a novel functional interaction between NTRK1 and YAP.

Kinase inhibitor screen identifies Ro 08-2750 as a potent inhibitor of CTGF promoter-driven luciferase activity. a Luciferase activity measurement in duplicate in response to 10 µm kinase inhibitor treatments. b Immunoblot analyses were performed with anti-YAP or anti-GAPDH antibodies. The samples were lysates from vector- or YAP-WT-transfected 293AD cells. GAPDH was used as the loading control. c Luciferase activity was measured in response to DMSO-, 5 or 10 μm Ro 08-2750-treated CTGF promoter luciferase, renilar- and YAP-WT-co-transfected 293AD cells. Error bars represent SD; **P< 0.01; ***P < 0.001 by two-tailed Student’s t test
NTRK1 inhibition suppresses the proliferation and migration of PANC1 and MDA-MB231 cells
Effects of NGF-NTRK1 on tumour cells have been previously reported, but the results are conflicting [18,19,20]. Therefore, we first used a cell enumeration approach to examine the effects of NTRK1 inhibition on two distinct cancer cell lines with well-defined YAP activity: PANC1 [21] (pancreatic) and MDA-MB-231 (breast) cancer cells [22]. After 24, 48, and 72 h of Ro 08-2750 treatment (5 and 10 μM), cell proliferation was decreased in a dose- and time-dependent manner compared to that in the control and DMSO groups (P < 0.05) (Fig. 2a). Next, we determined the effects of NTRK1 inhibition on colony formation in PANC1 and MDA-MB231 cells. As expected, Ro 08-2750 decreased both the size and number of colonies (Fig. 2b). Additionally, the effects of Ro 08-2750 treatment on the cell migration potential of PANC1 and MDA-MB-231 cells were investigated using wound-healing assays. Ro 08-2750-treated cells failed to migrate after 24 h, whereas the mock-treated control cells filled the wound gap well (Fig. 2c). To corroborate the NTRK1 inhibition effects, we employed two additional NTRK1 small-molecule inhibitors GNF 5837 and AG 879, respectively. GNF 5837 and AG 879 treatment inhibited CTGF promoter reporter activity (Supplementary Fig. S2). Also, AG 879 treatment decreased MDA-MB-231 cell proliferation and migration (Supplementary Fig. S3B, C).

NTRK1 inhibition suppresses the cell proliferation and migration of PANC1 and MDA-MB231 cells. a Cell numbers were counted in DMSO-, 5 or 10 μm Ro 08-2750-treated PANC1 and MDA-MB231 cells every 24 h for 72 h. The experiment was repeated independently three times. Error bars represent SD; *P < 0.05, **P < 0.01 by two-tailed Student’s t test. b Representative images of colony formation assay for DMSO- or 10 μm Ro 08-2750-treated PANC1 and MDA-MB231 cells for three weeks (left panel). Quantifications of colony formation assay from three independent experiments (right panel). Error bars represent SD; **P < 0.01 by two-tailed Student’s t test. c Representative images of wound healing assay for DMSO- or 10 μm Ro 08-2750-treated PANC1 and MDA-MB231 cells for 24 h (left panel). Quantifications of wound closure estimation from three independent experiments (right panel). Error bars represent SD; *P < 0.05, **P < 0.01 by two-tailed Student’s t test
Effects of NTRK1 inhibition on the Hippo pathway
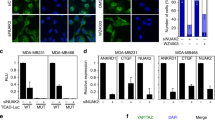
To test whether Ro 08-2750 activates the Hippo pathway, we measured the active forms of p-MST1/2 and p-LATS1, and the inactive form of p-YAP (S127). As shown in Fig. 3a, we found that although Ro 08-2750 treatment induced LATS1 and YAP1 phosphorylation, there were no notable effects on MST1/2 phosphorylation. Interestingly, Ro 08-2750 treatment decreased the relative nuclear YAP protein level and increased the cytoplasmic YAP level (Fig. 3b). In addition, the expression of several YAP target genes (e.g., ADMTS1, ANKRD1 and CYR61) was suppressed by Ro 08-2750 treatment in MDA-MB-231 and PANC1 cells (Fig. 3c). Furthemore, AG 879 treatment of MDA-MB-231 cells exhibited comparable effects (Supplementary Fig. S3A). Taken together, these results suggest that NTRK1 inhibition activates LATS1 and suppresses YAP transcriptional co-activator function(s).

