
Abstract
A two-neuron model of ventral tegmental area (VTA) opioid function classically involves VTA GABA neuron regulation of VTA dopamine neurons via a mu-opioid receptor dependent inhibitory circuit. However, this model predates the discovery of a third major type of neuron in the VTA: glutamatergic neurons. We found that about one-quarter of VTA neurons expressing the mu-opioid receptor are glutamate neurons without molecular markers of GABA co-release. Glutamate-Mu opioid receptor neurons are largely distributed in the anterior VTA. The majority of remaining VTA mu-opioid receptor neurons are GABAergic neurons that are mostly within the posterior VTA and do not express molecular markers of glutamate co-release. Optogenetic stimulation of VTA glutamate neurons resulted in excitatory currents recorded from VTA dopamine neurons that were reduced by presynaptic activation of the mu-opioid receptor ex vivo, establishing a local mu-opioid receptor dependent excitatory circuit from VTA glutamate neurons to VTA dopamine neurons. This VTA glutamate to VTA dopamine pathway regulated dopamine release to the nucleus accumbens through mu-opioid receptor activity in vivo. Behaviorally, VTA glutamate calcium-related neuronal activity increased following oral oxycodone consumption during self-administration and response-contingent oxycodone-associated cues during abstinent reinstatement of drug-seeking behavior. Further, chemogenetic inhibition of VTA glutamate neurons reduced abstinent oral oxycodone-seeking behavior in male but not female mice. These results establish 1) a three-neuron model of VTA opioid function involving a mu-opioid receptor gated VTA glutamate neuron pathway to VTA dopamine neurons that controls dopamine release within the nucleus accumbens, and 2) that VTA glutamate neurons participate in opioid-seeking behavior.
Similar content being viewed by others

Introduction
The United States continues to struggle with an opioid epidemic involving unprecedented numbers of opioid-related deaths [1]. Further, both prescription and synthetic opioid-related overdoses have been exacerbated by the COVID19 pandemic [2]. Understanding the neuronal cell-types and circuits affected by opioids may provide new insights into mitigating the severity of the opioid epidemic. Heroin, morphine, fentanyl, oxycodone, and other addicting opioids bind to the µ-opioid receptor (MOR) in the central nervous system [3]. Genetic ablation of MOR in mice results in resistance to the rewarding effects of morphine as well as naloxone-precipitated withdrawal [4], indicating that the MOR significantly contributes to rewarding and aversive components of opioid addiction. While neurons expressing the MOR are located throughout the brain, MOR binding within the ventral tegmental area (VTA) is necessary and sufficient for opioid reward. In rodents, activation of the MOR in the VTA is highly rewarding [5] and blocking the MOR in the VTA prevents the reinforcing aspects of opioids [6] as well as the self-administration of opioids [7]. Thus, mu-opioid receptors within the VTA are thought to play a critical role in opioid addiction.
Thirty years ago, a two-neuron model of VTA opioid function was proposed [8]. At this time, two types of VTA neurons were known: neurons that release the neuromodulator dopamine and neurons that release the inhibitory neurotransmitter GABA. Electrophysiological studies of VTA neurons suggested that VTA GABA neurons express the MOR and directly regulate VTA dopamine neurons via presynaptic actions [8]. The two-neuron model of opioid addiction has been highly replicated to show that MOR activation: 1) inhibits VTA GABA neurons and terminals resulting in the disinhibition of VTA dopamine neurons to cause opioid reward, and 2) blockade of the MOR in opioid-dependent subjects activates VTA GABA neurons to engender the aversive effects of opioid withdrawal [9,10,11,12,13]. The two neuron model results in downstream release of dopamine to the nucleus accumbens, although there has been conflicting evidence on the role of accumbal dopamine in opioid-induced locomotion, seeking, or self-administration behavior [14,15,16].
While highly influential, recent investigations suggest a need to revise the two-neuron model of VTA opioid function. Electrophysiological recording studies have shown that only a subset of VTA GABA neurons show MOR-elicited electrophysiological responses suggestive of MOR-expression [17, 18]. Additional work heavily implicates rostromedial tegmental area GABA neurons in opioid signaling and behavior [19]. A subset of VTA glutamate neurons, discovered fifteen years after the development of the two-neuron model of VTA opioid function [20], expresses MOR mRNA and shows MOR-elicited electrophysiological responses supportive of MOR-expression [21, 22]. Further, VTA glutamate receptor activation is required for the increased firing of dopamine neurons in response to morphine [23] and heroin self-administration [24]. VTA neurons expressing the vesicular glutamate transporter 2 (VGluT2) establish local circuits to VTA dopamine neurons as well as distal projections to the medial shell of the nucleus accumbens, and manipulation of this accumbal circuitry affects both reward-related and aversion-related behaviors in mice [25,26,27]. While VTA glutamate neurons have sparse projections to accumbens core [26, 28], VTA VGluT2 neurons may still influence core dopamine release via local glutamatergic circuits. Whole-cell recordings have also found that MOR activation affects ionotropic and metabotropic-mediated glutamatergic input to putative VTA dopamine neurons [29, 30]. These findings suggest the existence of a MOR-regulated glutamatergic projection to VTA dopamine neurons, but do not establish the source of such inputs. Together, these findings indicate that the current understanding of the VTA cell-types and circuits regulated by the MOR are not wholly understood. We aimed to identify the composition of MOR-expressing VTA cell-types and evaluate the role of VTA VGluT2 neurons in MOR-regulated dopaminergic responses as well as their roles in opioid-seeking behavior.
We found that VTA MOR-expressing cell-types are largely divided between GABA neurons defined by the vesicular GABA transporter (VGaT) and glutamate neurons defined by the VGluT2. VTA VGaT+MOR+ neurons predominate the posterior VTA while the majority of anterior VTA MOR-expressing neurons are VGluT2+MOR+ neurons. Both MOR cell-types largely lack markers of glutamate and GABA co-transmission (VGluT2+VGaT+), suggesting that each VTA MOR subpopulation is a subtype of VTA VGluT2−VGaT+ or VGluT2+VGaT− neurons. Further, we identified a local circuit from VTA VGluT2 neurons to VTA dopamine neurons that is regulated by the MOR using ex vivo slice electrophysiology. In vivo, this MOR-regulated VTA VGluT2 to VTA dopamine circuit modulates the release of dopamine in the nucleus accumbens. Finally, we demonstrated that VTA VGluT2 neurons are activated by oxycodone consumption and in males are necessary for cued reinstatement of opioid-seeking behavior. Together, we identify an unexpected MOR-regulated mesolimbic circuitry from VTA VGluT2+MOR+ neurons that participates in opioid-seeking behavior.
Materials and methods
Full methods, including histology, in situ hybridization, whole-cell electrophysiology, and in vivo VTA glutamate neuron stimulation and optical dopamine recordings in accumbens, are in Supplemental.
Animals
Male and female C57BL/6J and VGluT2-IRES::Cre mice (Slc17a6tm2(cre)Lowl/J; Stock #016963) were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred at the University of Colorado. Mice were maintained in a colony room with a 12‐h light/dark cycle with access to food and water ad libitum. Mice were between 8–20 weeks at the time of surgical procedures. All animal procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the University of Colorado Boulder Institutional Animal Care and Use Committee. Ex vivo electrophysiological experiments were approved by George Washington University Institutional Animal Care and Use Committee.
