
Abstract
EGFR ligands (e.g., EGF and TGFA) have been shown to be clinically associated with poor survival in lung cancer. Since TGFA itself initiates autochthonous tumors in liver, breast, and pancreas but not in the lung in transgenic mice in vivo, it would appear that an EGFR ligand may not initiate but rather promote lung cancer. However, it has not been proven in vivo whether lung cancer is promoted by an EGFR ligand. Using transgenic mouse models conditionally expressing EGFRL858R or KrasG12D with TGFA (an EGFR ligand) in lung epithelium, we determined that TGFA promoted the growth of EGFRL858R-lung tumors in airway regions but not that of KrasG12D-lung tumors. Analysis of TCGA datasets identified ΔNp63 and AGR2 as potential key tumor-promoting regulators, which were highly induced in the TGFA-induced EGFRL858R-lung tumors. The expression of AGR2 was positively correlated with the expression of TGFA in human EGFR-mutant lung adenocarcinomas. The expression of TGFA in human EGFR-mutant lung adenocarcinomas but not in the EGFR wild-type lung adenocarcinoma was associated with poor survival. These results suggest that targeting EGFR ligands may benefit patients who carry EGFR-mutant lung tumors but will not benefit patients with KRAS-mutant lung tumors.
Similar content being viewed by others


Introduction
In vitro, wild-type epidermal growth factor receptor (EGFR) requires epidermal growth factor (EGF) to transform NIH3T3 fibroblast cells to anchorage independence [1], indicating a dependency on an EGFR ligand for EGFR to activate its biological activity. However, the discovery of EGFR mutations in a portion of lung cancer cases [2,3,4] led to the finding that mutant EGFR is sufficient to induce anchorage-independent growth of both NIH3T3 and transformed tracheobronchial epithelial cells without an EGFR ligand, indicating that mutant EGFR functions as an oncogene independently of its ligand in lung cancer [5]. Although Meyerson and colleagues showed that mutant EGFR does not require an EGFR ligand (EGF) for anchorage-independent cell growth [5], Settleman and colleagues showed that EGF increased the cell number of tyrosine kinase inhibitor (TKI)-resistant EGFR-mutant lung cancer cell lines (H1975 and H1650; wild-type KRAS) but not that of KRAS-mutant lung cancer cell lines (H358 and H1734; wild-type EGFR) in vitro [6], suggesting that EGFR ligands are capable of promoting growth of specific types of lung cancer cells, including EGFR TKI-resistant lung cancer cells. In order to understand the role of an EGFR ligand in lung in vivo, Korfhagen and colleagues developed conditional transgenic mice whose lung epithelial cells induce the expression of transforming growth factor alpha (TGFA), an EGFR ligand [7, 8]. Although an EGFR ligand transformed NIH3T3 cells in vitro [1], the increased expression of TGFA alone in lung induced lung fibrosis but not lung tumors in vivo [7, 8], suggesting that an EGFR ligand is not sufficient to initiate the formation of a lung tumor in vivo, which is distinct from the findings that TGFA alone induced tumors in liver, breast, and pancreas in vivo [9,10,11]. Varmus and colleagues have shown that the conditional expression of two major lung oncogenes, mutant EGFR (EGFRL858R) or mutant KRAS (KrasG12D) [12, 13], in lung epithelial cells was sufficient to initiate lung tumors in vivo [14, 15]. Although the in vitro anchorage-independent cell growth assays and in vivo mouse studies suggest that EGFR ligands may not be required for lung tumor initiation [5, 14, 15], the expression of TGFA (an EGFR ligand) in lung adenocarcinoma is clinically associated with poor survival [16], again suggesting a potential role of an EGFR ligand as a tumor promoter. Recently, Rudensky and colleagues reported a “loss-of-function” study in which conditional deletion of Areg (an EGFR ligand) in T cells reduced the growth of transplantable lung tumor cells (LLC lung tumor cells that harbor KrasG12C; NrasQ61H and EO771 breast tumor cells) in mice [17, 18], suggesting that EGFR ligands promote the growth of metastatic lung tumors in vivo. These findings are relevant to the ongoing clinical trial of a vaccine (CIMAvax-EGF) that targets an EGFR ligand in lung cancer patients. An initial clinical trial with CIMAvax-EGF had promising results [19] and is now being followed by a clinical trial in the United States (NCT02955290). However, it is not yet known which oncogene-driven autochthonous lung tumors will respond to an EGFR ligand-targeted therapy. Identification of driver oncogenes that are promoted by EGFR ligands in vivo is critical in evaluating the efficacy of CIMAvax-EGF in the ongoing clinical trial. In the present study, in order to understand the role of an EGFR ligand in different types of lung tumorigenesis in vivo, we employed a “gain-of-function” approach that conditionally induced the expression of TGFA (an EGFR ligand) in lung epithelial cells in the presence of mutant EGFR or mutant Kras using transgenic mouse models and assessed whether TGFA promotes the growth of EGFR-mutant and/or Kras-mutant autochthonous lung tumors in vivo. Our data indicate that EGFR ligands worsen the EGFR-mutant lung tumorigenesis by acting on specific cell types, including airway epithelial cells and fibroblasts, and altering their cell lineage in vivo.
Results
TGFA (an EGFR ligand) induces the growth of EGFR-mutant tumor cells
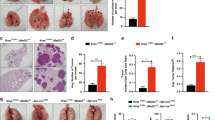
In order to understand the role of an EGFR ligand in lung cancer in vivo, we generated triple-transgenic mice that conditionally induce the expression of either EGFRL858R or KRASG12D along with TGFA in lung epithelial cells upon doxycycline administration (Figs. 1a and 2a and Supplementary Figure S1). The triple-transgenic mice were generated by crossing three different mice carrying different alleles, Scgb1a1-rtTA and [tetO]-EGFRL858R [15] or [tetO]-Kras4bG12D [14] (hereafter [tetO]-KrasG12D) with [tetO]-TGFA [8], resulting in Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA mice and Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA mice (Supplementary Table S1). Scgb1a1 of Scgb1a1-rtTA is derived from rat, which drives the expression of transgenes in the two cell populations (airway club cells and alveolar type 2 cells but not airway basal cells) in mice [20]. The effect of TGFA on EGFRL858R- or KRASG12D-lung tumorigenesis in vivo was demonstrated by the reduction in survival of the mice. TGFA significantly reduced the survival and the body weight of the mice that express EGFRL858R (Fig. 1b and Supplementary Figure S2; median 32 days for Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA mice vs 118 days for Scgb1a1-rtTA;[tetO]-EGFRL858R mice or 248 days for Scgb1a1-rtTA;[tetO]-TGFA mice; p < 0.05 compared to Scgb1a1-rtTA;[tetO]-EGFRL858R mice or Scgb1a1-rtTA;[tetO]-TGFA mice) while TGFA did not influence the survival of the mice that express KRASG12D (Fig. 2b and Supplementary Figure S3). As previously reported [14, 15], histological analysis indicated that the expression of either EGFRL858R or KRASG12D in lung epithelial cells induced lung tumors (adenoma and/or adenocarcinoma). Of note, EGFRL858R induced lung tumors in alveolar regions (Fig. 1c, d) while KRASG12D induced lung tumors in both airway and alveolar regions (Fig. 2c, d). As also previously reported [7, 8], TGFA alone induced fibrosis in pleural regions but did not induce the formation of a lung tumor (Figs. 1c and 2c). Unexpectedly, when EGFRL858R was co-expressed with TGFA, lung tumors driven by EGFRL858R were significantly progressed in airway regions accompanied with pleural fibrosis (Fig. 1c, far right panels, 1d). This is in contrast to lung tumors occurring mainly in alveolar regions when EGFRL858R alone is expressed (Fig. 1c, second panels from the left, and 1d). Consistent with the histology, EGFR downstream signaling pathways, which were assessed by the expression of phospho-ERK, phospho-STAT3, and phospho-AKT [15], were highly activated in the progressed EGFRL858R tumors in airway regions but not in alveolar regions (Supplementary Figures S4a and S5). This result suggests that TGFA promotes EGFRL858R-lung tumors derived from airway club cells but not from alveolar type 2 cells, which may cause airway obstruction that is relevant to the shortened survival in addition to the reduced weight. In contrast, TGFA did not influence the growth and location of KRASG12D-lung tumors in airway and alveolar regions (Fig. 2c, d), which reflects the unchanged survival (Fig. 2b and Supplementary Figure S3). This suggests that further activation of EGFR by TGFA does not influence the EGFR downstream pathways when KRAS harbors an oncogenic mutation in lung cancer. This result is in contrast to the data from KrasG12D-driven pancreatic tumors that were promoted by TGFA in an autochthonous transgenic mouse model [21].