NTRK1 inhibition suppresses YAP activity in PANC1 and MDA-MB231 cells. a Immunoblot analyses were performed with anti-pLATS1 (S909), anti-LATS1, anti-pYAP (S127), anti-YAP, anti-MST1 (Thr183), anti-MST1 or anti-GAPDH antibodies. The samples were lysates from no treatment, DMSO-, 5 or 10 μm Ro 08-2750-treated PANC1 and MDA-MB231 cells for 24 h. GAPDH was used as the loading control. b Representative images of YAP immunofluorescence staining in DMSO- or 10 μm Ro 08-2750-treated PANC1 cells for 24 h. Three independent experiments were performed. Scale bar = 20 µm. c mRNA expression (qRT-PCR) of YAP target genes CYR61, ADMTS1 and ANKRD1 in PANC1 cells, and CYR61 and ANKRD1 in MDA-MB231 cells that are treated with DMSO or 10 μm Ro 08-2750. GAPDH was used as an internal control. Three independent experiments were performed. Error bars represent SD; *P < 0.05; **P < 0.01 by two-tailed Student’s t test
YAP-S127A eliminates the effect of NTRK1 inhibition
Because LATS1-mediated Serine 127 phosphorylation led to YAP exclusion from nucleus, we generated a YAP (S127A) mutant resistant to LATS1 phosphorylation [7]. Subsequently, to determine whether NTRK1 inhibition affects constitutively active YAP, we transduced MDA-MB-231 with a vector control or YAP (S127A) (Fig. 4a). We found that the treatment of MDA-MB-231 cells with Ro 08-2750 decreased the proliferation in the control cells but not in the cells expressing the active form of YAP (Fig. 4b; Supplementary Fig. 4A). Similarly, wound-healing assays revealed that cell migration was suppressed by Ro 08-2750 treatment in the control cells, but this effect was eliminated by YAP-S127A expression (Fig. 4c; Supplemental Fig. 4b).

The effect of NTRK1 inhibition is eliminated by YAP-S127A. a Immunoblot of YAP expression in MDA-MB231 cells transduced with the empty vector control (pBabe-puro) or the pBabe-YAP-S127A construct. GAPDH was used as the loading control. b Cell numbers were counted in DMSO- or 10 μm Ro 08-2750-treated MDA-MB231 cells for 24 or 48 h. Three independent experiments were performed. Error bars represent SD; *P < 0.05 by two-tailed Student’s t test. c Representative images of wound-healing assay for DMSO- or 10 μm Ro 08-2750-treated MDA-MB231 cells for 24 h (left panel). Quantifications of wound closure estimation from three independent experiments (right panel). Error bars represent SD; *P < 0.05 by two-tailed Student’s t test
NTRK1 stimulation by NGF promotes cancer cell growth and migration
The results presented thus far indicate that NTRK1 inhibition activates LATS1 and inactivates YAP function. To test the converse hypothesis, i.e., whether NGF stimulation inactivates LATS1 and activates YAP, we first treated PANC1 and MDA-MB-231 cells with NGF to activate NTRK1 [23]. NGF treatment decreased the p-YAP and p-LATS1 levels in a time-dependent manner (Fig. 5a). The expression of YAP target genes (CTGF, CYR61 and ANKRD1) was upregulated after NGF treatment (Fig. 5b). In addition, proliferation was significantly higher in NGF-treated cells than in control cells, and this effect was completely reversed by the YAP small-molecule inhibitor Verteporfin or siYAP (Fig. 5c, d). Interestingly, using immunostaining, we found that YAP translocated to the nucleus following NGF treatment (Supplementary Fig. S5). In accordance with this result, colony formation and wound-healing assays also showed that NGF treatment potentiated cancer cell growth and migration (Fig. 5e).

