
Abstract
Microvascular pathology and ischemic lesions contribute substantially to neuronal dysfunction and loss that lead to Alzheimer disease (AD). To facilitate recovery, the brain stimulates neovascularization of damaged tissue via sprouting angiogenesis, a process regulated by endothelial cell (EC) sprouting and the EphB4/ephrinB2 system. Here, we show that in cultures of brain ECs, EphB4 stimulates the VE-cadherin/Rok-α angiogenic complexes known to mediate sprouting angiogenesis. Importantly, brain EC cultures expressing PS1 FAD mutants decrease the EphB4-stimulated γ-secretase cleavage of ephrinB2 and reduce production of the angiogenic peptide ephrinB2/CTF2, the VE-cadherin angiogenic complexes and EC sprouting and tube formation. These data suggest that FAD mutants may attenuate ischemia-induced brain angiogenesis. Supporting this hypothesis, ischemia-induced VE-cadherin angiogenic complexes, levels of neoangiogenesis marker Endoglin, vascular density, and cerebral blood flow recovery, are all decreased in brains of mouse models expressing PS1 FAD mutants. Ischemia-induced brain neuronal death and cognitive deficits also increase in these mice. Furthermore, a small peptide comprising the C-terminal sequence of peptide ephrinB2/CTF2 rescues angiogenic functions of brain ECs expressing PS1 FAD mutants. Together, our data show that PS1 FAD mutations impede the EphB4/ephrinB2-mediated angiogenic functions of ECs and impair brain neovascularization, neuronal survival and cognitive recovery following ischemia. Furthermore, our data reveal a novel brain angiogenic mechanism targeted by PS1 FAD mutants and a potential therapeutic target for ischemia-induced neurodegeneration. Importantly, FAD mutant effects occur in absence of neuropathological hallmarks of AD, supporting that such hallmarks may form downstream of mutant effects on neoangiogenesis and neuronal survival.
Similar content being viewed by others

Introduction
A large amount of evidence links brain vascular dysfunction to the onset of Alzheimer’s disease (AD). A strong association between cognitive decline and cerebrovascular abnormalities is supported by data that AD brains exhibit vascular pathology [1, 2] with changes in the microvasculature preceding neurodegenerative changes and cognitive decline [3,4,5]. Vascular damage of the brain induces ischemia, which causes further tissue damage (infarction) and it has been suggested that clinical expression of AD is partly determined by the presence of comorbid conditions such as brain infarcts associated with ischemia [6,7,8,9]. One of the protective mechanisms of the brain, which acts as a reparative function against ischemic insults is the neovascularization of affected tissue, a mechanism regulated by endothelial cells (ECs) [10]. Dysfunction of this EC-based process can lead to decreased neovascularization and tissue repair activity following toxic insults thus compromising the integrity and plasticity of brain vasculature and, ultimately, the neuronal health and function.
Angiogenesis, a process that modulates the structure and function of the vascular system, is controlled by several growth factors including FGF, VEGF, angiopoietin, and the ephrinB2 (efnB2)/EphB4 bidirectional ligand/receptor system [11, 12]. EphB4 receptors bind specifically to efnB2 ligands at the surface of ECs and stimulate cell sprouting, a function critical to insult-induced angiogenesis [13,14,15]. Sprouting of ECs is regulated by the recruitment of Raf-1 and Rok-α (Rho effector) kinases to vascular endothelial (VE)-cadherin-containing adherens junctions (AJ) [16]. The EphB4/efnB2 system regulates ischemia-induced brain angiogenesis and its angiogenic functions are regulated by Presenilin1 (PS1)/γ-secretase in vitro [15, 17, 18]. Specifically, binding of EphB4 to efnB2 stimulates the PS1/γ-secretase cleavage of efnB2 producing peptide efnB2/CTF2 that stimulates formation of Raf-1/Rok-a/VE-cadherin angiogenic complexes and sprouting of microvascular ECs [15, 19]. PS1 FAD mutants are responsible for most cases of familial AD (FAD) and have been shown to reduce the ε-cleavage of many type I tramnsmembrane proteins including efnB2 [15, 20,21,22,23]. Here, we asked whether such mutants affect the EphB4-regulated angiogenic functions of ECs and ischemia-induced brain neovascularization and repair. We found that EphB4-induced angiogenic functions of brain ECs such as sprouting, tube formation, and angiogenic complexes are decreased by PS1 FAD mutants. Furthermore, such mutants decrease the EphB4-induced γ-secretase cleavage of efnB2 and production of the angiogenic peptide efnB2/CTF2. We also show that PS1 FAD mutants decrease the ischemia-induced brain angiogenic complexes, neovascularization, and cerebral blood flow (CBF) restoration while increasing neuronal death and cognitive decline of FAD mouse models. Together, our data indicate that PS1 FAD mutants render the brain vulnerable to toxic insults by compromising brain vasculature repair mechanisms and suggest a novel theory by which FAD mutants may promote neurodegeneration.
Materials and methods
Time line of in vivo experiments
Animals
The PS1 M146V knock in (KI) [24], PS1 I213T KI [25], and PS1 knockout (KO) [26] mice have been described. We used 3–4-month-old male mice (C57B/6). All animal experiments were performed in accordance with the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee and with national regulations and policies. For the control group, age matched WT mice were used for each experiment. Investigators were not blinded to the mouse genotype and/or group allocation with the exception of the experimenters that performed the stereologic analysis and the measurement of NeuN-positive neurons.
Materials and antibodies
Anti-Collagen IV antibody (AB756P) was from EMD Millipore (Billerica, MA) and antibodies against GFAP (ab4674), NeuN (ab134014), and Rok-α (71598) were from Abcam (Cambridge, MA). Anti-VE cadherin (H-72, sc28644), Rok-α (D2, sc365275), and ephrinB2 (C18, sc910) antibodies were from Santa Cruz (Dallas, TX) and antibodies against Rok-α (PLA0013) and Von Willebrand Factor (VWF, AB7356) from Sigma. Anti-GAPDH (2118S) and anti-myc (9B11, 2276) antibodies were from Cell Signaling Technologies (Beverly, MA). Recombinant mouse EphB4-Fc and human Fc IgG and antibody against endoglin (CD-105; AF1320-SP) were from R & D Systems (Minneapolis, MN). Anti-Fc (rabbit anti-human IgG, Fcγ fragment specific, 309-005-008) was from Jackson immune research. 1,1′-Dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine-acetylated low density lipoprotein from human plasma (Dil-Ac-LDL) was purchased from Alfa Aesar Co. γ-Secretase inhibitors IX (DAPT) and X (L685,458) were from Calbiochem (Billerica, MA) and RO4929097 was from Selleck Chemicals.
Middle cerebral artery occlusion (MCAO)
Experiments were carried out in adult male mice weighing 18–33 g, no formal randomization was performed but all littermates of different genotypes were subjected to MCAO the same day to have a better distribution of covariates among the different genotypes. Focal cerebral ischemia was induced using an occluding silicon-coated intraluminal suture as described [27,28,29,30,31]. Successful application of MCAO was determined by Laser Doppler flowmetry. Only mice in which CBF was dropped to >70% of the baseline (before MCAO) after the occlusion were used for experiments. An additional criterion was that the infarcted area volume should be 25–45% of the ipsilateral hemisphere volume as determined by Diffusion-weighted Imaging (DWI). Lesion size was confirmed with TTC (2,3,5-triphenyltetrazolium chloride) staining. Isolated brains are sectioned coronally into five 1 mm slices using a mouse brain matrix (Brain Tree Science). Slices are immediately stained with TTC (Sigma) for 30 min and fixed with 4% PFA to detect infarct area. At least three biological replicates (from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Magnetic resonance imaging (MRI)
After inducing MCAO, brains were imaged using perfusion MRI (pMRI) to measure CBF at 1, 5, and 15 days post MCAO and T2 and DTI (Diffusion Tensor Imaging) to detect brain lesion, edema and anatomical changes. Imaging was performed at Mount Sinai imaging facility using a 7-Tesla, 200 mm diameter horizontal bore magnet interfaced to an SMIS console.
Before imaging, each mouse was anesthetized with intraperitoneal injection of a mixture of Ketamine (120 mg/kg) and xylazine (20 mg/kg), and a 1.0 mm diameter intravenous catheter was inserted into the tail vein for the contrast agent (0.1 mL (0.2 mL/kg) of Gd-DTPA) (Magnevist)). The tail vein was chosen as an easily accessible large vein for catheterization, and to ensure a well-defined cerebral contrast bolus.
During scanning, animals were anesthetized and maintained at 15% isofluorane in NO2 (75%) and O2 (23%) throughout the scan time.
After a localizer, we manually shimmed the brain region for optimal signal and signal distortion. First a dual echo T2-weighted scan was obtained to verify the ischemic lesion using a dual echo spin echo with TR = 2000ms, TE = 13/65 ms, FOV = 20 mm, matrix slice = 256 × 256, 16 slices, slice thickness 0.5 mm. These data were used for co-registration and identification of the location of the perfusion scans.
Baseline scans were acquired for 20 s after which the full dose of 0.15 mL bolus was injected. Scanning continued for another 3 min. Perfusion MRI protocol is as follows: T2* weighted Fast Low-Angle Shot sequence with the following parameters: TR = 1000 ms, TE = 9 ms, matrix = 64 × 64, FOV = 20 mm, slice thickness = 0.5 mm, 16 coronal slices. In addition, diffusion-weighted imaging was obtained using a Pulse Gradient Spin Echo Sequence with the following parameters: TR = 4000 ms, TE = 22.7 ms, matrix size = 128 × 128, FOV = 16 mm, slice thickness 0.6 mm. Images were acquired every second for 2 min, with injection of the contrast bolus beginning. After the pMRI data were acquired a final set of post contrast T1-weighted images were collected to complete the MRI data acquisition. The total acquisition time for each mouse, including set up, was 1–1.5 h.
