
Abstract
The transcriptional co-activators Yes-associated protein (YAP) and PDZ-binding domain (TAZ) are the known downstream effectors of the Hippo kinase cascade. YAP/TAZ have been shown to play important roles in cellular growth and differentiation, tissue development and carcinogenesis. Recent studies have found that, in addition to the Hippo kinase cascade, multiple non-Hippo kinases also regulate the YAP/TAZ cellular signaling and produce important effects on cellular functions, particularly on tumorigenesis and progression. In this article, we will review the multifaceted regulation of the YAP/TAZ signaling by the non-Hippo kinases and discuss the potential application of the non-Hippo kinase-regulated YAP/TAZ signaling for cancer therapy.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
YAP/TAZ, the mammalian homologues of Yorkie in Drosophila melanogaster, are vital effectors of the Hippo pathway to regulate cell proliferation and tissue growth [1]. As transcriptional co-activators, YAP/TAZ are found to mediate both differentiation and immortalization signaling pathways upon interaction with functionally differentiated transcription factors, such as TEADs, p53/p73, SMAD and RUNX2 [2]. For example, YAP interacts with TEAD and β-catenin-TCF3 to induce Oct4 transcription thus maintain stemness or binding to p53/p73 which can promote BMP4 expression during differentiation [3]. YAP/TAZ may participate in immortalization by activation transcription of CDK6 and hTERT [4, 5], the two key proteins that are involved in immortalization. Thus, YAP/TAZ have multifaceted effects on differentiation and immortalization dependent on their transcriptional context. Apparently, differences between differentiation and immortalization regulated by the YAP/TAZ signaling pathways are determined by the distinct transcription factors and the specific downstream target genes activated by YAP/TAZ.
Dysregulation of YAP/TAZ signaling frequently observed in various cancers, commonly related to uncontrolled YAP/TAZ activity. Inappropriate activation of YAP/TAZ results in up-regulated expression of target genes that have profound effects on cell proliferation, migration, metabolism and tumorigenesis [6]. It is known that YAP/TAZ are regulated by the Hippo kinase cascade. Recently, mounting research reports found that many kinases that are not in canonical Hippo kinase cascade regulate the YAP/TAZ signaling. We categorize these kinases as non-Hippo kinases. Most of non-Hippo kinases have function in other signaling pathways, such as mTOR signaling, MAPK and NF-κB signaling pathway. Simultaneously, they play indispensable roles in regulating the YAP/TAZ signaling in response to various stimulations or in specific contexts [7,8,9]. The phosphorylation of YAP/TAZ conducted by non-Hippo kinases occurs not only in the cytoplasm, but also in the nucleus. These non-Hippo kinases directly phosphorylate YAP/TAZ at specific residues to modulate the protein abundance or transcriptional activity of YAP/TAZ or indirectly regulate YAP/TAZ signaling by interacting with the Hippo kinase cascade.
In this review, we will briefly overview the canonical Hippo-YAP/TAZ signaling and focus on the non-Hippo kinase-regulated YAP/TAZ signaling. We will introduce regulation of YAP/TAZ by non-Hippo kinases in three aspects: (1) direct activation and inhibition of the YAP/TAZ signaling; (2) indirect regulation of the YAP/TAZ signaling; and (3) application of the non-Hippo kinase-regulated YAP/TAZ signaling for cancer therapy.
Overview of canonical hippo-YAP/TAZ signaling
The Hippo signaling pathway was initially identified in a genetic screen searched for overgrowth mutants in Drosophila and found to play important roles in modulating both cell cycle and survival [1, 2]. In mammals, core components of the Hippo signaling consist of Mammalian Ste20-like kinase 1/2 (MST1/2, the homologues to Hpo in Drosophila) with their adaptor protein Salvador homologue 1 (SAV1, the homologue to Sav in Drosophila), and large tumor suppressor kinase 1/2 (LATS1/2, the homologues to Wts in Drosophila) with adaptor protein Mps one binder kinase activator 1 (MOB1, the homologue to Mats in Drosophila) [10].
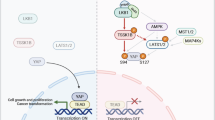
As downstream effectors of the Hippo signaling, YAP and TAZ (Yorkie in Drosophila) are transcriptional co-activators which shuttle between the cytoplasm and the nucleus [2]. Mechanistically, activation of MST1/2 initiates the canonical Hippo signaling (Fig. 1). The activation of MST1/2 is induced by autophosphorylation/trans-autophosphorylation or upstream kinases on Thr183 and Thr180 of the activation loop [11]. As adaptor protein, SAV1 forms a heterodimer with MST1 or MST2 through SARAH domains. Two SAV1-MST1/2 heterodimers form a heterotetramer via binding of SAV1 WW domains to stabilize MST1/2 trans-autophosphorylation status [12]. MST1/2 are the canonical upstream kinases activating LATS1/2. It has been proved that activation of LATS1/2 by MST1/2 contains three events [2, 13] (I) WWC proteins (WWC1/2/3) modulate the interaction between SAV1 and LATS1/2, in turn, SAV1 recruits MST1/2 to phosphorylate LATS1/2 at hydrophobic motif, (II) MST1/2 phosphorylate MOB1 on Thr12 and Thr35 to potentiate the interaction between MOB1 and LATS1/2, (III) the binding of MOB1 to LATS1/2 stimulates the conformational change in LATS1/2, resulting in autophosphorylation of the LATS1/2 activation loop for its full activation. Additionally, the mitogen-activated protein kinase kinase kinase kinases (MAP4Ks) phosphorylate and activate LATS1/2 as well [14]. Activated LATS1/2 phosphorylate downstream effectors YAP/TAZ, resulting in YAP/TAZ cytoplasmic retention via binding to 14-3-3 protein. YAP has five of the LATS phosphorylation consensus HXRXXS motifs, the serine residue in these five motifs, i.e. S61, S109, S127, S164 or S397, is phosphorylated by LATS. It is noted that the phosphorylation of YAP at S127 or TAZ at S89 by LATS promotes YAP/TAZ cytoplasmic sequestration by binding to 14-3-3. Phosphorylation of YAP/TAZ by LATS1/2 primes them for further phosphorylation by casein kinase 1 (CK1), causing ubiquitination and degradation of YAP/TAZ via a ubiquitin-proteasome system (UPS) [15, 16]. Once the Hippo kinase cascades are inhibited, YAP/TAZ translocate into the nucleus, interact with the TEA domain family members (TEAD1-4) transcription factors, induce the transcription of the target genes and produce promoting effects on cell growth, survival and tumorigenesis [2].

The core components and regulation of Hippo-YAP/TAZ signaling. When Hippo signaling is activated, the core components of Hippo pathway are phosphorylated sequentially, phosphorylated YAP/TAZ are separated in the cytoplasm by binding to 14-3-3 protein. Further phosphorylation induced by CK1 promotes YAP/TAZ proteasome degradation. YAP/TAZ are dephosphorylated once Hippo signaling is inhibited, resulting in YAP/TAZ translocate to the nucleus and then induce target gene expression by binding to TEADs
The non-hippo kinases that activate YAP/TAZ
In this section, we will address the non-Hippo kinases that directly activate YAP/TAZ (Table 1). The mechanisms underlying the activation of YAP/TAZ by non-Hippo kinases are summarized as following four types: (I) to disturb LATS phosphorylation at YAP S127 and TAZ S89 by occupying adjacent phosphorylation residues; (II) to obliterate the interaction between YAP/TAZ and 14-3-3 protein; (III) to block YAP/TAZ proteasome degradation executed by E3 ubiquitin ligase such as β-TrCP and CRL4DCAF12; and (IV) to stabilize YAP/TAZ protein in both cytoplasm and nucleus.
CDKs
The phosphorylation of YAP/TAZ is observed during mitosis and is vital for normal mitotic progression. Abnormal activation of YAP contributes to mitotic defects owing to dysregulated spindle checkpoint, and leads to oncogenic phenotypes [17, 18]. There are several cyclin-dependent kinases (CDKs) reported to phosphorylate and activate YAP/TAZ.