b-NGF treatment promotes cell growth and migration. a Immunoblot analyses were performed with anti-pLATS1 (S909), anti-LATS1, anti-pYAP (S127), anti-YAP, anti-MST1 (Thr183), anti-MST1 or anti-GAPDH antibodies. The samples were lysates from PANC1 or MDA-MB231 cells in serum-free medium for 24 h without or with 200 ng/mL b-NGF treatment for 5, 15 or 30 min. GAPDH was used as the loading control. b mRNA expression (qRT-PCR) of YAP target genes CTGF and CYR61 in PANC1 cells, and in CTGF, CYR61 and ANKRD1 in MDA-MB231 cells that were treated without or with 200 ng/mL b-NGF for 30 min. Three independent experiments were performed. GAPDH was used as an internal control. Error bars represent SD; **P < 0.01 by two-tailed Student’s t test. c Cell numbers were counted in PANC1 or MDA-MB231 cells under the condition of control, 200 ng/mL b-NGF or 200 ng/mL b-NGF together with 1 µm verteporfin treatment every 24 h for 72 h. Three independent experiments were performed. Error bars represent SD; **P < 0.01 by two-tailed Student’s t test. d Cell numbers were counted in PANC1 or MDA-MB-231 cells under the condition of siControl or siYAP together with 200 ng/mL b-NGF every 24 h for 72 h. Inset: Immunoblot analyses were performed with anti-YAP or anti-GAPDH antibodies. The samples were lysates from siControl or siYAP-transfected PANC1 or MDA-MB231 cells. GAPDH was used as the loading control. Three independent experiments were performed. Error bars represent SD; *P < 0.05, **P < 0.01, ***P < 0.001 by two-tailed Student’s t test. e Representative images of wound-healing assay for DMSO- or 200 ng/mL b-NGF-treated PANC1 or MDA-MB231 cells in serum-free medium for 24 h (left panel). Quantifications of wound closure estimation from three independent experiments (right panel). Error bars represent SD; **P < 0.01 by two-tailed Student’s t test
NTRK1 knockdown inhibits cell proliferation and migration
To investigate the functional consequences of NTRK1 gene expression, we knocked down NTRK1 using siRNA (siNTRK1) in PANC1 and MDA-MB-231 cancer cells. Knocking down NTRK1 dramatically reduced cell proliferation as measured by cell proliferation and colony formation assays (Fig. 6a–d). Furthermore, the perturbation of NTRK1 inhibited cell migration (Fig. 6e) and increased the levels of the active form of p-LATS1 and the inactive form of YAP (pYAP-S127) (Fig. 6a). Consistent with these findings, reduction of NTRK1 diminished its effects in YAP-S127 A-transformed MDA-MB-231 cells (Supplementary Fig. S6).