MRI analysis
For pMRI analysis, from the dynamic pMRI data set, relative cerebral blood volume (rCBV) values were calculated in regions of interest (ROIs) within lesions using standard algorithms based on intravascular injection dilution theory, with baseline subtraction to correct for contrast leakage [32]. CBV was proportional to the area under the corrected concentration-time curve, and was expressed relative to CBV measured in contralateral side of brains. At least three biological replicates (from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Culture of primary mouse cortical endothelial cells (pCEC)
pCECs from WT, I213T and M146V heterozygous and homozygous KI mouse brains at embryonic day 15.5 (E15.5) were isolated as previously described [33], and genotyped for M146V and I213T mutations [34]. Dissociated brain cells were plated onto collagen I coated plates at a density of ~2 × 104 cells per well in a 12-well plate. Cells were maintained in endothelial cell medium; MCDB-131 complete medium (VEC Technologies, NY) supplemented with ECGS (354006, Corning, NY) to a final concentration of 40 μg/mL and EGF to a final concentration of 0.01 μg/mL (Corning). Purity of the cultures was assessed with flow cytometry (FACS). ECs were incubated with fluorescent Dil-Ac-LDL (Alfa Aesar Co), trypsinized and analyzed by flow cytometry. We saw that 99.55% (SEM = 0.18%, n = 8) of the cells were Dil-Ac-LDL positive. To verify that cells internalizing Dil-Ac-LDL are endothelial, we performed ICC using anti-VWF and anti-endoglin (anti-CD105) antibodies (Supplementary Fig. S1). The above showed that our cultures contain more than 99% ECs. All cell cultures were tested for mycoplasma contamination.
Microcarrier bead sprouting assay
In vitro sprouting and quantitation were performed as described [15] with some modifications. Cells grown in ECGM were loaded on Cytodex 3 microcarrier (MC) beads (C3275, Sigma, MO) at 30 cells per bead. Beads had been recoated with 10 μg/mL Rat Collagen I (354236, Corning, NY) solution. pCECs were stimulated with 4 μg/mL Fc or EphB4-Fc. The number of capillary sprouts exceeding in length the diameter of the MC bead (~175 μm) and containing at least 3 nuclei (as visualized by Hoechst 33258 staining) was determined for 20 MC beads. Cells were photographed on an Olympus IX70 microscope with Retiga Exi camera. At least three biological replicates (cell cultures from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Tube formation assay
Formation of capillary-like structures was assessed in a 48-well plate using growth factor-reduced Matrigel (BD Biosciences, NJ) as described [35]. pCECs were grown in ECGM, labeled with 0.3 μM Calcein AM for 30 min at 37 °C, trypsinized and plated (24,000 cells/well) on top of Matrigel (200 μL/well) in tube formation medium (3:1 RPMI:MCDB-131). Cells were stimulated with 2 μg/mL of clustered Fc or EphB4-Fc. The highest number of tubes was detected at 3.5–4.5 h following EphB4-Fc stimulation, the 3.5-h time point was used for quantifications. Microphotographs of Calcein AM (for increased contrast: excitation: 485 nm, emission: 520 nm) overlapped with phase contrast (for better visualization of thin segments, and to identify the middle of the well) were obtained with an Olympus IX70 microscope with Retiga Exi camera. Five pictures per well captured most of the well surface and contained all formed tubes (48-well plate), the pictures were “stitched” together by Microsoft Image Composite Editor (Microsoft Research Computational Photography) and the total number of tubes were counted with ImageJ. At least three biological replicates (cell cultures from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Protein isolation, immunoprecipitation, and Western blotting
pCECs from embryo brains were rinsed twice with PBS and lysed in Triton-X buffer (10 mM Hepes, pH 7.4, 150 mM NaCl, 2 mM CaCl2, 0.02% (w/v) NaN3, 1% (v/v) Triton X-100, complete protease inhibitor cocktail (Roche Applied Science)). Immunoprecipitations (IPs) were carried out using specific antibodies and antigens were detected on Western Blot as described [19]. Semi-quantification of protein detection was done by image analysis with ImageJ.
Solid phase proximity ligation assay (spPLA)
We applied spPLA to detect protein complexes in lysates of mouse brains with lysis buffer (25 mM Tris-HCl (pH 7.4) 137 mM NaCl, 3 mM KCl, 0.5% IGEPAL CA-630, 1 mM sodium orthovanadate, 20 mM sodium fluoride, 25 mM sodium phyrophosphate, 50% glycerol, and proteinase inhibitor cocktail (Roche). Protein concentration in the lysate was adjusted to 1 mg/mL. Anti-VE cadherin rabbit polyclonal antibody (Santa Cruz Biotechnologies, sc28644) was biotinylated with ChromaLink One-shot antibody Biotinylation kit (solulink). Antibodies were labeled with DBCO-PEG4-NHS ester using click chemistry reaction (uptima); anti-VE cadherin was labeled with oligo DNA probe A or probe B; anti-Rock2 (Abcam, ab71598) was labeled with oligo DNA probe B according to the manufacturer’s protocol. After incubating biotinylated VE-cadherin antibody with microparticles, lysates were incubated with these microparticles. Probe A and probe B-labeled antibodies were diluted to spPLA buffer (1 mM d-biotin, 1 mg/mL BSA, 0.05% Tween-20, 100 nM goat IgG, 0.1 mg/mL Salmon sperm DNA and 5 mM EDTA in PBS). After incubation with lysates, the microparticles were incubated with diluted probe A and probB-labeled antibodies. After incubation, the region annealed by probe A and probe B was extended by DNA polymerase and these products were quantified by real time PCR. Negative controls omit one of the probe-labeled antibodies or include lysis buffer without the mouse brain extract.
At least three biological replicates (brains from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Transduction with viral constructs and transient transfection of cells
Transduction of pCECs with pMX viral vector was done as described previously [36]. Cells were transduced with murine full length efnB2 triple tagged with myc for detection purposes or with efnB2/CTF2 also tagged with triple myc [36]. HEK293 cells (ATCC) overexpressing efnB2-myc3 were grown in Dulbecco’s modified Eagle’s medium (DMEM) plus 10% fetal bovine serum (FBS). Cells were transiently transfected with 1 μg of cDNA using the Lipofectamine 3000 Reagent (Invitrogen) according to manufacturer’s protocol. The following constructs were used: FCbAIGW vector (Addgene), WT-PS1, PS1 M146V, and PS1 I213T (Genscript) in FCbAIGW vector. Cell cultures were tested for mycoplasma contamination. At least three biological replicates (from different cell cultures). Refer to the figure legend for n corresponding to number of biological replicates.
Tissue processing
Adult mice were anesthetized with 150 mg/kg ketamine and 30 mg/kg xylazine 1 and 30 days post MCAO and were sacrificed by transcardial perfusion with cold 4% paraformaldehyde in PBS. After perfusion, brains were removed and post-fixed in 4% paraformaldehyde for 48 h at 4 °C, transferred to 30% sucrose in PBS, and stored at 4 °C until sectioning. 50 μm-thick coronal sections were cut using a Leica VT1000 S Vibratome (Leica, Wetzlar, Germany) and stored in 0.02% sodium azide in PBS until used.
Immunohistochemisty
Immunohistochemical staining was performed on free-floating sections. Immunofluorescence staining for Collagen IV was performed on pepsin-digested tissue as described [37]. The sections were washed with dH2O for 10 min and treated with 1 mg/mL pepsin (Dako, CA) in 3% acetic acid for 15 min at 37 °C. After washing in dH2O three times, sections were blocked with PBS/0.1% tritonX-100/5% goat serum (PBS-TGS) for 2 h and primary antibodies against Collagen IV and GFAP were applied overnight in PBS-TGS at room temperature. Following washing in PBS for 1 h, staining was detected by incubation with species-specific AlexaFlour secondary antibody conjugates (1:1000; Invitrogen, CA) for 3 h in PBS-TFS. Sections were mounted on slides using Vectashield antifade mounting medium including DAPI (Vector Laboratories, CA).
Stereologic analysis
To measure the total length of vessels a stereologic analysis was performed as described previously [37, 38]. Briefly, every 10th section was chosen, beginning with a random section between 1 and 6 and immunostained for collagen IV and GFAP as described above. To reduce bias, all analyses were performed blind to genotype. GFAP was used to define ROI using a Zeiss Axioplan 2 microscope and StereoInvestigator software (v11.11.3; MBF Bioscience, Williston, VT). The ROI in our experiments covered areas of the striatum and occasionally the cortex. Area of GFAP immunostaining determined the ROI boundaries and was outlined with free hand tool of stereoinvestigator. Vascular area and length was then examined in ROI on collagen IV immunostaining using the software’s Spaceballs probe 21–24 and a 40× oil Zeiss Plan NEOFLUAR objective with a 1.3 NA. The dimensions of the random sampling grid used were set at x = 310 μm, y = 300 μm, and a hemisphere with a radius of 15 μm was used. A 0.5-μm guard zone was set at the top and bottom of the tissue to avoid counting in the compromised cut surface. Sampling grids were systematically and randomly placed by the stereology software throughout the ROI. At least three biological replicates (from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Measurement of NeuN-positive neurons
Neurons were stained with anti-NeuN and lesion area was identified with anti-GFAP staining. The number of NeuN-immunolabelled neurons in lesion area and contralateral area were counted in every 10th section using the Stereoinvestigator software. Images were obtained with a Zeiss Axioplan 2 microscope. Under low magnification, the boundary of lesion area was identified with GFAP staining and the boundary contour in lesion area was drawn using the software-pointing device. The same drawn area was used in the contralateral lateral area of the section. A systematic-random generated sampling grid was placed over the contour area, containing an average number of 162 20 µm × 20 µm square counting frames per animal (200 × 200 µm with a 2 μm guard zone and a dissector height of 25 µm). Only NeuN stained neurons within the counting frame and no contact with exclusion lines were counted using a ×40 objective lens. The total number of neurons per lesion size was estimated using the optical fractionator. At least three biological replicates (from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Behavioral tests
The behavioral tests include two locomotion and two memory tests, performed in the following order before and after MCAO: Open field test, rotarod test, Y maze spontaneous alternation and novel object recognition (NOR). We first ensured that all MCAO surgery underwent in a similar fashion among all experimental groups with ischemic deficits, neurological deficits and infarct volumes. One type of behavior test was tested per day and each mouse was tested once in each test. All tested animals were video-recorded with a digital camera (Logitec), tracked and analyzed with Ethovision XT tracking system software (Noldus information technology Inc). Habituation was done in the testing room for 30 min at the beginning of each test day. General motivation to explore and motor function were measured in an open field before assessing the functional cognition test (NOR and Y maze). For open field test, time of movement, number of entry into the center, and total distance of movement were measured [39]. After the functional cognition test, rotarod test was used as motor skill learning test. NOR test was done as described [40, 41]. For Y maze test mice were placed in one of arm of a Y shaped chamber and mouse entries in each arm of a Y shaped chamber were measured during 10 min. The percentage of alternations was calculated. For neurological scoring, all items on the global neurological scale are counted 24 h following MCAO with a point system adapted from a previous study as follows: 1 point for torso flexion to the right in the air; 2 points for deficit in gripping with a paw; 3 points for circling with the front paws when suspended from the tail; 4 points for spontaneous circling on the floor; 5 points for irresponsiveness to stimuli. The sum of all points 32 was used as the score of motor deficit. At least three biological replicates (from different animals). Refer to the figure legend for n corresponding to number of biological replicates.
