
Abstract
Despite the substantial heritability of antisocial behavior (ASB), specific genetic variants robustly associated with the trait have not been identified. The present study by the Broad Antisocial Behavior Consortium (BroadABC) meta-analyzed data from 28 discovery samples (N = 85,359) and five independent replication samples (N = 8058) with genotypic data and broad measures of ASB. We identified the first significant genetic associations with broad ASB, involving common intronic variants in the forkhead box protein P2 (FOXP2) gene (lead SNP rs12536335, p = 6.32 × 10−10). Furthermore, we observed intronic variation in Foxp2 and one of its targets (Cntnap2) distinguishing a mouse model of pathological aggression (BALB/cJ strain) from controls (BALB/cByJ strain). Polygenic risk score (PRS) analyses in independent samples revealed that the genetic risk for ASB was associated with several antisocial outcomes across the lifespan, including diagnosis of conduct disorder, official criminal convictions, and trajectories of antisocial development. We found substantial genetic correlations of ASB with mental health (depression rg = 0.63, insomnia rg = 0.47), physical health (overweight rg = 0.19, waist-to-hip ratio rg = 0.32), smoking (rg = 0.54), cognitive ability (intelligence rg = −0.40), educational attainment (years of schooling rg = −0.46) and reproductive traits (age at first birth rg = −0.58, father’s age at death rg = −0.54). Our findings provide a starting point toward identifying critical biosocial risk mechanisms for the development of ASB.
Similar content being viewed by others


Introduction
Antisocial behaviors (ASB) are disruptive acts characterized by covert and overt hostility and violation of the rights and safety of others [1]. The emotional, social, and economic costs incurred by victims of ASB are far-reaching, ranging from victims’ psychological trauma to reduced productivity when victims miss work to costs incurred by taxpayers in order to staff and run a justice system [2, 3]. ASB has been recognized not merely as a social problem, but also as a mental health economic priority [4]. In addition to causing harm to others, those with ASB are themselves at elevated risk of criminal convictions as well as mental health and substance abuse problems [5]. Moreover, given the relative stability of ASB [6], it is important to also examine personality traits potentially tied to overt behaviors. Previous meta-analyses demonstrated that the Five-Factor Model of personality (FFM), particularly the domains of Agreeableness, Conscientiousness, and Neuroticism, is potentially critical for better illuminating the correlates and causes of ASB [7, 8]. Given all this, it is a research imperative to illuminate the mechanisms underlying the pathogenesis, emergence, and persistence of ASB.
Toward this end, statistical genetic studies have consistently revealed the relevance of environmental and genetic risk factors in the genesis of inter-individual differences in ASB. Family studies—mostly conducted in samples of European ancestry—have demonstrated a considerable heritable component for ASB, with estimates of ~50% [9] across studies. The increasing availability of genome-wide data along with data on dimensional ASB measures facilitates in building more advanced explanatory models aimed at identifying trait-relevant genetic variants, that could serve as moderators of socio-environmental factors and vice versa. Moreover, while heritability estimates can differ across subtypes of ASB (e.g., significantly higher twin-based heritability estimates for aggressive forms (65%) versus non-aggressive, rule-breaking forms (48%) of ASB [10]), these subtypes are genetically correlated (rg = 0.38) [11].
Measuring antisocial behavior, a broad view
Considering multiple forms of ASB together increases power of genetic analysis and may improve our ability to detect new genetic variants. Here, we thus examine a broadly defined construct of ASB, an approach that has successful precedents. Large-scale genomic studies have indicated substantial genetic overlap among psychiatric disorders [12]. A recent genome-wide meta-analysis across eight neuropsychiatric disorders revealed extensive pleiotropic genetic effects (N = 232,964 cases and 494,162 controls) [13, 14]. The study found that 109 out of the total 146 contributing loci were associated with at least two psychiatric disorders, suggesting broad liability to these conditions. Moreover, the Externalizing Consortium recently conducted a multivariate analysis of large-scale genome-wide association studies (GWAS) of seven externalizing-related phenotypes (N = ~1.5 million) and found 579 genetic associations with a general liability to externalizing behavior [15]. Although these very large multivariate approaches are crucial in enhancing genetic discovery across phenotypes, they do not detect all the genetic variation relevant to individual disorders. Since ASB is a critical issue for psychiatry and for society, the present study uniquely focuses on (severe) forms of ASB and persistence over the lifespan. To do so, we initiated the Broad Antisocial Behavior Consortium (BroadABC) to perform large-scale meta-analytical genetic analyses utilizing a broad range of phenotypic ASB measures (e.g., conduct disorder symptoms, aggressive behavior, and delinquency). In our first meta-analysis [16], we demonstrated that effect sizes for SNPs with suggestive evidence of association with ASB were small, as anticipated for most polygenic traits. Still, we found that the collective effect across all of the included variants (typically referred to as “SNP heritability”) explained roughly 5% of the total variation in ASB [16], which is in line with meta-analyses of the ACTION [17] and EAGLE [18] consortium.
To date, however, no previous GWAS meta-analysis targeting broad ASB detected SNPs or genes that are well-replicated. The polygenic architecture of ASB underscores the importance of employing very large samples to yield sufficient power to detect genetic loci of small effect size. Therefore, we substantially boost statistical power by quadrupling the sample size and adding new cohorts to the BroadABC consortium. Since ASB is a critical issue for psychiatry and for society, the present study uniquely focuses on (severe) forms of ASB and persistence over the lifespan.
In our meta-analysis, we also include the results of a GWAS study of disruptive behavior disorders (DBDs) in the context of attention-deficit/hyperactivity disorder (ADHD), which identified three genome-wide significant loci for DBDs [19]. The present study considers multiple measures of ASB in people with and without psychiatric diagnoses across 28 samples to reveal the genetic underpinnings of ASB phenotypes typically studied in psychology, psychiatry, and criminology. These larger samples allow well-powered genetic correlation analyses and improved polygenic risk scores (PRS). Five independent cohorts (total N = 8058) were employed to validate the ASB PRS in different populations, at different developmental stages, and for different ASB phenotypes. Moreover, we conducted a follow-up analysis by using a mouse model of pathological aggression. Since ASB is known to correlate phenotypically with an array of cognitive and health problems [20,21,22,23], we tested for genetic overlap between ASB and a range of other traits and disorders, including anthropometric, cognitive, reproductive, neuropsychiatric, and smoking.