Knockdown of NTRK1 inhibits cell proliferation and migration. a Immunoblot analyses were performed with anti-NTRK1, anti-pLATS1 (S909), anti-LATS1, anti-pYAP (S127), anti-YAP, anti-MST1 (Thr183), anti-MST1 or anti-GAPDH antibodies. The samples were lysates from siCon or siNTRK1- transfected PANC1 or MDA-MB231 cells. GAPDH was used as the loading control. (b) Cell numbers were counted in siCon or siNTRK1-transfected PANC1 or MDA-MB231 cells every 24 h for 72 h. Three independent experiments were performed. Error bars represent SD; *P < 0.05 by two-tailed Student’s t test. (c) Quantifications of colony formation assay for siCon or siNTRK1-transfected PANC1 or MDA-MB231 cells for three weeks. Three independent experiments were performed. Error bars represent SD; **P < 0.01 by two-tailed Student’s t test. (d) Representative images of 24-h wound healing assay for siCon or siNTRK1-transfected PANC1 or MDA-MB231 cells (left panel). Quantifications of wound closure estimation from three independent experiments (right panel). Error bars represent SD; **P < 0.01 by two-tailed Student’s t test
NTRK1 knockdown inhibits tumour growth in mouse xenograft model
Next, to determine whether NTRK1 suppression affects xenograft tumour growth, we first transduced PANC1 and MDA-MB231 cells with lentiviral shCon or shNTRK1 constructs. As expected, we observed an increase in p-LATS1 and p-YAP in the NTRK1 knockdown cells (Fig. 7a). Likewise, when these cells were treated with NGF, the previously observed decrease in p-YAP and p-LATS1 protein levels was eliminated, further corroborating the critical role of NTRK1 in mediating the effects of NGF via the Hippo pathway (Fig. 7b). Finally, we subcutaneously injected PANC1 and MDA-MB231 cells that had been transduced with either shCon or shNTRK1s into SCID mice and monitored tumour growth. As shown in Fig. 7c, tumour growth was inhibited in mice transplanted with NTRK1 knockdown cells compared to that in mice transplanted with control cells (Fig. 7c). Immunoblots using the shCon- or shNTRK1-derived samples confirmed a marked reduction in YAP protein levels in the NTRK1 knockdown tumours (Fig. 7d).

Knockdown of NTRK1 suppresses tumour growth in vivo. a Immunoblot analyses were performed with anti-NTRK1, anti-pLATS1 (S909), anti-LATS1, anti-pYAP (S127), anti-YAP or anti-GAPDH antibodies. The samples were lysates from PANC1 or MDA-MB231 cells transduced with shCon or shNTRK1 hairpins. GAPDH was used as the loading control. (b) Immunoblot analyses were performed with anti-NTRK1, anti-pLATS1 (S909), anti-LATS1, anti-pYAP (S127), anti-YAP or anti-GAPDH antibodies. The samples were lysates from PANC1 or MDA-MB231 cells transduced with shCon or shNTRK1 hairpins under the condition of 24-h serum deprivation with or without 200 ng/mL b-NGF treatment for 30 min. GAPDH was used as the loading control. c Representative primary tumour images of shCon- or shNTRK1-transduced PANC1 or MDA-MB-231 cells subcutaneously injected into SCID mice (upper panel). Quantifications of primary tumour weight were measured 6 weeks after the subcutaneous injection of shCon- or shNTRK1-transduced PANC1 or MDA-MB-231 cells into SCID mice. n = 5 mice per group; error bars represent SD; ***P < 0.001 by two-tailed Student’s t test. d Immunoblot analyses were performed with anti-NTRK1, anti-YAP or anti-GAPDH antibodies. The samples were lysates from primary tumours that were injected with shCon- or shNTRK1-transduced PANC1 or MDA-MB-231 cells in SCID mice. GAPDH was used as the loading control. e Schematic model of the effect of NTRK1 regulation on YAP function
Discussion
The core kinase cascade of the Hippo pathway consists of LATS1/2, the adaptor protein MOB1, and Ste20-like protein kinase Mst1/2 andthe WW domain-containing protein WW45 [1]. Hippo signalling is controlled by a complex network of upstream components and mechanisms, many of which are involved in the regulation of cell adhesion and cell polarity [15]. For example, signalling from G-protein-coupled receptors and their ligands has been recently established to regulate YAP activity [24].
The control of YAP nuclear localization represents an exciting area in the design of new modalities for therapeutic intervention. For example, Liu-Chittenden et al. [25] identified verteporfin as a potent inhibitor of YAP1/TEAD interactions, and this compound inhibited YAP-induced liver growth in YAP-overexpressing or neurofibromin 2 (NF2)/merlin-inactivated mice. Likewise, Song et al. [26] identified a small molecule (CA3) that dramatically inhibited YAP1/TEAD transcriptional activity and oesophageal adenocarcinoma cell growth. Additionally, porphyrin- and dipyrrin-related derivatives have been reported to directly target TAZ or YAP proteins and inhibit TEAD transcriptional activity [27].