TGFA promotes growth of EGFR-mutant lung tumors in airway regions but not in alveolar regions. a Schematic view of the transgenic mouse model that conditionally induces the expression of EGFRL858R along with TGFA in lung epithelial cells, including airway club cells and alveolar type 2 cells, using tet-inducible system upon doxycycline administration. b Kaplan–Meier survival analysis using Prism 7 indicates that co-expression of EGFRL858R along with TGFA (Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA) significantly shortened survival of the mice compared to the expression of either EGFRL858R (Scgb1a1-rtTA;[tetO]-EGFRL858R) or TGFA (Scgb1a1-rtTA;[tetO]-TGFA) alone (p-value compared to EGFRL858R or TGFA alone; Gehan–Breslow–Wilcoxon test). c Co-expression of EGFRL858R along with TGFA (Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA) in lung epithelium induced lung tumors in airway regions while the expression of EGFRL858R alone (Scgb1a1-rtTA;[tetO]-EGFRL858R) induced lung tumors in alveolar lesions but not in airway regions. The expression of TGFA alone (Scgb1a1-rtTA;[tetO]-TGFA) induced fibrosis in pleural regions but not lung tumors. Histology was assessed by hematoxylin and eosin staining (H&E). The expression of human EGFR, EGFRL858R, and TGFA was confirmed by immunohistochemistry. Scale bar; 50 μm. d The number of EGFRL858R-lung tumors located in alveolar and airway regions is shown. Lung tumors on representative sections from 10 mice of either Scgb1a1-rtTA;[tetO]-EGFRL858R (EGFRL858R) or Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA (EGFRL858R;TGFA) were counted. Results are expressed as mean ± SD for each group. #p < 0.05 (Student’s t-test [two-tailed and unpaired])

TGFA does not promote growth of KRAS-mutant lung tumors. a Schematic view of the transgenic mouse model that conditionally induces the expression of KRASG12D along with TGFA in lung epithelial cells, including airway club cells and alveolar type 2 cells, using a tet-inducible system upon doxycycline administration. b Kaplan–Meier survival analysis using Prism 7 indicates that TGFA does not influence the survival of the mice whose lung tumors are induced by KRASG12D after doxycycline administration (Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA vs Scgb1a1-rtTA;[tetO]-KrasG12D or Scgb1a1-rtTA;[tetO]-TGFA; no significant difference p > 0.05; Gehan–Breslow–Wilcoxon test). c TGFA did not influence the growth of KRASG12D-mutant lung tumors in airway and alveolar regions (see Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA vs Scgb1a1-rtTA;[tetO]-KrasG12D). Histology was assessed by hematoxylin and eosin staining (H&E). The expression of human EGFR, EGFRL858R, and TGFA was assessed by immunohistochemistry. Scale bar; 50 μm. d The number of KRASG12D-lung tumors located in alveolar and airway regions is shown. Lung tumors on representative sections from nine mice of either Scgb1a1-rtTA;[tetO]-KrasG12D (KrasG12D) or Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA (KrasG12D;TGFA) were counted. TGFA did not alter the number of KRASG12D-lung tumors grown in airway or alveolar regions. Results are expressed as mean ± SD for each group.
ΔNp63 is induced in EGFRL858R-airway lung tumors driven by TGFA
The EGFRL858R-airway lung tumors promoted by TGFA protruded into airway lumina (Fig. 1c), a phenotype of which is similar to the airway epithelial cell hyperplasia seen in the conditional Pten-deleted mice [22, 23] and in mice whose Hippo-Yap pathway is conditionally activated in airway epithelial cells, including airway basal cells and club cells [24]. Thus, we hypothesized that Pten and/or components of the Hippo-Yap pathway might be altered in TGFA-driven EGFRL858R-airway lung tumors. However, immunohistochemical analysis indicated that the expression of PTEN and nuclear YAP1 (activated form) was not altered in the TGFA-driven EGFRL858R-airway lung tumors compared to other mouse groups (Supplementary Figures S4b and S5). The mRNA expression of Pten and Ctgf (a consensus downstream target of the Hippo-Yap pathway [25]) was not altered either (Supplementary Figure S4c), suggesting that EGFRL858R-airway lung tumor growth induced by TGFA does not occur via the PTEN or the Hippo-Yap pathway. Rajagopal and colleagues reported that ΔNp63 (an isoform of TP63 [TP63 for human and Trp63 for mouse]; a marker for basal stem cells and squamous cell carcinoma [26]) was required for the YAP1-mediated airway epithelial cell hyperplasia, suggesting that ΔNp63 is the downstream effector for the Hippo-Yap pathway to induce the cell hyperplasia [24] though ΔNp63 is regulated by not only the Hippo-Yap pathway but also by other signaling pathways [27]. Thus, we sought to determine whether ΔNp63 was induced in the TGFA-driven EGFRL858R-airway lung tumors. Notably, immunohistochemical analysis indicated that ΔNp63 was expressed only in EGFRL858R-airway lung tumor cells (Fig. 3a, top far right panel) but not in other lung tumor cells, including EGFRL858R-alveolar lung tumor cells and KRASG12D-lung tumor cells. The specific expression of ΔNTrp63 in the TGFA-driven EGFRL858R-airway lung tumors was further confirmed at the mRNA level (Fig. 3b). These results suggest that airway club cells expressing EGFRL858R adopt a basal and/or squamous cell carcinoma-like program in the presence of TGFA, which is similar to the finding that the overexpressed YAP1 in airway club cells induced airway epithelial cell hyperplasia accompanied by the induction of basal stem cell markers, including ΔNp63 [24].