Results
Meta-analysis on broad ASB identifies association with common variants in FOXP2
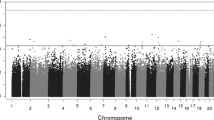
After quality control and imputation to the Haplotype Reference Consortium (HRC) or 1000 Genomes Project reference panel (see Online Methods), 85,359 individuals from 28 cohorts and a maximum of 7,392,849 variants were available for analysis. We carried out a pooled-sex GWAS meta-analysis for the broad ASB phenotype with METAL [24] and found one genome-wide significant locus, on chromosome 7 (chromosome band 7q31.1, Fig. 1a and Supplementary Table 3). The top lead SNP was rs12536335 (p = 6.32 × 10−10; Fig. 1b, c), located in an intronic region upstream of one of the transcriptional start-sites for the forkhead box protein P2 (FOXP2) gene [25, 26]. Consistent with this finding, a gene-based association test carried out with MAGMA [27], identified a significant association for FOXP2 (p = 7.43 × 10−7, Supplementary Note 3, Supplementary Fig. 1, and Supplementary Table 6). The FOXP2 gene has been related to the development of speech and language [28], yet is also implicated in a wide range of other traits and diagnoses [29] (see Fig. 1d). MAGMA generalized gene-set and tissue-specific gene-set analyses (sex-combined) yielded no significant gene-sets after Bonferroni-correction for multiple testing. The top gene-set for generalized gene-set analysis was activated NTRK2 signals through RAS signaling pathway (Supplementary Table 7), while the top tissue-specific gene expression was the hypothalamus (Supplementary Table 8). We next ran sex-specific GWAS meta-analyses. These analyses did not identify SNPs that reached genome-wide significance (Supplementary Tables 4 and 5).

a Manhattan plot of the GWAS meta-analysis, showing the negative log10-transformed p value for each SNP. SNP two-sided p values from a linear model were calculated using METAL [24], weighting SNP associations by sample size. b Regional association plot around chromosome 7:114043159 with functional annotations of SNPs in LD of lead SNP rs12536335 (shown in purple). The plot displays GWAS p value plotted against its chromosomal position, where colors represent linkage disequilibrium and r2 values with the most significantly associated SNP. c The plot displays CADD scores (Combined Annotation Dependent Depletion) and RegulomeDB scores of these SNPs. d PheWAS plot showing the significance of associations of common variation in the FOXP2 gene with a wide range of traits and diagnoses based on MAGMA gene-based tests (with Bonferroni-corrected p value: 1.05e−5), as obtained from GWASAtlas (https://atlas.ctglab.nl).
Mouse model of pathological aggression
Whole-genome sequencing analysis of SNVs in aggressive antisocial BALB/cJ mice compared to BALB/cByJ mice controls revealed differences between these lines located in introns of Foxp2 (rs241912422) and Cntnap2 (rs212805467; rs50446478; rs260305923; rs242237534), a well-studied neural target of this transcription factor.
Heritability and polygenic scoring
SNP heritability
To assess the proportion of variance in liability for broad ASB explained by all measured SNPs, we computed the SNP-based heritability (h2SNP) through LD score regression (LDSC) [30]. The h2SNP was estimated to be 3.4% (s.e. = 1.2%) in the quantitative BroadABC data, 24.8% (s.e. = 3.1%) in the Psychiatric Genetics Consortium/iPSYCH case-control data (with a prevalence estimate of 2% in the population) and 7.7% (s.e. = 1.1%) in the combined meta-analysis.
Polygenic risk scoring in five independent cohorts
To assess how well the PRS derived from our ASB GWAS meta-analysis predicts other measures of ASB, we carried out PRS analyses in five independent cohorts, none of which appeared in the GWAS meta-analysis (Fig. 2 and Supplementary Note 7).

PRSs are shown for broad ASB associated with childhood ASB in the Dunedin Longitudinal Study (A), with externalizing behavior in the E-Risk Study (B), with Conduct Disorder (C) and Oppositional Defiant Disorder (D) in the Philadelphia Neurodevelopmental Cohort Study, with ASB in the Quebec Longitudinal Study of Children’s Development Study (E), and with time-aggregated ASB in the Quebec Newborn Twin Study (F). Asterisks (*) show statistical significance after applying a Bonferroni correction on the 22 tested phenotypes at p < 0.0023.
Dunedin Longitudinal Study
In New Zealand, participants were derived from the Dunedin Longitudinal Study [31] (N = 1037, assessed 14 times from birth to age 45 years). We tested nine phenotypes and found significant associations with the BroadABC-based PRS for two: childhood ASB and official-records of juvenile convictions. Although not surviving Bonferroni adjustment, we found nominal significant (p < 0.05) association with the BroadABC-based PRS for eight phenotypes. We did not find evidence for a PRS association with partner violence. Lastly, we compared individuals grouped into the following four distinct developmental trajectories of ASB using general growth mixture modeling: low ASB across childhood through adulthood, childhood-limited ASB, adolescent-onset ASB, and life-course persistent (LCP) ASB [32]. Individuals following the LCP antisocial trajectory were characterized by the highest levels of genetic risk (see Supplementary Fig. 2); the nominally significant higher PRS of the LCP trajectory group compared to the low ASB group (p = 0.032 and p = 0.049, for p value thresholds 0.05 and 0.1 respectively) did not survive Bonferroni adjustment. For a full report of the findings in the Dunedin cohort, see Supplementary Table 9 and Supplementary Note 8.
Environmental Risk Longitudinal Twin Study (E-Risk)
In England and Wales, participants were included from the E-Risk Study (N = 2232, assessed five times from birth to age 18 years). We tested eight phenotypes and found significant associations for seven. PRS analyses revealed significant associations with parent- and teacher-reported ASB up to age 12 years, conduct disorder diagnosis up to age 12 years, with the externalizing spectrum at age 18 years, and with official records of criminal convictions up to age 22 years. For a full report of the findings in the E-risk Study, see Supplementary Table 10 and Supplementary Note 8.
Philadelphia Neurodevelopmental Cohort (PNC)
In the United States, participants were included from the PNC Study (N = 4201). We tested two phenotypes and found significant associations for both. We found that higher PRS for ASB were associated with symptom counts of both conduct disorder (p < 0.0001, delta R2 = 1.0%, Supplementary Table 11) and oppositional defiant disorder (p < 0.0001, delta R2 = 0.4%, Supplementary Table 12).
Quebec Longitudinal Study of Children’s Development (QLSCD)
In Canada, participants were included from the QLSCD study (N = 599). We tested one phenotype and did not find a significant association (p > 0.05, Supplementary Table 13) between PRS and the score on a self-report questionnaire related to conduct disorder, delinquency, and broad ASB in young adults (age range = 18–19 years).
Quebec Newborn Twin Study (QNTS)
In Canada, participants were derived from the QNTS study (N = 341). We tested two phenotypes and found a significant association for one. We computed a factor score based upon five teacher-rated assessments of ASB in youngsters during primary school (age range = 6–12 years). We found that higher PRS were associated with a higher factor score of ASB (p = 0.001, for p value thresholds 0.4, adjusted delta R2 = 3.9%, Supplementary Table 14). We failed to find evidence for an association between PRS and self-reported ASB in young adults (p > 0.05).
