
Abstract
The chromatin state is finely tuned to regulate function and specificity for transcription factors such as oestrogen receptor alpha (ER), which contributes to cell growth in breast cancer. ER transcriptional potential is mediated, in large part, by the specific associated proteins and co-factors that interact with it. Despite the identification and characterisation of several ER coregulators, a complete and systematic view of ER-regulating chromatin modifiers is lacking. By exploiting a focused siRNA screen that investigated the requirement for a library of 330 chromatin regulators in ER-mediated cell growth, we find that the NuRD and coREST histone deacetylation complexes are critical for breast cancer cell proliferation. Further, by proteomic and genomics approaches, we discover the transcription factor TRPS1 to be a key interactor of the NuRD and coREST complexes. Interestingly, TRPS1 gene amplification occurs in 28% of human breast tumours and is associated with poor prognosis. We propose that TRPS1 is required to repress spurious binding of ER, where it contributes to the removal of histone acetylation. Our data suggest that TRPS1 is an important ER-associated transcriptional repressor that regulates cell proliferation, chromatin acetylation and ER binding at the chromatin of cis-regulatory elements.
Similar content being viewed by others

Introduction
The transcription factor oestrogen receptor alpha (ER) is known to be a primary driver in several breast cancer subtypes and is present in >75% of all diagnosed cases [1]. Activation of ER by its ligand oestrogen leads to receptor dimerisation and this subsequently elicits transcriptional change by directly binding to chromatin [2]. ER activation in breast cancer models lead to both activation and repression of genes [3]. In breast cancer, ER is routinely targeted therapeutically, using direct ER antagonists such as Tamoxifen or by inhibiting oestrogen synthesis using aromatase inhibitors (AIs) [1]. Although these treatments that target ER activity have dramatically improved patient survival, response to therapy is variable and resistance to hormonal therapy is common. Understanding the fundamental components of the ER complex and the factors involved in mediating gene expression or repression is essential for understanding treatment response differences and for developing newer treatment strategies.
Recent studies have highlighted the importance of epigenetic regulation in cancer [4, 5]. Chromatin modifiers can affect post-translational modifications of histones that are associated with regulating chromatin accessibility in active regions. Chromatin accessibility at ER-binding regions is thought to be regulated prior to ER activation through the help of pioneering transcription factors such as FOXA1 and others [6]. This suggests that chromatin remodelling and epigenetic mechanisms regulate and predetermine ER-binding capabilities. This is particularly important given that ER-binding patterns are known to vary substantially within tumours from women who have distinct treatment responsiveness and clinical outcome [7].
Aberrant transcriptional activity in cancer is often linked with mutations in chromatin regulators [8]. For example, the H3K4 histone methyltransferase MLL2 and MLL3 are mutated in 7.4% and 12.3% of breast cancers, respectively [9]. We have recently identified a role for MLL3 in regulating ER activity [10, 11], suggesting that MLL3 mutations may be directly influencing the transcriptional activity of ER activity in cancer. Indeed, several chromatin regulators have been shown to influence ER activity. Components of the SWI/SNF complex such as BRG1 and BRM have been shown to regulate ER activity [12, 13] and are often deregulated in cancer [14]. Chromatin modifiers and readers are becoming viable drug targets and are currently the subject of intensive and promising pharmacological and clinical studies [15]. In vitro, Histone Deacetylase (HDAC) inhibition or altering the methylation status of cancer lines have been shown to reverse resistance to tamoxifen [16]. Recent studies targeting the ER cofactor BRD4 have demonstrated promising results in both ER-negative and relapsed ER-positive breast cancer [17,18,19]. The basis of ER gene regulation is a dynamic and complex one and an understanding of the epigenetic mechanisms underpinning ER function is essential for exploiting chromatin regulatory proteins as putative drug targets.
In this study, we take a multifaceted approach, involving small interfering RNA (siRNA) screens, genomic and proteomics approaches to identify the key chromatin regulators involved in modulating histone acetylation and ER function at enhancer elements within breast cancer cells. Our findings reveal a critical nexus involving the NuRD and coREST complex, which results in the discovery of TRPS1 as a direct modulator of enhancer potential.
Results
We employed a high-throughput siRNA screen assay to identify chromatin regulators implicated in ER-positive breast cancer cell growth. Coupled with this genetic approach, we also undertook a proteomic screening approach called rapid IP-mass spectrometry of endogenous protein (RIME) [20], allowing us to identify ER interactors by pulldown and proteomics analysis (Fig. 1a) while simultaneously revealing insight into which of these factors have a functional role in breast cancer cell growth.