Stereotactic surgery
Male and female VGluT2-IRES::Cre mice were anesthetized with 1–3% Isoflurane and secured in a stereotactic frame. AAV8-hSyn-DIO-hM4Di-mCherry (Addgene, Cambridge, MA), AAV5-hSyn-DIO-GFP (Addgene), or AAV1-Syn-FLEX-GCaMP6m (Addgene), AAV8-hSyn-FLEX-CoChR-GFP (UNC Vector Core), or AAV8-nEF-Con/Foff-ChRmine-oScarlet (Addgene) was injected into VTA (AP: −3.2 mm; ML: 0.0 mm; DV: −4.3 mm). All AAVs were injected at 2–5 ×1012 titer. Injection volume (500 nL) and flow rate (100 nL/min) were controlled with a microinjection pump (Micro4; World Precision Instruments, Sarasota, FL). Following injection, the needle was left in place for an additional 10 min for virus diffusion. Nucleus accumbens surgeries followed the same protocol with the modification of virus: AAV9-hSyn-GRABDA1h (coordinates AP: 1.34, ML: 1.1 DV: −4.5). For GCaMP recordings, mice were additionally implanted with an optic fiber (400 μm core diameter, 0.66 NA; Doric Lenses, Quebec, Canada) dorsal to VTA (AP: −3.2 mm relative to bregma; ML: −1.0 mm at 9.5°; DV: −4.2 mm) and for GRABDA recordings implanted dorsal to nucleus accumbens (AP: 1.34, ML: 1.1 DV: −4.3) that was secured with screws and dental cement. For ChRmine stimulation, mice were implanted with an optic fiber (200 μm core diameter, Doric) dorsal to VTA (AP: −3.2 mm; ML: −1.0 mm at 9.5°; DV: −4.2 mm) that was secured with screws and dental cement. Mice were allowed to recover for 3–4 weeks before experimentation.
Slice electrophysiology
For electrophysiological recordings, a single slice was transferred to a chamber perfused at a rate of 1.5–2.0 ml/min with heated (28–32 °C) artificial cerebrospinal fluid (aCSF, in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgCl2 6H2O, 11 glucose, 26 NaHCO3, 2.4 CaCl2, saturated with 95% O2 and 5% CO2. 100 µM picrotoxin was included in all recordings to pharmacologically isolate EPSCs. Non-GFP expressing cells were visually identified in the lateral VTA and selected for recordings. Slices were investigated under fluorescence prior to recording and slices with cell bodies expressing GFP outside of the VTA were not used. Whole-cell patch-clamp recordings were performed using a Sutter IPA amplifier (1 kHz low-pass Bessel filter and 10 kHz digitization) using SutterPatch software (Sutter Instruments). Voltage-clamp recordings at −70 mV were made using glass patch pipettes with resistance 2–4 MOhms, filled with internal solution containing (in mM): 135 K-gluconate, 10 HEPES, 4 KCl, 4 Mg-ATP, 0.3 Na3-GTP, pH 7.35, 275–285 mOsm. 0.4% biocytin was included in the internal solution for all recordings. Series resistance was monitored throughout voltage clamp recordings and recordings in which the series resistance changed more than 20% and/or exceed 20 MOhms were not included in the analysis. Junction potential (~16.5 mV, calculated according to the stationary Nernst–Planck equation [31] using LJPcalc software (https://swharden.com/LJPcalc)) was not corrected for.
EPSCs were optically evoked using an optical fiber (Plexon, Inc, Dallas, TX) coupled to a 465 nm LED and driver (Plexon, Inc, Dallas, TX). Light pulses (0.5–4 ms) were triggered by TTL pulses from the SutterPatch software. EPSCs were stimulated every 30 s to avoid desensitization of CoChR. The amplitude of the first detectable peak after optical stimulation was measured for each EPSC. After a stable 10 min baseline of EPSCs was collected, DAMGO (500 nM, Bio-Techne, Minneapolis, MN) was bath applied for 10 min. Data was analyzed offline using SutterPatch software and Graphpad Prism 9.0. To create timecourses, EPSC amplitudes were binned in 2-min bins and normalized to the last 5 min of the baseline. To determine average amplitudes pre- and post- DAMGO, EPSC amplitudes were averaged over the last 5 min of the baseline and the last 5 min of DAMGO application. Representative traces are an average of 10 consecutive sweeps immediately preceding application or removal of DAMGO. Synaptic latency was determined by measuring the delay between initiation of optical stimulation and onset of the first synaptic current.
Self-administration
Training commenced as previously described [32]. Briefly, mice trained on an FR1 schedule of reinforcement for water over three days and an FR2 schedule of reinforcement for water over three additional days. Within the active port, a cue light was illuminated until reward was earned. At reward, the cue light was terminated, a 7 kHz tone was sounded together over 10 s, and 20 µL reward was delivered. After 10 s, the cue light was re-illuminated. For oral oxycodone mice, FR2 oxycodone administration followed an escalating concentration schedule (0.05, 0.1, 0.3, 0.5, 1.0 mg/mL) for three days per dose and 3 h sessions per day under the same reward delivery conditions. For water mice, the same number of self-administration sessions were made as oxycodone mice, but the reward was always water vehicle. After three days at the highest dose, or the matched number of training days for water mice, all mice began extinction where active and inactive nose pokes were counted but had no programmed consequence. Extinction sessions were 1 h and criterion for extinction was calculated as 30% of the average total active pokes for each mouse at their highest dose. Mice were required to emit less than or equal to this active poke criterion for two consecutive days before shifting to cued reinstatement testing. The following day after reaching extinction criterion, single active nose pokes activated the cue previously associated with oxycodone reward consisting of a 10‐s 7 kHz tone together with the termination of a light within the active port. Inactive nose pokes were counted without other programmed consequence. Mice remained in the cue phase for 1 h following the first earned cue presentation. One hour prior to self-administration, extinction, or reinstatement, mice were separated and fed without access to water.
GCaMP recordings
Male and female VGluT2-IRES::Cre mice were injected in VTA with AAV1-Syn-FLEX-GCaMP6m at least four weeks prior to behavioral testing. GCaMP6m recordings were performed while mice operantly earned oral oxycodone (1 mg/mL) and during cued reinstatement testing. GCaMP6m was excited at two wavelengths (465 nm and 405 nm isosbestic control) with amplitude-modulated signals from two light-emitting diodes reflected off dichroic mirrors and then coupled into an optic fiber [33]. Signals from GCaMP and the isosbestic control channel were returned through the same optic fiber and acquired with a femtowatt photo-receiver (Newport, Irvine, CA), digitized at 1 kHz, and then recorded by a real-time signal processor (Tucker Davis Technologies). Behavioral timestamps for active pokes, inactive pokes, magazine entry, and cue presentation were digitized in Synapse by TTL input from MED-Associates. Analysis of the recorded calcium signal was performed using custom-written MATLAB scripts available at www.root-lab.org/code. Signals (465 nm and 405 nm) were downsampled (10X) and peri-event time histograms (PETHs) were created trial-by-trial between −10 s and +30 s surrounding each event. For each trial, data was detrended by regressing the isosbestic control signal (405 nm) on the GCaMP signal (465 nm) and then generating a predicted 405 nm signal using the linear model generated during the regression. The predicted 405 nm channel was subtracted from the 465 nm signal to remove movement, photo-bleaching, and fiber bending artifacts (ΔF). Baseline normalized maximum z-scores were taken from −3 to 0 s prior to programmed time stamps of interest (active/inactive poke, cue, reward entry) and maximum z-scores were taken from 0 to 3 s following behavioral events. AUC analysis per animal were normalized to the baseline 3 s prior to the behavioral event and 3 s following programmed behavioral events.
Chemogenetic inhibition of VTA glutamate neurons during oxycodone self-administration and reinstatement
Male and female VGluT2-IRES::Cre mice were injected in VTA with AAV8-hSyn-DIO-hM4Di-mCherry or AAV5-hSyn-DIO-GFP at least four weeks prior to behavioral testing. Mice self-administered oral oxycodone or water as described. However, after three days of self-administration at the highest dose, mice received an IP injection of a behaviorally subthreshold [34] dose of clozapine (0.1 mg/kg) ten minutes prior to self-administration. This dose has previously been shown to decrease VTA VGluT2 neuronal activity and block the involvement of VTA VGluT2 neurons in the repercussions of inescapable stress in both male and female mice [33]. After the clozapine session, three additional days of training at 1 mg/mL commenced to compare self-administration before and after chemogenetic manipulation of VTA VGluT2 neurons. After self-administration, active responding was extinguished as described and the following day after mice met extinction criterion, an IP subthreshold dose of clozapine (0.1 mg/kg) was given ten minutes prior to cued reinstatement testing. Two days after reinstatement testing mice were given IP injection of clozapine (0.1 mg/kg) and placed in an open field (Any Box, Stoelting). Mice with hM4Di-mCherry expression significantly expressed outside the A10 region were removed from analysis. AnyMaze tracking was used to evaluate distance traveled, maximum distance, and average speed per mouse.