Genetic correlations through LD score regression
ASB is known to correlate with an array of phenotypes [20,21,22]. At the same time there has been a growing availability of publicly accessible genetic data across these phenotypes. To test whether these phenotypic associations are also reflected in genetic correlations we performed analyses with LDSC in a selection of 73 traits and diagnoses (Supplementary Table 15 and Fig. 3). We found strong correlations between ASB and reproductive traits (e.g., younger age of first birth (rg = −0.58, s.e. = 0.06, p = 2.93 × 10−15)), cognitive traits (e.g., fewer years of schooling (rg = −0.49, s.e. = 0.06, p = 1.94 × 10−10)), anthropometric traits (e.g., increased waist-to-hip ratio (rg = 0.32, s.e. = 0.05, p = 5.59 × 10−6)), neuropsychiatric traits (e.g., more depressive symptoms (rg = 0.63, s.e. = 0.07, p = 2.45 × 10−16)) and smoking related traits (e.g., ever smoked (rg = 0.54, s.e. = 0.08, p = 1.48 × 10−7)). It is important to emphasize here that correlation, even when genetic, does not imply causation.

For traits with significant correlations with ASB in multiple studies (see Supplementary Table 15), we report the most recent study here.
Discussion
Our GWAS meta-analysis of broad ASB in 85,359 individuals from population cohorts and those with a clinical diagnosis related to ASB, revealed one novel associated locus on chromosome 7 (7:114043159, rs12536335), residing in the forkhead box P2 (FOXP2) gene. The lead SNP is relatively proximal (~14 kb upstream) to an important enhancer region located 330 kb downstream of the first transcriptional start site (TSS1) of the gene [26]. This SNP is also in the vicinity (~8 kb upstream) of a second transcriptional start site (TSS2) of FOXP2 that can drive expression of alternative transcripts. The FOXP2 gene is expressed in sensory, limbic, and motor circuits of the brain, as well as the lungs, heart, and gut [26]. It encodes a transcription factor that acts as a regulator of numerous target genes and has been implicated in multiple aspects of brain development (e.g., neuronal growth, synaptic plasticity) [33]. FOXP2 was first identified two decades ago when rare heterozygous mutations of the gene were linked to a monogenic disorder involving speech motor deficits, accompanied by impairments in expressive and receptive language [34, 35]. Nevertheless, there is scant evidence that common FOXP2 variants contribute to inter-individual differences in language function [36, 37]. Though prior behavioral research [38,39,40] reported a link between language problems and ASB, it is premature to over-interpret the FOXP2 findings here. SNPs at this locus have been associated, through GWAS, with a range of externalizing traits, including ADHD [41], cannabis use disorder [42], and generalized risk tolerance [43]. Given the involvement of SNPS at this locus in different behavioral traits and diagnoses, and considering the small effect sizes, it is clear the association of FOXP2 variation with ASB has limited explanatory value on its own. That said, nothing yet precludes the possibility that this SNP may help to yield deeper insights once placed in broader context by future research.
In the present study we also compared the BALB/cJ strain, a mouse model of pathological aggression, to BALB/cByJ controls, and found intronic variants in Foxp2 and one of its downstream targets, Cntnap2. Previous studies in human cellular models have shown that the protein encoded by FOXP2 can directly bind to regulatory regions in the CNTNAP2 locus to repress its expression [44]. Interestingly, mice with cortical-specific knockout of Foxp2 have been reported to show abnormalities in social behaviors [45]. Although these findings may indicate that the intronic SNVs are relevant to the behavioral differences between the strains, further evidence is needed to show that the variants actually have functional relevance for the mouse phenotype. Future studies may utilize complementary data comparing gene expression in the two mouse lines or could investigate functional impact (e.g., do they map to credible enhancer regions, are they likely to alter binding for transcription factors?) of the SNVs identified.
Contrary to previous BroadABC GWAS analyses, we did not find evidence for sex-specific genetic effects in the present study. Although we did have access to sex-specific data in considerable subsets (N = 22,322 males, N = 26,895 females), the power to detect new variants employing such sample sizes is still limited. Compared to our previous study, we found that the variance explained in independent samples by PRS based on the resulting summary statistics has substantially increased from 0.21 to 3.9%. Essentially, we found consistent links of our ASB PRS with multiple antisocial phenotypes at different developmental stages, from different reporting sources, and reflecting measurements from different disciplines (psychology, psychiatry, criminology). These links were found in individuals from New Zealand, Britain, the United States, and Canada, born as much as 30 years apart. We also show that our ASB PRS were more strongly associated with more severe and persistent types of ASB.
Notwithstanding the increase of effect size of the PRS, and calculations yielding a more precise estimate, the variance explained by the PRS was still relatively small, which was expected in light of the low SNP heritability. Given the highly polygenic architecture of ASB, contributing SNPs have low average effect sizes, thus leading to limited predictive power in independent samples. New PRS methods along with further increasing sample sizes will likely further increase the amount of variance accounted for by the PRS. Moreover, the association may be enhanced by improving the quality of phenotype measurements, which is reflected by our PRS results demonstrating the most robust association with high-quality measurement of ASB (using a factor score based upon multiple assessments). Aggregating data from measurements across ages, as opposed to the measures assessed at a single time point, can lead to more reliable trait measures and to better prediction [46]. Phenotypically, adding more extreme ASB phenotypes to the GWAS meta-analysis might also lead to more explained variance. In addition, the inclusion of clinical samples displaying extreme ASB phenotypes (e.g., [multiple] homicide, sexual assaults, etc.) in GWAS studies could help ensuring the generalizability of genetic findings to forensic populations. Thus, future efforts of the BroadABC will continue to focus on more severe forms of ASB and its persistence across the lifespan. Moreover, by considering genetically correlated traits through multi-trait GWAS methods [47] and multi-trait PRS methods [48] it might be possible to boost power for discovery through GWAS meta-analysis and PRS prediction. Lastly, a major limitation of the present study is that our GWAS results are limited to individuals of European ancestry. This Eurocentric bias may lead to more accurate predictions in individuals with European ancestry, compared to non-Europeans, thus potentially increasing disparities in outcomes related to ASB [49, 50]. To realize the full and equitable potential of polygenic risk, future genetic studies on ASB should also include non-European samples.
Developmental criminological research findings, such as the influential developmental taxonomy theory by Moffitt [51, 52], have suggested the existence of distinctive offending patterns across the life-course [53]. These developmental trajectories of ASB are thought to have different underlying etiological processes, with relatively more variance explained by genetic factors for life-course-persistent offending as compared to the more socially influenced adolescence-limited offending. Barnes et al. have previously produced evidence that heritability estimates were not uniform across different offending groups, suggesting that the causal processes may vary across offending patterns [54, 55]. In the present study we found a trend of higher PRS for ASB showing a stronger association with the life-course-persistent trajectory of ASB as compared to the low ASB group. The life-course-persistent trajectory is also known to be associated with profound brain alterations and diminished neurological health [56]. These findings are important since they have the capacity to help improve the current understanding of downstream neurobiological mechanisms relevant to the etiology of antisocial development [56]. Sufficiently powered future studies should thus aim to further elucidate the genetic risk and protective factors that underlie different offending trajectories [57].