Statistical analysis
Sample sizes to ensure adequate power (80%) were estimated with G*Power [42]. Data are expressed as mean values ± standard error of means (SEM) except if otherwise indicated in the figure legend. Statistical significance testing was performed with GraphPad Software. For group comparisons (>2) one-way analysis of variance (ANOVA) was used. Where decisions for statistical significance need to take into account multiple pairwise comparisons among groups, we use single-step multiple comparison methods when available (e.g., post hoc pairwise Tukey’s test), otherwise, we adjust the p values for multiple comparisons (e.g., Bonferroni adjustment of post hoc pairwise logrank tests). To test whether there was a proportional increase of CBF with days post-MCAO, we performed linear regression and looked for significance with a positive slope. To test for difference in the kinetics of edema resolution we performed survival analysis with a logrank test (Mantel–Cox method). To test treatment response when the data did not originate from normally distributed values, we used the Wilcoxon Signed Rank Test against theoretical median of 100% (no response). All statistical tests are two-sided unless otherwise specified in the figure legend. p < 0.05 was considered significant (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Results
PS1 FAD mutants inhibit EphB4-stimulated angiogenic functions and complexes of primary cortical endothelial cell (pCEC) cultures
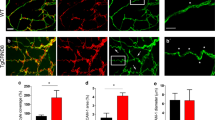
We asked whether EphB4 stimulates angiogenic functions of brain pCECs and whether these functions are affected by PS1 FAD mutants. Figure 1a–d shows that EphB4, known to bind cell surface efnB2, stimulates sprouting and tube formation of pCEC cultures prepared from WT mouse brains but not of pCEC cultures from mouse brains expressing PS1 FAD mutant M146V or I213T. It is known that angiogenic functions such as sprouting of lung and adrenal microvascular ECs are mediated by factor-stimulated molecular complexes between VE-cadherin and Rok-α kinase [16, 19] and we obtained data that EphB4 increases the VE-cadherin/Rok-α complex of pCEC cultures prepared from WT brains (Fig. 1e, lanes 1–2) but not of pCEC cultures prepared form mouse brains expressing PS1 FAD mutant M146V or I213T (Fig. 1e, f). Together our data show that EphB4 stimulates angiogenic functions of WT brain ECs but not of brain ECs expressing PS1 FAD mutant M146V or I213T. Furthermore, our data indicate that PS1 FAD mutants inhibit the EphB4-dependent angiogenic functions of brain ECs by blocking formation of the EphB4-stimulated angiogenic complexes and suggest that PS1 FAD mutants may also affect insult-stimulated brain angiogenesis in vivo.

PCECs from WT or KI mice expressing mutant PS1, WT/M146V or WT/I213T (heterozygous) and M146V/M146V or I213T/I213T (homozygous), were grown on microcarrier beads (a) or placed in Matrigel (b). Cells were stimulated with (2 μg/mL) Fc or EphB4-Fc and sprouting (c) or meshes/loops (d) were measured as in [19]. EphB4-Fc increases sprouting and mesh/loop count (tube network formation), shown as % of Fc, only in WT cells but not in cells expressing mutants M146V or I213T (heterozygous or homozygous). Scale bar 400 μm. ns: not significant, *p < 0.05, Wilcoxon Signed Rank Test against theoretical median of 100 (no response), n = 8A representative mesh/loop is shown in (b), in WT + EphB4-Fc panel, in yellow color. PCECs from WT or mice expressing mutant PS1, WT/M146V or PS1 WT/I213T were treated with either Fc or EphB4-Fc. Cells were extracted and IPed with anti-Rok-α antibodies and the presence of VE-cadherin was detected in the IPs as described [19] (e) EphB4-Fc increases the Rok-α/VE-cadherin complexes in WT but not in PS1 M146V or PS1 I213T cells. f Quantification of experiments shown in (e). EphB4-Fc significantly increases the VE-cadherin/Rok-α complexes (if no change from Fc the value is ratio of 1) only in WT cells. **p < 0.01, Wilcoxon signed rank test against theoretical median of 1, n = 8, 8 and 7 for WT, WT/M146V and WT/I213T respectively, ns: not significant. PS1/γ-secretase promote EphB4-Fc-induced angiogenesis of pCECs in vitro. g Representative photomicrographs (composite of Hoechst in blue and grayscale phase contrast) showing EphB4-Fc-induced sprouting from WT pCEC in the absence or presence of γ-secretase inhibitor RO4929097. Quantification of cell sprouting is shown in (h). i Representative photomicrographs (composite of Calcein AM and phase contrast) of EphB4-Fc-induced capillary-like tube (network) formation of pCEC from WT (Psen1WT/WT), PS1 hemizygous (Psen1WT/KO) and PS1 knockout (Psen1 KO/KO) embryonic mouse brains. Clustered Fc is used as control. Scale bar 400um. Quantification of pCEC tube formation is shown in (j) and was performed as described in “Methods”; Tukey boxplot, ns: not significant, *p < 0.05, Wilcoxon Signed Rank Test against theoretical median of 100 (no response), n = 8.
The EphB4-stimulated PS1/γ-secretase processing of efnB2 is inhibited by PS1 FAD mutants M146V and I213T
It has been reported that protein efnB2 is processed by PS1-, but not PS2-, containing γ-secretase complexes [43]. To examine whether the γ-secretase function of PS1 is involved in the EphB4-induced angiogenic functions of pCECs, we used cell cultures prepared from WT or PS1 KO mouse brains. We found that treatment of WT pCEC cultures with γ-secretase inhibitors decreases the EphB4-induced sprouting (Fig. 1g, h). Similarly, pCEC cultures from brains of PS1 KO mice fail to respond to EphB4 by stimulating tube formation (Fig. 1i, j). Our data indicate that the PS1-dependent γ-secretase function is pivotal to the EphB4-stimulated sprouting and tube formation of pCECs. Interestingly absence of even one allele of PS1 (PS1 ± cells) also reduces the EphB4-stimulated tube formation of pCECs (Fig. 1i, j) showing that both alleles of WT PS1 are necessary for efficient angiogenic response to EphB4. It has been shown that the γ-secretase cleavage of efnB2 produces cytosolic angiogenic peptide efnB2/CTF2 that regulates angiogenic functions of ECs such as sprouting and tube formation [19] and that PS1 FAD mutants decrease the γ-secretase activity-mediated ε-cleavage of γ-secretase substrates including efnB2 [15, 20,21,22,23]. We thus asked whether PS1 FAD mutants decrease the EphB4-stimulated γ-secretase cleavage of efnB2 thus reducing formation of efnB2/CTF2. We found that although EphB4-Fc stimulates production of efnB2/CTF2 in cells expressing WT PS1 (Fig. 2a, b, lanes 1–2), production of efnB2/CTF2 is significantly reduced in cells expressing PS1 FAD mutants M146V or I213T (Fig. 2b, lanes 3–6 and graph below Fig. 2b), indicating that these mutants reduce the EphB4-stimulated γ-secretase cleavage of efnB2 and production of efnB2/CTF2. Together our data indicate that PS1 FAD mutants inhibit angiogenic functions of pCECs by decreasing production of the angiogenic peptide efnB2/CTF2.