ΔNp63 is induced in EGFR-mutant airway lung tumors promoted by TGFA. a The expression of ΔNp63 in lungs of the transgenic mice assessed by immunohistochemistry is shown. ΔNp63 (a marker for airway basal cells and lung squamous cell carcinoma cells) was induced in TGFA-driven EGFRL858R-airway lung tumor cells derived from airway club cells. ΔNp63 was not expressed in EGFRL858R-alveolar lung tumor cells, KRASG12D-lung tumors cells, TGFA-driven pleural fibroblast cells, or normal lung epithelial cells (except airway basal cells; not shown here). Scale bar; 50 μm. b The mRNA expression of ΔNTrp63 (ΔNp63) from whole lungs of the transgenic mice (n = 3 for each group) is shown. The expression of ΔNTrp63 from lungs of Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA (EGFRL858R;TGFA) was significantly higher than that of either Scgb1a1-rtTA;[tetO]-EGFRL858R (EGFRL858R), Scgb1a1-rtTA;[tetO]-TGFA (TGFA), or littermate controls (Control). There was no significant change in the expression of ΔNTrp63 in the lungs of the mouse groups with genotypes of Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA (KrasG12D;TGFA), Scgb1a1-rtTA;[tetO]-KrasG12D (KrasG12D), or Scgb1a1-rtTA;[tetO]-TGFA (TGFA). Results are expressed as mean ± SEM for each group. *p < 0.05 (Student’s t-test [two-tailed and unpaired]). c Pearson correlation of ΔNp63 with EGFR ligands (EPGN, TGFA, BTC, EGF, EREG, HBEGF, and AREG) was performed using mRNA expression datasets from TCGA lung adenocarcinoma cases, including cases with EGFR mutations or KRAS mutations. Rank orders for each EGFR ligand were also obtained. Both analyses were performed as described in Materials and methods
Next, taking advantage of TCGA database [12], we sought to determine whether there is a possibility that in human lung cancer ΔNp63 is induced by EGFR ligands (including TGFA) only in EGFR-mutant lung adenocarcinoma but not in KRAS-mutant lung adenocarcinoma. We retrieved the RNA-seq data from TCGA cases of lung adenocarcinoma [12] that harbor mutations and/or copy number alterations in EGFR or KRAS, and assessed whether ΔNp63 is correlated with EGFR ligands in EGFR-mutant lung adenocarcinoma or in KRAS-mutant lung adenocarcinoma. A positive correlation of EGFR ligands (EPGN and EREG) and ΔNp63 was seen in EGFR-mutant lung adenocarcinoma but not in KRAS-mutant lung adenocarcinoma (Pearson correlation > 0.3; Fig. 3c). Of note, since 13–55% of lung adenosquamous carcinomas, the majority of whose lung tumors express ΔNp63, harbor EGFR mutations (Supplementary Table S2), EGFR ligands may be positively correlated with ΔNp63 in lung adenosquamous carcinoma, which implies that EGFR ligands may induce ΔNp63 in EGFR-mutant lung adenosquamous carcinoma.
AGR2 is induced only in EGFRL858R-airway lung tumors driven by TGFA
In analyzing the TCGA datasets, we also aimed to identify genes in an unbiased fashion that are highly correlated with TGFA only in EGFR-mutant lung adenocarcinoma but not in KRAS-mutant lung adenocarcinoma. Among each group of the top 100 genes highly correlated with TGFA in EGFR- or KRAS-mutant lung adenocarcinoma (Supplementary Table S3), 3 genes (PROM2, CARD10, and PRNP) were highly correlated with TGFA in both groups and they were excluded from further analysis (Fig. 4a and Supplementary Table S3). Among the 97 genes that are highly correlated with TGFA only in EGFR-mutant lung adenocarcinoma, Gene Ontology analysis using the ToppGene Suite (https://toppgene.cchmc.org) indicated that 3 genes (AGR2, ADORA1, and PLAUR) are known to be involved in positive regulation of the EGFR signaling pathway (GO:0045742; Fig. 4a), suggesting that these 3 genes may be induced by TGFA in the EGFRL858R-airway lung tumors and may also contribute to the promotion of the EGFRL858R-airway lung tumor growth. Since lung tumors express not only TGFA but also other EGFR ligands, including EGF, AREG, BTC, EREG, HBEGF, and EPGN, these EGFR ligands may also contribute to the induction of the 3 genes (AGR2, ADORA1, and PLAUR) in EGFR-mutant lung tumors. Thus, we assessed the correlation of all of the EGFR ligands with the 3 genes (AGR2, ADORA1, and PLAUR) in EGFR-mutant or KRAS-mutant lung adenocarcinoma cases. Notably, AGR2, a disulfide isomerase that promotes lung tumorigenesis [28], was positively correlated with 3 (TGFA, BTC, and EGF; Pearson correlation > 0.3) out of the 7 EGFR ligands in EGFR-mutant adenocarcinoma cases but not in KRAS-mutant lung adenocarcinoma cases (Fig. 4b). In contrast, ADORA1 and PLAUR were positively correlated with EGFR ligands in both EGFR-mutant and KRAS-mutant lung adenocarcinoma cases (Pearson correlation > 0.3; Fig. 4b). These analyses suggest that AGR2 is a more specific downstream target of EGFR ligands in EGFR-mutant lung adenocarcinoma than ADORA1 and PLAUR. Consistent with the analyses using human TCGA datasets, AGR2 was significantly expressed in EGFRL858R-airway lung tumors induced by TGFA but not in normal lung, TGFA-induced fibrotic lungs, EGFRL858R-alveolar lung tumors, or KRASG12D-lung tumors in mice (Fig. 4c, top far right panel). The specific expression of Agr2 in the mouse lungs that co-express EGFRL858R and TGFA was also confirmed at the mRNA level (Fig. 4d). These results suggest that AGR2 may contribute to the growth of EGFRL858R-airway lung tumors induced by TGFA.

AGR2 is induced in EGFR-mutant airway lung tumors promoted by TGFA. a Top 100 genes highly correlated with TGFA in EGFR-mutant or KRAS-mutant lung adenocarcinoma were extracted from TCGA database as described in Materials and methods. Three genes overlapped between EGFR-mutant and KRAS-mutant lung adenocarcinomas. The Gene Ontology (GO) analysis indicated that AGR2, ADORA1, and PLAUR that belong to the positive regulation of EGFR signaling pathway were included in the top 100 genes highly correlated with TGFA in EGFR-mutant but not that in KRAS-mutant lung adenocarcinoma. b Pearson correlation of AGR2, PLAUR, and ADORA1 with EGFR ligands (EPGN, TGFA, BTC, EGF, EREG, HBEGF, and AREG) was performed using mRNA expression datasets from TCGA lung adenocarcinoma cases, including cases with EGFR mutations or KRAS mutations. Rank orders for each EGFR ligand were also obtained. Both analyses were performed as described in Materials and methods. c The expression of AGR2 in lungs of the transgenic mice assessed by immunohistochemistry is shown. AGR2 (a disulfide isomerase) was induced in TGFA-driven EGFRL858R-airway lung tumor cells but not expressed in EGFRL858R-alveolar lung tumor cells, KRASG12D-lung tumors cells, TGFA-driven pleural fibroblast cells, or normal lung epithelial cells. Scale bar; 50 μm. d The mRNA expression of Agr2 from whole lungs of the transgenic mice (n = 3 for each group) is shown. The expression of Agr2 from lungs of Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA (EGFRL858R;TGFA) was significantly higher than that of Scgb1a1-rtTA;[tetO]-EGFRL858R (EGFRL858R), Scgb1a1-rtTA;[tetO]-TGFA (TGFA), or littermate controls (Control). Such specific induction of Agr2 in the lungs of Scgb1a1-rtTA;[tetO]-KrasG12D;[tetO]-TGFA (KrasG12D;TGFA) mice was not observed compared to mouse groups of Scgb1a1-rtTA;[tetO]-KrasG12D (KrasG12D) and Scgb1a1-rtTA;[tetO]-TGFA (TGFA). Results are expressed as mean ± SEM for each group. *p < 0.05 (Student’s t-test [one-tailed and unpaired])
AGR2 is required for tumorigenicity of EGFR-mutant lung adenocarcinoma in vitro
In order to address whether ΔNp63 and AGR2, both of which are induced by TGFA in EGFR-mutant lung adenocarcinoma, are functionally involved in tumorigenesis, we sought to assess their functions by a loss-of-function study using short hairpin RNA (shRNA) in vitro. ΔNp63 and AGR2 were previously shown to promote tumorigenesis in EGFR wild-type lung cancer cell lines [26, 28]; however, their roles in EGFR-mutant lung cancer cell lines have not been determined. Although ΔNp63 is expressed in a portion of human lung adenocarcinoma, squamous cell carcinoma, and adenosquamous cases in vivo [12, 29] (Supplementary Table S2), conventionally available EGFR-mutant lung cancer cell lines (e.g., H1975, H1650, and HCC827) hardly express endogenous ΔNp63 [30]. Thus, the in vitro study using shRNA here focuses on determining the role of AGR2 in the EGFR-mutant lung adenocarcinoma cells. We confirmed significant suppression of AGR2 by two independent shRNAs in H1975 cells (Supplementary Figure S6a). Importantly, anchorage-independent growth and migration of H1975 cells were significantly suppressed (Supplementary Figure S6b and S6c), indicating that AGR2 contributes to the malignant tumorigenesis of EGFR-mutant lung adenocarcinoma. Notably, TGFA-mediated promotion of anchorage-independent growth and migration of H1975 cells was also suppressed in the presence of shRNA targeting AGR2 (Supplementary Figure S6d and S6e). Contrary to the in vivo results (Fig. 4), TGFA did not further induce AGR2 mRNA in H1975 cells (AGR2 proficient cells) in vitro (data not shown). These results suggest that AGR2 may be required but not sufficient for the TGFA-mediated promotion of EGFR-mutant lung tumorigenesis.