Our genetic correlation analyses confirmed previously reported [16, 23, 58] correlations between ASB and a wide range of traits and diagnoses. The relatively small GWAS sample size of some traits, however, coupled with wide confidence intervals (such as agreeableness, rg = −0,81, s.e. = 0.47) calls for larger samples in order to achieve more precise estimates concerning the genetic overlap between personality and ASB. It seems worthwhile to mention again here that partially overlapping genetic architectures do not provide causal insights of any kind. In this case they merely signify the presence of some potentially shared biological mechanisms linking the conditions [59]. One can reasonably conclude, though, there are likely common underlying genetic factors which operate to increase a general vulnerability to a range of psychopathologies. These comorbid effects are in line with findings in the Dunedin Study demonstrating that life-course-persistent offenders are characterized by several pathological risk factors, related to domains of parenting, neurocognitive development, and temperament [52]. This signifies the importance of investigating pleiotropy and considering the complex etiology of the broader ASB phenotype. Large-scale collaborations, such as the BroadABC, will facilitate the expansion of epidemiological studies capable of further exploring the interaction of genetic risk and socio-environmental risks, and how these contribute to the multifaceted origin of ASB.
Methods
Samples
The meta-analysis included 21 new discovery samples of the BroadABC with GWAS data on a continuous measure of ASB, totaling 50,252 participants: The National Longitudinal Study of Adolescent to Adult Health [60] (ADH), Avon Longitudinal Study of Parents and Children [61,62,63], Brain Imaging Genetics [64], CoLaus|PsyCoLaus [65], Collaborative Study on the Genetics of Alcoholism [66], Finnish Twin Cohort [67] (FinnTwin), The Genetics of Sexuality and Aggression [68], Minnesota Center for Twin and Family Research [69], Phenomics and Genomics Sample [70], eight samples of the QIMR Berghofer Medical Research Institute (QIMR; 16Up project [16UP [71]], Twenty-Five and Up Study [25UP [72]], Genetics of Human Agency [73], Prospective Imaging Study of Ageing [74], Semi-Structured Assessment for the Genetics of Alcoholism SSAGA Phase 2 [SS2 [75]], Genetic Epidemiology of Pathological Gambling [GA [76]], Twin 89 Study [T89 [77]], and Nicotine Study [NC [78]]), Spit for Science [79] (S4S), two samples (from different genotype platforms) of the Twin Early Development Study [80], and the TRacking Adolescents’ Individual Lives Survey [81].
We complemented the above data with GWAS summary statistics on case-control data on DBDs from the recently published Psychiatric Genetics Consortium/iPSYCH consortium meta-analysis, which included data from seven cohorts (Cardiff sample, CHOP cohort, IMAGE-I & IMAGE-II samples, Barcelona sample, Yale-Penn cohort, and the Danish iPSYCH cohort), totaling 3802 cases and 31,305 controls [19].
We observed a high genetic correlation between the 21 meta-analyzed BroadABC samples and the 7 Psychiatric Genetics Consortium/iPSYCH samples, with the “Effective N” as weight (rg = 0.93, p = 9.04 × 10−8), indicating strong overlap of genetic effects. Hence, we continued with the combined 28 samples (N = 85,359) for all analyses.
All included studies were approved by local ethics committees, and informed consent was obtained from all of the participants. All study participants were of European ancestry. Full details on demographics, measurements, sample analysis, and quality control are provided in Supplementary Table 1.
Genome-wide association analysis and quality control of individual cohorts
In all 28 discovery samples, genetic variants were imputed using the reference panel of the HRC or the 1000G Phase 1 version 3 reference panel. The regression analyses were adjusted for age at measurement, sex, and the first ten principal components. To harmonize the imputation, data preparation, and genome-wide association (GWA) analyses, a specific analysis protocol (Supplementary Note 1) was followed in the 18 BroadABC discovery samples. Further details on the genotyping (platform and quality control criteria), imputation, and GWA analyses for each cohort are provided in Supplementary Table 2.
Two semi-independent analysts (JJT and EU) performed stringent within-cohort quality control, filtering out poor performing SNPs. SNPs were excluded if they met any of the following criteria: study-specific minor allele frequency (MAF) corresponding to a minor allele count < 100, poor imputation quality ((INFO/R2) score <0.6), and/or Hardy–Weinberg equilibrium p < 5 × 10−6. Moreover, we excluded SNPs and indels that were ambiguous (A/T or C/G with MAF > 0.4), duplicated, monomorphic, multiallelic, or reference-mismatched (Supplementary Note 2 and Supplementary Table 17). Then, we visually inspected the distribution of the summary statistics by creating quantile–quantile plots and Manhattan plots for the cleaned summary statistics from each cohort (Supplementary Notes 4–6). Discrepancies between the results files of the two semi-independent analysts were examined and errors corrected.
Meta-analyses on combined and sex-specific samples
A meta-analysis of the GWAS results of the 28 discovery samples was performed through fixed-effects meta-analysis in METAL, using SNP p values weighted by effective sample size. We meta-analyzed the BroadABC data (N = 50,252) with case-control data of the Psychiatric Genetics Consortium/iPSYCH consortium meta-analysis (N cases = 3802, N controls = 31,305, N effective = 6323) in METAL, leading to a total effective sample size of 56,575. Since some individuals participating in the eight QIMR studies may be related to some degree, we first meta-analyzed those samples with the sample overlap option in METAL before meta-analyzing those results with the rest of the samples. Although throughout the paper we report the total sample size (N = 85,359), we used the effective sample size (the METAL output) to calculate the PRS, SNP-based heritability (h2SNP) and genetic correlations. After combining all cleaned GWAS data files, meta-analysis results were filtered to exclude any variants with N < 30,000. Consequently, we removed 2,134,049 SNPs, resulting in 7,392,849 SNPs available for analysis. To investigate sex-specific genetic effects, we also ran the meta-analysis in the datasets for which we had sex-specific data (N = 50,252). However, sex-specific SNP heritabilities, as estimated with LD Score Regression, were small and non-significant (3.7% (s.e. = 2.2%) for males and 1.0% (s.e. = 1.8%) for females). Due to the non-significant sex-specific heritability estimates, the genetic correlation of male and female ASB could not be estimated reliably and no sex-specific follow-up analyses were conducted.
Whole-genome sequencing based on genetic differences between the BALB/c strains
Through whole-genome sequencing, we identified single nucleotide variants that distinguish aggressive BALB/cJ mice from control BALB/cByJ strains [82]. Sequencing libraries were prepared from high-quality genomic DNA using the TruSeq DNA PCR-Free kit (Illumina) and ultra-deep whole-genome sequencing (average 30X read-depth across the genome) was performed on a HiSeq X Ten System (Illumina). We developed an efficient data processing and quality control pipeline. Briefly, raw sequencing data underwent stringent quality control and was aligned to either the mm10 (BALB/cJ versus BALB/cByJ strain comparison). Isaac [83] was used to align reads and call single nucleotide variations (SNVs). We excluded SNVs that were covered by less than 20 reads, and that were not present in both animals from the same strain. SnpEff [84] was used to annotate SNVs and explore functional effects on gene function. SNVs differing between the two strains were annotated to a total of 1573 genes, which were subdivided into three different categories (intronic/exonic non-coding and synonymous variants (1422 genes), untranslated regions (90 genes), missense mutations and splicing variants (61 genes)).