In this study, we performed a targeted kinase inhibitor screen in human cancer cells to identify novel non-canonical Hippo pathway kinases that control cell proliferation and survival. Our investigations began with the unexpected observation that pharmacological inhibition of NTRK1 suppressed the reporter activity of a YAP canonical target gene (CTGF). This finding suggested a novel crosstalk between NTRK1 and YAP. A subsequent analysis of the subcellular localization of YAP further revealed that NTRK1 inhibition augmented YAP cytoplasmic localization and that pharmacological or genetic inhibition of NTRK1 suppressed the transcriptional expression of YAP target genes. Notably, although the total LATS1, MST and YAP protein levels were not affected, LATS1 and YAP phosphorylation was induced by NTRK1 inhibition. Consistent with these results, the direct stimulation of NTRK1 by NGF led to decreased p-YAP and p-LATS protein levels and YAP target gene expression, as well as cell proliferation and migration. Future molecular studies are needed to understand how NTRK1 activation alters the downstream signalling events in the Hippo pathway. Nonetheless, functionally associating NTRK1 with the Hippo signalling pathway reveals an important and new aspect of NGF growth factor signalling that regulates cancer cell growth in vitro and promotes tumour growth in mice.
In summary, our data recognize the importance of NTRK1-mediated post-transcriptional control of YAP that promotes cancer cell proliferation and tumourigenesis (Fig. 7e). When NGF drives NTRK1 activation, YAP co-activates the transcriptional expression of survival genes implicated in cell proliferation. On the other hand, inhibiting NTRK1 modulates LATS1/2 phosphorylation and YAP cytoplasmic sequestration. Collectively, our data expand our knowledge of how Hippo signalling interacts with other oncogenic pathways. Moreover, our findings support the previously ill-defined capacity of NGF signalling to contribute to the acquisition of tumourigenic potential, thus indicating that further exploration and investigation of therapeutics targeting this pathway is warranted.
Materials and methods
Cell culture, siRNA transfection and viral transductions
293AD, MDA-MB-231 and PANC1 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% foetal bovine serum (FBS). MDA-MB-231 and PANC1 cells were obtained from the American Type Culture Collection (ATCC, VA). 293AD cells were kindly provided by Daniel Haber (MGH, Boston). All cell cultures were authenticated by short tandem repeat profiling and routinely tested for mycoplasma.
ON-TARGETplus Non-targeting siRNAs (D-001810-0×) and SMARTPOOL ON-TARGETplus NTRK1 siRNAs (L-003159-00-0005) or YAP1 siRNA (L-012200-00-0005) targeting human NTRK1 or YAP1 were acquired from Dharmacon (Lafayette, CO). DharmaFECT 1 Transfection Reagent (T2001-02) has been used to transfect the siRNAs at a final concentration of 20 nmol/L. Cell lysates or functional assays were performed ~ 72 h after siRNA transfection.
The human YAP wild-type and YAP-S127A expression constructs were previously described [6, 7]. Lentivirus and retrovirus packaging, cell transduction and puromycin cell selection were performed as previously described [28].