AGR2 is highly expressed in human EGFR-mutant lung adenocarcinoma whose TGFA is highly expressed
In order to assess whether the expression of ΔNp63 and/or AGR2 is influenced by the expression of TGFA in EGFR-mutant lung adenocarcinoma at the protein level, we performed immunohistochemistry using human primary lung tumor specimens that harbor EGFR mutations (Supplementary Table S4). ΔNp63 was localized in the nucleus while AGR2 and TGFA were mainly localized in the cytoplasm (Fig. 5a, right panels). We separated each EGFR-mutant lung adenocarcinoma specimen into two groups (low vs high, separated by a 10% cutoff in tumor field) based on the expression of ΔNp63, AGR2, and TGFA (Fig. 5a). As shown in Fig. 5b, the expression of AGR2 was significantly positively correlated with the expression of TGFA at the protein level (two-tailed Fisher’s exact test: p = 0.0144), suggesting that TGFA induces the expression of AGR2 in human EGFR-mutant lung adenocarcinoma in vivo. However, such positive correlation of ΔNp63 with TGFA was not observed (two-tailed Fisher’s exact test: p = 0.4; Fig. 5b). Since the analysis of TCGA datasets indicated that EPGN and EREG (EGFR ligands) correlated with ΔNp63 more than TGFA (Fig. 3c), EPGN and EREG might be major inducers of ΔNp63. Further immunohistochemical analysis detecting EPGN and EREG with a larger number of EGFR-mutant lung adenocarcinoma cases is required to elucidate the regulation of ΔNp63 by EGFR ligands in EGFR-mutant lung adenocarcinoma.

TGFA is significantly associated with poor survival of patients whose lung adenocarcinoma harbor EGFR mutations but not EGFR wild-type lung adenocarcinoma. a Shown is immunohistochemical analysis of TGFA, AGR2, and ΔNp63 on primary lung adenocarcinoma harboring EGFR mutations. The criteria of low or high expression was determined as being below or above 10% of their expression in the tumor field. Scale bar; 50 μm. b The ratio of the expression of AGR2 high or AGR2 low was shown in TGFA low or TGFA high group (upper panels) in primary lung adenocarcinomas harboring EGFR mutations. The expression of AGR2 was significantly correlated with TGFA (two-tailed Fisher’s exact test, p < 0.05). The ratio of the expression of ΔNp63 high or ΔNp63 low was also shown in TGFA low or TGFA high group (lower panels). c Kaplan–Meier survival analysis using cases of lung cancer patients whose lung adenocarcinomas harbor EGFR mutations was performed based on the expression of TGFA detected by immunohistochemistry. The criteria were determined as described in a. The expression of TGFA in EGFR-mutant lung adenocarcinoma was significantly associated with worsened survival and recurrence-free survival (p < 0.05; Gehan–Breslow–Wilcoxon test). d Kaplan–Meier survival analysis using cases of lung cancer patients whose lung adenocarcinomas harbor wild-type EGFR was performed as described in c. The expression of TGFA in EGFR wild-type lung adenocarcinoma did not influence overall survival or recurrence-free survival (p > 0.05; Gehan–Breslow–Wilcoxon test)
TGFA associates with poor survival in EGFR-mutant lung adenocarcinoma but not EGFR wild-type lung adenocarcinoma
In order to further determine the importance of TGFA in EGFR-mutant lung adenocarcinoma, we assessed the overall survival and recurrence-free survival of lung cancer patients whose resected lung tumors carry EGFR mutations based on the expression of TGFA, which was detected by immunohistochemistry (Fig. 5a and Supplementary Table S4). As shown in Fig. 5c, TGFA was significantly associated with poor survival in both overall survival and recurrence-free survival in EGFR-mutant lung adenocarcinoma cases. However, such association was not observed in EGFR wild-type lung adenocarcinoma cases (Fig. 5d). These results suggest that TGFA (an EGFR ligand) promotes tumorigenesis of EGFR-mutant lung adenocarcinoma but not EGFR wild-type lung adenocarcinoma (e.g., KRAS-mutant lung adenocarcinoma), which is consistent with the mouse study.
Discussion
EGFR ligands have been known to be associated with poor survival in lung cancer in a portion of patients’ cohorts, thus the role of such ligands in lung tumorigenesis has been eagerly assessed by multiple studies. Such studies, which have been performed by in vitro assays, indicated that the presence of mutations and/or alterations in EGFR is critical for tumorigenic effects by EGFR ligands; however, such tumorigenic effects by an EGFR ligand have not previously been assessed by an in vivo study. Here we demonstrated a role of TGFA (an EGFR ligand) in lung tumorigenesis using transgenic mice that develop autochthonous lung tumors in vivo. TGFA promoted lung fibrosis and the growth of EGFR-mutant lung tumors in airway regions accompanied by the expression of ΔNp63 and AGR2, which are, to our knowledge, in vivo-specific downstream targets of ligand-activated mutant EGFR. TGFA did not influence the growth of Kras-mutant lung tumors. The expression of TGFA was associated with poor survival of human patients whose lung tumors harbor EGFR mutations. These results indicate that EGFR ligands bind to mutant EGFR in airway cells, which turns on the EGFR signaling pathway, thereby promoting the growth of EGFR-mutant lung tumors.