Polygenic risk score analyses
PRS were created for ASB using all available SNPs of the discovery dataset [85, 86]. PRS were computed as the weighted sum of the effect-coded alleles per individual. We calculated the PRS for subjects of five independent datasets, selected for their detailed phenotypes related to antisocial outcomes: (1) the Dunedin Study [46], (2) the E-risk study [87], (3) the Philadelphia Neurodevelopmental Cohort [88], (4) the Quebec Longitudinal Study of Child Development [89], and (5) the Quebec Newborn Twin Study [90]. All individuals were of European ancestry. To maintain uniformity across target cohorts, we adhered to the following parameters: Clumping was performed by removing markers in linkage disequilibrium, utilizing the following thresholds: maximum r2 = 0.2, window size = 500 kb. We excluded variants within regions of long-range LD [91] (including the Major Histocompatibility Complex, see Supplementary Table 16 for exact regions). Second generation PLINK [92] was employed to construct PRS for each phenotype, at the following 11 thresholds: p < 1 × 10−6, p < 1 × 10−4, p < 1 × 10−3, p < 1 × 10−2, p < 0.05, p < 0.1, p < 0.2, p < 0.3, p < 0.4, p < 0.5, p < 1.0. There are minor differences in the thresholding approach across the independent cohorts, i.e.,: while constructing the PRS for both ASB phenotypes, the Philadelphia Neurodevelopmental Cohort Study considered all aforementioned thresholds except p < 1 × 10−2, while the other cohorts considered all thresholds except p < 1.0 (see Supplementary Tables 9–14 for the exact thresholds). To correct for multiple testing, we applied a Bonferroni correction on the 22 tested phenotypes (α = 0.00227).
Genetic correlation analysis
To estimate the genetic correlation between ASB and a range of other phenotypes, we employed Linkage Disequilibrium Score Regression (LDSC) [30] through the LD Hub web portal (http://ldsc.broadinstitute.org/ldhub/) [93]. LD Hub, which is a centralized database of summary-level GWAS results, enables the screening of hundreds of traits. To maximize statistical power among the most likely candidate phenotypes, we focused on domains of traits that have been previously reported to be comorbid with ASB. Genetic correlations of ASB were thus calculated by selecting 68 phenotypes of health, physiological, personality, disorder and disease relevant outcomes in LD Hub. In addition, we manually ran genetic correlations analyses for a selection of traits that were not included in the LD Hub framework. Given their relevance for this study, we employed LDSC to examine the genetic correlation of ASB with FFM domains agreeableness, openness to experience and conscientiousness, by using data of the Genetics of Personality Consortium [94]. Similarly, we used data of the most recent GWAS meta-analysis on neuroticism [95] to compute the genetic correlation with ASB. Lastly, while excluding the iPSYCH/PGC samples (given the extensive sample overlap), we computed the genetic correlation of ASB with ADHD [41]. We corrected for multiple testing by applying a Bonferroni correction on the 73 tested genetic correlations (α = 0.0007).
References
Allen JJ, Anderson CA. Aggression and violence: definitions and distinctions. In: The Wiley handbook of violence and aggression, Peter Sturmey (Editor-in-Chief), John Wiley & Sons, New Jersey. American Cancer Society; 2017. p. 1–14.
Rivenbark JG, Odgers CL, Caspi A, Harrington H, Hogan S, Houts RM, et al. The high societal costs of childhood conduct problems: evidence from administrative records up to age 38 in a longitudinal birth cohort. J Child Psychol Psychiatry. 2018;59:703–10.
Brewin CR, Andrews B, Rose S, Kirk M. Acute stress disorder and posttraumatic stress disorder in victims of violent crime. Am J Psychiatry. 1999;156:360–6.
Eme R. Male life-course-persistent antisocial behavior: the most important pediatric mental health problem. Arch Pediatrics Adolesc Med. 2010;164:486–7.
Abram KM, Zwecker NA, Welty LJ, Hershfield JA, Dulcan MK, Teplin LA. Comorbidity and continuity of psychiatric disorders in youth after detention: a prospective longitudinal study. JAMA Psychiatry. 2015;72:84–93.
Loeber R. The stability of antisocial and delinquent child behavior: a review. Child Dev. 1982;53:1431–46.
Jones SE, Miller JD, Lynam DR. Personality, antisocial behavior, and aggression: a meta-analytic review. J Crim Justice. 2011;39:329–37.
Miller JD, Lynam D. Structural models of personality and their relation to antisocial behavior: a meta-analytic review*. Criminology. 2001;39:765–98.
Polderman TJC, Benyamin B, de Leeuw CA, Sullivan PF, van Bochoven A, Visscher PM, et al. Meta-analysis of the heritability of human traits based on fifty years of twin studies. Nat Genet. 2015;47:702–9.
Burt SA. Are there meaningful etiological differences within antisocial behavior? Results of a meta-analysis. Clin Psychol Rev. 2009;29:163–78.
Burt SA, Klump KL. Etiological distinctions between aggressive and non-aggressive antisocial behavior: results from a nuclear twin family model. J Abnorm Child Psychol. 2012;40:1059–71.
Smoller JW, Andreassen OA, Edenberg HJ, Faraone SV, Glatt SJ, Kendler KS. Psychiatric genetics and the structure of psychopathology. Mol Psychiatry. 2019;24:409–20.
Lee PH, Anttila V, Won H, Feng Y-CA, Rosenthal J, Zhu Z, et al. Genomic relationships, novel loci, and pleiotropic mechanisms across eight psychiatric disorders. Cell. 2019;179:1469–82.e11.
Abdellaoui A, Smit DJA, van den Brink W, Denys D, Verweij KJH. Genomic relationships across psychiatric disorders including substance use disorders. Drug Alcohol Depend. 2021;220:108535.
Linnér RK, Mallard TT, Barr PB, Sanchez-Roige S, Madole JW, Driver MN, et al. Multivariate genomic analysis of 1.5 million people identifies genes related to addiction, antisocial behavior, and health. BioRxiv. 2020:2020.10.16.342501.
Tielbeek JJ, Johansson A, Polderman TJC, Rautiainen M-R, Jansen P, Taylor M, et al. Genome-wide association studies of a broad spectrum of antisocial behavior. JAMA Psychiatry. 2017;74:1242–50.
Ip H, Laan C, Brikell I, Sánchez-Mora C, Nolte I, St. Pourcain B, et al. Genetic association study of childhood aggression across raters, instruments and age. 2019.
Pappa I, Pourcain BS, Benke K, Cavadino A, Hakulinen C, Nivard MG, et al. A genome-wide approach to children’s aggressive behavior: the EAGLE consortium. Am J Med Genet Part B Neuropsychiatr Genet. 2016;171:562–72.
Demontis D, Walters RK, Rajagopal VM, Waldman ID, Grove J, Als TD, et al. Risk variants and polygenic architecture of disruptive behavior disorders in the context of attention-deficit/hyperactivity disorder. Nat Commun. 2021;12:576.
Jackson DB, Vaughn MG. Promoting health equity to prevent crime. Prev Med. 2018;113:91–4.
Burt SA, Donnellan MB. Personality correlates of aggressive and non-aggressive antisocial behavior. Pers Individ Differ. 2008;44:53–63.