Statistics
Tests were conducted in SPSS (IBM) or Prism (GraphPad Software; San Diego, CA). GCaMP data was analyzed by comparing the maximum GCaMP z-score during baseline, 3 s prior to event compared to the maximum z-score following the following events 3 s following event initiation: active poke, magazine entry, and cue presentation during reinstatement using Wilcoxon sign ranked tests. Identical analysis was performed on AUC statistics from the same behavioral timepoints. Max GRABDA normalized z-score values were averaged across for all trials per frequency and experimental condition (saline versus oxycodone), with 3 days between each stimulation condition. For GRABDA recordings, maximum fluorescent baselines or stimulated peaks did not differ between male or female mice, therefore data was collapsed across sex. Repeated measured ANOVA compared fluorescent change across experimental conditions per frequency. A between-subjects t-test analyzed the difference active and inactive pokes during cued reinstatement. A repeated measures ANOVA was used to compare baseline oxycodone consumption to hM4Di test day consumption and to post-test baseline. Separate analyses were performed for each sex. For all ANOVAs, if the assumption of sphericity was not met (Mauchley’s test), the Greenhouse-Geisser correction was used and Sidak-adjusted pairwise comparisons followed up significant main effects or interactions. Slice electrophysiology data were analyzed using paired t-test.
Results
Cell-type specific expression of VTA MOR and topographic distribution
Given that expression of the MOR by VTA GABA neurons is well-established, and that VTA VGluT2+VGaT+ neurons are a subset of VTA GABA neurons [35], we determined which VGluT2 and/or VGaT-expressing cell-types express the MOR across the VTA. Thin sections of the VTA from wildtype C57 mice were examined for the expression of transcripts encoding VGluT2 (for the detection of VTA glutamate-releasing neurons), VGaT (for the detection of VTA GABA-releasing neurons), and Oprm1 (for the detection of MOR-expressing neurons) (Fig. 1A–C). To determine a topographical distribution of the MOR on VTA VGluT2+ and VGaT+ neurons, VTA sections prior to the full emergence of the interpeduncular nucleus were designated as anterior VTA while those afterwards were designated as posterior VTA. In anterior VTA, 58 ± 8.50% of MOR-neurons expressed VGluT2 without VGaT, 28 ± 6.99% expressed VGaT without VGluT2, and 10 ± 1.99% triple expressed the MOR with VGluT2 and VGaT transcripts (Fig. 1D). In posterior VTA, the distribution of MOR expressing cell-types shifted, with a predominance of MOR-neurons expressing VGaT without VGluT2 (86.5 ± 3.80%), and only 10 ± 3.39% of MOR-neurons expressing VGluT2 without VGaT, and 1.41 ± 0.94% of MOR-neurons expressing both VGluT2 and VGaT (Fig. 1E).

A VTA section at approximately −3.28 mm posterior from bregma labeled for RNA transcripts encoding VGluT2 (green), VGaT (red), Oprm1 (white) mRNAs. Dotted blue line represents VTA border. B Example VTA neuron co-expressing VGluT2 and Oprm1 without VGaT mRNA (purple arrow) as well as a VGluT2 and VGaT co-expressing neuron lacking Oprm1 mRNA (yellow arrow). C Example VTA neurons that co-express VGaT and Oprm1 mRNA (blue arrow) or express VGaT without Oprm1 mRNA. D. Anterior distribution of VTA Oprm1+ neurons. E Posterior distribution of VTA Oprm1+ neurons. F Anterior percentage of Oprm1+ neurons per cell-type. G Posterior percentage of Oprm1+ neurons per cell-type. All values represent the mean ± SEM. Scale bar in A is 500 µm and scale bar in subpanel B is 10 µm and applies to C–F.
We also identified the percent of each glutamatergic and GABAergic VTA cell-type that expresses the MOR within the anterior and posterior VTA. Within the anterior VTA, about 40% of VGluT2+VGaT− neurons expressed MOR (Fig. 1F). Similarly, about 40% of anterior VGluT2−VGaT+ neurons expressed MOR. Anterior VGluT2+VGaT+ neurons less frequently expressed the MOR (Fig. 1F). In the posterior VTA, most VGluT2−VGaT+ neurons expressed the MOR (80.22 ± 4.84%) while glutamatergic cell-types expressing the MOR were less frequent (20.15 ± 2.81% for VGluT2+VGaT− neurons and 11.47 ± 3.44% for VGluT2+VGaT+ neurons) (Fig. 1G). Together, in anterior VTA, MOR is mostly expressed in VGluT2+VGaT− neurons and this population of MOR-expressing neurons represents about 40% of anterior VTA VGluT2+VGaT− neurons. In posterior VTA, MOR is predominantly expressed in VGluT2−VGaT+ neurons, and this population of MOR-expressing neurons represents 80% of posterior VTA VGluT2−VGaT+ neurons. Thus, VGluT2+MOR+ neurons and VGaT+MOR+ neurons are genetically distinct subsets of VGluT2+VGaT− or VGluT2−VGaT+ neurons, respectively. There were no significant differences between male and female VTA cell-type distributions (Supplementary Fig. 1). The anterior and posterior distribution of all counted MOR-expressing (Oprm1+; Fig. 2A–D) or lacking (Oprm1−; Fig. 2E–G) VTA cell-types from all animals were registered to the Allen Atlas with SHARCQ [36] and displayed with brainrender [37].

All counted neurons from all animals were registered to the Allen atlas using SHARCQ [37] and plotted with brainrender [38]. A Oprm1+VGluT2+VGaT− neurons. B Oprm1+VGluT2−VGaT+ neurons. C Oprm1+VGluT2+VGaT+ neurons. D Oprm1+VGluT2−VGaT− neurons. E Oprm1−VGluT2+VGaT− neurons. F Oprm1−VGaT+VGluT− neurons. G Oprm1−VGluT2+VGaT+ neurons.
VTA VGluT2 synapses onto VTA dopamine neurons are regulated by the MOR
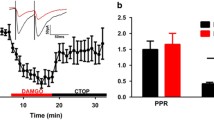
Based on the MOR-regulated VTA GABA to VTA dopamine neuron microcircuit [8], we hypothesized that VTA VGluT2+MOR+ neurons establish MOR-sensitive local glutamatergic synapses onto VTA dopamine neurons. To test this hypothesis, we expressed the blue light-gated ion channel CoChR [38] tethered to GFP in VTA VGluT2 neurons and recorded from non GFP-expressing lateral VTA neurons ex vivo; Fig. 3A). Consistent with previous literature, the lateral VTA contained primarily axon terminals from VTA VGluT2+ neurons whereas their somata were located primarily in the midline nuclei of the VTA. To determine the identity of the recorded postsynaptic neurons, we examined whether biocytin-filled neurons co-expressed tyrosine hydroxylase (TH) immunoreactivity. Of twelve sections with a recorded VTA neuron, we recovered the biocytin-expressing neuron in five sections and all co-expressed tyrosine hydroxylase (TH) immunoreactivity (Fig. 3B). Consistent with prior reports [39, 40], we found that optical stimulation of VTA VGluT2-CoChR neurons resulted in EPSCs on local VTA neurons (Fig. 3C). Application of the MOR-selective agonist DAMGO (500 nM) significantly reduced currents elicited by activation of VTA VGluT2-CoChR neurons (Baseline amplitude = 49.71 ± 8.6 pA, DAMGO amplitude = 31.08 ± 7.1 pA, p = 0.014) (Fig. 3D). In all neurons that showed a DAMGO-induced reduction in VGluT2-CoChR currents, the paired-pulse ratio significantly increased following DAMGO application (Baseline PPR = 0.87 ± 0.1, DAMGO PPR = 1.18 ± 0.1, p = 0.003) (Fig. 3E), consistent with a presynaptic site of MOR activation. Together these results indicate that VTA VGluT2+MOR+ neurons establish MOR-regulated local glutamatergic synapses onto VTA dopamine neurons (Fig. 3F). These EPSCs exhibited onset latencies of 2.8 ± 0.29 ms, consistent with a monosynaptic input [41, 42] (Fig. 2G).