a HEK293 cells overexpressing efnB2 in retroviral pMX vector [36] were transiently transfected with WT PS1 in FCbAIGW vector and treated with 2 μg/mL EphB4-Fc or Fc in the presence or absence of γ-secretase inhibitor L685,458 (1 μM) as indicated in the figure. Cells were extracted in 1% SDS buffer [19] and full length efnB2 (FL-efnB2), CTF1 and CTF2 fragments of efnB2 (efnB2/CTF1 and efnB2/CTF2) were detected on WB (arrows) with anti-efnB polyclonal antibody (C18, Santa Cruz,). EphB4-Fc increases the accumulation of cytoplasmic efnB2 fragment (efnB2/CTF2, lane 2, indicated with a star). EfnB2/CTF2 accumulation is inhibited by γ-secretase inhibitor L685,458 (lane 4) showing that this is the fragment produced by the PS1/γ-secretase cleavage of efnB2 [15]. b HEK293 cells overexpressing efnB2 were transiently transfected with either WT or mutant PS1 M146V or I213T in FCbAIGW vector. Cells were treated with EphB4-Fc or Fc as in (a). Cells were extracted and full length efnB2 and CTF1 and CTF2 fragments were detected on WB as in (a). Arrows show the fragments. CTF2 amount in each treatment was normalized to full length efnB2 (quantified by a lower exposure image within the linear detection range) under the same treatment and expressed as % of Fc. EphB4-Fc increases the accumulation of efnB2/CTF2 in WT cells (lane 2, indicated with a star) but not in mutant M146V- (lane 4) or I213T- (lane 6) expressing cells. Graph: EphB4-Fc significantly increases the accumulation of efnB2/CTF2 only in WT cells, *p < 0.02, one-way ANOVA with post hoc Tukey’s, n = 3. Peptide NCB-2 increases sprouting of PS1 FAD pCECs. c PCECs were treated with vehicle or control ΔTAT peptide or NCB-2 peptide all fluorescently labeled with 5-FAM (fluorescein) for detection purposes. Following treatment, pCECs were fixed and observed in an Olympus IX70 microscope with Retiga Exi camera to detect fluorescence. NCB-2 is introduced in the pCECs (third panel, fluorescence detection in cells) but not the control peptide ΔTAT which cannot penetrate the cell membrane (second panel, no fluorescence detection in cells). d PCECs from WT or PS1 M146V or PS1 MI213T mutant mouse brains were treated with vehicle (NT), NCB-2 or control peptides ΔTAT or ΔPDZ. Cells were allowed to attach on beads and sprouts were measured as in Fig. 1. NCB-2 increases sprouting in WT and PS1 FAD pCECs. Presence of γ-secretase inhibitor does not inhibit the NCB-2-induced sprouting of WT ECs. None of the control peptides increased sprouting. ***p < 0.001 one-way ANOVA with post hoc Tukey’s, n = 8.
Peptide NCB-2 potently increases sprouting of PS1 FAD mutant ECs
To further identify the amino acid sequence involved in the sprouting angiogenic functions of efnB2/CTF2, we prepared a small peptide (NCB-2) based on the primary sequence of efnB2/CTF2. This peptide comprises the ten C-terminal amino acid sequence of efnB2/CTF2 including the PDZ-binding domain of efnB2, and increases neuronal chemotaxis via efnB2 reverse signaling [44]. NCB-2 is fused to the TAT sequence at its N-terminus to facilitate membrane penetration [45] and is also tagged with 5-FAM (fluorescein) for detection purposes (GenSript). NCB-2 penetrates the cell membrane entering into the cells as detected by fluorescence (Fig. 2c, panels NCB-2). We found that NCB-2 stimulates sprouting of WT EC cultures (Fig. 2d and Supplementary Fig. S2a, panels a, b) indicating that the last ten amino acids of peptide efnB2/CTF2 are sufficient to induce sprouting. Importantly, NCB-2 also stimulates sprouting of ECs expressing PS1 FAD mutant M146V or I213T to a similar extent as it stimulates sprouting of WT cells (Fig. 2d) showing that this peptide rescues angiogenic functions of FAD mutant-expressing brain ECs that have decreased ability to sprout in response to EphB4 (Fig. 1) suggesting NCB-2 acts downstream of EphB4. Furthermore, γ-secretase inhibitors do not affect the NCB-2-stimulated sprouting indicating that NCB-2 acts downstream of the γ-secretase cleavage activity that produces efnB2/CTF2 (Fig. 2d). For controls, we used peptide (ΔPDZ) that lacks the four C-terminal amino acids of the PDZ-binding domain and peptide ΔTAT which lacks the TAT sequence and therefore is not able to penetrate the cell membrane entering into the cells (Fig. 2c, panels ΔΤΑΤ). These peptides do not increase sprouting of pCECs (Fig. 2d). Together our data show that peptide NCB-2 is sufficient to stimulate sprouting of pCECs independent of γ-secretase activity and that the peptide’s PDZ-binding domain is necessary for the sprouting activity of NCB-2. Furthermore, this peptide rescues the angiogenic functions of pCECs expressing PS1 FAD mutants. Our data also show that while γ-secretase mediates cleavage of efnB2 and production of efnB2/CTF2, it acts upstream of the functional sites of efnB2/CTF2 and NCB-2. In support, exogenous peptide efnB2/CTF2 potently stimulates sprouting of PS1 KO pCECs (Supplementary Fig. S2b, c).
PS1 FAD mutant M146V prolongs ischemia-induced brain edema
To test whether PS1 FAD mutants affect the ability of the brain to repair tissue damage, we induced focal ischemia in mouse brains by transient MCAO [46]. MCAO-induced brain edema, which predicts the final infarct size [47], was detected using MRI (DWI imaging). We found that the size of brain edema was not affected by M146V mutant compared with WT one day following MCAO (Fig. 3a), an observation confirmed in an independent mouse cohort with TTC staining (Supplementary Fig. S3a), however the duration of the edema significantly increased in mutant mice. It was detected up to 15 days post MCAO in PS1 M146V brains but it was not detected longer than 5 days following MCAO in WT brains (Fig. 3b). Our data show that PS1 FAD mutant M146V prolongs the ischemia-induced edema in brain following MCAO suggesting that this mutant increases vulnerability of the brain to ischemic lesions.

MCAO was performed in WT and PS1 FAD mutant (WT/M146V and M146V/M146V) mice and brain edema was detected with MRI 1, 5, and 15 days post MCAO and quantified using in-house software. Edema size was normalized to whole brain size in each animal. a In T2 images, there were no statistically significant differences in edema size among WT, WT/M146V, and M146V/M146V mice 1 day after MCAO. Bars represent mean, error bars represent ± SE. n = 6, 4, and 6 independent animals for WT, WT/M146V, and M146V/M146V respectively, n.s: not significant. b Edema in mutant mouse brains endures significantly longer compared with WT mice. The figure shows brain edema survival curves for WT and FAD mice (WT/M146V and M146V/M146V). We first performed survival analysis of the edema with a logrank test (Mantel–Cox method) for differences in the kinetics of edema resolution among WT and mutant mice which showed a statistically significant difference in survival (of edema) than what would be expected if all genotypes were to perform similarly (p < 0.05). We then did a follow-up logrank analysis for all genotype pairs which showed that the edema in mutant mice resolved significantly slower when compared with WT mice (Bonferroni-adjusted p is <0.01 and <0.05 for WT/M146V and M146V/M146V respectively) whereas there was no significant difference between WT/M146V and M146V/M146V (Bonferroni-adjusted p is >0.2), n = 9, 5 and 11 independent animals for WT, WT/M146V, and M146V/M146V, respectively. Overall, brain edema in WT/M146V and M146V/M146V mice remained up to 15 days after MCAO while in WT mice it was not detected anymore.
Ischemia-induced neovascularization and CBF restoration are decreased in brains of PS1 FAD mice
To test whether PS1 FAD mutants decrease the ability of the brain to respond to ischemia by stimulating neovascularization and CBF restoration, WT and M146V mice were subjected to MCAO, a procedure known to stimulate angiogenesis [48,49,50,51,52] and thus tissue healing [48, 49, 53, 54]. Thirty days later, brains were fixed and sections were stained for collagen IV, a marker of blood vessels [55] (Fig. 4a–c). The size of lesioned areas was defined by measuring the MCAO-induced brain scar following GFAP staining and total blood vessel length in the penumbra area of the lesion (ipsilateral) was estimated using Stereoinvestigator software and normalized to the corresponding length of vessels in the contralateral (control) side. We found no significant difference in the lesion sizes of WT and FAD mice (Fig. 4d and Supplementary Fig. S3a). Brains expressing FAD mutants however showed decreased total blood vessel length compared with WT controls in the penumbra of lesioned area (Fig. 4e). This indicates that MCAO-induced angiogenesis/neovascularization decreases in the brain of mice expressing PS1M146V, an outcome believed to impair brain’s ability to heal after ischemia. There was no significant difference in total vascular staining between WT and mutant animals in the control (contralateral) side of the brains used to normalize the data showing that the effect of PS1 mutant in total vessel length appears only after the applied ischemic stress (Supplementary Fig. S3b). To test whether pCEC viability under ischemia is affected by PS1 FAD mutants we subjected cultures of primary cortical ECs from WT and mutant mice to oxygen-glucose deprivation (OGD), a stress that mimics components of in vivo ischemia [56]. Viability of pCEC was determined using the MTT assay [57]. We found that M146V does not affect OGD-induced death of pCEC (Fig. 4f) an observation consistent with our hypothesis that the decreased collagen IV staining observed in M146V mice after MCAO is not due to increased death of mutant ECs but rather to impaired angiogenesis. Further support for this conclusion is provided by the expression of the neovascularization marker endoglin (CD105) [58,59,60] which, following MCAO, increases in WT brains but not in brains expressing FAD mutants (Fig. 4g, h). Since CD105 increases in ECs during angiogenic sprouting [61,62,63,64] our data indicate that PS1 FAD mutants inhibit the insult-induced neovascularization by targeting angiogenic sprouting of the brain. Together our data suggest that PS1 FAD mutants impair brain’s vascular response to ischemia.