Piquero AR, Daigle LE, Gibson C, Piquero NL, Tibbetts SG. Research note: Are life-course-persistent offenders at risk for adverse health outcomes? J Res Crime Delinq. 2007;44:185–207.
Tielbeek JJ, Boutwell BB. Exploring the genomic architectures of health, physical traits and antisocial behavioral outcomes: a brief report. Front Psychiatry. 2020;11:539.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
MacDermot KD, Bonora E, Sykes N, Coupe A-M, Lai CSL, Vernes SC, et al. Identification of FOXP2 truncation as a novel cause of developmental speech and language deficits. Am J Hum Genet. 2005;76:1074–80.
Becker M, Devanna P, Fisher SE, Vernes SC. Mapping of human FOXP2 enhancers reveals complex regulation. Front Mol Neurosci. 2018;11:47.
de Leeuw CA, Mooij JM, Heskes T, Posthuma D. MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput Biol. 2015;11:e1004219.
den Hoed J, Fisher SE. Genetic pathways involved in human speech disorders. Curr Opin Genet Dev. 2020;65:103–11.
Watanabe K, Stringer S, Frei O, Umićević Mirkov M, de Leeuw C, Polderman TJC, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet. 2019;51:1339–48.
Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Patterson N, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet. 2015;47:291–5.
Poulton R, Moffitt TE, Silva PA. The Dunedin multidisciplinary health and development study: overview of the first 40 years, with an eye to the future. Soc Psychiatry Psychiatr Epidemiol. 2015;50:679–93.
Odgers CL, Moffitt TE, Broadbent JM, Dickson N, Hancox RJ, Harrington H, et al. Female and male antisocial trajectories: from childhood origins to adult outcomes. Dev Psychopathol. 2008;20:673–716.
den Hoed J, Devaraju K, Fisher SE. Molecular networks of the FOXP2 transcription factor in the brain. EMBO Rep. 2021;22:e52803.
Lai CSL, Fisher SE, Hurst JA, Vargha-Khadem F, Monaco AP. A forkhead-domain gene is mutated in a severe speech and language disorder. Nature. 2001;413:519–23.
Lai CSL, Gerrelli D, Monaco AP, Fisher SE, Copp AJ. FOXP2 expression during brain development coincides with adult sites of pathology in a severe speech and language disorder. Brain. 2003;126:2455–62.
Uddén J, Hultén A, Bendtz K, Mineroff Z, Kucera KS, Vino A, et al. Toward robust functional neuroimaging genetics of cognition. J Neurosci. 2019;39:8778–87.
Deriziotis P, Fisher SE. Speech and language: translating the genome. Trends Genet. 2017;33:642–56.
Warr-Leeper G, Wright NA, Mack A. Language disabilities of antisocial boys in residential treatment. Behav Disord. 1994;19:159–69.
Cohen NJ, Menna R, Vallance DD, Barwick MA, Im N, Horodezky NB. Language, social cognitive processing, and behavioral characteristics of psychiatrically disturbed children with previously identified and unsuspected language impairments. J Child Psychol Psychiatry. 1998;39:853–64.
Brownlie E, Beitchman J, Escobar M, Young A, Atkinson L, Johnson C, et al. Early language impairment and young adult delinquent and aggressive behavior. J Abnorm Child Psychol. 2004;32:453–67.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Johnson EC, Demontis D, Thorgeirsson TE, Walters, RK, Polimanti, R, et al. A large-scale genome-wide association study meta-analysis of cannabis use disorder. Lancet Psychiatry. 2020;7:1032–45.
Karlsson Linnér R, Biroli P, Kong E, Meddens SFW, Wedow R, Fontana MA, et al. Genome-wide association analyses of risk tolerance and risky behaviors in over 1 million individuals identify hundreds of loci and shared genetic influences. Nat Genet. 2019;51:245–57.
Vernes SC, Newbury DF, Abrahams BS, Winchester L, Nicod J, Groszer M, et al. A functional genetic link between distinct developmental language disorders. N Engl J Med. 2008;359:2337–45.
Medvedeva VP, Rieger MA, Vieth B, Mombereau C, Ziegenhain C, Ghosh T, et al. Altered social behavior in mice carrying a cortical Foxp2 deletion. Hum Mol Genet. 2019;28:701–17.
Caspi A, Houts RM, Ambler A, Danese A, Elliott ML, Hariri A, et al. Longitudinal assessment of mental health disorders and comorbidities across 4 decades among participants in the Dunedin Birth Cohort Study. JAMA Netw Open. 2020;3:e203221.
Turley P, Walters RK, Maghzian O, Okbay A, Lee JJ, Fontana MA, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50:229–37.
Maier RM, Zhu Z, Lee SH, Trzaskowski M, Ruderfer DM, Stahl EA, et al. Improving genetic prediction by leveraging genetic correlations among human diseases and traits. Nat Commun. 2018;9:989.
Whose genomics? Nat Hum Behav. 2019;3:409–10.
Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51:584–91.
Moffitt TE. Adolescence-limited and life-course-persistent antisocial behavior: a developmental taxonomy. 1993.
Moffitt TE. Male antisocial behaviour in adolescence and beyond. Nat Hum Behav.2018;2:177–86.
Jennings WG, Reingle JM. On the number and shape of developmental/life-course violence, aggression, and delinquency trajectories: a state-of-the-art review. J Crim Justice. 2012;40:472–89.
Barnes JC, Beaver K, Boutwell B. Examining the genetic underpinnings of Moffitt’s developmental taxonomy: a behavioral genetic analysis. Criminology. 2011;49:923–54.
Motz RT, Barnes JC, Caspi A, Arseneault L, Cullen FT, Houts R, et al. Does contact with the justice system deter or promote future delinquency? Results from a longitudinal study of British adolescent twins. Criminology. 2020;58:307–35.
Carlisi CO, Moffitt TE, Knodt AR, Harrington H, Ireland D, Melzer TR, et al. Associations between life-course-persistent antisocial behaviour and brain structure in a population-representative longitudinal birth cohort. Lancet Psychiatry. 2020;7:245–53.
Wertz J, Caspi A, Belsky DW, Beckley AL, Arseneault L, Barnes JC, et al. Genetics and crime: integrating new genomic discoveries into psychological research about antisocial behavior. Psychol Sci. 2018;29:791–803.
Tielbeek JJ, Barnes JC, Popma A, Polderman TJC, Lee JJ, Perry JRB, et al. Exploring the genetic correlations of antisocial behaviour and life history traits. BJPsych Open. 2018;4:467–70.
Martin J, Taylor MJ, Lichtenstein P. Assessing the evidence for shared genetic risks across psychiatric disorders and traits. Psychol Med. 2018;48:1759–74.
Harris KM, Halpern CT, Whitsel EA, Hussey JM, Killeya-Jones LA, Tabor J, et al. Cohort profile: the national longitudinal study of adolescent to adult health (add health). Int J Epidemiol. 2019;48:1415–1415k.
Boyd A, Golding J, Macleod J, Lawlor DA, Fraser A, Henderson J, et al. Cohort profile: the ‘children of the 90s’-the index offspring of the Avon longitudinal study of parents and children. Int J Epidemiol. 2013;42:111–27.