Cyclin-dependent kinase 1 (CDK1) belongs to CDKs cell-cycle-related subfamilies, is a mitotic CDK activated during G2/M and sufficient for driving the cell cycle. Cdk1 is the only Cdk that is able to govern the cell cycle in mouse embryos even when all interphase Cdks are absent [19]. In response to anti-mitotic drugs such as taxol and nocodazole which arrest cells in G2/M, YAP is observed phosphorylated independent of Hippo kinases [17]. By administration of taxol and specific kinase inhibitors, CDK1 was identified executing the phosphorylation of YAP during G2/M arrest. CDK1 phosphorylates YAP at T119, S289 and S367 in vitro and at T119, S289 in cells. The phosphorylation mediated by CDK1 is critical for cell migration and invasion driven by YAP [17]. Besides direct phosphorylation, CDK1 may enhance YAP activity via phosphorylating vestigial-like protein 4 (VGLL4). VGLL4 is an antagonist of YAP via binding to TEAD competitively [20]. CDK1 phosphorylates VGLL4 and reduces the affinity of VGLL4 binding to TEAD [21]. Consistently, expression of the YAP-TEAD target gene CTGF is reduced in cells overexpressing the CDK1-phosphorylation-defective mutant of VGLL4. Hence, CDK1 acts as a positive regulator in the YAP-mediated oncogenic functions.
Cyclin-dependent kinase 8 (CDK8) belongs to the CDKs transcriptional subfamilies and is a homologue to yeast protein Srb10. CDK8 functions as an enzymatic module of the Mediator complex and involves in basal transcription process [19, 22]. Previous study found that CDK8 displays oncogenic properties in colon tumorigenesis [23]. Knockout of CDK8 leads to elevated phosphorylation of YAP at S127. Consistently, expression of the YAP target gene CTGF is suppressed in a LATS-independent manner [24]. Furthermore, CDK8 was identified directly phosphorylates YAP at T119, S128, S289 and S367 in vitro. In mitotic-arrest cells treated with nocodazole, CDK8 contributes to hyper-phosphorylation of YAP at S128 along with phosphorylation of T119 and S289 [24]. As S128 is adjacent to the key LATS-phosphorylation site S127, phosphorylation of S128 elevates the YAP transcriptional activity. In turn, YAP activity is essential for the CDK8-driven tumorigenesis. Taken together, CDK8 modulates YAP activity as a positive activator via two mechanisms: (I) to reduce the phosphorylation of YAP at S127; and (II) directly to phosphorylate YAP at T119, S128, S289 and S367, which is vital for YAP activation [24].
Cyclin-dependent kinase 7 (CDK7) is a member of CDKs transcriptional subfamilies, the same as CDK8. CDK7 has essential roles in mediating both the cell cycle via functioning as a CDK-activating kinase and the transcription via assembly of transcriptional initiation factor II-H (TFIIH) [19, 25]. Recent research reports evaluated a functional connection of CDK7 to YAP, given that both CDK7 and YAP are co-localized in nucleus [26, 27]. Immunohistochemistry (IHC) staining in human malignant pleural mesothelioma (MPM) tissue showed a positive correlation between expression of CDK7 and YAP. Additionally, inhibition of CDK7 promotes YAP degradation thus diminishes the protein level of YAP in MPM cells [26]. Further investigation found that CDK7 phosphorylates YAP/TAZ in the nucleus and prevents YAP/TAZ from proteasomal degradation mediated by CRL4DCAF12 E3 ubiquitin ligase complex. CDK7 phosphorylates YAP at S128 and TAZ at S90, resulting in enhancement of the YAP/TAZ protein stability and activity [27]. When treating cells with THZ1, a pharmacological inhibitor of CDK7, the level of YAP protein and the expression of the YAP target genes are repressed accordantly [26, 27]. Moreover, growth of tumor in the triple-negative breast cancer (TNBC) xenograft mice models and overgrowth of liver induced by the YAP dysregulation are suppressed upon administration of THZ1 [27].
mTOR
mTOR is a serine/threonine kinase highly conserved from yeast to mammals. Both mTOR and Hippo signaling modulate cell growth and organ size. Dysregulation of these two pathways may lead to tumorigenesis. mTOR has two multiprotein complex forms, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), with different subunits and sensitivity to rapamycin [28]. Previous study has shown that activation of mTORC1 up-regulates YAP protein expression in both cytoplasm and nucleus, in conjunction with enhanced YAP transcriptional activity [29]. mTORC2 has the similar effects on YAP. Sciarretta et al. [30] reported that mTORC2 plays a role as the upstream kinase of MST1 in heart, phosphorylates MST1 at S438 in SARAH domain, thus abrogates MST1 homodimerization and activation. Most recent research found that mTORC2 interacts with YAP via Sin1 and directly phosphorylates YAP at S436 independent of the Hippo kinases [7]. The phosphorylation of YAP at S436 by mTORC2 elevates the YAP protein level and transcriptional activity and strengthens the interaction between YAP and TEAD. YAP promotes glioblastoma (GBM) cell proliferation, migration and invasion while activated by mTORC2. Moreover, the positive correlation between expression of mTORC2 and YAP in GBM patient tumor samples was observed by IHC staining [7].
MAPKs
Besides mTOR kinases, mitogen-activated protein kinases (MAPKs) also play important roles in modulating YAP/TAZ activity. Previous studies have demonstrated that MAPKs are involved in mediating the mechanical tension-induced YAP activity [8]. A recent report found that ERK phosphorylates YAP at S274 and S352 (conserved in human S289 and S367) during metaphase in ERBB2-overexpressed cardiomyocytes in a mouse heart failure model study, the phosphorylation of YAP on these sites by ERK elevates activity of YAP and expression of the YAP target genes [31]. Besides direct phosphorylation, ERK2 interacts with and phosphorylates 14-3-3 protein at S37, releases YAP from the YAP/14-3-3 complex, and promotes translocation of YAP into the nucleus in response to hypoxia [32].
Nemo-like kinase (NLK), the ortholog of Nemo in Drosophila, is an atypical MAPK. The kinase domain of NLK displays 45% identity to that of ERK and 38% identity to CDK1 [33]. In response to osmotic stress, NLK phosphorylates and activates YAP [34]. NLK phosphorylates YAP at S128 that is adjacent to the key LATS-phosphorylation site S127, suppresses interaction between YAP and 14-3-3, and enhances YAP nuclear translocation [34].
MK5, another member of atypical MAPKs and also known as MAPKAPK5, has been identified as an activator of YAP [33, 35]. MK5 physically binds to YAP in a kinase-dependent manner. The interaction between MK5 and YAP reduces the proteasomal degradation of YAP induced by CK1 due to disrupting interaction between YAP and β-TrCP [35].
SFKs
The activation of YAP/TAZ is regulated by tyrosine kinases as well. YAP was initially identified as the interactive protein of Yes, a Src family tyrosine kinase (SFK) [36]. SFKs are non-receptor protein tyrosine kinases and function in a wide range of cellular processes. SFKs share similar structural architecture. In addition to Yes, Src and LCK also interact with YAP [37,38,39]. Yes binds to YAP via its Src homology domain 3 (SH3) domain and phosphorylates YAP at Y407 in a kinase-dependent manner [36]. Similar to Yes, Src phosphorylates and activates YAP [37]. SFKs are activated in cholangiocarcinoma (CCA) cells because of the up-regulated platelet derived growth factor receptor (PDGFR) signaling [38]. In CCA cells, activated SFKs induce tyrosine phosphorylation of YAP at Y407, promote association of YAP with transcription factor TBX5, elevate expression of the YAP target gene Mcl-1, and enhance survival of CCA cells [38]. It has been identified that LCK is the kinase phosphorylates YAP at Y407 in CCA [39].
The non-hippo kinases that inactivate YAP/TAZ
YAP/TAZ play fundamental roles in cellular biological processes. Meanwhile, the activity of YAP/TAZ is inhibited to sustain cellular homeostasis when cells are response to nutrient shortage, viral infections or inflammatory cytokines such as TNFα and IL-1β. The inhibitory regulation of YAP/TAZ by non-Hippo kinases is normally via following three aspects (Table 2): (I) phosphorylation of YAP at S127 and TAZ at S89 to promote the proteasomal degradation of YAP/TAZ; (II) disruption of interaction between YAP/TAZ and TEAD to down-regulate the transcriptional activity of YAP/TAZ; and (III) induction of the YAP/TAZ degradation through lysosomes.