WT, M146V heterozygous (WT/M146V), and M146V homozygous (M146V/M146V) mice were subjected to transient focal cerebral ischemia for 50 mins. Brains were isolated and sectioned 30 days later. Brain sections of WT (a), M146V heterozygous (b), or homozygous (c) mice were stained with anti-collagen IV and anti-GFAP antibodies. Lesion areas were defined by GFAP staining and an identical area in the contralateral side was used as control for normalization of the values (outlined in (a)–(c) left). Scale bar: 1 mm. Collagen IV staining of the lesion area (ipsilateral side) is shown in (a)–(c) (right). Scale bar: 50 μm. d MCAO-induced scar area (ipsilateral side) was determined by GFAP staining. No significant difference in the size of the area between WT and M146V mutant mice was observed. One-way ANOVA with post hoc Tukey’s, n = 4, 5, and 4 for animals for WT, WT/M146V and M146V/M146V respectively, n.s: not significant. e Quantification of collagen IV immunostaining in lesion area shows that staining increases in WT mice 30 days post MCAO but not in PS1 M146V mutants, either heterozygous or homozygous, suggesting that brain neovascularization does not increase in mutant M146V after ischemic insult as efficiently as in WT. **p < 0.002, one-way ANOVA with post hoc Tukey’s, n = 4, n.s: not significant. f WT and M146V pCECs were subjected to OGD. Cells were incubated under Normoxic condition (37oC, 5% CO2 and 95% air) in normal medium or Hypoxic condition (37 oC, 5% CO2, 2% O2, 93% N2) in glucose- and serum-free medium for 6 h following 12-h incubation in normal conditions. Viability was determined with MTT. Mutant cells do not die more than WT cells under OGD. One-way ANOVA, n = 3, n.s: not significant. g WT or PS1 M146V KI mice were subjected to MCAO and CD105 levels were detected in brain extracts 1, 3, and 30 days later with WB. h CD105 levels increase in WT brains 30 days post MCAO indicating increased neovascularization but not in M146V mutant mice. Values are normalized to CD105 levels in WT, 1 day post MCAO. **p < 0.01, one-way ANOVA with post hoc Tukey’s, n = 4 except for n = 3 for WT at 30 days post MCAO, n.s: not significant. WT and PS1 WT/M146V (heterozygous) mice were subjected to MCAO for 50 min. Brain perfusion was examined on an MRI imager at 1, 5, 10, and 15 days after ischemic insult. i Relative cerebral blood volume (rCBV) map of WT mouse brain using perfusion MRI (pMRI). rCBV map (arrow) overlaid onto anatomical T2 image of brain. j Quantification shows that CBF significantly increases at 15 days post MCAO in WT mice but not in mutant mice (n = 6, 4, 6 for WT, WT/M146V, and M146V/M146V, respectively). **p < 0.01, linear regression, ns: not significant. Values are expressed as % of CBF in WT at day 1 post MCAO. Normalization of CBF in lesion areas was done using the CBF in control (contralateral) side. k, l NeuN-immunopositive neurons were counted in brain sections of WT and M146V mutant mice. Number of NeuN-containing neurons in the MCAO-induced lesion area (as defined by GFAP staining) was normalized to number of NeuN-immunopositive neurons in an identical area of the contralateral side of each section (outlined in the NeuN + GFAP panels on the left). Scale bar for the NeuN+GFAP pictures: 1 mm and for the GFAP or NeuN pictures: 50 μm. The number of NeuN-expressing neurons in lesion (ipsilateral) of PS1 M146V brains after MCAO is significantly lower compared with WT. *p < 0.02 one-way ANOVA with post hoc Tukey’s, n = 3. m Novel object recognition (NOR) test: WT, heterozygous (WT/M146V), or homozygous (M146V/M146V) mice were subjected to NOR test as described in material and methods. PS1 M146V mice have decreased performance compared with WT mice following MCAO, *p < 0.03, one-way ANOVA, n = 7 for WT, n = 8 for WT/M146V, and n = 6 for M146V/M146V. n Y maze test: Mice were subjected to Y maze test as described in materials and methods. PS1 M146V mice have decreased performance compared with WT mice following MCAO, **p < 0.002, one-way ANOVA, n = 7 for WT, n = 8 for WT/M146V and n = 6 for M146V/M146V. o Open field test: Mice were subjected to open field test. No significant difference in motor coordination or in motivation was observed between WT and mutant mice following MCAO. One-way ANOVA, n = 6 for WT, n = 9 for WT/M146V, and n = 7 for M146V/M146V. n.s: not significant. p Rotarod test: Mice were subjected to rotarod test. No significant difference was observed in motor coordination between WT and mutant mice following MCAO. One-way ANOVA, n = 7 for WT, n = 8 for WT/M146V, and n = 7 for M146V/M146V n.s: not significant. All behavioral experiments were performed pre- and post-MCAO. All data were analyzed and normalized to pre MCAO scores. There were no differences in baseline behavior scores among groups.
CBF is decreased following brain ischemia. Its restoration is a marker of recovery of normal vascular function and an indirect measure of angiogenesis in the brain [65, 66]. CBF in lesion area, as defined by T2 weighted MRI imaging (Fig. 4i), was measured and quantified with perfusion MRI (T7). One day after MCAO, CBF decreases in both WT and FAD mouse brains to similar levels (Supplementary Fig. S3c). Fifteen days later CBF is significantly increased in WT mice, however no significant increase is observed in mice expressing M146V (Fig. 4j). CBF levels did not significantly differ between WT and FAD animals before ischemic insult or in the control (contralateral) side of the brain (Supplementary Fig. S3c) suggesting that the effects of the PS1 FAD mutants on CBF appear only after the toxic insult. Combined these results indicate that FAD mutants impair the ischemia-induced angiogenic activity of the brain rendering it more vulnerable to insults.
Neuronal death and cognitive decline following MCAO increase in brains of PS1 FAD mice
Brain ischemia induces neuronal loss. To test whether ischemia-induced neuronal death is affected by PS1 FAD mutants, WT and KI mice expressing mutant M146V were subjected to MCAO. Thirty days later, brains were fixed and prepared sections stained with NeuN. Neurons were counted in the penumbra of lesion as defined by GFAP staining and number of NeuN-positive neurons was estimated using Stereoinvestigator software and normalized to the corresponding number of NeuN-positive neurons in the contralateral (control) side. We found fewer NeuN-positive neurons in the lesion area in brains expressing PS1 M146V compared with WT (Fig. 4k, l) despite that the remaining lesion areas determined by GFAP staining, were of comparable size in all mice brains (Fig. 4d). Similar data were obtained in mice expressing PS1 FAD mutant I213T (Supplementary Fig. S3d). The effect of PS1 M146V on neuronal death appeared only in the brain area affected by ischemia (ipsilateral), as there was no significant difference in the number of neurons in the area not affected by ischemia (contralateral) of WT and PS1 M146V brains (Supplementary Fig. S3e).Taken together, our data suggest that PS1 FAD mutations increase neuronal vulnerability to MCAO-induced ischemia-reperfusion injury (IR-I).
IR-I induces behavioral deficits in mice [67,68,69]. These deficits correlate with neuronal death caused by IR-I [67, 68, 70]. Since expression of PS1 M146V mutant correlates with decreased NeuN-positive neurons following MCAO (Fig. 4k, l), we asked whether mutant mice have poorer post-MCAO cognitive outcomes compared with WT mice. To this end, we used behavioral assessments in mice before MCAO and 15 days following MCAO. Neurological scoring was tested based on previously published neurological scales [71, 72]. Recognition memory was tested using NOR test and the Y maze test was used to evaluate spatial learning and memory [40, 41, 73,74,75]. Locomotor function and motivation were tested using the open field and rotarod tests as previously described [39, 76]. We found that both homozygous and heterozygous PS1 M146V mice showed a higher decrease in post-MCAO performance in NOR and Y maze tests compared with WT (Fig. 4m, n). These outcomes are consistent with the decreased numbers of NeuN-positive neurons in the FAD brains following IR-I (Fig. 4k, l). No significant difference was observed between WT and mutant mice in open field and rotarod tests following MCAO demonstrating that motor coordination, balance or anxiety were not differentially affected in these mice (Fig. 4o, p). Similar results were obtained with I213T mutant mice (Supplementary Fig. S3f, g). In support of the hypothesis that behavioral deficits in FAD mice are not due to defects in locomotive capacity, no post-MCAO differences in either global neurologic function or focal neurological deficits between PS1 WT and M146V or I213T mice were observed (Supplementary Fig. S4). The above show that mice expressing PS1 FAD mutants suffer greater IR-I-induced memory deficits compared with WT mice, consistent with increased brain neuronal loss in the FAD mice.
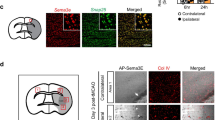
PS1 M146V inhibits ischemia-induced brain angiogenic complexes
Since ischemia induces angiogenesis and neovascularization, a process known to be mediated by VE-cadherin angiogenic complexes, we asked whether ischemia also induces such complexes in the brain and whether this induction is affected by PS1 FAD mutant M146V. Using spPLA [77,78,79] in brain extracts from WT and mutant mice subjected to MCAO, we found that ischemia increases the VE-cadherin/Rok-α complexes in WT brains but has little effect on these complexes in brains heterozygous for FAD mutant M146V (Fig. 5) indicating that FAD mutants compromise the ischemia-induced angiogenic response of the brain by decreasing the induction of VE-cadherin angiogenic complexes. Combined with literature data that the EphB4/efnB2 system is required for brain’s angiogenic response to ischemia [17, 18], our findings that VE-cadherin/Rok-α angiogenic complexes are stimulated by EphB4 in brain ECs (Fig. 1) and by ischemia in the brain (Fig. 5) support the conclusion that the EphB4/efnB2 system mediates brain’s angiogenic response to ischemia by stimulating these complexes. No difference in these complexes was observed between WT and PS1 FAD mouse embryo brains in the absence of ischemia (Supplementary Fig. S5a, b) indicating that the FAD mutants target the ischemia-induced complex formation. Together, our data show that PS1 FAD mutants impair formation of ischemia-induced brain angiogenic complexes suggesting a mechanism by which these mutants impair brain’s neovascularization in response to ischemia.