Northstone K, Lewcock M, Groom A, Boyd A, Macleod J, Timpson N, et al. The Avon longitudinal study of parents and children (ALSPAC): an update on the enrolled sample of index children in 2019. Wellcome Open Res. 2019;4:51.
Fraser A, Macdonald-Wallis C, Tilling K, Boyd A, Golding J, Davey Smith G, et al. Cohort profile: the Avon longitudinal study of parents and children: ALSPAC mothers cohort. Int J Epidemiol. 2013;42:97–110.
Franke B, Vasquez AA, Veltman JA, Brunner HG, Rijpkema M, Fernández G. Genetic variation in CACNA1C, a gene associated with bipolar disorder, influences brainstem rather than gray matter volume in healthy individuals. Biol Psychiatry. 2010;68:586–8.
Preisig M, Waeber G, Vollenweider P, Bovet P, Rothen S, Vandeleur C, et al. The PsyCoLaus study: methodology and characteristics of the sample of a population-based survey on psychiatric disorders and their association with genetic and cardiovascular risk factors. BMC Psychiatry. 2009;9:9.
The collaborative study on the genetics of alcoholism. Alcohol Health Res World. 1995;19:228–36.
Kaprio J. The Finnish Twin cohort study: an update. Twin Res Hum Genet. 2013;16:157–62.
Johansson A, Jern P, Santtila P, von der Pahlen B, Eriksson E, Westberg L, et al. The genetics of sexuality and aggression (GSA) twin samples in Finland. Twin Res Hum Genet. 2013;16:150–6.
Miller MB, Basu S, Cunningham J, Eskin E, Malone SM, Oetting WS, et al. The Minnesota center for twin and family research genome-wide association study. Twin Res Hum Genet. 2012;15:767–74.
Rujescu D, Hartmann AM, Giegling I, Konte B, Herrling M, Himmelein S, et al. Genome-wide association study in vestibular neuritis: involvement of the host factor for HSV-1 replication. Front Neurol. 2018;9:591.
Mitchell BL, Kirk KM, McAloney K, Wright MJ, Davenport TA, Hermens DF, et al. 16Up: outline of a study investigating wellbeing and information and communication technology use in adolescent twins. Twin Res Hum Genet. 2020;23:345–57.
Mitchell BL, Campos AI, Rentería ME, Parker R, Sullivan L, McAloney K, et al. Twenty-Five and Up (25Up) study: a new wave of the Brisbane longitudinal twin study. Twin Res Hum Genet. 2019;22:154–63.
Morosoli JJ, Colodro-Conde L, Barlow FK, Medland SE. Investigating perceived heritability of mental health disorders and attitudes toward genetic testing in the United States, United Kingdom, and Australia. Am J Med Genet B Neuropsychiatr Genet. 2021. https://doi.org/10.1002/ajmg.b.32875.
Lupton MK, Robinson GA, Adam RJ, Rose S, Byrne GJ, Salvado O, et al. A prospective cohort study of prodromal Alzheimer’s disease: prospective imaging study of ageing: genes, brain and behaviour (PISA). Neuroimage Clin. 2021;29:102527.
Madden PA, Bucholz KK, Dinwiddie SH, Slutske WS, Bierut LJ, Statham DJ, et al. Nicotine withdrawal in women. Addiction. 1997;92:889–902.
Slutske WS, Meier MH, Zhu G, Statham DJ, Blaszczynski A, Martin NG. The Australian Twin Study of Gambling (OZ-GAM): rationale, sample description, predictors of participation, and a first look at sources of individual differences in gambling involvement. Twin Res Hum Genet. 2009;12:63–78.
Knopik VS, Heath AC, Madden PAF, Bucholz KK, Slutske WS, Nelson EC, et al. Genetic effects on alcohol dependence risk: re-evaluating the importance of psychiatric and other heritable risk factors. Psychol Med. 2004;34:1519–30.
Saccone SF, Pergadia ML, Loukola A, Broms U, Montgomery GW, Wang JC, et al. Genetic linkage to chromosome 22q12 for a heavy-smoking quantitative trait in two independent samples. Am J Hum Genet. 2007;80:856–66.
Dick DM, Nasim A, Edwards AC, Salvatore JE, Cho SB, Adkins A, et al. Spit for Science: launching a longitudinal study of genetic and environmental influences on substance use and emotional health at a large US university. Front Genet. 2014;5:47.
McAra L, McVie S. Youth crime and justice: key messages from the Edinburgh study of youth transitions and crime. Criminol Crim Justice. 2010;10:179–209.
Oldehinkel AJ, Rosmalen JG, Buitelaar JK, Hoek HW, Ormel J, Raven D, et al. Cohort profile update: the TRacking Adolescents’ Individual Lives Survey (TRAILS). Int J Epidemiol. 2015;44:76–76n.
Jager A, Amiri H, Bielczyk N, van Heukelum S, Heerschap A, Aschrafi A, et al. Cortical control of aggression: GABA signalling in the anterior cingulate cortex. Eur Neuropsychopharmacol. 2020;30:5–16.
Raczy C, Petrovski R, Saunders CT, Chorny I, Kruglyak S, Margulies EH, et al. Isaac: ultra-fast whole-genome secondary analysis on Illumina sequencing platforms. Bioinformatics. 2013;29:2041–3.
Cingolani P, Platts A, Wang LL, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92.
Choi SW, Mak TS-H, O’Reilly PF. Tutorial: a guide to performing polygenic risk score analyses. Nat Protoc. 2020;15:2759–72.
Wray NR, Lin T, Austin J, McGrath JJ, Hickie IB, Murray GK, et al. From basic science to clinical application of polygenic risk scores: a primer. JAMA Psychiatry. 2020. https://doi.org/10.1001/jamapsychiatry.2020.3049.
Wertz J, Agnew-Blais J, Caspi A, Danese A, Fisher HL, Goldman-Mellor S, et al. From childhood conduct problems to poor functioning at age 18 years: examining explanations in a longitudinal cohort study. J Am Acad Child Adolesc Psychiatry. 2018;57:54–60.e4.
Satterthwaite TD, Connolly JJ, Ruparel K, Calkins ME, Jackson C, Elliott MA, et al. The Philadelphia neurodevelopmental cohort: a publicly available resource for the study of normal and abnormal brain development in youth. Neuroimage. 2016;124:1115–9.
Orri M, Boivin M, Chen C, Ahun MN, Geoffroy M-C, Ouellet-Morin I, et al. Cohort profile: Quebec longitudinal study of child development (QLSCD). Soc Psychiatry Psychiatr Epidemiol. 2021;56:883–94.
Boivin M, Brendgen M, Dionne G, Ouellet-Morin I, Dubois L, Pérusse D, et al. The Quebec Newborn Twin Study at 21. Twin Res Hum Genet. 2019;22:475–81.