NDR1/2
Nuclear Dbf2-related kinase 1 and 2 (NDR1/2) are the closest homologs of LATS1/2 in the AGC (protein kinase A (PKA)/PKG/PKC-like) family of serine/threonine protein kinases. NDR1/2 directly phosphorylate YAP at S61, S109, S127 and S164 in vitro [40]. All these four sites are phosphorylated by LATS1/2 as well [15]. Through direct phosphorylation, NDR1/2 inhibit nuclear localization of YAP and expression of the YAP target genes in human colorectal cancer cells, thus repress colon cancer cell proliferation [40].
AMPK
The AMP-activated protein kinase (AMPK) is a trimeric serine/threonine protein kinase and the major cellular sensor in response to energy stress [41]. AMPK signaling pathway interplays with mTOR signaling pathway to ensure cellular energy homeostasis. mTORC1 and mTORC2 function as the positive regulators and promote cell growth in response to nutrient abundance, whereas AMPK as the negative regulator that inhibits cell growth under condition of nutrient shortage [41]. Contrary roles in controlling cellular energy status of mTORC1/mTORC2 and AMPK are consistent with their modulation of the YAP activity. As mentioned before, mTORC1/mTORC2 phosphorylate and activate YAP [7, 29], while AMPK negatively regulates YAP activity. Under energy stress, the phosphorylation of YAP S61, S94, T119 and S127 is elevated [42, 43]. S127 of YAP is phosphorylated by LATS and phosphorylation of S61, S94 and T119 is dependent on AMPK [43, 44]. YAP S94 is a critical residue for interaction with TEAD [45]. Phosphorylation of YAP S94 by AMPK disrupts interaction of YAP with TEAD upon shortage of energy [42]. In addition to S94, S61 and T119 are also the AMPK phosphorylation sites. Phosphorylation of S61 by AMPK also inhibits the YAP transcriptional activity, however, the mechanism underlying the inhibition remains unknown [43].
TAK1 and IKK
TAK1 or MAP3K7, initially identified as TGF-β-activated kinase 1, is a member of mitogen-activated protein kinase kinase kinases (MAP3Ks) family. TAK1 functions in both the nuclear factor-κB (NF-κB) and the MAPK signaling pathways [46]. Because of cellular and microenvironmental complexity, TAK1 was observed to produce opposite cellular effects in different tumor types or pathological stages [46]. TAK1 also produces the similar effects on regulation of YAP activity. For example, TAK1 promoted the formation of the YAP/TAZ-TRAF6 complex thus prevented YAP/TAZ from proteasomal degradation via ubiquitination of K63 [47]. Thus, it was proposed that TAK1 stabilizes YAP/TAZ protein and enhances YAP/TAZ activity [47, 48]. However, it was also observed that TAK1 regulated YAP activity by direct phosphorylation in response to inflammatory cytokines, such as TNFα and IL-1β [9]. TAK1 interacts with and directly phosphorylates YAP at multiple sites including S127, induces YAP degradation via the β-TrCP-mediated ubiquitination, thus represses YAP activity [9].
The IκB kinase (IKK) complex contains kinase subunits IKKα, IKKβ and regulatory subunit IKKγ (Nemo), or IKK-related kinases (TBK1 and IKKε). IKK complex plays essential roles in innate immunity via activating the NF-κB and IRF signaling [49]. It has been reported that IKKε interacts with and directly phosphorylates YAP at S419, induces YAP degradation through lysosomes, resulting in relief of the repression effect of YAP on antiviral immunity upon viral stimulation [50]. Thus, IKKε modulates YAP activity by phosphorylation as a negative regulator.
MEKK3 and MEKK5
MEKK3 (mitogen-activated kinase kinase kinase 3, MAP3K3) is a serine/threonine protein kinase belongs to MAP3K family. MEKK3 has multiple functions including phosphorylation of IKK and activation of the NF-κB signaling [49]. Emerging evidence elucidated MEKK3 has multifaceted roles in modulating YAP activity [51]. Upon stimulation, MEKK3 phosphorylates and activates LATS1/2 in MST1/2 and MAP4Ks independent manners. Moreover, MEKK3 directly interacts with and phosphorylates YAP at S371, resulting in inhibitory regulation of YAP transcriptional activity [52].
MEKK5 (mitogen-activated kinase kinase kinase 5, also ASK1) is a member of MAP3K family. Our previous study has demonstrated that MEKK5 plays inhibitory role in regulation of E3 ligase NEDD4-mediated lung cancer cell migration [53]. Moreover, we identified that MEKK5 participates in regulation of TAZ signaling, MEKK5 interacts with and inactivates TAZ, leads to TAZ cytoplasmic retention in a kinase-dependent manner [54].
GSK3
Glycogen synthase kinase 3 (GSK3) is a serine/threonine kinase that implicated in a number of cellular functions. Similar to β-catenin, which is phosphorylated by GSK3 and subsequently recognized by β-TrCP, TAZ is directly phosphorylated by GSK3 at S58 and S62 that compose its N-terminal phosphodegron, resulting in TAZ degradation by UPS [55]. Meanwhile, Azzolin et al. found that GSK3 destabilizes TAZ in a kinase-dependent but indirect manner [56]. In this vein, GSK3 phosphorylates and endows with β-catenin as a bridge between TAZ and β-TrCP, thus fosters TAZ proteasomal degradation. Collectively, by direct and indirect effects, GSK3 functions as an inhibitory regulator of TAZ.
The non-hippo kinases that regulate YAP/TAZ indirectly
In addition to direct activation or inhibition by phosphorylation of YAP/TAZ, some non-Hippo kinases regulate the YAP/TAZ signaling without direct phosphorylation of YAP/TAZ. These kinases modulate the YAP/TAZ signaling through phosphorylation or regulation of assembly of the Hippo cascade components, thus effect the Hippo signaling and YAP/TAZ (Table 3).
LKB1 and AMPK-related kinase
The liver kinase B1 (LKB1) is a serine/threonine protein kinase ubiquitously expressed. LKB1 is considered as a tumor suppressor that governs cell proliferation, metabolism and polarity [57]. Knockdown of LKB1 is found to decrease phosphorylation of YAP and up-regulate expression of the YAP target genes [58, 59]. The inactivation effect of LKB1 on YAP activity partially depends on its substrates, MARKs. MARK1, 3 and 4 function as the activators of the Hippo kinase, thus inhibit YAP activity [59]. In addition, AMPK is a well-known substrate of LKB1 and the inhibitory regulation of YAP by LKB1 relies on AMPK phosphorylation as well [60].
NUAK2 (also known as SNARK) is a serine/threonine protein kinase, belongs to AMPK kinase family and is regulated by LKB1. NUAK2 participates in various cellular processes including proliferation, migration, cell adhesion and metabolism [61]. It has been observed that NUAK2 responds to nutrient stress similar to AMPK [62]. Intriguingly, a positive feed-forward loop between YAP/TAZ and NUAK2 was identified. NUAK2 is the target gene of YAP/TAZ and mediates the YAP-dependent cancer cell and tumor growth [63, 64]. Inhibition of NUAK2 activity by its specific inhibitors represses the YAP-mediated cancer cell or organ growth [63]. NUAK2 regulates the YAP/TAZ activity by phosphorylation and inhibition of LATS [64]. NUAK2 interacts with LATS and phosphorylates LATS at S613 and T246. Because LATS S613 is adjacent to MOB1-LATS binding site, phosphorylation at S613 by NUAK2 inhibits LATS activity and the YAP phosphorylation by LATS [64]. Given the vital roles of NUAK2 in promoting the YAP/TAZ oncogenic function, inhibitors of NUAK2 may be applied for cancer therapy.
PDK1
PDK1 (3-phosphoinositide-dependent protein kinase-1) modulates YAP/TAZ activity as well. Fan and colleagues [65] identified PDK1 interacts with MST and LATS via SAV1 in serum-starved confluent MCF-10 A cells. PDK1 forms a complex with active Hippo components in response to serum-starvation, thus impairs nuclear translocation of YAP. PDK1 is activated and recruited to plasma membrane upon EGF stimulation, resulting in the dissociation of PDK1 from the Hippo kinase complex and inactivation of LATS. As a consequence, YAP is trans-localized in nucleus. The effect of PDK1 on YAP is independent of the PI3K/AKT signaling [65]. Subsequent research found that Src might be the upstream kinase of PDK1 in this regulation [66].