VE-cadherin/Rok-α complexes were measured with spPLA in brain extracts of WT and PS1 M146V mutant mice, which had been subjected to MCAO. IR-I significantly increases complexes at 3, 7 and 15 days post MCAO only in WT and not in PS1 FAD brains. ****p < 0.0001 one-way ANOVA with post hoc Tukey’s, n = 5, 4, 3, 4, 3 for WT and 4, 6, 6, 3, 5 for WT/M146V mice.
Discussion
Here, we present evidence that following ischemic insult, mouse brains expressing PS1 FAD mutants show decreased neovascularization compared with WT. We also obtained evidence that these mutants decrease the ischemia-stimulated sprouting angiogenesis marker CD105 suggesting that FAD mutants decrease neovascularization by targeting sprouting angiogenesis. This conclusion is further supported by our data that the FAD mutants decrease angiogenic functions of brain ECs such as sprouting, tube formation and angiogenic complexes, all known to play pivotal roles in sprouting angiogenesis and neovascularization [80]. Combined with the requirement of both WT PS1 alleles for efficient tube formation in vitro (Fig. 1) our data support the hypothesis that PS1 FAD mutants cause the loss of PS1 functions in sprouting angiogenesis.
It is known that the EphB4/efnB2 system regulates sprouting angiogenesis [81,82,83], and that EphB4 stimulates the PS1/γ-secretase processing of efnB2 and the product of this processing promotes sprouting of ECs [19]. Here, we show that PS1 FAD mutants decrease the EphB4-stimulated pCEC sprouting, tube formation and VE-cadherin/Rok-α angiogenic complexes, steps known to regulate angiogenesis. Furthermore, we found that following IR-I the angiogenic complexes, neovascularization and CBF increase in WT but not in FAD mutant mouse brains. Combined with evidence that IR-I induces brain neovascularization in an EphB4/efnB2-dependent manner and that neovascularization is mediated by members of angiogenic complexes [16,17,18], our observations indicate that PS1 FAD mutants decrease ischemia-induced neovascularization by impairing the EphB4/efnB2-regulated angiogenic complexes of brain ECs.
Our data shows that following IR-I, neovascularization in brains expressing PS1 FAD mutants decreases and consequently the restoration of blood flow in the injured area is impaired. Interestingly impairment of vascular function in mutant brains manifests only after the ischemic insult suggesting that the effects of PS1 FAD mutants on vascular functions appear only following toxic stress. This is consistent with the fact that patients with FAD mutations survive until the adult life without demonstrating brain malfunctions at an earlier age. The problems may arise at a later age as stresses like IR-I become more frequent. It is now known that brain ischemic episodes are much more frequent than originally believed. It has been found that in addition to major ischemic episodes there are numerous silent ones, about five times more frequent than major ones [84], and that about one-third of aged people suffer silent ischemic episodes [85]. Reduced ability of the brain to repair damage caused by multiple ischemic insults could lead to neuronal dysfunction and dementia.
IR-I is frequent in AD patients. Neuropathological post-mortem examinations of AD brains have shown that about 30% of patients have evidence of post-ischemic injury [86, 87]. It has been suggested that IR-I in hippocampus aggravates cognitive impairment by increasing neuronal death, deposition of Aβ peptide [88, 89], and pathological phosphorylation of tau protein [90]. Additional work indicates an association between brain IR-I and sporadic AD [84, 88,89,90,91,92,93,94,95] supporting the hypothesis that IR-I is a significant contributor to AD [96,97,98,99,100]. Our findings are therefore relevant to AD and suggest that PS1 mutants render the brain vulnerable to toxic insults such as ischemic stroke, decreasing the brain’s capacity to respond to these insults by neovascularization and restoration of normal blood flow in the injured areas. This may be due to impairment of the molecular mechanism of angiogenesis regulated by protein complexes between VE-cadherin and Rok-α. Increased vascular vulnerability of the brain could then result in increased neuronal death and cognitive decline. In support of this hypothesis, our data show that following IR-I, NeuN-positive neurons in the injured area of FAD mutant-expressing brains are significantly decreased compared with neurons of WT brains. Furthermore, IR-I-induced cognitive decline is more severe in mice expressing FAD mutants compared with WT mice. Interestingly, brain edema and tissue scars formed in the injured areas are of the same size in both WT and mutant mice, excluding the possibility that the decreased number of NeuN-positive neurons measured in the mutant brains is due to a different lesion size. However, the duration of edema is significantly longer in FAD mutant brains indicating that blood supply is restricted significantly longer in the lesioned areas of these brains, increasing the possibility of neuronal dysfunction and death.
To explore the mechanism by which PS1 FAD mutants M146V and I213T decrease the EphB4-stimulated angiogenic functions of ECs, we asked whether these mutants decrease the EphB4-stimulated proteolytic cleavage of efnB2 by PS1/γ-secretase. It has been reported by several investigators that PS1 FAD mutants decrease the PS1/γ-secretase cleavage of several substrates at the ε site including N-cadherin, Notch1, EphB2 receptor and efnB2 ligand [15, 20,21,22,23, 101], and we show here that these mutants decrease the EphB4-stimulated production of angiogenic peptide efnB2/CTF2 suggesting that they reduce angiogenic functions of brain ECs and neovascularization by decreasing peptide efnB2/CTF2. Overexpression of efnB2/CTF2 in pCECs that lack PS1 (PS1 KO) strongly increases the sprouting of these cells confirming that the angiogenic function of efnB2/CTF2 is independent of γ-secretase activity (Supplementary Fig. S2c).
To identify the specific efnB2/CTF2 domain that mediates the angiogenic activity of efnB2/CTF2, we constructed peptide NCB-2 that comprises the ten C-terminal residues of efnB2/CTF2 including the PDZ-binding motif plus the adjacent six residues. We found that this peptide stimulates significantly the sprouting of both WT and PS1 FAD mutant-expressing ECs suggesting NCB-2 rescues angiogenic functions of ECs from the FAD mutant-induced activity reduction. Combined with our data that these mutants decrease the cellular levels of efnB2/CTF2 while concomitantly decreasing the angiogenic functions of brain ECs in response to EphB4, this implies that the limiting factor for sprouting in PS1 FAD pCECs is the decreased production of EphB4-stimulated efnB2/CTF2. Interestingly the NCB-2 peptide-stimulated sprouting of brain ECs was at similar or higher levels compared with the induction by efnB2/CTF2 (Supplementary Fig. S2c) suggesting that the last ten amino acids of efnB2/CTF2 are sufficient to fully exert its sprouting angiogenic function. In addition, γ-secretase inhibitors did not inhibit the peptide-associated angiogenic sprouting of the cells indicating that the peptide does not require the cleavage of other angiogenic factor receptors that are γ-secretase substrates, such as Notch1 or VEGFR [102, 103] in order to increase sprouting.
Our finding that PS1 FAD mutants target efnB2-mediated angiogenesis is novel and suggests impaired efnB2 angiogenic functions in FAD. It will be interesting to test whether the same efnB2-mediated mechanism is impaired in sporadic AD.
In summary, our data reveal a novel molecular mechanism by which PS1 FAD mutants decrease the restorative capacity of brain against toxic insults such as IR-I, increasing neuronal death and cognitive decline. These mutants decrease the γ-secretase processing of efnB2 and production of the angiogenic peptide efnB2/CTF2 thus inhibiting formation of the angiogenic complexes in response to EphB4 or to ischemia. This impedes brain’s angiogenic response rendering it more vulnerable to ischemia, increasing neuronal death and cognitive decline. We found that administration of peptide NCB-2 that derives from efnB2/CTF2 strongly increases angiogenic sprouting of brain ECs expressing FAD mutants. These findings suggest that this peptide is a promising agent that could lead to pharmaceutical intervention promoting angiogenic functions in the brain and decreasing toxicity-induced neuronal death and degeneration. Importantly, the effects of PS1 FAD mutants on the ischemia-stimulated angiogenesis occur in the absence of amyloid plaques and neurofibrillary tangles [104, 105] indicating that PS1 FAD mutants can affect brain functions in the absence of neuropathological hallmarks of AD [106]. Combined with failed clinical trials targeting AD neuropathological markers, these observations support the hypothesis that in FAD, neuropathological hallmarks may form downstream from mutant effects on brain vasculature and neuronal survival.
References
Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26:573–8.
Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611.
de la Torre JC. Alzheimer’s disease is a vasocognopathy: a new term to describe its nature. Neurol Res. 2004;26:517–24.
Gorelick PB. Risk factors for vascular dementia and Alzheimer disease. Stroke. 2004;35:2620–2.
Zlokovic BV. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005;28:202–8.
Pimentel-Coelho PM, Rivest S. The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer’s disease. Eur J Neurosci. 2012;35:1917–37.
van Rooden S, Goos JD, van Opstal AM, Versluis MJ, Webb AG, Blauw GJ, et al. Increased number of microinfarcts in Alzheimer disease at 7-T MR imaging. Radiology. 2014;270:205–11.
Smith EE, Schneider JA, Wardlaw JM, Greenberg SM. Cerebral microinfarcts: the invisible lesions. Lancet Neurol. 2012;11:272–82.
Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277:813–7.
Beck H, Plate KH. Angiogenesis after cerebral ischemia. Acta Neuropathol. 2009;117:481–96.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Brantley-Sieders DM, Chen J. Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis. 2004;7:17–28.
Blits-Huizinga CT, Nelersa CM, Malhotra A, Liebl DJ. Ephrins and their receptors: binding versus biology. IUBMB Life. 2004;56:257–65.
Palmer A, Zimmer M, Erdmann KS, Eulenburg V, Porthin A, Heumann R, et al. EphrinB phosphorylation and reverse signaling: regulation by Src kinases and PTP-BL phosphatase. Mol Cell. 2002;9:725–37.
Georgakopoulos A, Litterst C, Ghersi E, Baki L, Xu C, Serban G, et al. Metalloproteinase/Presenilin1 processing of ephrinB regulates EphB-induced Src phosphorylation and signaling. EMBO J. 2006;25:1242–52.