Price AL, Weale ME, Patterson N, Myers SR, Need AC, Shianna KV, et al. Long-range LD can confound genome scans in admixed populations. Am J Hum Genet. 2008;83:132–5.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Zheng J, Erzurumluoglu AM, Elsworth BL, Kemp JP, Howe L, Haycock PC, et al. LD Hub: a centralized database and web interface to perform LD score regression that maximizes the potential of summary level GWAS data for SNP heritability and genetic correlation analysis. Bioinformatics. 2017;33:272–9.
de Moor MHM, Costa PT, Terracciano A, Krueger RF, de Geus EJC, Toshiko T, et al. Meta-analysis of genome-wide association studies for personality. Mol Psychiatry. 2012;17:337–49.
Nagel M, Jansen PR, Stringer S, Watanabe K, de Leeuw CA, Bryois J, et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat Genet. 2018;50:920–7.
Acknowledgements
GM-V was supported by a Doctoral Research Scholarship from the FRQSC. MB, IO-M, and J-PG are supported by the Canada Research Chair Program. KR is supported by a Sir Henry Wellcome Postdoctoral Fellowship. ADB and DD were supported by grants from the Lundbeck Foundation (R102-A9118, R155-2014-1724 and R248-2017-2003), the EU FP7 Program (Grant No. 602805, “Aggressotype”) and H2020 Program (Grant No. 667302, “CoCA”), NIMH (1U01MH109514-01. JL, AM, and KR acknowledge the following funders: Academy of Finland, the Signe and Ane Gyllenberg foundation, Juho Vainio foundation, Yrjö Jahnsson foundation, Jalmari and Rauha Ahokas foundation, Sigrid Juselius Foundation and The Finnish Society of Sciences and Letters. DD is supported by NIH R01 AA015416 (Finnish Twin Study), P50 AA022537 (Alcohol Research Center), R25 AA027402 (VCU GREAT), R34 AA027347 (Personalized Risk Assessment), R01 AA028064 (Parental Marital Discord, PI: JS), and U10 AA008401 (COGA) from the National Institute on Alcohol Abuse and Alcoholism (NIAAA), and by R01 DA050721 (Externalizing Consortium) from the National Institute on Drug Abuse (NIDA). BF, MK, and NRM were also supported by funding from the European Community’s Horizon 2020 Programme (H2020/2014–2020) under grant agreements no. 728018 (Eat2beNICE) and no. 847879 (PRIME). They also received relevant funding from the Netherlands Organization for Scientific Research (NWO) for the Dutch National Science Agenda NeurolabNL project (grant 400-17-602). HMS and MRM are members of the Medical Research Council (MRC) Integrative Epidemiology Unit at the University of Bristol (MC_UU_00011/7). This work is also supported by the NIHR Biomedical Research Centre at University Hospitals Bristol NHS Foundation Trust and the University of Bristol. AW has been supported by funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 602768 for the ACTION consortium. JK has been supported by the Academy of Finland Academy professorship program (grants 265240 and 263278). LC-C was supported by a QIMR Berghofer Research Fellowship. JJM was supported by a QIMR Berghofer International PhD Scholarship. ML was funded by an NHMRC Boosting Dementia Leadership Fellowship (APP1140441). SEM and NGM were funded by NHMRC investigator grants (APP1172917 and APP1172990). PJ has been supported by Academy of Finland (Grants no. 319403, 284385 and 274521) and the Stiftelsen för Åbo Akademi Foundation. MK was supported by a personal Rubicon grant (no. 45219212) from the Dutch Research Council. Additional acknowledgements for each study cohort are described in Supplementary Table 1. We acknowledge the work of all members of the Spit for Science Working Group. Spit for Science Director: Danielle M. Dick. Registry management: Kimberly Pedersen, Zoe Neale, Nathaniel Thomas. Data cleaning and management: Amy E. Adkins, Nathaniel Thomas, Zoe Neale, Kimberly Pedersen, Thomas Bannard and Seung B. Cho. Data collection: Amy E. Adkins, Peter Barr, Holly Byers, Erin C. Berenz, Erin Caraway, Seung B. Cho, James S. Clifford, Megan Cooke, Elizabeth Do, Alexis C. Edwards, Neeru Goyal, Laura M. Hack, Lisa J. Halberstadt, Sage Hawn, Sally Kuo, Emily Lasko, Jennifer Lend, Mackenzie Lind, Elizabeth Long, Alexandra Martelli, Jacquelyn L. Meyers, Kerry Mitchell, Ashlee Moore, Arden Moscati, Aashir Nasim, Zoe Neale, Jill Opalesky, Cassie Overstreet, A. Christian Pais, Kimberly Pedersen, Tarah Raldiris, Jessica Salvatore, Jeanne Savage, Rebecca Smith, David Sosnowski, Jinni Su, Nathaniel Thomas, Chloe Walker, Marcie Walsh, Teresa Willoughby, Madison Woodroof and Jia Yan. Genotypical data processing and cleaning: Cuie Sun, Brandon Wormley, Brien Riley, Fazil Aliev, Roseann Peterson and Bradley T. Webb.
Author information
Authors and Affiliations
Consortia
Contributions
Study concept and design: JJT, TJCP, and DP. Acquisition, analysis, or interpretation of data: EU, BSW, LC-C, EG, TTM, BEL, PRJ, AJ, HMS, GP, GRBS, AGA, KR, BK, MK, AMH, JES, IMN, DD, ALKM, SAB, JES, KS, RP, KMH, SV, MM, WGI, NRM, JM, JFV, BLM, JJM, TFMA, IO-M, RET, SMC, JPG, MRB, GD, FV, and MKL. Drafting of the manuscript: JJT, TJCP, PRJ, BBB, TEM, AC, and DP. Study supervision: TJCP, NGM, EC, KR, JGE, JL, CAH, AJO, HS, HL, MP, AW, EV, PJ, YL, DR, IG, TP, JK, KPH, MRM, GM-V, RP, EV, BBB, FA, DD, AP, SVF, ADB, SEM, BF, MB, JBP, JCG, JCB, SEF, TEM, AC, and DP. All authors critically evaluated and revised the manuscript. JJT had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Corresponding author
Ethics declarations
Competing interests
BF has received educational speaking fees from Medice.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tielbeek, J.J., Uffelmann, E., Williams, B.S. et al. Uncovering the genetic architecture of broad antisocial behavior through a genome-wide association study meta-analysis. Mol Psychiatry 27, 4453–4463 (2022). https://doi.org/10.1038/s41380-022-01793-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01793-3
- Springer Nature Limited
This article is cited by
-
Examining intergenerational risk factors for conduct problems using polygenic scores in the Norwegian Mother, Father and Child Cohort Study
Molecular Psychiatry (2024)
-
Update on Antisocial Personality Disorder
Current Psychiatry Reports (2024)
-
Hypothalamic subunit volumes and relations to violence and psychopathy in male offenders with or without a psychotic disorder
European Archives of Psychiatry and Clinical Neuroscience (2024)
-
Dissecting the polygenic contribution of attention-deficit/hyperactivity disorder and autism spectrum disorder on school performance by their relationship with educational attainment
Molecular Psychiatry (2024)
-
Resting heart rate and antisocial behaviour: a Mendelian randomisation study
Scientific Reports (2023)