RNAi gene target sequences are summarized below (5′−3′ direction):
shNon-target-Con: CAACAAGATGAAGAGCACCAA;
shNTRK1-A: AGTCAGCCACGGTGATGAAAT;
shNTRK1-B: TATCTACAGCACCGACTATTA;
Quantitative real-time PCR (qRT-PCR)
Total RNA was prepared using Trizol Reagent (Life Technologies; MA) and quantitative real-time PCR was performed using standard protocols. GAPDH expression was used as the internal normalization control. QRT-PCR primer sequences are as follows:
CTGF-F: 5′-GGAAATGCTGCGAGGAGTGG-3′;
CTGF-R: 5′-GAACAGGCGCTCCACTCTGTG-3′;
CYR61-F: 5′-CACACCAAGGGGCTGGAATG-3′;
CYR61-R: 5′-CCCGTTTTGGTAGATTCTGG-3′;
ANKRD1-F: 5′-GCCAAAGACAGAGAAGGAGATAC-3′;
ANKRD1-R: 5′-GAGATCCGCGCCATACATAAT-3′;
ADMTS1-F: 5′-TCACCACAGCCCATGAATTAG-3′;
ADMTS1-R: 5′-GGAATCCTGGTTCACACCATTA-3′;
GAPDH-F: 5′-GTGAAGGTCGGAGTCAACGG-3′;
GAPDH-R: 5′-GAGGTCAATGAAGGGGTCATTG-3′
Immunoblot and antibodies
RIPA lysis buffer (Boston Bio-Products, MA) was used for protein extraction from cell lines. The Halt™ Protease Inhibitor Cocktail, EDTA free (100 × ) (Life Technology; MA), was added before harvesting protein lysates. Forty micrograms of protein lysates was separated by SDS-PAGE electrophoresis, transferred to PVDF membranes (EMD Millipore, MA) and blocked with non-fat milk for 1 h. Primary antibodies were prepared in 5% BSA or non-fat milk and incubated with PVDF membrane at 4 °C overnight. The HRP-conjugated goat anti-mouse (Bio-Rad # 1706516) or goat anti-rabbit (Bio-Rad # 1706515) secondary antibodies were incubated with the membrane for 2 h. ECL Plus Western Blotting Detection Reagents (GE Healthcare; PA) were used for protein detection.
Anti-NTRK1 (#2505), anti-YAP (#14074), anti-phospho-YAP (Ser127) (#13008), anti-LATS1 (#9153), anti-phospho-LATS1 (Ser909) (#9157), anti-MST1 (#14946) and anti-phospho-MST1/2 (T183/T180) (#3681) antibodies was purchased from Cell Signalling Technologies (Beverly, MA); anti-GAPDH (#Y1041) antibody from Ubiquitin-Proteasome Biotechnologies (Aurora; CO); and anti-Flag M2 antibody from Sigma-Aldrich (#F1804-1MG).
Immunofluorescence assay
Immunofluorescence staining was performed as previously described [29].
Luciferase reporter assay
CTGF promoter reporter luciferase reporter construct was kindly provided by Dr. Camargo. Briefly, luciferase reporter, Renilla and YAP1 wild-type constructs were co-transfected into 293AD cells with X-tremeGENE 9 DNA Transfection Reagent following the manufacturer’s protocol (Roche). Firefly and Renilla luciferase activities were detected by Dual-Luciferase Reporter Assay (Promega; WI). Each sample was performed in triplicate and a minimum of three independent experiments were performed.
Cell proliferation and colony formation assay
MDA-MB231 and PANC1 cells were seeded in 35-mm dishes at a concentration of 2 × 104 cells/mL. Cells were treated with DMSO, Ro 08-2750 (5 and 10 μm), or 200 ng/mLbNGF for 24, 48 and 72 h. Post treatment, cell numbers were counted using a hemocytometer. For colony formation assays, ~200 cells/well were plated into a six-well plate, images were taken and colonies were counted after 3 weeks. The NTRK1 inhibitor was added every day. Each accumulation of more than 50 cells was counted as a positive colony. Each sample was performed in triplicate and three independent experiments were performed.
Wound-healing assay
MDA-MB-231 or PANC1 cells grown to confluency were scratched and grown for 24 h in a medium containing +/−10% FBS (for b-NGF treatment experiments). Images were taken 24 h later. Each sample was performed in triplicate and three independent experiments were performed.
In vivo tumour xenograft assay
In vivo tumour growth assays were performed as previously described [30]. Female NOD/SCID mice of 6–8 weeks were obtained from RPCI. Animal experiments were performed under the rules provided by Declaration of Helsinki and approved by the Institutional Animal Care and Use Committee of the RPCI (Buffalo, NY).