Wimmer R, Cseh B, Maier B, Scherrer K, Baccarini M. Angiogenic sprouting requires the fine tuning of endothelial cell cohesion by the Raf-1/Rok-alpha complex. Dev Cell. 2012;22:158–71.
Xing S, Pan N, Xu W, Zhang J, Li J, Dang C, et al. EphrinB2 activation enhances angiogenesis, reduces amyloid-beta deposits and secondary damage in thalamus at the early stage after cortical infarction in hypertensive rats. J Cereb Blood Flow Metab. 2018;39:1776–89.
Ghori A, Freimann FB, Nieminen-Kelha M, Kremenetskaia I, Gertz K, Endres M, et al. EphrinB2 activation enhances vascular repair mechanisms and reduces brain swelling after mild cerebral Ischemia. Arterioscler Thromb Vasc Biol. 2017;37:867–78.
Warren NA, Voloudakis G, Yoon Y, Robakis NK, Georgakopoulos A. The product of the gamma-secretase processing of ephrinB2 regulates VE-cadherin complexes and angiogenesis. Cell Mol Life Sci. 2018;75:2813–26.
Marambaud P, Wen PH, Dutt A, Shioi J, Takashima A, Siman R, et al. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell. 2003;114:635–45.
Moehlmann T, Winkler E, Xia X, Edbauer D, Murrell J, Capell A, et al. Presenilin-1 mutations of leucine 166 equally affect the generation of the Notch and APP intracellular domains independent of their effect on Abeta 42 production. Proc Natl Acad Sci USA. 2002;99:8025–30.
Heilig EA, Gutti U, Tai T, Shen J, Kelleher RJ 3rd. Trans-dominant negative effects of pathogenic PSEN1 mutations on gamma-secretase activity and Abeta production. J Neurosci. 2013;33:11606–17.
Litterst C, Georgakopoulos A, Shioi J, Ghersi E, Wisniewski T, Wang R, et al. Ligand binding and calcium influx induce distinct ectodomain/gamma-secretase-processing pathways of EphB2 receptor. J Biol Chem. 2007;282:16155–63.
Guo Q, Fu W, Sopher BL, Miller MW, Ware CB, Martin GM, et al. Increased vulnerability of hippocampal neurons to excitotoxic necrosis in presenilin-1 mutant knock-in mice. Nat Med. 1999;5:101–6.
Nakano Y, Kondoh G, Kudo T, Imaizumi K, Kato M, Miyazaki JI, et al. Accumulation of murine amyloidbeta42 in a gene-dosage-dependent manner in PS1 ‘knock-in’ mice. Eur J Neurosci. 1999;11:2577–81.
Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–39.
Kim JH, Yenari MA, Giffard RG, Cho SW, Park KA, Lee JE. Agmatine reduces infarct area in a mouse model of transient focal cerebral ischemia and protects cultured neurons from ischemia-like injury. Exp Neurol. 2004;189:122–30.
Yenari MA, Palmer JT, Sun GH, de Crespigny A, Mosely ME, Steinberg GK. Time-course and treatment response with SNX-111, an N-type calcium channel blocker, in a rodent model of focal cerebral ischemia using diffusion-weighted MRI. Brain Res. 1996;739:36–45.
Hara H, Huang PL, Panahian N, Fishman MC, Moskowitz MA. Reduced brain edema and infarction volume in mice lacking the neuronal isoform of nitric oxide synthase after transient MCA occlusion. J Cereb Blood Flow Metab. 1996;16:605–11.
Huang Z, Huang PL, Ma J, Meng W, Ayata C, Fishman MC, et al. Enlarged infarcts in endothelial nitric oxide synthase knockout mice are attenuated by nitro-L-arginine. J Cereb Blood Flow Metab. 1996;16:981–7.
Kitagawa K, Matsumoto M, Yang G, Mabuchi T, Yagita Y, Hori M, et al. Cerebral ischemia after bilateral carotid artery occlusion and intraluminal suture occlusion in mice: evaluation of the patency of the posterior communicating artery. J Cereb Blood Flow Metab. 1998;18:570–9.
Shen Q, Duong TQ. Magnetic resonance imaging of cerebral blood flow in animal stroke models. Brain Circ. 2016;2:20–7.
Kumar TP, Vasudevan A. Isolation and culture of endothelial cells from the embryonic forebrain. J Vis Exp. 2014;23:e51021.
Baki L, Neve RL, Shao Z, Shioi J, Georgakopoulos A, Robakis NK. Wild-type but not FAD mutant presenilin-1 prevents neuronal degeneration by promoting phosphatidylinositol 3-kinase neuroprotective signaling. J Neurosci. 2008;28:483–90.
Ponce ML. Tube formation: an in vitro matrigel angiogenesis assay. Methods Mol Biol. 2009;467:183–8.
Georgakopoulos A, Xu J, Xu C, Mauger G, Barthet G, Robakis NK. Presenilin1/gamma-secretase promotes the EphB2-induced phosphorylation of ephrinB2 by regulating phosphoprotein associated with glycosphingolipid-enriched microdomains/Csk binding protein. FASEB J. 2011;25:3594–604.
Gama Sosa MA, Gasperi RD, Rocher AB, Wang AC, Janssen WG, Flores T, et al. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am J Pathol. 2010;176:353–68.
Schmitz C, Hof PR. Design-based stereology in neuroscience. Neuroscience. 2005;130:813–31.
Tatem KS, Quinn JL, Phadke A, Yu Q, Gordish-Dressman H, Nagaraju K. Behavioral and locomotor measurements using an open field activity monitoring system for skeletal muscle diseases. J Vis Exp. 2014;29:51785.
Riddle MC, McKenna MC, Yoon YJ, Pattwell SS, Santos PM, Casey BJ, et al. Caloric restriction enhances fear extinction learning in mice. Neuropsychopharmacology. 2013;38:930–7.
Leger M, Quiedeville A, Bouet V, Haelewyn B, Boulouard M, Schumann-Bard P, et al. Object recognition test in mice. Nat Protoc. 2013;8:2531–7.
Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39:175–91.
Barthet G, Shioi J, Shao Z, Ren Y, Georgakopoulos A, Robakis NK. Inhibitors of gamma-secretase stabilize the complex and differentially affect processing of amyloid precursor protein and other substrates. FASEB J. 2011;25:2937–46.
Yu Y, Liu M, Ng TT, Huang F, Nie Y, Wang R, et al. PDZ-reactive peptide activates Ephrin-B reverse signaling and inhibits neuronal chemotaxis. ACS Chem Biol. 2016;11:149–58.
Vives E, Brodin P, Lebleu B. A truncated HIV-1 Tat protein basic domain rapidly translocates through the plasma membrane and accumulates in the cell nucleus. J Biol Chem. 1997;272:16010–7.
Liu F, McCullough LD. Middle cerebral artery occlusion model in rodents: methods and potential pitfalls. J Biomed Biotechnol. 2011;2011:464701.
Lu M, Mitsias PD, Ewing JR, Soltanian-Zadeh H, Bagher-Ebadian H, Zhao Q, et al. Predicting final infarct size using acute and subacute multiparametric MRI measurements in patients with ischemic stroke. J Magn Reson Imaging. 2005;21:495–502.
Chu M, Hu X, Lu S, Gan Y, Li P, Guo Y, et al. Focal cerebral ischemia activates neurovascular restorative dynamics in mouse brain. Front Biosci (Elite Ed). 2012;4:1926–36.
Martin A, Mace E, Boisgard R, Montaldo G, Theze B, Tanter M, et al. Imaging of perfusion, angiogenesis, and tissue elasticity after stroke. J Cereb Blood Flow Metab. 2012;32:1496–507.
Shin HY, Kim JH, Phi JH, Park CK, Kim JE, Kim JH, et al. Endogenous neurogenesis and neovascularization in the neocortex of the rat after focal cerebral ischemia. J Neurosci Res. 2008;86:356–67.
Frontczak-Baniewicz M, Walski M. New vessel formation after surgical brain injury in the rat’s cerebral cortex I. Formation of the blood vessels proximally to the surgical injury. Acta Neurobiol Exp (Wars). 2003;63:65–75.
Gertz K, Kronenberg G, Kalin RE, Baldinger T, Werner C, Balkaya M, et al. Essential role of interleukin-6 in post-stroke angiogenesis. Brain. 2012;135:1964–80.
Seevinck PR, Deddens LH, Dijkhuizen RM. Magnetic resonance imaging of brain angiogenesis after stroke. Angiogenesis. 2010;13:101–11.
Zhang ZG, Zhang L, Jiang Q, Chopp M. Bone marrow-derived endothelial progenitor cells participate in cerebral neovascularization after focal cerebral ischemia in the adult mouse. Circ Res. 2002;90:284–8.
Baluk P, Morikawa S, Haskell A, Mancuso M, McDonald DM. Abnormalities of basement membrane on blood vessels and endothelial sprouts in tumors. Am J Pathol. 2003;163:1801–15.
Singh G, Siddiqui MA, Khanna VK, Kashyap MP, Yadav S, Gupta YK, et al. Oxygen glucose deprivation model of cerebral stroke in PC-12 cells: glucose as a limiting factor. Toxicol Mech Methods. 2009;19:154–60.
Huang Q, Voloudakis G, Ren Y, Yoon Y, Zhang E, Kajiwara Y, et al. Presenilin1/gamma-secretase protects neurons from glucose deprivation-induced death by regulating miR-212 and PEA15. FASEB J. 2018;32:243–53.
Muramatsu R, Takahashi C, Miyake S, Fujimura H, Mochizuki H, Yamashita T. Angiogenesis induced by CNS inflammation promotes neuronal remodeling through vessel-derived prostacyclin. Nat Med. 2012;18:1658–64.