Receptor tyrosine kinases (RTKs)
Mounting evidence has demonstrated that receptor tyrosine kinases (RTKs) are associated with the Hippo signaling pathway and regulate YAP/TAZ activity.
The epidermal growth factor receptor (EGFR, also known as ERBB1 and HER1), a member of the ERBB tyrosine kinase family, is ubiquitously expressed in epithelial and fibroblast cells. The EGFR signaling pathway is well studied and known as a driver of tumorigenesis in human non-small cell lung cancer (NSCLC) and breast cancer [67, 68]. EGFR is activated by its ligands such as EGF, TGFα, epiregulin and neuregulin [69]. In head and neck squamous cell carcinomas (HNSCC) cells, the EGFR signaling activates YAP/TAZ independent of MST1/2. Further study found that EGFR phosphorylates MOB1 at Y95, Y114 and Y117 upon EGF stimulation or by overexpression of EGFR or EGFR active mutants [70]. Although interaction of MOB1 with LATS1 is not disrupted by the tyrosine phosphorylation, activity of LATS1 is reduced and expression of the YAP/TAZ target genes is elevated. Treatment of HNSCC cells with the EGFR inhibitor erlotinib enhanced phosphorylation of YAP and repressed expression of the YAP target genes [70]. It has been found that the EGFR signaling promotes the YAP/TAZ activity through activation of PI3K and PDK1 in hepatocellular carcinoma (HCC) cells. The oncogenic effect of EGFR in HCC is partially mediated by the YAP/TAZ signaling [71, 72].
HER2, another member of the ERBB tyrosine kinase family, was reported to reprogram normal cells into tumor-initiating cells by regulating cell’s mechanical properties by activation of Ras and Rac1 [73]. Mechanical signals transducted by the HER2-Ras-Rac1 cascade activates YAP/TAZ activity, and transcription of oncogenic genes. Meanwhile, YAP/TAZ sustain HER2 and Ras induced oncogenic reprogramming [73]. With the crucial roles played in mechanotransduction, YAP/TAZ function as key mediators in mechanosignaling.
Neurotrophic receptor tyrosine kinase 1 (NTRK1, also known as TRKA), a member of the tropomyosin receptor kinase family, was initially identified in colon carcinoma. Expression of NTRK1 and its ligand nerve growth factor (NGF) in nervous system were found a few years later [74]. The pro-oncogenic role of NTRK1 has been demonstrated in multiple tumor types, and NTRK1 promotes tumorigenesis mostly by activation of the MAPK and PI3K signaling [74]. Recently, a report has shown that NTRK1 promotes tumorigenesis through regulating the Hippo-YAP signaling [75]. Inhibition of NTRK1 induces activation of LATS that leads to increase in phosphorylation at S127 and cytoplasmic localization of YAP. Consistently, stimulation of NTRK1 with NGF suppresses the YAP phosphorylation, enhances YAP activity, and promotes cancer cell proliferation and migration [75].
ROCK1/2
Rho-associated protein kinase 1 and 2 (ROCK1 and ROCK2) are serine/threonine kinases that belong to AGC family and initially identified as downstream effectors of small GTPase RhoA. Plenty of evidence has demonstrated that YAP/TAZ nuclear localization and transcriptional activity are regulated by ROCK1/2 [76, 77]. Depletion of ROCK2 severely reduced YAP oncogenic activity [76]. Moreover, pharmacological inhibition of ROCK1 markedly impaired TEAD/YAP transcription that is mediated by mechano-signaling [77].
Targeting the YAP/TAZ signaling by kinase inhibitors in cancer therapy
Inhibitors of the PI3K signaling
In MCF-10 A breast cancer cells, inhibition of PI3K with wortmannin/LY294002 or PDK1 with BX795 impairs nuclear localization of YAP, while the AKT inhibitor VIII has an insignificant effect [65, 66]. The similar results were obtained in HCC, PI3K and PDK1 inhibitors abolish the EGFR-induced reduction of pYAP-S127 [71], suggesting that inhibitors of the PI3K-PDK1 axis may be applied for targeting the YAP signaling in cancer therapy.
Inhibitors of the src family kinases (SFKs)
SFKs have been demonstrated to enhance the YAP-oncogenic activity, not only via activating the PI3K-PDK1 signaling, but also through directly phosphorylation and stabilization of YAP/TAZ [37, 66]. PP2 is a selective inhibitor of SFKs, treatment with PP2 in human renal proximal tubule epithelial (HK-2) and MCF-10 A cells elevated phosphorylation of YAP at S127 and reduced YAP activity [66, 78]. In triple-negative breast cancer MDA-MB231 and MDA-MB468 cells, PP2 suppresses cell viability, migration and invasion, suggesting that PP2 may have a therapeutic effect on the YAP/TAZ-dependent cancer [79].
Dasatinib is an SFK inhibitor approved by FDA and EMA for treatment of Philadelphia chromosome-positive (Ph +) chronic myeloid leukemia (CML) and Ph + acute lymphoblastic leukemia (ALL) [80]. Recent reports have demonstrated that overexpression of Yes contributes to tumor growth and metastasis in NSCLC both in vitro and in vivo. By inhibiting Yes kinase activity, dasatinib represses the tyrosine phosphorylation of YAP, impairs the nuclear translocation of YAP, and reduces expression of the YAP target genes [81]. Datasinib significantly inhibits proliferation of MDA-MB-231 cells that are the YAP/TAZ-dependent breast cancer cells [82]. Consistently, tumors with activated YAP have high sensitivity to dasatinib [83]. In a CCA patient-derived xenograft (PDX) model, treatment with dasatinib elevates cancer cell death and decreases tumor volume in mice [39]. The inhibitory effects of dasatinib on cancer cell proliferation partially rely on its inhibition of the YAP/TAZ signaling, suggesting that dasatinib has potential therapeutic efficacy for the YAP/TAZ-driven cancer. However, some studies have shown that treatment with dasatinib alone did not have a significant effect on growth of the PDX tumors in vivo [84, 85]. Treatment of cancer patients with dasatinib failed in lung cancer clinical trial [86]. Combination with other anti-cancer drugs may be an approach for application of dasatinib for cancer therapy.
Inhibitors of EGFR
EGFR is frequently mutated to drive lung and breast cancer tumorigenesis and progression. EGFR tyrosine kinase inhibitors (TKIs) have been used as the first-line treatment for patients with advanced EGFR-mutant NCSLC [87]. Currently, the third-generation EGFR-TKI osimertinib has been demonstrated superior over the first-generation (erlotinib and gefitinib) and the second-generation EGFR-TKIs (afatinib) with a lower rate of serious adverse events and the similar safety profile [88]. Inhibition of EGFR with the TKI erlotinib enhances YAP phosphorylation at S127 and reduces expression of the YAP target genes in HNSCC cells bearing the EGFR mutation [70]. However, clinical trials frequently observed resistance to treatment with EGFR-TKIs in NSCLC patients [89]. Some studies have shown that YAP plays a role in resistance to EGFR-TKIs [90, 91]. YAP signaling is activated in the EGFR-TKI-resistant cells and knockout or inhibition of YAP enhances the sensitivity of cancer cells to EGFR-TKIs [92].
Inhibitors that disrupt interaction of YAP/TAZ with TEAD
Verteporfin (VP) is a small molecular weight chemical and used to antagonize YAP/TAZ binding to TEAD [93]. However, application of VP for therapy of the YAP-driven cancer is limited due to the off-target effect [94].
Approaches with combination of inhibitors
In preclinical studies, inhibition of YAP/TAZ with VP combined with EGFR-TKIs has shown more effective in suppressing proliferation of cancer cells [92]. In combination of the EGFR-TKI gefitinib with simvastatin that inhibits YAP, the anti-tumor effect is more significant than the single agent administration alone in HCC cells [71]. Trametinib is a MEK inhibitor has been approved for treatment of BRAF-mutant melanoma and applied for multiple cancer therapy [95]. Treatment of HCC cells with simvastatin and trametinib significantly enhanced the cytostatic effects in the cell proliferation and colony formation assays [71]. MYF-01-37 is a newly developed TEAD inhibitor that covalently binds to TEAD, thus disrupts interaction between YAP and TEAD [96]. In a recent study inhibition of both the EGFR/MEK and the YAP signaling pathways overcomes dormant therapy-resistant cancer cells by promoting cellular apoptosis. Treatment of NSCLC cells with MYF-01-37, osimertinib (EGFR-TKI), and trametinib (MEK inhibitor) results in complete repression of YAP activity and a significant increase in apoptosis [96].