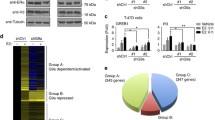
siRNA and proteomics screen to identify ER repressors. a Outline of screening approach for identifying novel ER regulatory proteins. b Graph representing changes in growth following siRNA knockdown for each chromatin modifier in MCF7 cells. Change in cell viability is represented on the Y axis as Z-score deviations from the mean. c Graph representing the RIME proteomic interactors of ER and TRPS1. Proteins identified as both a RIME interactor and a hit based on the siRNA screen have been coloured by their (siRNA screen) Z-score values
We systematically performed a siRNA-based genetic screen for 332 chromatin regulators using the ER-positive breast cancer cell line MCF7. This screen has been previously validated and used to identify chromatin regulators in human epidermal stem cells [21]. Asynchronous MCF7 cells were transfected and subsequently grown for 5 days after which cell viability was assessed using a cellular ATP assay. A pool of three individual siRNAs were used against each target factor and four replicates were performed. Transfection plates were verified to have no position based artefacts. Analysis of results indicated that siRNA treatments resulted in both increased and decreased cell proliferation (Fig. 1b) when compared with non-targeting (NT) control siRNA. BRCA1, BRD4, CHAF1A, EP400, MLL2, SMNDC1 and TDRD7 were among the top 10% of genes where siRNA knockdown led to growth inhibition. As such, these represent genes that are required for cell viability and proliferation. Several of these factors such as BRCA1 and BRD4 have been previously linked with the direct regulation of ER, strengthening the validity of our screen [22, 23]. More recently, MLL2 has also been implicated as an important factor in shaping the chromatin landscape for ER binding [24]. Interestingly, siRNA knockdown of several transcriptional repressors such as the members of the NuRD and coREST complexes: CHD3, CHD4, LSD1, MTA1, MTA2, RBBP7 also led to decreased growth (Fig. 1b). This is in contrast to their perceived role as inhibitors of transcription and cell growth and shows that each of these putative co-repressor proteins are required for cell proliferation. Given that multiple components of the NuRD and coREST complex were identified, there is a clear need for these complexes in ER-driven cancer cell growth.
We performed parallel proteomics experiments with the aim of differentiating direct ER linked chromatin regulators from secondary or ER-independent regulatory complexes. Immunoprecipitation based mass spectrometry experiments (RIME) of ER [20, 25] were performed and ER associated chromatin complexes were compared with factors identified in the siRNA screen (Fig. 1c). We found CHD4, LSD1, MTA2 and RBBP7 to be interacting with ER and required for sustained cell growth, as assessed in the siRNA library screening approach. Thus, members of NuRD or coREST complexes were identified from two independent screens to be both essential factors for cell proliferation and components of the ER complex (Fig. 1, Table S1 and Figure S1A).
We hypothesised that for ER to initiate both activation and repression of transcription, sequence specific transcriptional repressors may be involved in effecting this distinction. As canonical components of the NuRD/coREST complexes do not contain sequence specific transcription factors, we analysed the ER RIME dataset for potential ER interacting transcriptional repressors that could potentially mediate the activity elicited by the NuRD/coREST complex. As a result, we identified the transcription factor Trichorhinophalangeal Syndrome I (TRPS1) to be a novel interactor of ER. TRPS1 is a repressive transcription factor possessing a GATA-like DNA-binding domain similar to GATA3, which has been extensively characterised as an important ER coregulator [26, 27]. Furthermore, TRPS1 interacts with CTBP2 [28] and with HDAC1 and HDAC2 [29], suggesting that TRPS1 is part of a transcriptional repression complex. In breast cancer, TRPS1 is expressed in the luminal compartment of the mammary gland [30] and as such, suggests that it may play a functional role with the ER complex in vivo. Analysis of the METABRIC dataset (997 breast cancers) indicated that TRPS1 tends to be more expressed in ER-positive breast cancer and in the luminal A and B subtypes (Fig. 3d). The TRPS1 genomic region is amplified in 575 tumours out of 2051 (28%) and this gene amplification is significantly associated with poor survival (log-rank test p-value: 1.923e-4). Moreover, a previous in silico analysis combining a siRNA screen several cancer genomics datasets predicted TRPS1 as an oncogene in breast cancer and TRPS1 overexpression increases colony formation in non-tumorigenic mammary gland cells MCF10A [31].