Due to the discovery of TKIs specifically binding to mutant EGFR, thereby blocking its oncogenic function [2,3,4], EGFR ligands as therapeutic targets have been almost forgotten. However, continuous recurrences of EGFR TKI-resistant lung adenocarcinoma have led to the need to investigate additional therapeutic targets, which includes EGFR ligand-targeted therapy such as CIMAvax-EGF [19]. Following the development of the mouse models mimicking primary EGFR-mutant lung cancer (EGFRL858R or EGFRΔL747-S752) by Politi et al. [15], additional mouse models mimicking recurrent EGFR-mutant lung cancer (EGFRT790M-L858R, EGFRT790M-ΔE746-A750, and/or EGFRL858R/T790M/C797S) have been developed and used for preclinical studies [31,32,33,34,35]. Although EGF was shown to increase the cell number of lung cancer cells that carry such recurrent EGFR mutations in vitro [6], the in vivo role of EGFR ligands in such EGFR-mutant lung cancer mouse models has not been investigated until the present study. Hence, the importance of EGFR ligands as therapeutic targets has not been properly assessed. Importantly, our study demonstrates that TGFA (an EGFR ligand) extensively induced the growth of EGFRL858R-lung tumors along with fibrosis, supporting the therapeutic approach targeting EGFR ligands. Further study with different combinations of triple-transgenic mice carrying the allele of ectopic TGFA with other TKI-naive or -resistant EGFR-mutant allele (EGFRΔL747-S752, EGFRT790M-L858R, EGFRT790M-ΔE746-A750, or EGFRL858R/T790M/C797S) will determine the in vivo role of TGFA in lung cancer driven by different types of EGFR mutations. Since hepatocyte growth factor or fibroblast growth factor (FGF) (but not EGF) conferred resistance to erlotinib (the first-generation EGFR TKI)-mediated cell death of EGFR-mutant lung adenocarcinoma cells in vitro [36], TGFA-mediated fibrosis associated with EGFR-mutant lung tumors in vivo may induce growth factors (e.g., FGF) that confer resistance to EGFR TKIs, including gefitinib, erlotinib, afatinib, and osimertinib. Testing these TKIs in triple-transgenic mice that conditionally express different types of mutant EGFR along with TGFA in lung epithelium will validate TGFA as a therapeutic target for recurrent EGFR-mutant lung cancer (e.g., osimertinib-resistant) in vivo.
Currently, there are seven EGFR ligands, including EGF, TGFA, HBEGF, BTC, AREG, EREG, and EPGN. In the present study, we employed a “gain-of-function” approach using the transgenic mouse system that carry tet-inducible TGFA, which allows the conditional induction of TGFA along with EGFRL858R or KRASG12D in lung epithelium. Although there are no transgenic mice that carry tet-inducible EGFR ligands other than TGFA to our knowledge, transgenic mice (Tg[CAG-Btc]) developed by Schneider et al. [37] that overexpress BTC (an EGFR ligand) ubiquitously, including lung, can be used to assess the role of BTC in EGFR-mutant or Kras-mutant lung cancers by crossing the mice with the double-transgenic mice that overexpress mutant EGFR or mutant KRAS. The roles of EGFR ligands can also be assessed by a “loss-of-function” approach using mice in which EGFR ligands are conditionally or permanently deleted. Rudensky and colleagues reported using mice in which Areg (an EGFR ligand) is conditionally deleted in T cells that AREG is required for growth of transplanted LLC lung tumor cells (KrasG12C; NrasQ61H) and EO771 breast tumor cells. Of note, AREG did not directly influence the tumorigenesis of LLC and EO771 cells, suggesting that AREG promotes the growth of metastatic lung or breast tumor cells through stromal cells [18]. Although TGFA did not promote the growth of primary autochthonous KrasG12D-lung tumors in our mouse model, different EGFR ligands may influence tumorigenesis depending on driver genes and/or location (e.g., primary vs metastatic). Additionally, the roles of EGFR ligands in other lung cancers driven by different driver oncogenes can also be assessed by crossing the tet-inducible TGFA transgenic mice or EGFR ligand-deleted mice with other lung cancer model mice, including [tetO]-BRAFV600E [38], [tetO]-PIK3CAH1047R [39], [tetO]-HER2YVMA [40], [tetO]-EML4-ALK [41], [tetO]-CRAFBxB [42], [tetO]- DDR2L63V [43], and [tetO]-KIF5B-RET [44]. A recent in vitro study by Lemmon and colleagues using MCF-7 breast adenocarcinoma cells indicated in vitro that EGF, TGFA, HBEGF, BTC, and AREG induced proliferation of MCF-7 breast adenocarcinoma cells while EREG and EPGN induced differentiation of MCF-7 cells [45], suggesting that each EGFR ligand may function differently in influencing the tumor morphology. Creation of new transgenic models overexpressing other EGFR ligands, including EGF, HBEGF, AREG, EREG, and EPGN will lead to comprehensive understanding of in vivo functions of EGFR ligands in lung tumorigenesis that is determined by not only lung tumor cells but also tumor-associated stromal cells, both of which may be influenced by EGFR ligands.
Although lung tumors were not initiated by TGFA itself in vivo [7, 8], TGFA alone did initiate tumors in liver, breast, and pancreas in vivo [9,10,11]. Importantly, TGFA promoted the growth of KrasG12D-driven pancreatic tumors [21], which is distinct from our present study demonstrating that TGFA promoted the growth of EGFRL858R-driven lung tumors but not that of KrasG12D-driven lung tumors (Figs. 1 and 2), indicating a tissue-specific role of TGFA. TGFA is also reported to morphologically convert acinar cells to duct-like cells in the pancreas [11], suggesting that TGFA can reprogram cell lineage. In our present study, TGFA induced ΔNp63-positive EGFRL858R-airway tumor cells that are likely to be derived from ΔNp63-negative airway club cells (Fig. 3a, b). Airway basal cells (ΔNp63-positive) are considered to be stem cells that produce differentiated airway epithelial cells (ΔNp63-negative), including club, ciliated and goblet cells, to maintain airway homeostasis [46]. However, upon loss of airway basal cells, airway club cells were shown to be dedifferentiated into airway basal cells in mice [47], indicating the plasticity of airway club cells. Indeed, overexpression of YAP1 in airway club cells converts ΔNp63-negative airway club cells into ΔNp63-positive airway basal cells, which in turn promotes airway squamous stratification [24]. Such plasticity of airway club cells has also been demonstrated in the transformation of airway club cells to adenosquamous lung tumors that harbor the characteristics of both adenocarcinoma and squamous carcinoma of the lung. The induction of mutant HER2 or the loss of Pten and Smad4 in airway club cells has been shown to lead to lung adenosquamous tumor development [40, 48]. Such plasticity may not be limited to airway club cells. Transdifferentiation of lung adenocarcinoma (ΔNp63-negative) to lung squamous and/or adenosquamous carcinoma (ΔNp63-positive) has been shown in alveolar regions of mouse lungs whose cells express mutant Kras and lack Stk11 [49,50,51], suggesting that alveolar type 2 cells might be the origin of the cells that develop lung squamous and/or adenosquamous carcinoma. In the present study, we demonstrated that mutant EGFR, co-expressed with TGFA, induced the development of ΔNp63-positive airway tumor cells (Fig. 3a, b), which suggests the possibility that mutant EGFR that normally drives lung adenocarcinoma (ΔNp63-negative) may drive lung adenosquamous carcinoma (ΔNp63-positive) in the presence of excessive and/or a particular EGFR ligand (e.g., TGFA). Importantly, EGFR mutations are frequently seen in human lung adenosquamous carcinoma (Supplementary Table S2), suggesting that our mouse model may recapitulate the initiation event of human lung adenosquamous carcinoma. However, since the mice did not fully develop the lung adenosquamous phenotype, additional genetic alteration may be required to fully mimic the pathogenesis of lung adenosquamous carcinoma. Further study investigating EGFR ligands in human lung adenosquamous carcinoma that carry EGFR mutations will lead to the understanding whether EGFR ligands contribute to the cell lineage alteration in lung cancer.