The SFKs inhibitor dasatinib in combination with EGFR-TKI erlotinib or afatinib has been used in phase I/II trial for lung cancer patients with the acquired EGFR-TKIs resistance. However, objective response in patients to the treatment in these clinical trials was not observed [97, 98]. The third-generation EGFR-TKI osimertinib is a preferred option for treatment of the EGFR-mutant NSCLC. When osimertinib combined with dasatinib, the anti-tumor effect was displayed in NSCLC patients with the EGFR-mutation [99], suggesting that combination of these two inhibitors is effective for NSCLC therapy.
Recent research revealed that oncogenic Hippo-YAP and PI3K signaling is elevated and activated in high-grade gliomas derived from the Pten/Tp53-lossed Olig1/2-expressing intermediate lineage precursors of mice. While targeting the YAP signaling with VP or the PI3K signaling with the analog of wortmannin PX-866 alone partially inhibited tumor cell proliferation, targeting both the YAP and PI3K signaling pathways by combination treatment with VP and PX-866 completely inhibited the growth of tumor cells [100], suggesting that targeting YAP signaling combined with other oncogenic signaling may significantly enhance cancer therapeutic efficacy.
Conclusion
As essential transcriptional co-activators, YAP/TAZ have pivotal functions in cellular homeostasis, tissue development and carcinogenesis. Dysregulation of YAP/TAZ activity resulting from either elevation of protein abundance or activation promotes tumorigenesis, cancer progression, and resistance to cancer therapy.
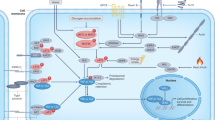
In response to multiple stimuli, several non-Hippo kinases directly phosphorylate YAP/TAZ at specific residues to sustain YAP/TAZ protein stability and abundance and facilitate the YAP/TAZ nuclear translocation. Under energy stress or viral infection, several non-Hippo kinases interact with and phosphorylate YAP/TAZ to disrupt their interaction with TEAD or promote their degradation, thus inhibit the YAP/TAZ signaling. In addition to direct interaction and phosphorylation, some non-Hippo kinases regulate YAP/TAZ activity through regulation of the Hippo kinase cascade, thus indirectly control YAP/TAZ activity. Taken together, regulation of the YAP/TAZ signaling by the non-Hippo kinases is multifaceted (Fig. 2), depending on cell type and signaling context.

Regulation of YAP/TAZ by non-Hippo kinases. Activation and inhibition of transcriptional co-activators YAP/TAZ are regulated by multiple non-Hippo kinases both in the cytoplasm and in the nucleus. Non-Hippo kinases induce serine/threonine phosphorylation and tyrosine phosphorylation on YAP specific residues. Generally, CDK7, CDK8 and NLK phosphorylate YAP/TAZ at S128 and S90 to disturb LATS phosphorylation at YAP S127 and TAZ S89, thus prevent YAP/TAZ from proteasomal degradation. SFKs (Src, Yes, LCK) phosphorylate YAP at Y407 to stabilize YAP protein and promote YAP translocation in the nucleus. MK5 physically binds to YAP to stabilize YAP protein as well. Phosphorylation of YAP at S436 by mTORC2 elevates interaction between YAP and TEADs; phosphorylation at T119, S289 and S367 conducted by CDK1, CDK8 and ERK1 (S289 and S367 only) up-regulate YAP transcriptional activity. To inactivate YAP/TAZ, NDR1/2 and TAK1 phosphorylate YAP at S127, IKK phosphorylates YAP at S419, thus induce YAP degradation by proteasomes or lysosomes. S94 phosphorylated by AMPK disrupt the interaction between YAP and TEAD to down-regulate the transcriptional activity of YAP. MAP3Ks, MEKK3, MEKK5 and GSK3 interact with and down-regulate YAP/TAZ activity as well. Some of the non-Hippo kinases regulate YAP/TAZ signaling in a Hippo-dependent manner. NUAK2 and NTRK1 target LATS, EGFR targets MOB1, mTORC2 targets MST1 to inhibit LATS full activation. LKB1 mediates downstream kinases AMPK and MARKs, PDK1 and upstream kinase Src regulate the assembly of Hippo cascade components to affect the Hippo signaling and YAP/TAZ
Targeting the YAP/TAZ or the non-Hippo kinases that regulate YAP/TAZ signaling for cancer therapy is still under development by disrupting interaction of YAP/TAZ with TEAD or inhibiting the non-Hippo kinases. Because of heterogeneity and complexity of genetic and epigenetic background of tumors, targeting the YAP/TAZ signaling alone may not be enough to inhibit tumor growth and effective for cancer therapy, particularly in the tumor with a multiple oncogenic signaling pathways. Therefore, targeting the YAP/TAZ signaling or the non-Hippo kinase regulated YAP/TAZ signaling combined with other oncogenic signaling may be an effective approach for targeted therapy of the YAP/TAZ-driven cancers in future.
Abbreviations
- ALL:
-
Acute lymphoblastic leukemia
- AMPK:
-
AMP-activated protein kinase
- CCA:
-
Cholangiocarcinoma
- CDK:
-
Cyclin-dependent kinase
- CK1:
-
Casein kinase 1
- CML:
-
Chronic myeloid leukemia
- EGFR:
-
Epidermal growth factor receptor
- GBM:
-
Glioblastoma
- GSK3:
-
Glycogen synthase kinase 3
- HCC:
-
Hepatocellular carcinoma
- HNSCC:
-
Head and neck squamous cell carcinomas
- IHC:
-
Immunohistochemistry
- IKK:
-
IκB kinase
- LATS1/2:
-
Large tumor suppressor kinase 1/2
- LKB1:
-
Liver kinase B1
- MAP4Ks:
-
Mitogen-activated protein kinase kinase kinase kinases
- MAPKs:
-
Mitogen-activated protein kinases
- MOB1:
-
Mps one binder kinase activator 1
- MPM:
-
Malignant pleural mesothelioma
- MST1/2:
-
Mammalian Ste20-like kinase 1/2
- mTOR:
-
Mammalian target of rapamycin
- NDR1/2:
-
Nuclear Dbf2-related kinase 1/2
- NGF:
-
Nerve growth factor
- NF-κB:
-
Nuclear factor-κB
- NLK:
-
Nemo-like kinase
- NSCLC:
-
Non-small cell lung cancer
- NTRK1:
-
Neurotrophic receptor tyrosine kinase 1
- PDGFR:
-
Platelet derived growth factor receptor
- PDK1:
-
3-phosphoinositide-dependent protein kinase-1
- PDX:
-
Patient-derived xenograft
- Ph +:
-
Philadelphia chromosome-positive
- ROCK1/2:
-
Rho-associated protein kinase 1 and 2
- RTK:
-
Receptor tyrosine kinase
- SAV1:
-
Salvador homologue 1
- SFK:
-
Src family tyrosine kinase
- SH3:
-
Src homology domain 3
- STK38:
-
Serine/threonine kinase 38
- TCF/LEF:
-
T-cell factor/lymphoid enhancer factor
- TEADs:
-
TEA domain family members
- TFIIH:
-
Transcriptional initiation factor II-H
- TKI:
-
Tyrosine kinase inhibitor
- TNBC:
-
Triple-negative breast cancer
- UPS:
-
Ubiquitin-proteasome system
- VGLL4:
-
Vestigial-like protein 4
References
Huang J, Wu S, Barrera J, Matthews K, Pan D (2005) The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell 122:421–434. https://doi.org/10.1016/j.cell.2005.06.007
Piccolo S, Dupont S, Cordenonsi M (2014) The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev 94:1287–1312. https://doi.org/10.1152/physrev.00005.2014
Papaspyropoulos A, Bradley L, Thapa A et al (2018) RASSF1A uncouples wnt from Hippo signalling and promotes YAP mediated differentiation via p73. Nat Commun 9:424. https://doi.org/10.1038/s41467-017-02786-5