Kinase inhibitor library screen
The kinase inhibitor screen was performed by the Small Molecule Screen Shared Resource at RPCI. YAP wild-type plasmid and CTGF promoter luciferase reporter were co-transfected into HEK293AD cells. Transfected cells were treated with 10 μM Tocriscreen Kinase Inhibitor Toolbox (Tocris Bioscience 3514) in a 384-well plate for 24 h in duplicate. Luciferase activity and cell survival were measured by luciferase assay (Promega; WI) and Resazurin cell enumeration assay (Thermo Fisher Scientific; MA). Luciferase activities and cell survival for each drug were compared with those of DMSO-treated controls,.
Statistical analysis
Statistical tests were performed using the SPSS statistics software package (SPSS, IL). All results are expressed as mean ± SD. *, P < 0.05; **, P < 0.01; ***, P < 0.
References
Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505.
Harvey K, Tapon N. The Salvador-Warts-Hippo pathway - an emerging tumour-suppressor network. Nat Rev Cancer. 2007;7:182–91.
Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–28.
Zanconato F, Cordenonsi M, Piccolo S. YAP/TAZ at the roots of cancer. Cancer Cell. 2016;29:783–803.
Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in The Cancer Genome Atlas. Cell. 2018;173:321–37 e10.
Overholtzer M, Zhang J, Smolen GA, Muir B, Li W, Sgroi DC, et al. Transforming properties of YAP, a candidate oncogene on the chromosome 11q22 amplicon. Proc Natl Acad Sci USA. 2006;103:12405–10.
Zhang J, Ji JY, Yu M, Overholtzer M, Smolen GA, Wang R, et al. YAP-dependent induction of amphiregulin identifies a non-cell-autonomous component of the Hippo pathway. Nat Cell Biol. 2009;11:1444–50.
Mohseni M, Sun J, Lau A, Curtis S, Goldsmith J, Fox VL, et al. A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol. 2014;16:108–17.
Niederhauser O, Mangold M, Schubenel R, Kusznir EA, Schmidt D, Hertel C. NGF ligand alters NGF signaling viap75(NTR) and trkA. J Neurosci Res. 2000;61:263–72.
Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–17.
Fiore M, Chaldakov GN, Aloe L. Nerve growth factor as a signaling molecule for nerve cells and also for the neuroendocrine-immune systems. Rev Neurosci. 2009;20:133–45.
Lange AM, Lo HW. Inhibiting TRK proteins in clinical cancer therapy. Cancers. 2018;10:105.
Nakagawara A. Trk receptor tyrosine kinases: a bridge between cancer and neural development. Cancer Lett. 2001;169:107–14.
Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246–57.
Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63–79.
Zhang J, Smolen GA, Haber DA. Negative regulation of YAP by LATS1 underscores evolutionary conservation of the Drosophila Hippo pathway. Cancer Res. 2008;68:2789–94.
Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & Dev. 2007;21:2747–61.
Aloe L, Rocco ML, Balzamino BO, Micera A. Nerve growth factor: role in growth, differentiation and controlling cancer cell development. J Exp Clin Cancer Res. 2016;35:116.
Descamps S, Pawlowski V, Revillion F, Hornez L, Hebbar M, Boilly B, et al. Expression of nerve growth factor receptors and their prognostic value in human breast cancer. Cancer Res. 2001;61:4337–40.
Descamps S, Toillon RA, Adriaenssens E, Pawlowski V, Cool SM, Nurcombe V, et al. Nerve growth factor stimulates proliferation and survival of human breast cancer cells through two distinct signaling pathways. J Biol Chem. 2001;276:17864–70.
Mello SS, Valente LJ, Raj N, Seoane JA, Flowers BM, McClendon J, et al. A p53 super-tumor suppressor reveals a tumor suppressive p53-Ptpn14-Yap axis in pancreatic cancer. Cancer Cell. 2017;32:460–73 e6.