A Experimental schematic: VGluT2-IRES::Cre mice received a Cre-dependent injection encoding CoChR-GFP in the medial VTA. Brains were extracted and slices were prepared for whole-cell recording. B Immunohistochemical labeling of VTA VGluT2-CoChR-GFP processes (green) proximal to biocytin-filled neuron (white) co-labeled with tyrosine hydroxylase (red). C Normalized EPSCs following VTA VGluT2 neuron optical stimulation. DAMGO (500 nM) wash at 10 min reduced optically-elicited EPSCs. D Recorded neurons significantly reduced EPSC in recorded neurons (n = 9/10), p = 0.014. E Significant increase in paired pulse ratio (PPR), p = 0.003, suggesting pre-synaptic mu-opioid receptor regulation. F Left: Representative trace, baseline (black), DAMGO (red); Right: Scaled trace, demonstrating PPR change. G EPSCs exhibited onset latencies of 2.8 ± 0.29 ms. Scale bar is 20 ms (horizontal) and 20 pA (vertical).
Nucleus accumbens dopamine release is regulated by a MOR-gated VTA VGluT2 neuron projection to VTA dopamine neurons
Having established a VTA VGluT2+MOR+ neuron microcircuit to VTA dopamine neurons, we aimed to test the hypothesis that this circuit influences dopamine release within the nucleus accumbens in vivo. To test this hypothesis, we expressed the red light-shifted ion channel ChRmine [43] in VTA VGluT2 neurons and in the same animals expressed the green light sensitive optical dopamine sensor GRABDA1h [44] in nucleus accumbens core (Fig. 4A; Supplementary Fig. 2). Given that VGluT2-expressing dopamine neurons co-release glutamate and dopamine in the accumbens shell, but not core [45], we targeted the accumbens core to maximize detection of dopamine from non-glutamatergic dopamine neurons. We then stimulated VTA VGluT2 neurons and recorded accumbal GRABDA activity in vivo following saline or oxycodone injection. ChRmine activation of VTA VGluT2 neurons resulted in increased GRABDA signal in nucleus accumbens core. While baseline GRABDA activity did not change between saline and oxycodone treatments (F(2,12) = 3.075, p = 0.084), accumbal GRABDA activity significantly differed between saline and oxycodone treatments following ChRmine stimulation of VTA VGluT2 neurons (F(2,12) = 5.168, p = 0.024). Posthoc contrast tests showed that oxycodone administration significantly reduced GRABDA activity following ChRmine activation of VTA VGluT2 neurons compared with each saline treatment (saline before oxycodone: F(1,6) = 10.412, p = 0.018; saline after oxycodone: F(1,6) = 8.847, p = 0.025) (Fig. 4B–E). Oxycodone administration was sufficient to reduce the VTA VGluT2+ stimulated released of DA to the nucleus accumbens core. We measured similar effect sizes for each stimulation frequency: 5 Hz = 1.23, 10 Hz = 1.28, 20 Hz = 1.00, 40 Hz = 0.96. Lastly, there was no significant difference between male or female evoked dopamine in the core following VTA VGluT2+ stimulation in either the presence oxycodone, p = 0.92, or the initial response with saline, p = 0.99.

A–E Surgical schematic: AAV-GRABDA was injected in the nucleus accumbens core and implanted with a recording fiber optic. Medial VTA was injected with AAV-FLEX-ChRmine and implanted with a stimulating optic fiber. Experimental timeline: Four weeks following injections and implantations, mice were tail-restricted to minimize movement-related GRABDA signaling and administered an IP injection of saline. Five optical trains of 589 nm light were delivered to VTA VGluT2 neurons at 5 Hz (B), 10 Hz (C), 20 Hz (D), 40 Hz (E) (order counterbalanced). Three days following saline pre-test, mice were administered an IP injection of oxycodone (10 mg/kg) and stimulated under the same conditions. Three days later, mice were given an IP saline injection and VTA VGluT2 neurons were stimulated once more.
VTA VGluT2 neurons are activated by opioid-seeking behavior
After establishing a MOR-mediated VTA VGluT2 to VTA dopamine neuron pathway regulating accumbal dopamine release, we aimed to test the involvement of VTA VGluT2 neurons in opioid-seeking behavior. To accomplish this, we expressed the calcium-sensor GCaMP6m in VTA VGluT2+ neurons and recorded population changes in intracellular calcium during oral oxycodone self-administration or cue-induced reinstatement of extinguished drug-seeking behavior (Fig. 5A; Supplementary Fig. 3). Mice trained on a fixed ratio 2 reinforcement schedule to operantly self-administer oral oxycodone. A tone cue was paired with drug delivery to establish a drug-cue association (Fig. 5B). Due to sex differences in self-administration of oral oxycodone [32] we separated female and male mice to evaluate sex as a biological variable. During self-administration, VTA VGluT2 GCaMP activity significantly increased from baseline following response-contingent presentation of the oxycodone-associated cues, F(1,14) = 22.87, p = 0.0003, for female mice: t(8) = 3.615, p = 0.0056, and male mice: t(8) = 3.148, p = 0.0142. Area under the curve (AUC) also significantly increased from baseline for oxycodone active poke F(1,14) = 15.41, p = 0.0015, for female mice: t(8) = 2.860, p = 0.0250, and male mice: t(8) = 2.691, p = 0.0348. There were no significant main effects of sex between maximum recorded fluorescence F(1,14) = 0.0023, p = 0.9622, or area under the curve F(1,14) = 0.01433, p = 0.9064. VTA VGluT2 GCaMP activity significantly increased from baseline following head entry into the oxycodone magazine where drug consumption occurred, F(1,14) = 40.21, p < 0.0001, for female mice: t(8) = 5.580, p = 0.0001, and male mice: t(8) = 3.562 p = 0.0062. AUC was also significantly different from baseline for oxycodone magazine entry, F(1,14) = 36.83, p < 0.0001, for female mice: t(8) = 5.120, p = 0.0003, and male mice: t(8) = 3.462, p = 0.0076 mice. There were no significant main effects of sex between maximum recorded fluorescence F(1,14) = 0.344, p = 0.566 or area under the curve, F(1,14) = 1.402, p = 0.2562 (Fig. 5C–H). After extinction of active responses in the absence of the oxycodone-associated cue, active responses under extinction conditions resulted in the presentation of the previously drug-paired tone cue. VTA VGluT2 calcium activity significantly increased from baseline following response-contingent presentation of the drug-associated cue, F(1,10) = 26.30, p = 0.0004, for female mice: t(7) = 4.749, p = 0.0016, and male mice: t(5) = 2.702, p = 0.0440. Cue reinstatement response AUC was also significantly different from baseline, F(1,10) = 20.33, p = 0.0011, female mice: t(7) = 3.634, p = 0.0091 and male mice: t(5) = 2.832, p = 0.0353. There were no significant main effect of sex for maximum fluorescence F(1,10) = 0.1345, p = 0.7215 or AUC F(1,10) = 0.03267, p = 0.8602 (Fig. 5I–K). Additionally there were no significant interactions between epoch and sex for active poke F(1,14) = 0.1088, p = 0.7464, magazine entry F(1,14) = 2.110, p = 0.1669, or cued reinstatement F(1,10) = 1.188, p = 0.3013 and no significant differences for extinction duration between groups, F(3,39) = 1.025, p = 0.3919.

A Surgical schematic: VGluT2-IRES::Cre male and female mice were injected in medial VTA with AAV-FLEX-GCaMP and were implanted with a fiber optic recording ferrule. B Experimental timeline: 4 weeks following viral injection and implant, mice were trained on a FR2 reinforcement schedule. Mice self-administered an escalating dose schedule of oxycodone (0, 0.5, 0.1, 0.3, 0.5, 1.0 mg/mL). VGluT2 calcium activity was recorded fiber photometrically at the highest dose. After reaching extinction criterion, mice were recorded during cued reinstatement. C Normalized z-score for female (green) and male (blue) mice following active poke (AP) for oxycodone reward during self-administration compared to baseline (BL). Mean represented by darker line and standard error represented by shaded line. D Maximum values for individual mice following active poke (0–3 s poke) compared to baseline. E AUC analysis for recorded signals (0–3 s following poke). F Normalized z-score of both sexes following oxycodone magazine entry (ME) where consumption occurs. G Maximum values for individual mice following magazine entry (0–3 s). H AUC analysis for recorded signals (0–3 following entry). I Normalized z-score of both sexes following cue presentation during cued reinstatement test (CR). J Maximum values for individual mice following cue presentation (0–3 s). K AUC for individual mice following cue presentation during reinstatement (0–3 s).