Xie Q, Chen J, Feng H et al (2013) YAP/TEAD-mediated transcription controls cellular senescence. Cancer Res 73:3615–3624. https://doi.org/10.1158/0008-5472.Can-12-3793
Zhang Q, Liu N, Bai J et al (2020) Human telomerase reverse transcriptase is a novel target of Hippo-YAP pathway. Faseb j 34:4178–4188. https://doi.org/10.1096/fj.201902147R
Harvey KF, Zhang X, Thomas DM (2013) The Hippo pathway and human cancer. Nat Rev Cancer 13:246–257. https://doi.org/10.1038/nrc3458
Holmes B, Benavides-Serrato A, Saunders JT, Kumar S, Nishimura RN, Gera J (2021) mTORC2-mediated direct phosphorylation regulates YAP activity promoting glioblastoma growth and invasive characteristics. Neoplasia 23:951–965. https://doi.org/10.1016/j.neo.2021.07.005
Liu Z, Wu H, Jiang K et al (2016) MAPK-Mediated YAP activation Controls Mechanical-Tension-Induced Pulmonary Alveolar Regeneration. Cell Rep 16:1810–1819. https://doi.org/10.1016/j.celrep.2016.07.020
Deng Y, Lu J, Li W et al (2018) Reciprocal inhibition of YAP/TAZ and NF-κB regulates osteoarthritic cartilage degradation. Nat Commun 9:4564. https://doi.org/10.1038/s41467-018-07022-2
Badouel C, Garg A, McNeill H (2009) Herding Hippos: regulating growth in flies and man. Curr Opin Cell Biol 21:837–843. https://doi.org/10.1016/j.ceb.2009.09.010
Deng Y, Pang A, Wang JH (2003) Regulation of mammalian STE20-like kinase 2 (MST2) by protein phosphorylation/dephosphorylation and proteolysis. J Biol Chem 278:11760–11767. https://doi.org/10.1074/jbc.M211085200
Lin Z, Xie R, Guan K, Zhang M (2020) A WW Tandem-Mediated Dimerization Mode of SAV1 essential for Hippo Signaling. Cell Rep 32:108118. https://doi.org/10.1016/j.celrep.2020.108118
Qi S, Zhu Y, Liu X et al (2022) WWC proteins mediate LATS1/2 activation by Hippo kinases and imply a tumor suppression strategy. Mol Cell. https://doi.org/10.1016/j.molcel.2022.03.027
Meng Z, Moroishi T, Mottier-Pavie V et al (2015) MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun 6:8357. https://doi.org/10.1038/ncomms9357
Hao Y, Chun A, Cheung K, Rashidi B, Yang X (2008) Tumor suppressor LATS1 is a negative regulator of oncogene YAP. J Biol Chem 283:5496–5509. https://doi.org/10.1074/jbc.M709037200
Zhao B, Li L, Tumaneng K, Wang CY, Guan KL (2010) A coordinated phosphorylation by lats and CK1 regulates YAP stability through SCF(beta-TRCP). Genes Dev 24:72–85. https://doi.org/10.1101/gad.1843810
Yang S, Zhang L, Liu M, Chong R, Ding SJ, Chen Y, Dong J (2013) CDK1 phosphorylation of YAP promotes mitotic defects and cell motility and is essential for neoplastic transformation. Cancer Res 73:6722–6733. https://doi.org/10.1158/0008-5472.Can-13-2049
Yang S, Zhang L, Chen X, Chen Y, Dong J (2015) Oncoprotein YAP regulates the spindle checkpoint activation in a mitotic phosphorylation-dependent manner through up-regulation of BubR1. J Biol Chem 290:6191–6202. https://doi.org/10.1074/jbc.M114.624411
Malumbres M (2014) Cyclin-dependent kinases. Genome Biol 15:122. https://doi.org/10.1186/gb4184
Koontz LM, Liu-Chittenden Y, Yin F, Zheng Y, Yu J, Huang B, Chen Q, Wu S, Pan D (2013) The Hippo effector yorkie controls normal tissue growth by antagonizing scalloped-mediated default repression. Dev Cell 25:388–401. https://doi.org/10.1016/j.devcel.2013.04.021
Zeng Y, Stauffer S, Zhou J, Chen X, Chen Y, Dong J (2017) Cyclin-dependent kinase 1 (CDK1)-mediated mitotic phosphorylation of the transcriptional co-repressor Vgll4 inhibits its tumor-suppressing activity. J Biol Chem 292:15028–15038. https://doi.org/10.1074/jbc.M117.796284
Carlsten JO, Zhu X, Gustafsson CM (2013) The multitalented mediator complex. Trends Biochem Sci 38:531–537. https://doi.org/10.1016/j.tibs.2013.08.007
Firestein R, Bass AJ, Kim SY et al (2008) CDK8 is a colorectal cancer oncogene that regulates beta-catenin activity. Nature 455:547–551. https://doi.org/10.1038/nature07179
Zhou J, Zeng Y, Cui L et al (2018) Zyxin promotes colon cancer tumorigenesis in a mitotic phosphorylation-dependent manner and through CDK8-mediated YAP activation. Proc Natl Acad Sci U S A 115:E6760–e9. https://doi.org/10.1073/pnas.1800621115
Fisher RP (2005) Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci 118:5171–5180. https://doi.org/10.1242/jcs.02718
Miao J, Kyoyama H, Liu L et al (2020) Inhibition of cyclin-dependent kinase 7 down-regulates yes-associated protein expression in mesothelioma cells. J Cell Mol Med 24:1087–1098. https://doi.org/10.1111/jcmm.14841
Cho YS, Li S, Wang X, Zhu J, Zhuo S, Han Y, Yue T, Yang Y, Jiang J (2020) CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev 34:53–71. https://doi.org/10.1101/gad.333146.119
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149:274–293. https://doi.org/10.1016/j.cell.2012.03.017
Liang N, Zhang C, Dill P et al (2014) Regulation of YAP by mTOR and autophagy reveals a therapeutic target of tuberous sclerosis complex. J Exp Med 211:2249–2263. https://doi.org/10.1084/jem.20140341
Sciarretta S, Zhai P, Maejima Y et al (2015) mTORC2 regulates cardiac response to stress by inhibiting MST1. Cell Rep 11:125–136. https://doi.org/10.1016/j.celrep.2015.03.010
Aharonov A, Shakked A, Umansky KB et al (2020) ERBB2 drives YAP activation and EMT-like processes during cardiac regeneration. Nat Cell Biol 22:1346–1356. https://doi.org/10.1038/s41556-020-00588-4
Jia Y, Li HY, Wang J, Wang Y, Zhang P, Ma N, Mo SJ (2019) Phosphorylation of 14-3-3ζ links YAP transcriptional activation to hypoxic glycolysis for tumorigenesis. Oncogenesis 8:31. https://doi.org/10.1038/s41389-019-0143-1
Cargnello M, Roux PP (2011) Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev 75:50–83. https://doi.org/10.1128/mmbr.00031-10
Hong AW, Meng Z, Yuan HX, Plouffe SW, Moon S, Kim W, Jho EH, Guan KL (2017) Osmotic stress-induced phosphorylation by NLK at Ser128 activates YAP. EMBO Rep 18:72–86. https://doi.org/10.15252/embr.201642681
Seo J, Kim MH, Hong H, Cho H, Park S, Kim SK, Kim J (2019) MK5 regulates YAP Stability and is a molecular target in YAP-Driven cancers. Cancer Res 79:6139–6152. https://doi.org/10.1158/0008-5472.Can-19-1339
Sudol M (1994) Yes-associated protein (YAP65) is a proline-rich phosphoprotein that binds to the SH3 domain of the yes proto-oncogene product. Oncogene 9:2145–2152
Taniguchi K, Wu LW, Grivennikov SI et al (2015) A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature 519:57–62. https://doi.org/10.1038/nature14228
Smoot RL, Werneburg NW, Sugihara T et al (2018) Platelet-derived growth factor regulates YAP transcriptional activity via src family kinase dependent tyrosine phosphorylation. J Cell Biochem 119:824–836. https://doi.org/10.1002/jcb.26246
Sugihara T, Werneburg NW, Hernandez MC et al (2018) YAP Tyrosine Phosphorylation and Nuclear localization in Cholangiocarcinoma cells are regulated by LCK and Independent of LATS Activity. Mol Cancer Res 16:1556–1567. https://doi.org/10.1158/1541-7786.Mcr-18-0158