To explore the putative functional role of TRPS1 in breast cancer cells, we assessed whether TRPS1 is required for cell proliferation. We performed Incucyte analysis of cell growth in MCF7 cells after TRPS1 depletion. We found that TRPS1 depletion slowed the growth rate of MCF7 cells, suggesting that TRPS1 is required for optimal cell proliferation (Fig. 2a).

Cell growth in TRPS1 depleted conditions and ChIP-seq of TRPS1 and novel TRPS1 interactors. a MCF7 cell growth assessed by Incucyte following knockdown of TRPS1. Average cell confluence was assessed every 3 h for a total of 200 h in siRNA control (blue dots) and siRNA TRPS1 (orange dots) conditions. Cells depleted of TRPS1 grow slower than control transfected cells. b Genome browser snapshot of ChIP-seq peaks for ER and TRPS1 in MCF7 cells at the TFF1 gene locus. c Heatmaps representing the ChIP-seq signals of ER, TRPS1, coREST, ZNF217, LSD1 and an IgG control at TRPS1 peaks in MCF7 cells
To explore the role of TRPS1 in breast cancer cells, we performed TRPS1 RIME experiments to explore the interactome and regulatory networks associated with this protein [20]. We conducted TRPS1 RIME experiments in MCF7, T47D and ZR75-1 ER-positive breast cancer cells using a commercial anti-TRPS1 antibody. TRPS1 was successfully immunoprecipitated in all three cell lines, confirming the validity of the experiment and the specificity of the TRPS1 antibody. TRPS1 was shown to interact with several components of the NuRD and coREST complexes and this result was consistent in all three cell lines and between replicates. TRPS1 was found to physically interact with CHD4, coREST, CTBP1, CTBP2, GATAD2B, HDAC2, LSD1, MBD3, MTA1, MTA2, RBBP4, RBBP7 and ZNF217 (Fig. 1c). We also identified transcription factors such as COUP-TFII, FOXA1 and GATA3, which are key ER complex components [26, 32, 33] to be TRPS1 interactors. Although the interaction between TRPS1 Ikaros domain and CTBP2 has been characterised previously [28], we confirmed this interaction with the coREST complex by a reciprocal RIME of the core coREST component ZNF217. The ZNF217 RIME identified ZNF217 and several coREST members (coREST, CTBP1, CTBP2, HDAC1, HDAC2, LSD1), along with TRPS1, which was the second most enriched protein after ZNF217 (Supplementary Figure 1C).
Based on the RIME data, we reasoned that the transcription factor TRPS1 recruits the HDAC complexes coREST and NuRD to the chromatin. Given that coREST and NuRD components were essential for cell proliferation based on the siRNA screen, the transcriptional role of TRPS1 may be a modulator of ER activity. In order to identify the genome-wide binding pattern of TRPS1 and to assess potential chromatin interplay with the ER complex, we performed chromatin immunoprecipitation (ChIP) sequencing of TRPS1 in MCF7 and ZR75-1 cells (two replicates for each cell line). Uniquely mapped reads were used to call peaks using MACS2 [34]. In total, we found 23,368 TRPS1 binding sites in MCF7 cells and 56,069 binding sites in ZR751 cells. By motif analysis, we found the GATA motif as the top motif enriched in TRPS1 peaks (Fig. 3b and Supplementary Figure S2). This is in agreement with the hypothesis that TRPS1 binds DNA through its GATA-like DNA binding domain. Strikingly, we found that TRPS1 co-occupies the chromatin with ER in a cell-type-specific manner (Figs. 2b, 3a). In MCF7 cells, ER bound 51% of TRPS1 peaks, whereas the reverse overlap comparison found 40 % of all ER peaks were occupied by TRPS1 (Fig. 3c).

TRPS1 and ER share binding regions in breast cancer. a Heatmaps representing the ChIP-seq signals of ER and TRPS1 in MCF7 and ZR751 cells showing common binding sites and cell line-specific binding sites. b Motif analysis of TRPS1 bound regions reveals GATA as the most enriched motif. c Overlap between ER and TRPS1-binding sites in MCF7 cells. d TRPS1 data in METABRIC cohort: TRPS1 expression level in 997 breast tumours based on ER positivity and PAM50 classification and breast-specific survival
As such, ChIP-seq analysis suggests a global functional connection between TRPS1 and the ER complex in breast cancer cells. To further explore the putative role of components discovered in our RIME and siRNA screens, we performed ChIP-seq of coREST, LSD1 and ZNF217 in MCF7 cells. IgG control ChIP-seq were included as negative controls. We could not validate CHD4 antibodies by RIME and therefore did not proceed with this specific factor any further. We found binding signal of coREST, LSD1 and ZNF217 at ER/TRPS1 binding sites and at TRPS1-only binding sites (Fig. 2c and Supplementary Figure 3). This result supports our hypothesis that TRPS1 is involved in the recruitment of transcription repressive proteins at regions that are bound by the ER complex.