VTA VGluT2 neuronal activity is required for cue-induced reinstatement of opioid-seeking behavior in male mice
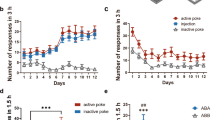
Given that VTA VGluT2 GCaMP signal was increased by opioid-seeking behavior, we expressed the inhibitory designer receptor hM4Di or a fluorophore control in VTA VGluT2 neurons. We delivered a behaviorally subthreshold dose of clozapine [34] that reduces hM4Di-VGluT2 neuronal activity [33], during oral oxycodone self-administration or cue-induced reinstatement (Fig. 6A). In male mice, chemogenetic inhibition of VTA VGluT2 neurons did not modify oxycodone consumption F(2,63) = 1.218, p = 0.3020 (Fig. 6B). However, inhibition of VTA VGluT2 neurons significantly reduced active responding during cue-induced reinstatement following extinction of opioid-seeking behaviors, F(1,52) = 8.437, p = 0.0054 (Fig. 6C). Inactive pokes were not changed by VGluT2 neuron inhibition in male mice (Fig. 6D), F(1,52) = 0.5148, p > 0.05. VTA VGluT2 neuron inhibition did not significantly alter oxycodone consumption, F(2,30) = 0.5864, p = 0.5626, or reinstatement-related active responding, F(1,36) = 2.684, p = 0.1101 (Fig. 6E, F). Additionally, inactive pokes were not altered by chemogenetic inhibition in female mice, F(1,37) = 0.8589, p = 0.3601 (Fig. 6G). The number of days to reach extinction criterion before reinstatement did not differ between groups, F(3,39) = 1.025, p = 0.3919 (Supplementary Fig. 4).

A Surgical schematic: VGluT2-IRES::Cre male and female mice were injected with AAV-FLEX-hM4Di or AAV-FLEX-GFP in the medial VTA. Experimental timeline: 4 weeks after viral expression, mice trained to orally administer drug on a FR2 reinforcement schedule and oxycodone dose was escalated. Behaviorally subthreshold clozapine (0.1 mg/kg) was administered for one day at the highest dose, followed by 3 more days of self-administration. Mice were next trained to extinguish active responses and after extinction criterion was met, were administered clozapine (IP) during cued reinstatement testing. Subpanel: male and female mice differentially consumed oxycodone. B Mg/kg oxycodone consumption for males, pre clozapine (3-day average), on clozapine day, and post clozapine (3-day average). C Recorded active pokes during cued reinstatement testing. VTA VGluT2 chemogenetic inhibition significantly reduced reinstatement behavior compared to GFP control mice. D Inactive pokes did not differ by experimental group. E Mg/kg oxycodone consumption for females. F Active pokes during cue reinstatement testing. hM4Di inhibition did not alter reinstatement behavior in female mice. G hM4Di inhibition did not alter inactive poke behavior.
An additional cohort of male and female VGluT2::Cre mice received identical intra-VTA hM4D or GFP injections and trained to orally self-administer water vehicle as a control for thirst behavior. Chemogenetic inhibition of VTA VGluT2 neurons did not significantly alter water self-administration, F(1,28) = 0.05567, p = 0.8152, or cue reinstatement in male or female mice following extinction, F(1,28) = 0.05420, p = 0.8176 (Supplementary Fig. 4), suggesting that VTA VGluT2 neurons participate in oxycodone-seeking behavior rather than water-seeking behavior. To test the possibility that the reduced reinstatement of male mice following chemogenetic inhibition of VTA VGluT2 neurons resulted from changes in locomotor behavior, mice were injected with 0.1 mg/kg clozapine, placed in an open field and tracked for the following locomotor parameters: average speed, distance traveled, and maximum speed. There were no differences between any groups for distance traveled F(3,56) = 0.6438, p = 0.5901, average speed F(3,56) = 0.6488, p = 0.5870, or maximum speed, F(3,56) = 1.152, p = 0.3362. Thus, reduced reinstatement of oxycodone-seeking behavior in male mice was not driven by changes in locomotor behavior (Supplementary Fig. 5).
Discussion
VTA VGluT2 neuron regulation of VTA dopamine neurons via a MOR-regulated local circuit
Understanding the circuit-level neural contributors to opioid self-administration and relapse are foundational to treating opioid use disorder. VTA MORs are required for the reinforcing effects of opioids [6], sufficient to induce reinstatement of extinguished drug-seeking behavior [46], and are canonically expressed in nondopaminergic GABA-releasing neurons [8]. Due to the cellular heterogeneity of VTA GABA neurons [35], we used in situ hybridization to determine the distribution of VTA VGluT2+VGaT−, VGluT2+VGaT+, and VGluT2−VGaT+ neurons that express the MOR. The different types of MOR-expressing neurons had distinct topographies across the anteroposterior VTA. Most anterior VTA MOR+ neurons expressed VGluT2 without VGaT while most posterior VTA MOR+ neurons expressed VGaT without VGluT2. The anterior VTA MOR-expressing VGluT2 neurons were about 40% of VGluT2+VGaT− neurons whereas the posterior VTA MOR-expressing VGaT neurons were more than four-fifths of VGluT2−VGaT+ neurons. These data may inform why VTA opioid function differs across the VTA anterior-posterior axis. For instance, while activation of the MOR is sufficient to induce reward selectively within the posterior VTA [47, 48], AMPA receptors within the anterior VTA, where VGluT2-expressing neurons are most dense [20, 49], are selectively necessary for morphine reward [50]. Further, overexpression of AMPA receptor in anterior VTA enhances morphine reward while overexpression in the posterior VTA causes morphine to be interpreted as aversive [51]. One limitation of our in situ hybridization experiment was the lack of detection of dopamine neurons. Given that a subpopulation of VGluT2+ neurons co-express TH to co-transmit glutamate and dopamine [20, 45, 49, 52], it is not clear whether VGluT2+TH− or VGluT2+TH+ neurons express MOR. Subsets of TH+ neurons show MOR-elicited electrophysiological responses supportive of MOR-expression [53] and further investigation will be necessary.
Based on the VTA MOR-regulated GABA projection to VTA dopamine neurons, we hypothesized that VTA VGluT2 projections to VTA dopamine neurons are also regulated by the MOR. Optogenetic stimulation of VTA VGluT2 neurons resulted in excitatory postsynaptic currents in VTA dopamine neurons, consistent with prior reports of a local glutamatergic pathway [27, 39, 40]. Activation of the MOR by the selective agonist DAMGO depressed the amplitude of VGluT2-CoChR-evoked EPSCs in TH-expressing neurons, indicating that MOR modulates VTA glutamatergic transmission to local dopamine neurons. Application of DAMGO also increased the paired-pulse ratio, indicating a decrease in release probability at these synapses [54]. Therefore, our results suggest that the MOR is expressed on presynaptic terminals of VTA VGluT2 projections to VTA dopamine neurons. Though we identified all biocytin-labeled neurons from recordings as expressing tyrosine hydroxylase, a limitation of this experiment was that we were unable to identify all recorded neurons and thus cannot exclude the possibility that non-dopamine neurons may also receive local MOR-gated inputs from VTA VGluT2+ neurons. Moreover, the local VGluT2+MOR+ projection identified here may be a subset of local projections from VGluT2+ neurons [39]. A second limitation of this experiment is we cannot exclude the possibility that there may also be input from transfected VGluT2-CoChR neurons nearby but outside the VTA.