Nunez-Gomez E, Pericacho M, Ollauri-Ibanez C, Bernabeu C, Lopez-Novoa JM. The role of endoglin in post-ischemic revascularization. Angiogenesis. 2017;20:1–24.
Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM, et al. Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res. 2011;31:2283–90.
Liu Z, Lebrin F, Maring JA, van den Driesche S, van der Brink S, van Dinther M, et al. ENDOGLIN is dispensable for vasculogenesis, but required for vascular endothelial growth factor-induced angiogenesis. PLoS ONE. 2014;9:e86273.
Barnett JM, Suarez S, McCollum GW, Penn JS. Endoglin promotes angiogenesis in cell- and animal-based models of retinal neovascularization. Investig Ophthalmol Vis Sci. 2014;55:6490–8.
Xu Y, Wang D, Zhao LM, Zhao XL, Shen JJ, Xie Y, et al. Endoglin is necessary for angiogenesis in human ovarian carcinoma-derived primary endothelial cells. Cancer Biol Ther. 2013;14:937–48.
Park S, Dimaio TA, Liu W, Wang S, Sorenson CM, Sheibani N. Endoglin regulates the activation and quiescence of endothelium by participating in canonical and non-canonical TGF-beta signaling pathways. J Cell Sci. 2013;126:1392–405.
Howard-Claudio C. MRI methods for the detection of angiogenesis. Appl Radiol. 2005;34:9–24.
Gupta RK, Haris M, Husain N, Husain M, Prasad KN, Pauliah M, et al. Relative cerebral blood volume is a measure of angiogenesis in brain tuberculoma. J Comput Assist Tomogr. 2007;31:335–41.
DeVries AC, Nelson RJ, Traystman RJ, Hurn PD. Cognitive and behavioral assessment in experimental stroke research: will it prove useful? Neurosci Biobehav Rev. 2001;25:325–42.
Murphy TH, Corbett D. Plasticity during stroke recovery: from synapse to behaviour. Nat Rev Neurosci. 2009;10:861–72.
Balkaya M, Krober JM, Rex A, Endres M. Assessing post-stroke behavior in mouse models of focal ischemia. J Cereb Blood Flow Metab. 2013;33:330–8.
Baron JC, Yamauchi H, Fujioka M, Endres M. Selective neuronal loss in ischemic stroke and cerebrovascular disease. J Cereb Blood Flow Metab. 2014;34:2–18.
Capdeville C, Pruneau D, Allix M, Plotkine M, Boulu RG. Naloxone effect on the neurological deficit induced by forebrain ischemia in rats. Life Sci. 1986;38:437–42.
Bozeat S, Gregory CA, Ralph MA, Hodges JR. Which neuropsychiatric and behavioural features distinguish frontal and temporal variants of frontotemporal dementia from Alzheimer’s disease? J Neurol Neurosurg Psychiatry. 2000;69:178–86.
Grayson B, Leger M, Piercy C, Adamson L, Harte M, Neill JC. Assessment of disease-related cognitive impairments using the novel object recognition (NOR) task in rodents. Behav Brain Res. 2015;285:176–93.
Dong YF, Kataoka K, Tokutomi Y, Nako H, Nakamura T, Toyama K, et al. Perindopril, a centrally active angiotensin-converting enzyme inhibitor, prevents cognitive impairment in mouse models of Alzheimer’s disease. FASEB J. 2011;25:2911–20.
Tajima H, Kawasumi M, Chiba T, Yamada M, Yamashita K, Nawa M, et al. A humanin derivative, S14G-HN, prevents amyloid-beta-induced memory impairment in mice. J Neurosci Res. 2005;79:714–23.
Lee BH, Kim J, Lee RM, Choi SH, Kim HJ, Hwang SH, et al. Gintonin enhances performance of mice in rotarod test: Involvement of lysophosphatidic acid receptors and catecholamine release. Neurosci Lett. 2016;612:256–60.
Darmanis S, Nong RY, Hammond M, Gu J, Alderborn A, Vanelid J, et al. Sensitive plasma protein analysis by microparticle-based proximity ligation assays. Mol Cell Proteom. 2010;9:327–35.
Lundberg M, Eriksson A, Tran B, Assarsson E, Fredriksson S. Homogeneous antibody-based proximity extension assays provide sensitive and specific detection of low-abundant proteins in human blood. Nucleic Acids Res. 2011;39:e102.
Assarsson E, Lundberg M, Holmquist G, Bjorkesten J, Thorsen SB, Ekman D, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS ONE. 2014;9:e95192.
Goodwin AM. In vitro assays of angiogenesis for assessment of angiogenic and anti-angiogenic agents. Microvasc Res. 2007;74:172–83.
Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, et al. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev. 1999;13:295–306.
Sawamiphak S, Seidel S, Essmann CL, Wilkinson GA, Pitulescu ME, Acker T, et al. Ephrin-B2 regulates VEGFR2 function in developmental and tumour angiogenesis. Nature. 2010;465:487–91.
Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–6.
Vermeer SE, Prins ND, den Heijer T, Hofman A, Koudstaal PJ, Breteler MM. Silent brain infarcts and the risk of dementia and cognitive decline. N Engl J Med. 2003;348:1215–22.
Fried LP, Borhani NO, Enright P, Furberg CD, Gardin JM, Kronmal RA, et al. The Cardiovascular Health Study: design and rationale. Ann Epidemiol. 1991;1:263–76.
Kalaria RN. The role of cerebral ischemia in Alzheimer’s disease. Neurobiol Aging. 2000;21:321–30.
Pluta R. Ischemia–reperfusion factors in sporadic Alzheimer’s disease. In: Welsh EM (ed). New research on Alzheimer’s disease. New York: Nova Science Publishers, Inc; 2006. p. 183–234.
Pluta R. The role of apolipoprotein E in the deposition of beta-amyloid peptide during ischemia-reperfusion brain injury. A model of early Alzheimer’s disease. Ann NY Acad Sci. 2000;903:324–34.
Pluta R, Jablonski M, Czuczwar SJ. Postischemic dementia with Alzheimer phenotype: selectively vulnerable versus resistant areas of the brain and neurodegeneration versus beta-amyloid peptide. Folia Neuropathol. 2012;50:101–9.
Wen Y, Yang SH, Liu R, Perez EJ, Brun-Zinkernagel AM, Koulen P, et al. Cdk5 is involved in NFT-like tauopathy induced by transient cerebral ischemia in female rats. Biochim Biophys Acta. 2007;1772:473–83.
Pluta R, Ulamek-Koziol M, Januszewski S, Scislewska M, Bogucka-Kocka A, Kocki J. Alzheimer’s factors in postischemic dementia. Rom J Morphol Embryol. 2012;53:461–6.
Pluta R, Kida E, Lossinsky AS, Golabek AA, Mossakowski MJ, Wisniewski HM. Complete cerebral ischemia with short-term survival in rats induced by cardiac arrest. I. Extracellular accumulation of Alzheimer’s beta-amyloid protein precursor in the brain. Brain Res. 1994;649:323–8.
Maslinska D, Laure-Kamionowska M, Taraszewska A, Deregowski K, Maslinski S. Immunodistribution of amyloid beta protein (Abeta) and advanced glycation end-product receptors (RAGE) in choroid plexus and ependyma of resuscitated patients. Folia Neuropathol. 2011;49:295–300.
Qi JP, Wu H, Yang Y, Wang DD, Chen YX, Gu YH, et al. Cerebral ischemia and Alzheimer’s disease: the expression of amyloid-beta and apolipoprotein E in human hippocampus. J Alzheimers Dis. 2007;12:335–41.
Wisniewski HM, Maslinska D. Beta-protein immunoreactivity in the human brain after cardiac arrest. Folia Neuropathol. 1996;34:65–71.
Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118:103–13.
de la Torre JC. Is Alzheimer’s disease preceded by neurodegeneration or cerebral hypoperfusion? Ann Neurol. 2005;57:783–4.
Niedermeyer E. Considerations of the ischemic basis and therapy of Alzheimer Disease. Clin EEG Neurosci. 2007;38:55–6.
Pluta R. Alzheimer lesions after ischemia-reperfusion brain injury. Folia Neuropathol. 2004;42:181–6.
Pluta R. From brain ischemia-reperfusion injury to possible sporadic Alzheimer’s disease. Curr Neurovasc Res. 2004;1:441–53.
Robakis NK, Georgakopoulos A. Allelic interference: a mechanism for trans-dominant transmission of loss of function in the neurodegeneration of familial Alzheimer’s disease. Neurodegener Dis. 2014;13:126–30.
De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, et al. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature. 1999;398:518–22.
Rahimi N, Golde TE, Meyer RD. Identification of ligand-induced proteolytic cleavage and ectodomain shedding of VEGFR-1/FLT1 in leukemic cancer cells. Cancer Res. 2009;69:2607–14.
Tanemura K, Chui DH, Fukuda T, Murayama M, Park JM, Akagi T, et al. Formation of tau inclusions in knock-in mice with familial Alzheimer disease (FAD) mutation of presenilin 1 (PS1). J Biol Chem. 2006;281:5037–41.
Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409–21.
Robakis NK. Mechanisms of AD neurodegeneration may be independent of Aβ and its derivatives. Neurobiol Aging 2011;32:372–9.
Acknowledgements
We thank Ms. Bridget Wicinski for help with stereologic design and microscopy analyses. We also thank Dr. Elodie Drapeau from department of Psychiatry at the Icahn School of Medicine at Mount Sinai for her assistance in behavioral tests. This work was supported by NIH Grants 2R01-NS047229, P50AG05138, and AG-008200.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Yoon, Y., Voloudakis, G., Doran, N. et al. PS1 FAD mutants decrease ephrinB2-regulated angiogenic functions, ischemia-induced brain neovascularization and neuronal survival. Mol Psychiatry 26, 1996–2012 (2021). https://doi.org/10.1038/s41380-020-0812-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-020-0812-7
- Springer Nature Limited