As the coREST and NuRD complexes have histone deacetylation activity, we assessed whether TRPS1 and its interacting chromatin regulators contribute to histone acetylation. As TRPS1 is potentially facilitating the HDAC activity of the coREST and NuRD complexes, we hypothesised that inhibition of TRPS1 would result in increased histone acetylation at the enhancer elements bound by ER, due to impaired deacetylation. We depleted TRPS1 by siRNA (Western blot validation is shown in Supplementary Figure S5) and mapped TRPS1 binding and H3K27ac enrichment by ChIP-seq in MCF7 cells, each of which was conducted in triplicate. These data were integrated with ER ChIP-seq analysis following TRPS1 depletion. As expected, the depletion of TRPS1 by siRNA results in substantially reduced TRPS1 binding (replicate 1: 11,254 peaks in siRNA control versus 4386 peaks in siRNA TRPS1; replicate 2: 36,547 peaks in siRNA control versus 583 peaks in siRNA TRPS1; replicate 3: 26,054 peaks in siRNA control versus 1997 peaks in siRNA TRPS1). Importantly, we found that upon TRPS1 depletion, 4793 ER peaks were weaker, 25,484 ER peaks did not change and interestingly, 18,400 ER peaks were stronger (Fig. 4a). Furthermore, we found that the stronger ER peaks following TRPS1 depletion were also TRPS1-binding sites under control condition (i.e., in siRNA non-targeted transfected conditions). At these regions where ER is stronger following TRPS1 silencing, TRPS1 was lost as expected and H3K27ac signal was increased (Fig. 4b, c). As such, at a subset of TRPS1-binding sites, the presence of TRPS1 decreases ER binding and histone acetylation. There was no significant change in H3K27ac at the ER peaks that are conserved in control and TRPS1 depletion conditions (Fig. 4d, e), implying that these regions are not influenced by TRPS1. This suggests that TRPS1 functions to restrict ER binding, as removal of TRPS1 results in an increased number of ER-binding sites, as well as a failure to deacetylate the H3K27ac chromatin mark at these regions. Overall, these data support the hypothesis that the presence of TRPS1 mediates histone deacetylation and that decreases in TRPS1 levels relieve this repression and culminate in increased ER binding and enhancer acetylation.


TRPS1 is an ER repressor. a Heatmaps representing siTRPS1-specific ER peaks and the ChIP-seq signal of ER, TRPS1 and H3K27ac in siNT (siRNA Non-Targeted) and siTRPS1 conditions in MCF7 cells. b Average signal at siTRPS1-specific ER peaks. c Second replicate of ChIP-seq H3K27ac showing the gain of H3K27ac at ER gained peaks after TRPS1 depletion. d Heatmaps representing the ChIP-seq signal of ER, TRPS1 and H3K27ac at ER lost peaks (siNT specific) and ER conserved peaks (siNT/siTRPS1 common) in control and TRPS1 depletion conditions. e Average ChIP-seq signal of ER, TRPS1 and H3K27ac at ER siNT/siTRPS1 common sites. f Proposed model for ER regulation by TRPS1: TRPS1 limits ER binding through chromatin deacetylation via NuRD and coREST complex
In order to characterise global transcriptional consequences of TRPS1 inhibition, we performed RNA-sequencing experiments in MCF7 cells transfected with siRNA against TRPS1 or a NT control siRNA for 48 h. The cells were also co-treated with estradiol (E2) or control (vehicle) for the last 12 h and four replicates were performed. Using BETA analysis combining siNT/siTRPS1 differential gene expression (expression fold change under 0.5 or above 1.5) and TRPS1 ChIP-seq data, we could identify 213 direct gene targets repressed by TRPS1 and 10 direct gene targets activated by TRPS1 (Supplementary Table 3). This suggests that TRPS1 is mostly a transcriptional repressor (examples of genes repressed by TRPS1 in Supplementary Figure 9). When TRPS1 is depleted, histone acetylation is increased at the 213 direct gene targets repressed transcriptionaly by TRPS1 (Supplementary Figure 7). With DESeq2, we identified 779 E2 upregulated genes and 570 E2 downregulated genes in control condition (expression fold change under 0.5 or above 1.5; adjusted p-value < 0.001). Among these genes, only 569 and 406 genes were still, E2 upregulated and E2 downregulated, respectively, after TRPS1 depletion (Supplementary Figure 6A). Overall, 27% of E2 upregulated genes were not activated in the absence of TRPS1, whereas 29% of E2 downregulated genes were no