VTA VGluT2+ regulation of NAcc dopamine following oxycodone administration
Having identified a local VTA VGluT2+MOR+ microcircuit to dopamine neurons, we hypothesized that this pathway belonged to a macrocircuit resulting in dopamine release within the nucleus accumbens. MOR agonists, such as heroin and morphine, cause the release of dopamine preferentially in the nucleus accumbens shell compared with the core [55, 56]. Consistent with this, evidence suggests that VTA GABA+MOR+ neuron inhibition by heroin preferentially disinhibits VTA dopamine neurons that project to medial shell [57]. A subset of VTA VGluT2 neurons co-releases glutamate and dopamine to the nucleus accumbens medial shell [45]. We biased our GRABDA recordings to the nucleus accumbens core, which does not receive projections from VTA VGluT2+TH+ neurons [26, 28, 58], and thus core dopamine results from the activation of VGluT2−TH+ neurons. Optogenetic activation of VTA VGluT2 neurons resulted in dopamine release within the accumbens core and the magnitude of this response was reduced by the MOR agonist oxycodone in vivo. These results suggest that VTA VGluT2+MOR+ neurons regulate local VTA dopamine neurons that project to the accumbens core.
While these results were consistent with our hypothesis, there are two important experimental limitations of this in vivo stimulation and recording experiment to consider. First, though our recording fibers targeted the accumbens core, GRABDA was expressed in some recordings within the medial accumbens shell and thus GRABDA recordings may have been influenced by dopaminergic projections to the shell that originate from VTA neurons that co-release dopamine and glutamate. If so, these glutamate and dopamine co-releasing neurons would have been directly activated by VTA ChRmine stimulation. Second, because oxycodone was systemically administered rather than locally injected in the VTA, it is certain that oxycodone’s MOR effects occurred within and outside the VTA. Thus, in addition to reducing local glutamate release onto VTA dopamine neurons, oxycodone binding to MOR in accumbens (and other sites) likely influenced the observed reduction in ChRmine-induced accumbal dopamine release. Some neurons that may participate in this effect may be MOR-expressing medium spiny neurons or several interneurons that regulate accumbal dopamine release [59,60,61,62,63,64,65,66].
Recently it has been shown that disinhibition of glutamate release is partly responsible for the activation of VTA dopamine neurons following morphine administration [23]. We found that MOR activation decreased EPSCs elicited by VTA VGluT2-CoChR neurons ex vivo, and reduced accumbal dopamine elicited by VTA VGluT2-ChRmine neurons in vivo. Therefore, it is unlikely that VTA VGluT2+MOR+ neurons play a role in exciting VTA dopamine neurons following activation of the MOR. Instead, MOR activity reduces glutamatergic drive from VTA VGluT2 neurons to VTA dopamine neurons. Glutamatergic inputs from the prefrontal cortex may excite VTA dopamine neurons following morphine administration [67].
Role of VTA VGluT2+ neurons in oral oxycodone seeking and reinstatement
We found that VTA VGluT2 neurons were significantly activated by different elements of oxycodone self-administration and abstinent drug-seeking (reinstatement). Population changes in VTA VGluT2 neuron calcium significantly increased following actions to acquire and consume oxycodone during self-administration and abstinent drug-seeking during reinstatement. Prior research has shown that subsets of VTA VGluT2 neurons are activated by reward-seeking actions, reward consumption, and reward-related cues [68, 69]. Because the drug-associated cue was delivered following nose poke responses to acquire drug, further research will be necessary to disentangle whether the drug-seeking action, the drug-associated cue, and/or the learned response-drug association, activate the same or different VTA VGluT2 neuron subtypes.
We have previously found that male mice consume less oxycodone (mg/kg) than female mice at the highest doses of self-administration [32]. We replicated this result and separated neuronal recording data between males and females to critically evaluate the potential contribution of VTA VGluT2 neurons to this behavioral difference. Neither phasic (maximum z-score fluorescent change) nor tonic (AUC) measures of activity following any analyzed event significantly differed between males or female mice. However, during cue-induced reinstatement testing, chemogenetic inhibition of VTA VGluT2 neurons significantly reduced reinstatement behavior for male but not female mice. These findings demonstrate that VTA VGluT2 neurons play a causal role in abstinent cue-induced drug-seeking (relapse) behavior in at least male mice. While it is plausible that DREADD function differed between sexes, the same dose of clozapine was shown previously to block the involvement of VTA VGluT2 neurons in the repercussions of inescapable stress in both male and female mice [33]. It is possible that despite no identified sex differences in MOR distribution or VGluT2 neuronal signaling of drug-seeking behavior that circuit level differences exist between males and female mice that contribute to this discrepancy. Relatedly, we have found divergent roles of sex hormones between sexes that contribute to oxycodone-related behavior in mice [32]. Afferent circuitry to VTA VGluT2 neurons [70] may also provide insight into this discrepancy and is of interest for future investigation.
For both male and female mice, chemogenetic inhibition of VTA VGluT2 neurons had no effect on self-administration behavior. One reason for this may be that the presence of oxycodone during self-administration already inhibited VTA VGluT2+MOR+ neurons and thus chemogenetic inhibition would be mitigated. It is also plausible that disinhibition of VTA dopamine neurons resulting from activation of the MOR on midbrain GABA neurons and terminals overshadowed the effects of chemogenetic inhibition of VTA glutamate neurons during oral oxycodone self-administration.
Extending the two-neuron model of opioid signaling
Canonically, the reinforcing effects of opioids are thought to result from opioid activation of the MOR on midbrain GABAergic somata or terminals that disinhibit VTA dopamine neurons [8]. This two-neuron model of opioid addiction has been challenged by findings demonstrating that: 1) about half of MOR-expressing axon terminals synapsing on TH-expressing dendrites are asymmetric (putative excitatory) or symmetric (putative inhibitory) [71]; 2) select subsets, rather than all VTA GABA or glutamate neurons, express the MOR [21, 22]; 3) glutamate transmission within the VTA plays an essential role in the disinhibition of VTA dopamine neurons following MOR activation [23] as well as heroin self-administration [24]; and 4) ablation of the MOR within central and peripheral VGluT2 neurons results in reduced oxycodone self-administration, place preference, locomotion, and withdrawal [72]. Our results extend these findings by demonstrating that a subset of VTA VGluT2 neurons expresses the MOR and that VTA VGluT2 neurons participate in a local circuit that drives MOR-gated accumbal dopamine release. We interpret these results to reflect that VTA dopamine neurons are regulated in a three-neuron model whereby 1) MOR activation results in increased mesolimbic dopamine release via disinhibition of VGaT+MOR+ neurons and 2) mesolimbic dopamine release is restrained by MOR-dependent depression of excitatory synaptic drive from local VTA VGluT2+MOR+ inputs onto accumbens-projecting VTA dopamine neurons (Supplementary Fig. 6). Due to the dense interconnected nature of VTA neurons, an additional MOR sensitive microcircuit between VGluT2+MOR+ and VGaT+MOR+ local populations may also play a regulatory role in subsequent dopamine release but has yet to be explored. Further research will also be needed to identify if the same or different VTA dopamine neurons are regulated by VTA VGaT+MOR+ or VGluT2+MOR+ neurons. However, because heroin preferentially activates VTA dopamine neurons projecting to the nucleus accumbens medial shell [57], and we detected MOR-regulated dopamine release in the nucleus accumbens core as a result of activating VTA VGluT2 neurons, we hypothesize that each VTA MOR+ cell-type regulates distinct accumbal dopamine release targets to differentially regulate self-administration versus abstinent drug-seeking (relapse/reinstatement). In this model, reduction of VGaT+MOR+ firing by MOR activation disinhibits shell-projecting dopamine neurons to drive opioid reinforcement. In addition, because D1 receptors in accumbens core, but not shell, are required for cue-induced reinstatement of opioid seeking [73], activation of VGluT2+MOR+ neurons results in the activation of core-projecting dopamine neurons to drive abstinent drug-seeking.
References
Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65:1445–52.
Ghose R, Forati AM, Mantsch JR. Impact of the COVID-19 pandemic on opioid overdose deaths: a spatiotemporal analysis. J Urban Health. 2022;99:316–27.
Zaki PA, Bilsky EJ, Vanderah TW, Lai J, Evans CJ, Porreca F. Opioid receptor types and subtypes: the delta receptor as a model. Annu Rev Pharm Toxicol. 1996;36:379–401.