In our present study, TGFA was associated with poor survival in patients who carry EGFR mutations but not those in patients who carry wild-type EGFR, which is consistent with the data obtained by our mouse model. Of note, although it was previously shown by Sugimachi and colleagues that TGFA was associated with poor survival in lung adenocarcinoma patients who resided in Japan [16], Rusch and colleagues did not observe such association in the US patients who had non-small-cell lung cancer (NSCLC), including lung adenocarcinoma, lung squamous cell carcinoma, and lung large-cell carcinoma [52]. Since these studies were performed before EGFR mutations were identified, it is unknown whether the association of TGFA with patients whose lung tumors carried EGFR mutations would influence the survival in the populations in Japan and the United States. Since lung adenocarcinoma patients in Japan carry EGFR mutations more frequently than those in the United States [13], the positive association of TGFA with poor survival in lung adenocarcinoma patients in Japan but not in the United States might be derived from the frequent number of patients who carry EGFR mutations in Japan but not in the United States. In addition to TGFA, EREG was also assessed by Kurie and colleagues as to whether EREG was associated with survival of NSCLC patients that resided in the United States. A trend was observed that EREG was associated with poor survival in the population; however, it was not statistically significant (p = 0.054) [53]. Selection of the NSCLC patients by tumor histology (e.g., adenocarcinoma) and/or mutation status of driver genes (e.g., EGFR) may provide a better prognosis value of EREG. Systematic staining of all of the EGFR ligands in larger cohorts of lung cancer specimens whose types of driver oncogenes are identified will provide precise prognosis and therapeutic values of EGFR ligands in lung cancer.
In summary, using in vivo autochthonous mouse models, we determined that TGFA, an EGFR ligand, promoted the growth of EGFR-mutant lung tumors but not that of Kras-mutant lung tumors. The EGFR-mutant lung tumors enhanced by TGFA induced the key regulators ΔNp63 and AGR2 that promote tumorigenesis. TGFA was associated with poor survival in EGFR-mutant lung adenocarcinoma in humans. Although EGFR TKI therapy has been shown to extend the survival of patients whose lung tumors carry EGFR mutations, such lung cancers recur by acquiring different mechanisms of resistance to the therapy. Our present study suggests that targeting EGFR ligands (e.g., by using vaccinations such as CIMAvax-EGF [19] or therapeutic antibodies against TGFA and EREG) may lead to therapeutic benefits for EGFR TKI-resistant EGFR-mutant lung cancers. The outcome of the current clinical trial using CIMAvax-EGF vaccine (NCT02955290) may be critically improved by selecting such patients whose lung tumors carry EGFR mutations (including EGFR TKI-resistant ones) but not KRAS mutations.
Materials and methods
Mice
[tetO]-TGFA mice were obtained from W. Hardie and T. Korfhagen at Cincinnati Children’s Hospital Medical Center and the University of Cincinnati College of Medicine, Cincinnati, OH [8] and crossed with Scgb1a1-rtTA;[tetO]-EGFRL858R or Scgb1a1-rtTA;[tetO]-Kras4bG12D [14, 15, 54] to develop Scgb1a1-rtTA;[tetO]-EGFRL858R;[tetO]-TGFA (FVB/N;B6;CBA) or Scgb1a1-rtTA;[tetO]-Kras4bG12D;[tetO]-TGFA (FVB/N). Transgenic mice were provided chow containing doxycycline (625 mg/kg chow) beginning at 4–5 weeks of age. Mouse maintenance and procedures were done in accordance with the institutional protocol guidelines of Cincinnati Children’s Hospital Medical Center Institutional Animal Care and Use Committee. Mice were housed in a pathogen‐free barrier facility in humidity and temperature‐controlled rooms on a 12:12‐h light/dark cycle and were allowed food and water ad libitum. Since all transgenic mice were healthy before doxycycline administration, all of them were enrolled for the study. For the survival study, at least 16 mice in each genotype were enrolled. For the histological study, at least 3 mice in each genotype that developed lung tumors were enrolled. Littermates were used for the comparison. See Supplementary Table S1 for further mouse information.
Histology and immunohistochemistry
Staining was performed using 5-μm paraffin-embedded lung sections as previously described [54]. The number of tumors per hematoxylin and eosin-stained section were counted from 9–10 mice of each group (see Supplementary Table S1 for details). Rabbit anti-EGFR (1:100; clone D38B1; cat#4267), rabbit anti-EGFRL858R (1:100; clone 43B2; cat#3197), rabbit anti-phospho-AKT (1:100; clone D9E; cat#4060), rabbit anti-phospho ERK1/2 (1:100; clone; D13.14.4E; cat#4370), rabbit anti-phospho-STAT3 (1:100; cat#9131), rabbit anti-PTEN (1:100; clone 138G6; cat#9559), and rabbit anti-YAP (1:100; clone D8H1X; cat#14074) from Cell Signaling Technology, Danvers, MA; rabbit anti-TGFA (1:200; HPA042297) from Atlas Antibodies, Stockholm, Sweden; rabbit anti-ΔNp63 (1:500; cat#619002) from BioLegend, San Diego, CA; and guinea pig anti-AGR2 (1:500; a gift from J. Whitsett) were used for immunohistochemistry as primary antibodies. Antigen retrieval was performed by incubating sections in pH 9.0 Tris-EDTA (for TGFA, EGFR, and EGFRL858R) or pH 6.0 citrate (for AGR2 and ΔNp63) at 112.5 °C for 15 min using either decloaking chamber (Biocare Medical) or microwave.
Taqman gene expression analysis
Taqman gene expression analysis with RNA was performed according to the manufacturer’s instructions (Thermo Fisher, Waltham, MA) with Taqman probes (Mm00507853_m1 for Agr2; Mm01169470_m1 for ΔNTrp63; Mm00477208_m1 for Pten; Mm01192533_g1 for Ctgf; cat# 4352933 for Actb for normalization; and Hs00608187_m1 for TGFA).
Analysis of TCGA-human lung adenocarcinoma samples
The RNA-seq data of human lung adenocarcinoma samples were downloaded from Broad Institute TCGA Genome Data Analysis Center (Firehose stddata_2016_01_28 run). Gene expression was quantified using RSEM [55] and normalized to a fixed upper quartile. Expression values <1 were set to 1 and all data were log2-transformed. A total of 33 lung adenocarcinoma samples were identified as having EGFR mutations [12] and they constituted the EGFR-mutant lung adenocarcinoma group in our analysis, while the KRAS-mutant lung adenocarcinoma group consisted of 75 lung adenocarcinoma samples with KRAS mutations [12]. The correlations of genes were measured using Pearson’s correlation. Genes expressed (log2-transformed expression > 0) in at least 10% of the samples in EGFR-mutant lung adenocarcinoma group or KRAS-mutant lung adenocarcinoma group were included in the correlation analysis. The rank values for ΔNp63, AGR2, PLAUR, and ADORA1 in Figs. 3c and 4b were the rank of ΔNp63, AGR2, PLAUR, and ADORA1 among all genes included in the correlation analysis with each of the EGFR ligands in EGFR-mutant lung adenocarcinoma or in KRAS-mutant lung adenocarcinoma. Take as an example, the value 60 in Fig. 4b means that AGR2 is the 60th top correlated gene with TGFA in 33 EGFR-mutant lung carcinoma samples.