Zhang L, Tang F, Terracciano L et al (2015) NDR functions as a physiological YAP1 kinase in the intestinal epithelium. Curr Biol 25:296–305. https://doi.org/10.1016/j.cub.2014.11.054
González A, Hall MN, Lin SC, Hardie DG (2020) AMPK and TOR: the Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab 31:472–492. https://doi.org/10.1016/j.cmet.2020.01.015
Mo JS, Meng Z, Kim YC, Park HW, Hansen CG, Kim S, Lim DS, Guan KL (2015) Cellular energy stress induces AMPK-mediated regulation of YAP and the Hippo pathway. Nat Cell Biol 17:500–510. https://doi.org/10.1038/ncb3111
Wang W, Xiao ZD, Li X, Aziz KE, Gan B, Johnson RL, Chen J (2015) AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol 17:490–499. https://doi.org/10.1038/ncb3113
DeRan M, Yang J, Shen CH et al (2014) Energy stress regulates hippo-YAP signaling involving AMPK-mediated regulation of angiomotin-like 1 protein. Cell Rep 9:495–503. https://doi.org/10.1016/j.celrep.2014.09.036
Li Z, Zhao B, Wang P, Chen F, Dong Z, Yang H, Guan KL, Xu Y (2010) Structural insights into the YAP and TEAD complex. Genes Dev 24:235–240. https://doi.org/10.1101/gad.1865810
Mukhopadhyay H, Lee NY (2020) Multifaceted roles of TAK1 signaling in cancer. Oncogene 39:1402–1413. https://doi.org/10.1038/s41388-019-1088-8
Santoro R, Zanotto M, Simionato F et al (2020) Modulating TAK1 expression inhibits YAP and TAZ oncogenic functions in pancreatic Cancer. Mol Cancer Ther 19:247–257. https://doi.org/10.1158/1535-7163.Mct-19-0270
Wang G, Sun Q, Zhu H, Bi Y, Zhu H, Xu A (2021) The stabilization of yes-associated protein by TGFβ-activated kinase 1 regulates the self-renewal and oncogenesis of gastric cancer stem cells. J Cell Mol Med 25:6584–6601. https://doi.org/10.1111/jcmm.16660
Liu F, Xia Y, Parker AS, Verma IM (2012) IKK biology. Immunol Rev 246:239–253. https://doi.org/10.1111/j.1600-065X.2012.01107.x
Wang S, Xie F, Chu F et al (2017) YAP antagonizes innate antiviral immunity and is targeted for lysosomal degradation through IKKɛ-mediated phosphorylation. Nat Immunol 18:733–743. https://doi.org/10.1038/ni.3744
Santoro R, Zanotto M, Carbone C, Piro G, Tortora G, Melisi D (2018) MEKK3 sustains EMT and stemness in pancreatic Cancer by regulating YAP and TAZ transcriptional activity. Anticancer Res 38:1937–1946. https://doi.org/10.21873/anticanres.12431
Lu J, Hu Z, Deng Y, Wu Q, Wu M, Song H (2021) MEKK2 and MEKK3 orchestrate multiple signals to regulate Hippo pathway. J Biol Chem 296:100400. https://doi.org/10.1016/j.jbc.2021.100400
Sun A, Zhu J, Xia S, Li Y, Wu T, Shao G, Yang W, Lin Q (2021) MEKK5 interacts with and negatively regulates the E3 ubiquitin ligase NEDD4 for mediating Lung Cancer Cell Migration. Life (Basel) 11:1153. https://doi.org/10.3390/life11111153
Han T, Gao J, Wang L et al (2020) ASK1 inhibits proliferation and migration of lung cancer cells via inactivating TAZ. Am J Cancer Res 10:2785–2799
Huang W, Lv X, Liu C, Zha Z, Zhang H, Jiang Y, Xiong Y, Lei QY, Guan KL (2012) The N-terminal phosphodegron targets TAZ/WWTR1 protein for SCFβ-TrCP-dependent degradation in response to phosphatidylinositol 3-kinase inhibition. J Biol Chem 287:26245–26253. https://doi.org/10.1074/jbc.M112.382036
Azzolin L, Zanconato F, Bresolin S, Forcato M, Basso G, Bicciato S, Cordenonsi M, Piccolo S (2012) Role of TAZ as mediator of wnt signaling. Cell 151:1443–1456. https://doi.org/10.1016/j.cell.2012.11.027
Boudeau J, Sapkota G, Alessi DR (2003) LKB1, a protein kinase regulating cell proliferation and polarity. FEBS Lett 546:159–165. https://doi.org/10.1016/s0014-5793(03)00642-2
Nguyen HB, Babcock JT, Wells CD, Quilliam LA (2013) LKB1 tumor suppressor regulates AMP kinase/mTOR-independent cell growth and proliferation via the phosphorylation of Yap. Oncogene 32:4100–4109. https://doi.org/10.1038/onc.2012.431
Mohseni M, Sun J, Lau A et al (2014) A genetic screen identifies an LKB1-MARK signalling axis controlling the Hippo-YAP pathway. Nat Cell Biol 16:108–117. https://doi.org/10.1038/ncb2884
Wang S, Ma K, Zhou C et al (2019) LKB1 and YAP phosphorylation play important roles in Celastrol-induced β-catenin degradation in colorectal cancer. Ther Adv Med Oncol 11:1758835919843736. https://doi.org/10.1177/1758835919843736
Zagórska A, Deak M, Campbell DG, Banerjee S, Hirano M, Aizawa S, Prescott AR, Alessi DR (2010) New roles for the LKB1-NUAK pathway in controlling myosin phosphatase complexes and cell adhesion. Sci Signal 3:ra25. https://doi.org/10.1126/scisignal.2000616
Lefebvre DL, Rosen CF (2005) Regulation of SNARK activity in response to cellular stresses. Biochim Biophys Acta 1724:71–85. https://doi.org/10.1016/j.bbagen.2005.03.015
Yuan WC, Pepe-Mooney B, Galli GG et al (2018) NUAK2 is a critical YAP target in liver cancer. Nat Commun 9:4834. https://doi.org/10.1038/s41467-018-07394-5
Gill MK, Christova T, Zhang YY et al (2018) A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat Commun 9:3510. https://doi.org/10.1038/s41467-018-05939-2
Fan R, Kim NG, Gumbiner BM (2013) Regulation of Hippo pathway by mitogenic growth factors via phosphoinositide 3-kinase and phosphoinositide-dependent kinase-1. Proc Natl Acad Sci U S A 110:2569–2574. https://doi.org/10.1073/pnas.1216462110
Kim NG, Gumbiner BM (2015) Adhesion to fibronectin regulates Hippo signaling via the FAK-Src-PI3K pathway. J Cell Biol 210:503–515. https://doi.org/10.1083/jcb.201501025
da Cunha Santos G, Shepherd FA, Tsao MS (2011) EGFR mutations and lung cancer. Annu Rev Pathol 6:49–69. https://doi.org/10.1146/annurev-pathol-011110-130206
Masuda H, Zhang D, Bartholomeusz C, Doihara H, Hortobagyi GN, Ueno NT (2012) Role of epidermal growth factor receptor in breast cancer. Breast Cancer Res Treat 136:331–345. https://doi.org/10.1007/s10549-012-2289-9
Freed DM, Bessman NJ, Kiyatkin A et al (2017) EGFR Ligands differentially stabilize receptor dimers to Specify Signaling Kinetics. Cell 171:683 – 95.e18 https://doi.org/10.1016/j.cell.2017.09.017
Ando T, Arang N, Wang Z et al (2021) EGFR regulates the Hippo pathway by promoting the tyrosine phosphorylation of MOB1. Commun Biol 4:1237. https://doi.org/10.1038/s42003-021-02744-4
Xia H, Dai X, Yu H, Zhou S, Fan Z, Wei G, Tang Q, Gong Q, Bi F (2018) EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis 9:269. https://doi.org/10.1038/s41419-018-0302-x
Moon H, Park H, Chae MJ, Choi HJ, Kim DY, Ro SW (2022) Activated TAZ induces liver cancer in collaboration with EGFR/HER2 signaling pathways. BMC Cancer 22:423. https://doi.org/10.1186/s12885-022-09516-1
Panciera T, Citron A, Di Biagio D et al (2020) Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat Mater 19:797–806. https://doi.org/10.1038/s41563-020-0615-x
Khotskaya YB, Holla VR, Farago AF, Mills Shaw KR, Meric-Bernstam F, Hong DS (2017) Targeting TRK family proteins in cancer. Pharmacol Ther 173:58–66. https://doi.org/10.1016/j.pharmthera.2017.02.006
Yang X, Shen H, Buckley B et al (2019) NTRK1 is a positive regulator of YAP oncogenic function. Oncogene 38:2778–2787. https://doi.org/10.1038/s41388-018-0609-1