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–23.
Bozarth MA, Wise RA. Neural substrates of opiate reinforcement. Prog Neuropsychopharmacol Biol Psychiatry. 1983;7:569–75.
Britt MD, Wise RA. Ventral tegmental site of opiate reward: antagonism by a hydrophilic opiate receptor blocker. Brain Res. 1983;258:105–8.
Zhang Y, Landthaler M, Schlussman SD, Yuferov V, Ho A, Tuschl T, et al. Mu opioid receptor knockdown in the substantia nigra/ventral tegmental area by synthetic small interfering RNA blocks the rewarding and locomotor effects of heroin. Neuroscience. 2009;158:474–83.
Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–8.
Laviolette SR, Gallegos RA, Henriksen SJ, van der Kooy D. Opiate state controls bi-directional reward signaling via GABAA receptors in the ventral tegmental area. Nat Neurosci. 2004;7:160–9.
Madhavan A, Bonci A, Whistler JL. Opioid-Induced GABA potentiation after chronic morphine attenuates the rewarding effects of opioids in the ventral tegmental area. J Neurosci. 2010;30:14029–35.
Madhavan A, He L, Stuber GD, Bonci A, Whistler JL. micro-Opioid receptor endocytosis prevents adaptations in ventral tegmental area GABA transmission induced during naloxone-precipitated morphine withdrawal. J Neurosci. 2010;30:3276–86.
Ting AKR, Vargas-Perez H, Mabey JK, Shin SI, Steffensen SC, van der Kooy D. Ventral tegmental area GABA neurons and opiate motivation. Psychopharmacology. 2013;227:697–709.
Taylor AM, Castonguay A, Ghogha A, Vayssiere P, Pradhan AA, Xue L, et al. Neuroimmune regulation of GABAergic neurons within the ventral tegmental area during withdrawal from chronic morphine. Neuropsychopharmacology. 2016;41:949–59.
Pettit HO, Ettenberg A, Bloom FE, Koob GF. Destruction of dopamine in the nucleus accumbens selectively attenuates cocaine but not heroin self-administration in rats. Psychopharmacology. 1984;84:167–73.
Badiani A, Belin D, Epstein D, Calu D, Shaham Y. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011;12:685–700.
Stinus L, Cador M, Le, Moal M. Interaction between endogenous opioids and dopamine within the nucleus accumbens a. Ann N Y Acad Sci. 1992;654:254–73.
Margolis EB, Toy B, Himmels P, Morales M, Fields HL. Identification of rat ventral tegmental area GABAergic neurons. PLoS One. 2012;7:e42365.
Matsui A, Jarvie BC, Robinson BG, Hentges ST, Williams JT. Separate GABA afferents to dopamine neurons mediate acute action of opioids, development of tolerance, and expression of withdrawal. Neuron. 2014;82:1346–56.
Matsui A, Williams JT. Opioid-sensitive GABA inputs from rostromedial tegmental nucleus synapse onto midbrain dopamine neurons. J Neurosci. 2011;31:17729–35.
Yamaguchi T, Sheen W, Morales M. Glutamatergic neurons are present in the rat ventral tegmental area. Eur J Neurosci. 2007;25:106–18.
Kudo T, Konno K, Uchigashima M, Yanagawa Y, Sora I, Minami M, et al. GABAergic neurons in the ventral tegmental area receive dual GABA/enkephalin-mediated inhibitory inputs from the bed nucleus of the stria terminalis. Eur J Neurosci. 2014;39:1796–809.
Miranda‐Barrientos J, Chambers I, Mongia S, Liu B, Wang HL, Mateo‐Semidey GE, et al. Ventral tegmental area GABA, glutamate, and glutamate‐GABA neurons are heterogeneous in their electrophysiological and pharmacological properties. Eur J Neurosci. 2021;54:4061–84.
Chen M, Zhao Y, Yang H, Luan W, Song J, Cui D, et al. Morphine disinhibits glutamatergic input to VTA dopamine neurons and promotes dopamine neuron excitation. Elife. 2015;4:e09275.
Xi ZX, Stein EA. Blockade of ionotropic glutamatergic transmission in the ventral tegmental area reduces heroin reinforcement in rat. Psychopharmacology. 2002;164:144–50.
Zell V, Steinkellner T, Hollon NG, Warlow SM, Souter E, Faget L, et al. VTA glutamate neuron activity drives positive reinforcement absent dopamine co-release. Neuron. 2020;107:864–73 e4.
Qi J, Zhang S, Wang HL, Barker DJ, Miranda-Barrientos J, Morales M. VTA glutamatergic inputs to nucleus accumbens drive aversion by acting on GABAergic interneurons. Nat Neurosci. 2016;19:725–33.
Dobi A, Margolis EB, Wang HL, Harvey BK, Morales M. Glutamatergic and nonglutamatergic neurons of the ventral tegmental area establish local synaptic contacts with dopaminergic and nondopaminergic neurons. J Neurosci. 2010;30:218–29.
Taylor SR, Badurek S, Dileone RJ, Nashmi R, Minichiello L, Picciotto MR. GABAergic and glutamatergic efferents of the mouse ventral tegmental area. J Comp Neurol. 2014;522:3308–34.
Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–36.
Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. J Neurophysiol. 2005;93:3086–93.
Marino M, Misuri L, Brogioli D. A new open source software for the calculation of the liquid junction potential between two solutions according to the stationary Nernst-Planck equation. Preprint at https://arxiv.org/abs/1403.3640. 2014.
Phillips AG, McGovern DJ, Lee S, Ro K, Huynh DT, Elvig SK, et al. Oral prescription opioid-seeking behavior in male and female mice. Addict Biol. 2020;25:e12828.
McGovern DJ, Ly A, Ecton KL, Huynh DT, Prévost ED, Gonzalez SC, et al. Ventral tegmental area glutamate neurons mediate nonassociative consequences of stress. Mol Psychiatry. 2022. https://doi.org/10.1038/s41380-022-01858-3. [Epub ahead of print].
Gomez JL, Bonaventura J, Lesniak W, Mathews WB, Sysa-Shah P, Rodriguez LA, et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017;357:503–07.
Root DH, Mejias-Aponte CA, Zhang S, Wang HL, Hoffman AF, Lupica CR, et al. Single rodent mesohabenular axons release glutamate and GABA. Nat Neurosci. 2014;17:1543–51.
Lauridsen K, Ly A, Prévost ED, McNulty C, McGovern DJ, Tay JW, et al. A Semi-Automated Workflow for Brain Slice Histology Alignment, Registration, and Cell Quantification (SHARCQ). eNeuro. 2022;9(2):ENEURO.0483-21.2022.
Claudi F, Tyson AL, Petrucco L, Margrie TW, Portugues R, Branco T. Visualizing anatomically registered data with brainrender. Elife. 2021;10:e65751.
Klapoetke NC, Murata Y, Kim SS, Pulver SR, Birdsey-Benson A, Cho YK, et al. Independent optical excitation of distinct neural populations. Nat Methods. 2014;11:338–46.
Wang HL, Qi J, Zhang S, Wang H, Morales M. Rewarding effects of optical stimulation of ventral tegmental area glutamatergic neurons. J Neurosci. 2015;35:15948–54.
Yoo JH, Zell V, Gutierrez-Reed N, Wu J, Ressler R, Shenasa MA, et al. Ventral tegmental area glutamate neurons co-release GABA and promote positive reinforcement. Nat Commun. 2016;7:13697.
Petreanu L, Mao T, Sternson SM, Svoboda K. The subcellular organization of neocortical excitatory connections. Nature. 2009;457:1142–5.
Piñol RA, Bateman R, Mendelowitz D. Optogenetic approaches to characterize the long-range synaptic pathways from the hypothalamus to brain stem autonomic nuclei. J Neurosci Methods. 2012;210:238–46.
Marshel JH, Kim YS, Machado TA, Quirin S, Benson B, Kadmon J, et al. Cortical layer-specific critical dynamics triggering perception. Science. 2019;365:eaaw5202.
Sun F, Zeng J, Jing M, Zhou J, Feng J, Owen SF, et al. A genetically encoded fluorescent sensor enables rapid and specific detection of dopamine in flies, fish, and mice. Cell. 2018;174:481–96.e19.