longer repressed upon TRPS1 silencing. Furthermore, the genes still E2 regulated upon TRPS1 depletion were also significantly less E2 activated or E2 repressed (Supplementary Figure 6B). This suggests that TRPS1 depletion directly influences ER activity and importantly, reorganises ER-binding events, characterised by a substantial increase in new ER-binding sites. We then investigated potential transcriptional activity linked to the 18,400 newly enriched ER-binding sites formed upon TRPS1 depletion. We identified 4036 genes promoters located at <30 kb from these gained peaks. Among these genes, 75 are E2 regulated in control, 61 are E2 regulated upon TRPS1 depletion and 86 are E2 regulated in both conditions. Thus, although TRPS1 prevents ER binding at several thousand sites, depletion of TRPS1 alone is not sufficient to alter transcriptional activity.
The TRPS1 gene is very often amplified in breast cancer (28% of 2051 cases in the METABRIC cohort and 20% of 963 patients in the TCGA cohort) and its amplification is associated with worse prognosis (Fig. 3d). The TRPS1 gene is located in the 8q arm, a chromosomal region, which is often amplified in breast cancer. Therefore, we cannot exclude that the worse prognosis associated with TRPS1 gene amplification is due to a more global effect of the 8q arm amplification. In the METABRIC dataset, we found that lower TRPS1 expression is associated with worse prognosis in all breast cancer (p-value = 0.005; Supplementary Figure 3) but this observation is not significant when considering ER-negative and ER-positive tumours separately. This might be because the TRPS1 gene is more likely to be expressed in ER-positive breast cancer that are of better prognosis than ER-negative breast cancer (Fig. 3d).
Discussion
Taken together, our data suggest that transcriptional repressors like NuRD and coREST complexes are needed for gene regulation by ER and for ER-mediated cell growth. At ER-binding sites, TRPS1 might participate in maintaining histone deacetylation through the recruitment of the repressing complexes together with other transcription factors (Fig. 4f). We hypothesise that this chromatin resetting is necessary to allow the recruitment of ER at the appropriate time and location within the genome [35, 36]. At other genomic regions, TRPS1-mediated histone deacetylation would prevent additional ER binding and therefore restricts the number of ER-binding regions and ER target genes. Our mechanistic insight, along with known consequences of TRPS1 genomic alterations in cancer, suggests TRPS1 is an important factor regulating ER and breast cancer progression.
Materials and methods
Biological material
MCF7, T47D and ZR75-1 human cell lines were obtained from ATCC and grown in Dulbecco’s modified Eagle’s medium (DMEM) or RPMI supplemented with 10% fetal bovine serum (FBS) at 37 °C in 5% CO2 in a humidified incubator. For proliferation assay, cells were seeded into 24-well dishes at a cell density of 2 × 104 per well and growth was monitored using the Essen Bioscience INCUCYTE machine (Ann Arbor, MI, USA). The cell lines were authenticated and tested negative for mycoplasma contamination by the CRUK CI Biorepository Core.
siRNA screen
The siRNA screen was conducted as previously published [21]. In total, 332 siRNAs were custom made from Ambion and targeted factors containing HD, BROMO, CHROMO, PWWP, tandem BRCT, TUDOR, BAH, MBT, SET (including DOT1L), JMJC, JMJN, PRMT, HAT, HDAC, SIRT, DNMT, MBD and SNF2 ATP-dependent remodelers.
For siRNA transfections, 22 million cells were harvested and resuspended in 1.98 mL of buffer SF (Amaxa nucleofection kit). In all, 165 μL of this was aliquoted into round bottom 96-well plates along with 2 μL of siRNA at 10 mM concentration. The mixture was then electroporated (Amaxa 96-well plate electroporater) and 180 μL of warm media was added to each well. To another round bottom 96-well plate contaning 190 μL of media, 60 μL of the electroporated mixture was added. In total, 10 μL of this was then transferred to flat bottom 96-well plates with an additional 100 μL of media. A final of 2400 cells were seeded in each well. Medium was changed the day after transfection for all wells and every alternate day thereafter for a total of 6 days. Cells were then analysed using a cell viability assay (CellTitre-glo, Promega).