Zucchini C, Manara MC, Cristalli C et al (2019) ROCK2 deprivation leads to the inhibition of tumor growth and metastatic potential in osteosarcoma cells through the modulation of YAP activity. J Exp Clin Cancer Res 38:503. https://doi.org/10.1186/s13046-019-1506-3
Esposito D, Pant I, Shen Y, Qiao RF, Yang X, Bai Y, Jin J, Poulikakos PI, Aaronson SA (2022) ROCK1 mechano-signaling dependency of human malignancies driven by TEAD/YAP activation. Nat Commun 13:703. https://doi.org/10.1038/s41467-022-28319-3
Kim DH, Choi HI, Park JS, Kim CS, Bae EH, Ma SK, Kim SW (2019) Src-mediated crosstalk between FXR and YAP protects against renal fibrosis. Faseb j 33:11109–11122. https://doi.org/10.1096/fj.201900325R
Mezquita B, Mezquita P, Pau M, Gasa L, Navarro L, Samitier M, Pons M, Mezquita C (2018) All-trans-retinoic acid activates the pro-invasive Src-YAP-Interleukin 6 axis in triple-negative MDA-MB-231 breast cancer cells while cerivastatin reverses this action. Sci Rep 8:7047. https://doi.org/10.1038/s41598-018-25526-1
McCafferty EH, Dhillon S, Deeks ED (2018) Dasatinib: a review in Pediatric Chronic myeloid leukemia. Paediatr Drugs 20:593–600. https://doi.org/10.1007/s40272-018-0319-8
Garmendia I, Pajares MJ, Hermida-Prado F et al (2019) YES1 drives Lung Cancer Growth and Progression and predicts sensitivity to Dasatinib. Am J Respir Crit Care Med 200:888–899. https://doi.org/10.1164/rccm.201807-1292OC
Oku Y, Nishiya N, Shito T, Yamamoto R, Yamamoto Y, Oyama C, Uehara Y (2015) Small molecules inhibiting the nuclear localization of YAP/TAZ for chemotherapeutics and chemosensitizers against breast cancers. FEBS Open Bio 5:542–549. https://doi.org/10.1016/j.fob.2015.06.007
Yang H, Sun B, Xu K et al (2021) Pharmaco-transcriptomic correlation analysis reveals novel responsive signatures to HDAC inhibitors and identifies Dasatinib as a synergistic interactor in small-cell lung cancer. EBioMedicine 69:103457. https://doi.org/10.1016/j.ebiom.2021.103457
Slemmons KK, Yeung C, Baumgart JT, Juarez JOM, McCalla A, Helman LJ (2020) Targeting Hippo-Dependent and Hippo-Independent YAP1 Signaling for the treatment of Childhood Rhabdomyosarcoma. Cancer Res 80:3046–3056. https://doi.org/10.1158/0008-5472.Can-19-3853
Rao G, Kim IK, Conforti F, Liu J, Zhang YW, Giaccone G (2018) Dasatinib sensitises KRAS-mutant cancer cells to mitogen-activated protein kinase kinase inhibitor via inhibition of TAZ activity. Eur J Cancer 99:37–48. https://doi.org/10.1016/j.ejca.2018.05.013
Kelley MJ, Jha G, Shoemaker D, Herndon JE 2nd, Gu L, Barry WT, Crawford J, Ready N (2017) Phase II study of Dasatinib in previously treated patients with Advanced Non-Small Cell Lung Cancer. Cancer Invest 35:32–35. https://doi.org/10.1080/07357907.2016.1253710
Besse B, Adjei A, Baas P et al (2014) 2nd ESMO Consensus Conference on Lung Cancer: non-small-cell lung cancer first-line/second and further lines of treatment in advanced disease. Ann Oncol 25:1475-84. https://doi.org/10.1093/annonc/mdu123
Soria JC, Ohe Y, Vansteenkiste J et al (2018) Osimertinib in untreated EGFR-Mutated Advanced Non-Small-Cell Lung Cancer. N Engl J Med 378:113–125. https://doi.org/10.1056/NEJMoa1713137
Cortot AB, Jänne PA (2014) Molecular mechanisms of resistance in epidermal growth factor receptor-mutant lung adenocarcinomas. Eur Respir Rev 23:356–366. https://doi.org/10.1183/09059180.00004614
Ghiso E, Migliore C, Ciciriello V et al (2017) YAP-Dependent AXL overexpression mediates resistance to EGFR inhibitors in NSCLC. Neoplasia 19:1012–1021. https://doi.org/10.1016/j.neo.2017.10.003
Nilsson MB, Sun H, Robichaux J et al (2020) A YAP/FOXM1 axis mediates EMT-associated EGFR inhibitor resistance and increased expression of spindle assembly checkpoint components. Sci Transl Med. https://doi.org/10.1126/scitranslmed.aaz4589
Park HS, Lee DH, Kang DH et al (2021) Targeting YAP-p62 signaling axis suppresses the EGFR-TKI-resistant lung adenocarcinoma. Cancer Med 10:1405–1417. https://doi.org/10.1002/cam4.3734
Liu-Chittenden Y, Huang B, Shim JS, Chen Q, Lee SJ, Anders RA, Liu JO, Pan D (2012) Genetic and pharmacological disruption of the TEAD-YAP complex suppresses the oncogenic activity of YAP. Genes Dev 26:1300–1305. https://doi.org/10.1101/gad.192856.112
Cunningham R, Hansen CG (2022) The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin Sci (Lond) 136:197–222. https://doi.org/10.1042/cs20201474
Khan ZM, Real AM, Marsiglia WM, Chow A, Duffy ME, Yerabolu JR, Scopton AP, Dar AC (2020) Structural basis for the action of the drug trametinib at KSR-bound MEK. Nature 588:509–514. https://doi.org/10.1038/s41586-020-2760-4
Kurppa KJ, Liu Y, To C et al (2020) Treatment-Induced Tumor Dormancy through YAP-Mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37:104–22e12. https://doi.org/10.1016/j.ccell.2019.12.006
Gold KA, Lee JJ, Harun N et al (2014) A phase I/II study combining erlotinib and dasatinib for non-small cell lung cancer. Oncologist 19:1040–1041. https://doi.org/10.1634/theoncologist.2014-0228
Creelan BC, Gray JE, Tanvetyanon T, Chiappori AA, Yoshida T, Schell MJ, Antonia SJ, Haura EB (2019) Phase 1 trial of dasatinib combined with afatinib for epidermal growth factor receptor- (EGFR-) mutated lung cancer with acquired tyrosine kinase inhibitor (TKI) resistance. Br J Cancer 120:791–796. https://doi.org/10.1038/s41416-019-0428-3
Kim C, Liu SV, Crawford J et al (2021) A phase I trial of Dasatinib and Osimertinib in TKI Naïve patients with Advanced EGFR-Mutant Non-Small-Cell Lung Cancer. Front Oncol 11:728155. https://doi.org/10.3389/fonc.2021.728155
Verma R, Chen X, Xin D, Luo Z, Ogurek S, Xin M, Rao R, Berry K, Lu QR (2023) Olig1/2-expressing intermediate lineage progenitors are predisposed to PTEN/p53-loss-induced gliomagenesis and harbor specific therapeutic vulnerabilities. Cancer Res. https://doi.org/10.1158/0008-5472.Can-22-1577
Acknowledgements
We thank Wannian Yang for very helpful discussions and critical reading of this manuscript. We apologize for not being able to include some original work due to space constraints.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81372208 and 81871888).
Author information
Authors and Affiliations
Contributions
JZ, TW and QL designed and organized the manuscript. JZ wrote the manuscript and prepared the figures. TW reviewed and edited the manuscript. JZ, WT and QL finalized the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors have no conflicts of interest to declare.
Ethical Approval
The review does not cover human participants and animal studies.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhu, J., Wu, T. & Lin, Q. Non-hippo kinases: indispensable roles in YAP/TAZ signaling and implications in cancer therapy. Mol Biol Rep 50, 4565–4578 (2023). https://doi.org/10.1007/s11033-023-08329-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11033-023-08329-0