Cell lines
Human lung cancer cell line H1975 (adenocarcinoma, EGFRL858R; EGFRT790M) was obtained from the American Type Culture Collection (ATCC; Manassas, VA). Mycoplasma contamination was not detected by the Universal Mycoplasma Detection Kit (cat # 30-1012, ATCC).
Western blotting
Immunoblot assays were performed using rabbit anti-AGR2 antibody (1:5000; clone D9V2F; cat# 13062, Cell Signaling Technology) and rabbit anti-ACTA1 (1:5,000; cat# A2066, Sigma-Aldrich, St Louis, MO) as described previously [56].
Short hairpin RNA
Human AGR2 (NM_006408) targeting shRNAs in MISSION pLKO.1-puro vector (non-targeted shRNA [sh02] and shAGR2 [#1, TRCN0000146445; #2, TRCN0000146537], originally made by Sigma-Aldrich) were obtained from Lenti-shRNA Library Core at Cincinnati Children’s Hospital Medical Center. Subsequently, lentivirus was produced by the Viral Vector Core at Cincinnati Children’s Hospital Medical Center. H1975 cells were infected with lentivirus carrying shRNA targeting human AGR2 and cells were selected by puromycin.
Soft agar colony formation assay
Cells were plated into 0.4% low-melting agarose gel (SeaPlaque AGAROSE, cat#50101, Lonza, Basel, Switzerland) at a density of 1 × 104 cells/well on top of 1% low-melting agarose gel in six-well plates. A volume of 1 ml of medium (RPMI1640 containing 10% fetal calf serum and 1% penicillin/streptomycin) was added once a week in order to prevent desiccation. Colonies were counted after 3 weeks using ImageJ (National Institutes of Health) and each experiment was performed twice independently in triplicate wells.
Cell migration (scratch) assay
Cells were plated at a density of 1 × 106 cells/well in six-well plates. Twenty-four hours after serum starvation, cells were treated with mitomycin C (cat#47589, MilliporeSigma, Billerica, MA) at a final concentration of 10 μg/ml for 3 h in order to inhibit cell proliferation prior to making scratches. Cell migration was assessed by measuring the invaded area of scratch at a time point of 0 and 12 h with ImageJ (National Institutes of Health). Each experiment was performed twice independently in triplicate wells.
Human specimens
Paraffin sections were obtained in accordance with institutional guidelines for use of human tissue for research purposes from Nagasaki University Graduate School of Biomedical Sciences, Nagasaki, Japan (approval# 05062433-2) and Kawasaki Medical School, Okayama, Japan (approval# 1310). Written informed consent was obtained from all participants. Patients’ information is summarized in Supplementary Table S4.
Statistical analysis
Prism 7 (GraphPad Software, La Jolla, CA) was used for Kaplan–Meier survival analysis. Statistical differences were determined using Gehan–Breslow–Wilcoxon test, Fisher’s exact test (two-tailed), Student’s t-test (two-tailed or one-tailed and unpaired), or Welch test (pairwise). The difference between two groups was considered significant when the p-value was <0.05.
References
Velu TJ, Beguinot L, Vass WC, Willingham MC, Merlino GT, Pastan I, et al. Epidermal-growth-factor-dependent transformation by a human EGF receptor proto-oncogene. Science. 1987;238:1408–10.
Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal growth factor receptor mutations in lung cancer. Nat Rev Cancer. 2007;7:169–81.
Gazdar AF, Minna JD. Deregulated EGFR signaling during lung cancer progression: mutations, amplicons, and autocrine loops. Cancer Prev Res (Phila). 2008;1:156–60.
Linardou H, Dahabreh IJ, Bafaloukos D, Kosmidis P, Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat Rev Clin Oncol. 2009;6:352–66.
Greulich H, Chen TH, Feng W, Jänne PA, Alvarez JV, Zappaterra M, et al. Oncogenic transformation by inhibitor-sensitive and -resistant EGFR mutants. PLoS Med. 2005;2:e313.
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7.
Korfhagen TR, Swantz RJ, Wert SE, McCarty JM, Kerlakian CB, Glasser SW, et al. Respiratory epithelial cell expression of human transforming growth factor-alpha induces lung fibrosis in transgenic mice. J Clin Invest. 1994;93:1691–9.
Hardie WD, Le Cras TD, Jiang K, Tichelaar JW, Azhar M, Korfhagen TR. Conditional expression of transforming growth factor-alpha in adult mouse lung causes pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004;286:L741–9.
Jhappan C, Stahle C, Harkins RN, Fausto N, Smith GH, Merlino GT. TGF alpha overexpression in transgenic mice induces liver neoplasia and abnormal development of the mammary gland and pancreas. Cell. 1990;61:1137–46.
Sandgren EP, Luetteke NC, Palmiter RD, Brinster RL, Lee DC. Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell. 1990;61:1121–35.
Wagner M, Lührs H, Klöppel G, Adler G, Schmid RM. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998;115:1254–62.
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50.
Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci. 2016;107:713–20.
Fisher GH, Wellen SL, Klimstra D, Lenczowski JM, Tichelaar JW, Lizak MJ, et al. Induction and apoptotic regression of lung adenocarcinomas by regulation of a K-Ras transgene in the presence and absence of tumor suppressor genes. Genes Dev. 2001;15:3249–62.
Politi K, Zakowski MF, Fan PD, Schonfeld EA, Pao W, Varmus HE. Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev. 2006;20:1496–510.
Tateishi M, Ishida T, Mitsudomi T, Sugimachi K. Prognostic implication of transforming growth factor alpha in adenocarcinoma of the lung—an immunohistochemical study. Br J Cancer. 1991;63:130–3.
Giannou AD, Marazioti A, Kanellakis NI, Giopanou I, Lilis I, Zazara DE, et al. NRAS destines tumor cells to the lungs. EMBO Mol Med. 2017;9:672–86.
Green JA, Arpaia N, Schizas M, Dobrin A, Rudensky AY. A nonimmune function of T cells in promoting lung tumor progression. J Exp Med. 2017;214:3565–75.
Rodriguez PC, Popa X, Martínez O, Mendoza S, Santiesteban E, Crespo T, et al. A phase III clinical trial of the epidermal growth factor vaccine CIMAvax-EGF as switch maintenance therapy in advanced non-small cell lung cancer patients. Clin Cancer Res. 2016;22:3782–90.
Perl AK, Zhang L, Whitsett JA. Conditional expression of genes in the respiratory epithelium in transgenic mice: cautionary notes and toward building a better mouse trap. Am J Respir Cell Mol Biol. 2009;40:1–3.
Siveke JT, Einwächter H, Sipos B, Lubeseder-Martellato C, Klöppel G, Schmid RM. Concomitant pancreatic activation of Kras(G12D) and Tgfa results in cystic papillary neoplasms reminiscent of human IPMN. Cancer Cell. 2007;12:266–79.
Yanagi S, Kishimoto H, Kawahara K, Sasaki T, Sasaki M, Nishio M, et al. Pten controls lung morphogenesis, bronchioalveolar stem cells, and onset of lung adenocarcinomas in mice. J Clin Invest. 2007;117:2929–40.