Four replicates of each experiment were performed and data obtained from the assay was initially screened for consistency between replicates. A Grubbs’ test was performed to identify and subsequently remove outliers among the four replicates.
siRNA assay for ChIP-seq and RNA-seq
Cells were transfected with siRNA using Lipofectamine2000 (Invitrogen). TRPS1 was silenced using a siRNA pool ON-TargetplusSMARTpool purchased from Dharmacon (catalogue number M-009644-01-0005). AllStars Negative Control siRNA (Qiagen) was used as a negative control.
ChIP-seq library preparation
The antibodies used for the ChIP-seq were anti-coREST (Abcam, ab32631), anti-ER (Santa-Cruz, ref. sc-543), anti-H3K27ac (Abcam, ref. ab4729), anti-LSD1 (Abcam, ab17721), anti-TRPS1 (R&D Systems, ref. AF4838), anti-ZNF217 (Santa-Cruz, sc-55351) and rabbit serum (sc-2027). ChIP were performed as previously described in Schmidt et al. [37], using 10 μg of antibody and 60 million cells. The ChIP-seq and the input libraries were prepared using the TruSeq ChIP Sample Prep Kit (Illumina, ref. IP-202-1012).
RNA-seq library preparation
For hormonal deprivation, MCF7 cells were cultured in phenol-red free DMEM supplemented with 5% charcoal-treated FBS. After 24 h, the cells were transfected with siRNA against TRPS1 or a control siRNA for 48 h. The cells were also co-treated with 100 nM estradiol or vehicle for the last 12 h. Four biological replicates were performed.
Western blots
Western blots were processed as previously described in Ross-Innes et al [38]. The antibodies used were anti-TRPS1 (R&D Systems, ref. AF4838) and anti-histone H3 (ab1791).
Rapid IP-mass spectrometry of endogenous protein
Rapid immunoprecipitation mass spectrometry experiments were performed as previously described [20]. The antibodies used were anti-TRPS1 (R&D Systems, ref. AF4838) and anti-ZNF217 (Abcam, ab48133). MS data were submitted to PRIDE Archive (Project PXD009863).
Bioinformatics
For ChIP-seq libraries, reads were mapped with Bowtie2 to the indexed reference genome UCSC hg19 downloaded from Bowtie2 website. Sam files were then sorted and converted in bam files with Samtools. Sorted bam files were then used for peak calling using MACS2 [34] with default parameters. Common peaks between two replicates were used for the Venn diagrams. Sequencing depth normalised heatmaps were generated as described previously [7]. For screen-shots, normalised bgr files were made, thanks to MACS2 with the command spmr (fragment pileup per million reads) and visualised on Integrative Genomics Viewer. Motif analysis was performed using the Cistrome SeqPos motif tool (version 1.0.0). The de novo motif search and the scan the TRANSFAC motifs was done on the 2000 strongest TRPS1 peaks in MCF7 cells in terms of MACS2 fold enrichment, 200 bp around the summits. RNA-seq libraries were made in four biological replicates. Reads were mapped with Tophat and processed with DESeq2. Significant differentially expressed genes were filtered with adjusted p-value <0.001 and fold change >1.5 or <0.5. To predict the direct gene targets of TRPS1, a BETA analysis was performed using the siNT/siTRPS1 differential gene expression and the TRPS1 ChIP-seq peaks (Wang et al. [39]). Sequencing data were submitted to European Nucleotide Archive (Project number PRJEB26463).
References
Johnston SRD, Dowsett M. Aromatase inhibitors for breast cancer: lessons from the laboratory. Nat Rev Cancer. 2003;3:821–31.
Thomas C, Gustafsson J-Å. The different roles of ER subtypes in cancer biology and therapy. Nat Rev Cancer. 2011;11:597–608.
Thomas C, Gustafsson J-Å. Progesterone receptor-estrogen receptor crosstalk: a novel insight. Trends Endocrinol Metab. 2015;26:453–4.
Rodríguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–9.
Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12–27.
Jozwik KM, Carroll JS. Pioneer factors in hormone-dependent cancers. Nat Rev Cancer. 2012;12:381–5.
Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin S-F, Palmieri C, Caldas C, Carroll JS. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93.
Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P. Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer. Nat Rev Genet. 2013;14:765–80.
Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y, Gräf S, Ha G, Haffari G, Bashashati A, Russell R, McKinney S, METABRIC Group, Langerød A, Green A, Provenzano E, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52.
Jozwik KM, Chernukhin I, Serandour AA, Nagarajan S, Carroll JS. FOXA1 directs H3K4 monomethylation at enhancers via recruitment of the methyltransferase MLL3. Cell Rep. 2016;17:2715–23.
Serandour AA, Avner S, Percevault F, Demay F, Bizot M, Lucchetti-Miganeh C, Barloy-Hubler F, Brown M, Lupien M, Métivier R, Salbert G, Eeckhoute J. Epigenetic switch involved in activation of pioneer factor FOXA1-dependent enhancers. Genome Res. 2011;21:555–65.
Belandia B, Orford RL, Hurst HC, Parker MG. Targeting of SWI/SNF chromatin remodelling complexes to estrogen-responsive genes. EMBO J. 2002;21:4094–103.
DiRenzo J, Shang Y, Phelan M, Sif S, Myers M, Kingston R, Brown M. BRG-1 is recruited to estrogen-responsive promoters and cooperates with factors involved in histone acetylation. Mol Cell Biol. 2000;20:7541–9.
Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014;30:356–63.
Helin K, Dhanak D. Chromatin proteins and modifications as drug targets. Nature. 2013;502:480–8.
Fortunati N, Bertino S, Costantino L, De Bortoli M, Compagnone A, Bandino A, Catalano MG, Boccuzzi G. Valproic acid restores ER alpha and antiestrogen sensitivity to ER alpha-negative breast cancer cells. Mol Cell Endocrinol. 2010;314:17–22.
Alluri PG, Asangani IA, Chinnaiyan AM. BETs abet Tam-R in ER-positive breast cancer. Cell Res. 2014;24:899–900.
Ladd B, Mazzola AM, Bihani T, Lai Z, Bradford J, Collins M, Barry E, Goeppert AU, Weir HM, Hearne K, Renshaw JG, Mohseni M, Hurt E, Jalla S, Bao H, Hollingsworth R, Reimer C, Zinda M, Fawell S, D’Cruz CM. Effective combination therapies in preclinical endocrine resistant breast cancer models harboring ER mutations. Oncotarget. 2016;7:54120–36.
Shu S, Lin CY, He HH, Witwicki RM, Tabassum DP, Roberts JM, Janiszewska M, Huh SJ, Liang Y, Ryan J, Doherty E, Mohammed H, Guo H, Stover DG, Ekram MB, Peluffo G, Brown J, D’Santos C, Krop IE, Dillon D, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529:413–7.
Mohammed H, D’Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JLL, Zwart W, Saadi A, Ross-Innes CS, Chin S-F, Menon S, Stingl J, Palmieri C, Caldas C, Carroll JS. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013;3:342–9.
Mulder KW, Wang X, Escriu C, Ito Y, Schwarz RF, Gillis J, Sirokmány G, Donati G, Uribe-Lewis S, Pavlidis P, Murrell A, Markowetz F, Watt FM. Diverse epigenetic strategies interact to control epidermal differentiation. Nat Cell Biol. 2012;14:753–63.
Kawai H, Li H, Chun P, Avraham S, Avraham HK. Direct interaction between BRCA1 and the estrogen receptor regulates vascular endothelial growth factor (VEGF) transcription and secretion in breast cancer cells. Oncogene. 2002;21:7730–9.
Nagarajan S, Hossan T, Alawi M, Najafova Z, Indenbirken D, Bedi U, Taipaleenmäki H, Ben-Batalla I, Scheller M, Loges S, Knapp S, Hesse E, Chiang C-M, Grundhoff A, Johnsen SA. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014;8:460–9.
Toska E, Osmanbeyoglu HU, Castel P, Chan C, Hendrickson RC, Elkabets M, Dickler MN, Scaltriti M, Leslie CS, Armstrong SA, Baselga J. PI3K pathway regulates ER-dependent transcription in breast cancer through the epigenetic regulator KMT2D. Science. 2017;355:1324–30.
Mohammed H, Taylor C, Brown GD, Papachristou EK, Carroll JS, D’Santos CS. Rapid immunoprecipitation mass spectrometry of endogenous proteins (RIME) for analysis of chromatin complexes. Nat Protoc. 2016;11:316–26.