Zhang S, Qi J, Li X, Wang HL, Britt JP, Hoffman AF, et al. Dopaminergic and glutamatergic microdomains in a subset of rodent mesoaccumbens axons. Nat Neurosci. 2015;18:386–92.
Stewart J. Reinstatement of heroin and cocaine self-administration behavior in the rat by intracerebral application of morphine in the ventral tegmental area. Pharm Biochem Behav. 1984;20:917–23.
Jhou TC, Xu SP, Lee MR, Gallen CL, Ikemoto S. Mapping of reinforcing and analgesic effects of the mu opioid agonist endomorphin-1 in the ventral midbrain of the rat. Psychopharmacology. 2012;224:303–12.
Zangen A, Ikemoto S, Zadina JE, Wise RA. Rewarding and psychomotor stimulant effects of endomorphin-1: anteroposterior differences within the ventral tegmental area and lack of effect in nucleus accumbens. J Neurosci. 2002;22:7225–33.
Yamaguchi T, Wang HL, Li X, Ng TH, Morales M. Mesocorticolimbic glutamatergic pathway. J Neurosci. 2011;31:8476–90.
Shabat-Simon M, Levy D, Amir A, Rehavi M, Zangen A. Dissociation between rewarding and psychomotor effects of opiates: differential roles for glutamate receptors within anterior and posterior portions of the ventral tegmental area. J Neurosci. 2008;28:8406–16.
Carlezon WA Jr., Haile CN, Coppersmith R, Hayashi Y, Malinow R, Neve RL, et al. Distinct sites of opiate reward and aversion within the midbrain identified using a herpes simplex virus vector expressing GluR1. J Neurosci. 2000;20:Rc62.
Yamaguchi T, Qi J, Wang HL, Zhang S, Morales M. Glutamatergic and dopaminergic neurons in the mouse ventral tegmental area. Eur J Neurosci. 2015;41:760–72.
Margolis EB, Hjelmstad GO, Fujita W, Fields HL. Direct bidirectional mu-opioid control of midbrain dopamine neurons. J Neurosci. 2014;34:14707–16.
Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–9.
Pontieri FE, Tanda G, Di Chiara G. Intravenous cocaine, morphine, and amphetamine preferentially increase extracellular dopamine in the "shell" as compared with the "core" of the rat nucleus accumbens. Proc Natl Acad Sci USA. 1995;92:12304–8.
Tanda G, Pontieri FE, Di Chiara G. Cannabinoid and heroin activation of mesolimbic dopamine transmission by a common mu1 opioid receptor mechanism. Science 1997;276:2048–50.
Corre J, van Zessen R, Loureiro M, Patriarchi T, Tian L, Pascoli V, et al. Dopamine neurons projecting to medial shell of the nucleus accumbens drive heroin reinforcement. Elife. 2018;7:e39945.
Hnasko TS, Hjelmstad GO, Fields HL, Edwards RH. Ventral tegmental area glutamate neurons: electrophysiological properties and projections. J Neurosci. 2012;32:15076–85.
Chuhma N, Mingote S, Moore H, Rayport S. Dopamine neurons control striatal cholinergic neurons via regionally heterogeneous dopamine and glutamate signaling. Neuron. 2014;81:901–12.
Elghaba R, Bracci E. Dichotomous effects of Mu opioid receptor activation on striatal low-threshold spike interneurons. Front Cell Neurosci. 2017;11:385.
Jabourian M, Venance L, Bourgoin S, Ozon S, Pérez S, Godeheu G, et al. Functional mu opioid receptors are expressed in cholinergic interneurons of the rat dorsal striatum: territorial specificity and diurnal variation. Eur J Neurosci. 2005;21:3301–9.
Miura M, Masuda M, Aosaki T. Roles of micro-opioid receptors in GABAergic synaptic transmission in the striosome and matrix compartments of the striatum. Mol Neurobiol. 2008;37:104–15.
Ponterio G, Tassone A, Sciamanna G, Riahi E, Vanni V, Bonsi P, et al. Powerful inhibitory action of mu opioid receptors (MOR) on cholinergic interneuron excitability in the dorsal striatum. Neuropharmacology. 2013;75:78–85.
Arttamangkul S, Platt EJ, Carroll J, Farrens D. Functional independence of endogenous μ- and δ-opioid receptors co-expressed in cholinergic interneurons. Elife. 2021;10:e69740.
Cachope R, Mateo Y, Mathur BN, Irving J, Wang HL, Morales M, et al. Selective activation of cholinergic interneurons enhances accumbal phasic dopamine release: setting the tone for reward processing. Cell Rep. 2012;2:33–41.
Gómez AA, Shnitko TA, Barefoot HM, Brightbill EL, Sombers LA, Nicola SM, et al. Local μ-opioid receptor antagonism blunts evoked phasic dopamine release in the nucleus accumbens of rats. ACS Chem Neurosci. 2019;10:1935–40.
Yang L, Chen M, Ma Q, Sheng H, Cui D, Shao D, et al. Morphine selectively disinhibits glutamatergic input from mPFC onto dopamine neurons of VTA, inducing reward. Neuropharmacology. 2020;176:108217.
Root DH, Estrin DJ, Morales M. Aversion or salience signaling by ventral tegmental area glutamate neurons. iScience. 2018;2:51–62.
McGovern DJ, Polter AM, Root DH. Neurochemical signaling of reward and aversion to ventral tegmental area glutamate neurons. J Neurosci. 2021;41:5471–86.
Faget L, Osakada F, Duan J, Ressler R, Johnson AB, Proudfoot JA, et al. Afferent inputs to neurotransmitter-defined cell types in the ventral tegmental area. Cell Rep. 2016;15:2796–808.
Garzón M, Pickel VM. Plasmalemmal mu-opioid receptor distribution mainly in nondopaminergic neurons in the rat ventral tegmental area. Synapse. 2001;41:311–28.
Reeves KC, Kube MJ, Grecco GG, Fritz BM, Muñoz B, Yin F, et al. Mu opioid receptors on vGluT2-expressing glutamatergic neurons modulate opioid reward. Addict Biol. 2021;26:e12942.
Bossert JM, Poles GC, Wihbey KA, Koya E, Shaham Y. Differential effects of blockade of dopamine D1-family receptors in nucleus accumbens core or shell on reinstatement of heroin seeking induced by contextual and discrete cues. J Neurosci. 2007;27:12655–63.
Acknowledgements
We thank Alysabeth Phillips and Declan Mulcahy for technical assistance.
Funding
This research was supported by the Webb-Waring Biomedical Research Award from the Boettcher Foundation (DHR), R01 DA047443 (DHR), F31 MH125569 (DJM), F31 MH132322 (AL), a 2020 NARSAD Young Investigator grant from the Brain and Behavior Research Foundation (DHR). Further support was provided by NIH Grants R00MH106757 and R01MH122712, a grant from the Margaret Q. Landenberger Foundation, and a 2019 NARSAD Young Investigator grant (to AMP). Some of the imaging work was performed at the BioFrontiers Institute Advanced Light Microscopy Core (RRID: SCR_018302). Laser scanning confocal microscopy was also performed on a Nikon A1R microscope supported by NIST-CU Cooperative Agreement award number 70NANB15H226. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Prism and Biorender were used to make figures and schematics.
Author information
Authors and Affiliations
Contributions
DJM, AMP, and DHR conceived and performed experiments, wrote the manuscript, and secured funding. CM, AL, EDP, BR performed experiments and contributed to the writing of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
McGovern, D.J., Polter, A.M., Prévost, E.D. et al. Ventral tegmental area glutamate neurons establish a mu-opioid receptor gated circuit to mesolimbic dopamine neurons and regulate opioid-seeking behavior. Neuropsychopharmacol. 48, 1889–1900 (2023). https://doi.org/10.1038/s41386-023-01637-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-023-01637-w
- Springer Nature Switzerland AG
This article is cited by
-
Distinct µ-opioid ensembles trigger positive and negative fentanyl reinforcement
Nature (2024)
-
Control of dopamine communication by opioids: glutamate enters the discussion
Neuropsychopharmacology (2023)