Davé V, Wert SE, Tanner T, Thitoff AR, Loudy DE, Whitsett JA. Conditional deletion of Pten causes bronchiolar hyperplasia. Am J Respir Cell Mol Biol. 2008;38:337–45.
Zhao R, Fallon TR, Saladi SV, Pardo-Saganta A, Villoria J, Mou H, et al. Yap tunes airway epithelial size and architecture by regulating the identity, maintenance, and self-renewal of stem cells. Dev Cell. 2014;30:151–65.
Zhao B, Ye X, Yu J, Li L, Li W, Li S, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–71.
Watanabe H, Ma Q, Peng S, Adelmant G, Swain D, Song W, et al. SOX2 and p63 colocalize at genetic loci in squamous cell carcinomas. J Clin Invest. 2014;124:1636–45.
Yoh K, Prywes R. Pathway regulation of p63, a director of epithelial cell fate. Front Endocrinol (Lausanne). 2015;6:51.
Fessart D, Domblides C, Avril T, Eriksson LA, Begueret H, Pineau R. et al. Secretion of protein disulphide isomerase AGR2 confers tumorigenic properties. eLife. 2016;5:e13887
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature. 2012;489:519–25.
Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, et al. A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2015;33:306–12.
Li D, Shimamura T, Ji H, Chen L, Haringsma HJ, McNamara K, et al. Bronchial and peripheral murine lung carcinomas induced by T790M-L858R mutant EGFR respond to HKI-272 and rapamycin combination therapy. Cancer Cell. 2007;12:81–93.
Regales L, Balak MN, Gong Y, Politi K, Sawai A, Le C, et al. Development of new mouse lung tumor models expressing EGFR T790M mutants associated with clinical resistance to kinase inhibitors. PLoS ONE. 2007;2:e810.
Zhou W, Ercan D, Chen L, Yun CH, Li D, Capelletti M, et al. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature. 2009;462:1070–4.
Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, et al. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–61.
Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–32.
Wilson TR, Fridlyand J, Yan Y, Penuel E, Burton L, Chan E, et al. Widespread potential for growth-factor-driven resistance to anticancer kinase inhibitors. Nature. 2012;487:505–9.
Schneider MR, Dahlhoff M, Herbach N, Renner-Mueller I, Dalke C, Puk O, et al. Betacellulin overexpression in transgenic mice causes disproportionate growth, pulmonary hemorrhage syndrome, and complex eye pathology. Endocrinology. 2005;146:5237–46.
Ji H, Wang Z, Perera SA, Li D, Liang MC, Zaghlul S, et al. Mutations in BRAF and KRAS converge on activation of the mitogen-activated protein kinase pathway in lung cancer mouse models. Cancer Res. 2007;67:4933–9.
Engelman JA, Chen L, Tan X, Crosby K, Guimaraes AR, Upadhyay R, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–6.
Perera SA, Li D, Shimamura T, Raso MG, Ji H, Chen L, et al. HER2YVMA drives rapid development of adenosquamous lung tumors in mice that are sensitive to BIBW2992 and rapamycin combination therapy. Proc Natl Acad Sci USA. 2009;106:474–9.
Chen Z, Sasaki T, Tan X, Carretero J, Shimamura T, Li D, et al. Inhibition of ALK, PI3K/MEK, and HSP90 in murine lung adenocarcinoma induced by EML4-ALK fusion oncogene. Cancer Res. 2010;70:9827–36.
Ceteci F, Xu J, Ceteci S, Zanucco E, Thakur C, Rapp UR. Conditional expression of oncogenic C-RAF in mouse pulmonary epithelial cells reveals differential tumorigenesis and induction of autophagy leading to tumor regression. Neoplasia. 2011;13:1005–18.
Xu C, Buczkowski KA, Zhang Y, Asahina H, Beauchamp EM, Terai H, et al. NSCLC driven by DDR2 mutation is sensitive to dasatinib and JQ1 combination therapy. Mol Cancer Ther. 2015;14:2382–9.
Huang Q, Schneeberger VE, Luetteke N, Jin C, Afzal R, Budzevich MM, et al. Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol Cancer Ther. 2016;15:2521–9.
Freed DM, Bessman NJ, Kiyatkin A, Salazar-Cavazos E, Byrne PO, Moore JO, et al. EGFR ligands differentially stabilize receptor dimers to specify signaling kinetics. Cell. 2017;171:683–95.
Hogan BL, Barkauskas CE, Chapman HA, Epstein JA, Jain R, Hsia CC, et al. Repair and regeneration of the respiratory system: complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell. 2014;15:123–38.
Tata PR, Mou H, Pardo-Saganta A, Zhao R, Prabhu M, Law BM, et al. Dedifferentiation of committed epithelial cells into stem cells in vivo. Nature. 2013;503:218–23.
Liu J, Cho SN, Akkanti B, Jin N, Mao J, Long W. ErbB2 pathway activation upon Smad4 loss promotes lung tumor growth and metastasis. Cell Rep. 2015;10:1599–1613.
Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448:807–10.
Han X, Li F, Fang Z, Gao Y, Li F, Fang R, et al. Transdifferentiation of lung adenocarcinoma in mice with Lkb1 deficiency to squamous cell carcinoma. Nat Commun. 2014;5:3261.
Zhang H, Fillmore Brainson C, Koyama S, Redig AJ, Chen T, Li S, et al. Lkb1 inactivation drives lung cancer lineage switching governed by Polycomb Repressive Complex 2. Nat Commun. 2017;8:14922.
Rusch V, Klimstra D, Venkatraman E, Pisters PW, Langenfeld J, Dmitrovsky E. Overexpression of the epidermal growth factor receptor and its ligand transforming growth factor alpha is frequent in resectable non-small cell lung cancer but does not predict tumor progression. Clin Cancer Res. 1997;3:515–22.
Zhang J, Iwanaga K, Choi KC, Wislez M, Raso MG, Wei W, et al. Intratumoral epiregulin is a marker of advanced disease in non-small cell lung cancer patients and confers invasive properties on EGFR-mutant cells. Cancer Prev Res (Phila). 2008;1:201–7.
Maeda Y, Tsuchiya T, Hao H, Tompkins DH, Xu Y, Mucenski ML, et al. Kras(G12D) and Nkx2-1 haploinsufficiency induce mucinous adenocarcinoma of the lung. J Clin Invest. 2012;122:4388–400.
Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinforma. 2011;12:323.
Maeda Y, Hunter TC, Loudy DE, Davé V, Schreiber V, Whitsett JA. PARP-2 interacts with TTF-1 and regulates expression of surfactant protein-B. J Biol Chem. 2006;281:9600–6.
Acknowledgements
We thank J. Whitsett, H. Varmus, K. Politi, T. Korfhagen, W. Hardie, J. Gokey, and M. Durbin for mice, materials, and discussion.
Funding
This study was supported by the American Lung Association (RG309608), Trustee Award Grant, CF-RDP Pilot & Feasibility Grant, Cincinnati Children’s Hospital Medical Center (YM), the Rotary Foundation Global Grant (KT), AMED (TF), and the Ministry of Education, Science and Culture, Japan (TT, TF, YN, and TN)
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Tomoshige, K., Guo, M., Tsuchiya, T. et al. An EGFR ligand promotes EGFR-mutant but not KRAS-mutant lung cancer in vivo. Oncogene 37, 3894–3908 (2018). https://doi.org/10.1038/s41388-018-0240-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0240-1
- Springer Nature Limited