Theodorou V, Stark R, Menon S, Carroll JS. GATA3 acts upstream of FOXA1 in mediating ESR1 binding by shaping enhancer accessibility. Genome Res. 2013;23:12–22.
Eeckhoute J, Keeton EK, Lupien M, Krum SA, Carroll JS, Brown M. Positive cross-regulatory loop ties GATA-3 to estrogen receptor alpha expression in breast cancer. Cancer Res. 2007;67:6477–83.
Perdomo J, Crossley M. The Ikaros family protein Eos associates with C-terminal-binding protein corepressors. Eur J Biochem. 2002;269:5885–92.
Wu L, Wang Y, Liu Y, Yu S, Xie H, Shi X, Qin S, Ma F, Tan TZ, Thiery JP, Chen L. A central role for TRPS1 in the control of cell cycle and cancer development. Oncotarget. 2014;5:7677–90.
Chen JQ, Bao Y, Litton J, Xiao L, Zhang H-Z, Warneke CL, Wu Y, Shen X, Wu S, Katz RL, Sahin A, Bondy M, Murray JL, Radvanyi L. Expression and relevance of TRPS-1: a new GATA transcription factor in breast cancer. Horm Cancer. 2011;2:132–43.
Sanchez-Garcia FL, Villagrasa P, Matsui J, Kotliar D, Castro VN, Akavia U-D, Chen B-J, Saucedo-Cuevas L, Rodriguez Barrueco R, Llobet-Navas D, Silva JM, Pe er D. Integration of genomic data enables selective discovery of breast cancer drivers. Cell. 2014;159:1461–75.
Hurtado A, Holmes KA, Ross-Innes CS, Schmidt D, Carroll JS. FOXA1 is a key determinant of estrogen receptor function and endocrine response. Nat Genet. 2011;43:27–33.
Kurihara I, Lee D-K, Petit FG, Jeong J, Lee K, Lydon JP, DeMayo FJ, Tsai M-J, Tsai SY. COUP-TFII mediates progesterone regulation of uterine implantation by controlling ER activity. PLoS Genet. 2007;3:e102.
Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 2008;9:R137.
Perissi V, Jepsen K, Glass CK, Rosenfeld MG. Deconstructing repression: evolving models of co-repressor action. Nat Rev Genet. 2010;11:109–23.
Cerase A, Pintacuda G, Tattermusch A, Avner P. Xist localization and function: new insights from multiple levels. Genome Biol. 2015;16:166.
Schmidt D, Wilson MD, Spyrou C, Brown GD, Hadfield J, Odom DT. ScChIP-seq: using high-throughput sequencing to discover protein-DNA interactions. Methods. 2009;48:240–8.
Ross-Innes CS, Stark R, Holmes KA, Schmidt D, Spyrou C, Russell R, Massie CE, Vowler SL, Eldridge M, Carroll JS. Cooperative interaction between retinoic acid receptor-alpha and estrogen receptor in breast cancer. Genes Dev. 2010;24:171–82.
Wang S, Sun H, Ma J, Zang C, Wang C, Wang J, Tang Q, Meyer CA, Zhang Y, Liu XS. Target analysis by integration of transcriptome and ChIP-seq data with BETA. Nature protocols. 2013;8:2502–2515.
Acknowledgements
We would like to thank Clive D’Santos and Chris Taylor from the CRUK CI Proteomics core, James Hadfield and the other members of the Genomics core, and Suraj Menon from the Bioinformatics core. Jason Carroll is supported by an ERC Consolidator grant and a Komen Scholar Award. We would like to acknowledge the support of the University of Cambridge, Cancer Research UK and Hutchison Whampoa Limited.
Author contributions
AAS, HM, KWM and JSC designed the project and wrote the manuscript. AAS, HM and AM performed the experiments and analysed the data.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
These authors contributed equally: A.A. Serandour and H. Mohammed.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Serandour, A.A., Mohammed, H., Miremadi, A. et al. TRPS1 regulates oestrogen receptor binding and histone acetylation at enhancers. Oncogene 37, 5281–5291 (2018). https://doi.org/10.1038/s41388-018-0312-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0312-2
- Springer Nature Limited
This article is cited by
-
TRPS1 maintains luminal progenitors in the mammary gland by repressing SRF/MRTF activity
Breast Cancer Research (2024)
-
TRPS1 regulates the opposite effect of progesterone via RANKL in endometrial carcinoma and breast carcinoma
Cell Death Discovery (2023)