Heidary Arash E, Shiban A, Song S, Attisano L. MARK4 inhibits Hippo signaling to promote proliferation and migration of breast cancer cells. EMBO Rep. 2017;18:420–36.
Benito-Gutierrez E, Garcia-Fernandez J, Comella JX. Origin and evolution of the Trk family of neurotrophic receptors. Mol Cell Neurosci. 2006;31:179–92.
Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. 2012;150:780–91.
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, et al. Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes & Dev. 2012;26:1300–5.
Song S, Xie M, Scott AW, Jin J, Ma L, Dong X, et al. A novel YAP1 inhibitor targets CSC-enriched radiation-resistant cells and exerts strong antitumor activity in esophageal adenocarcinoma. Mol Cancer Ther. 2018;17:443–54.
Gibault F, Bailly F, Corvaisier M, Coevoet M, Huet G, Melnyk P, et al. Molecular features of the YAP inhibitor verteporfin: synthesis of hexasubstituted dipyrrins as potential inhibitors of YAP/TAZ, the downstream effectors of the Hippo pathway. ChemMedChem. 2017;12:954–61.
Li YW, Guo J, Shen H, Li J, Yang N, Frangou C, et al. Phosphorylation of Tyr188 in the WW domain of YAP1 plays an essential role in YAP1-induced cellular transformation. Cell Cycle. 2016;15:2497–505.
Wilson KE, Li YW, Yang N, Shen H, Orillion AR, Zhang J. PTPN14 forms a complex with Kibra and LATS1 proteins and negatively regulates the YAP oncogenic function. J Biol Chem. 2014;289:23693–700.
Zhang Y, Shen H, Withers HG, Yang N, Denson KE, Mussell AL, et al. VGLL4 selectively represses YAP-dependent gene induction and tumorigenic phenotypes in breast cancer. Sci Rep. 2017;7:6190.
Acknowledgements
We would like to thank Dr. Haber who kindly provided us 293AD cells and Dr. Camargo who kindly provided us the CTGF promoter luciferase reporter construct. Also, we thank the Small Molecule Screen, Flow and Image Cytometry, Laboratory Animal and Experimental Tumour Models Shared Resources/Facilities of RPCI. This work was supported by the National Natural Science Foundation of China 81672921; Innovation Capacity Support Plan of Shaanxi Province, Grant/Award Number:2018TD-002 (to SH). This work was supported by the Roswell Park Cancer Institute and National Cancer Institute (NCI) Grant #P30 CA016056, Roswell Park Alliance Foundation, National Cancer Institute (NCI) R01 CA207504 and the American Cancer Society Research Scholar Grant RSG-14-214-01-TBE (to JZ).
Author contributions
XY, HS, CF and JZ designed the experiments. XY, HS, BB, YC, NY, AM, MC, CF, SH and JZ carried out the experiments and analysed the data. CF, LK and JZ wrote the paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Yang, X., Shen, H., Buckley, B. et al. NTRK1 is a positive regulator of YAP oncogenic function. Oncogene 38, 2778–2787 (2019). https://doi.org/10.1038/s41388-018-0609-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0609-1
- Springer Nature Limited
This article is cited by
-
Non-hippo kinases: indispensable roles in YAP/TAZ signaling and implications in cancer therapy
Molecular Biology Reports (2023)
-
Computing microRNA-gene interaction networks in pan-cancer using miRDriver
Scientific Reports (2022)
-
Genome-wide analysis identifies critical DNA methylations within NTRKs genes in colorectal cancer
Journal of Translational Medicine (2021)
-
Coregulation of pathways in lung cancer patients with EGFR mutation: therapeutic opportunities
British Journal of Cancer (2021)
-
ALKBH5 suppresses tumor progression via an m6A-dependent epigenetic silencing of pre-miR-181b-1/YAP signaling axis in osteosarcoma
Cell Death & Disease (2021)